Targeting Dendritic Cell Function during Systemic Autoimmunity to Restore Tolerance

Abstract
:1. Introduction
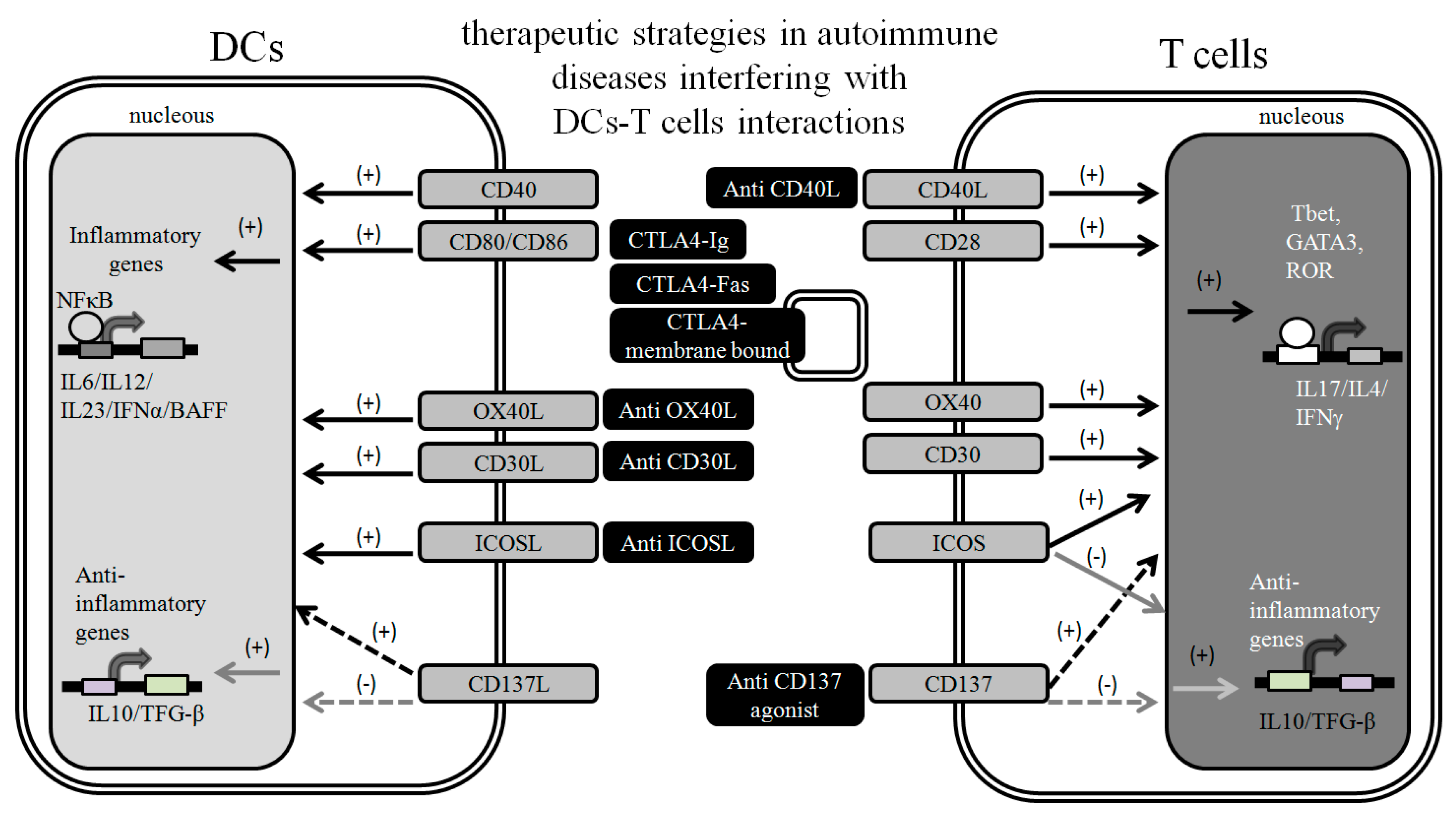
2. Targeting DC-T Cell Interactions to Prevent Autoimmunity

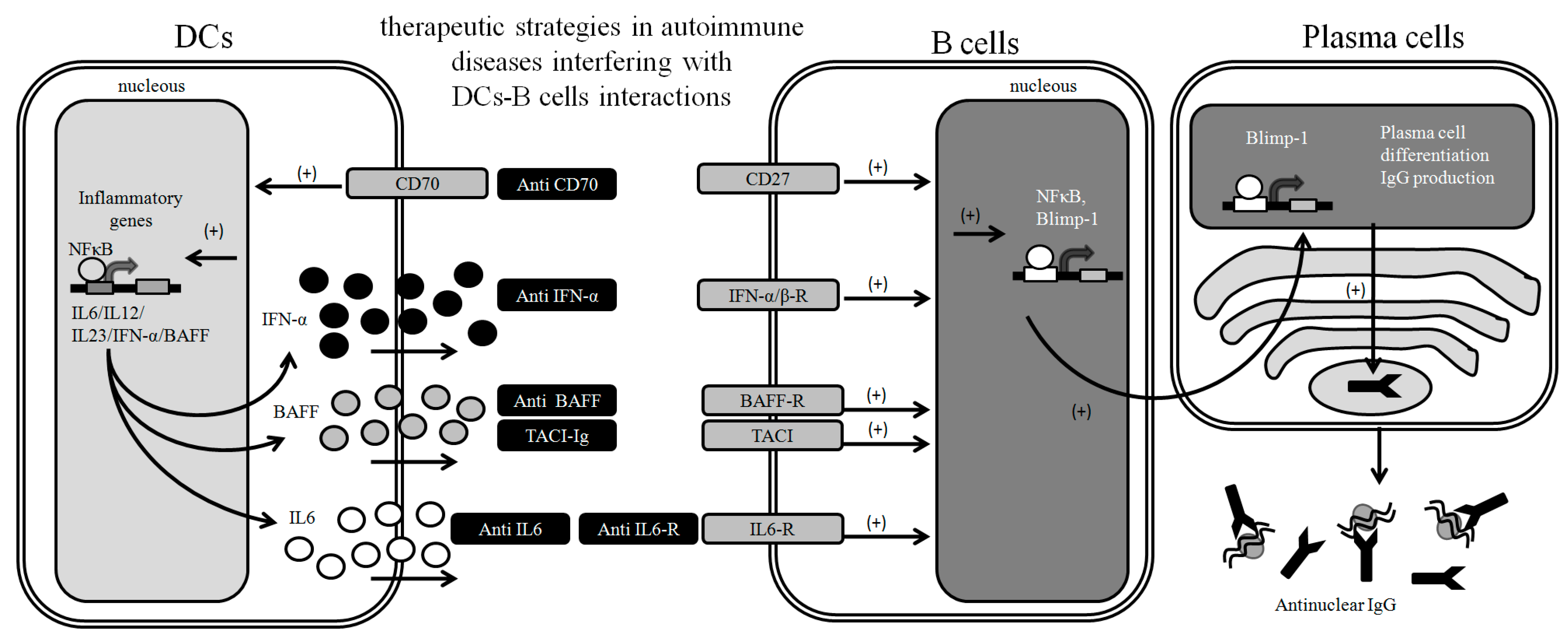
3. Targeting DC-B Cell Interactions to Prevent Autoimmunity

4. DC Abnormalities in Human Autoimmune Diseases
5. DC Maturation Stimuli during Autoimmunity
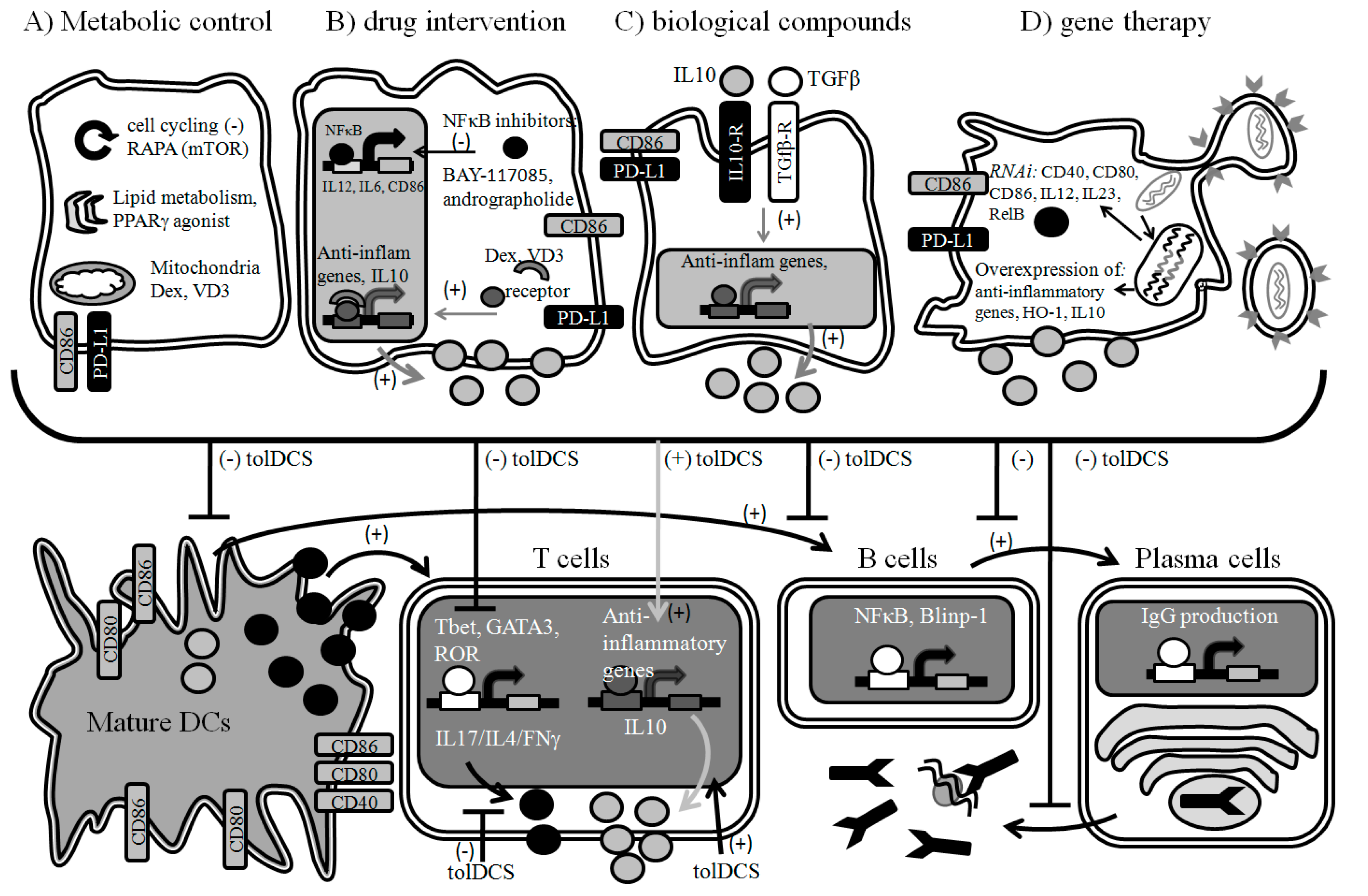
6. Designing New Therapies Based on Tolerogenic DCs
6.1. Metabolic Control


{kind=link}
{kind=link}
{kind=link}
| Agent | Protocol | Type of Tolerogenic Response | Targeted Disease | Reference | |||||
|---|---|---|---|---|---|---|---|---|---|
| Species | Differentiation | Relevant Antigen | Type of Study | ||||||
| Dexamethasone and Vitamin D3 | human | Blood monocytes, GM-CSF and IL-4, 5–6 days | alloantigen | in vitro; pre-clinical | Maturation-resistant phenotype, IL10/IL12; Impact in metabolism (lipids, glucose and oxidative phosphorylation); Migratory phenotype alterations; Reduce T cell priming and allospecific T cell response | Immune-mediated diseases; Prevention of graft rejection; Rheumatoid arthitis; Sjogren syndrome | Ferreira
et al., 2011 [141]; Volchenkov et al., 2013 [148]; Volchenkov et al., 2013 [150]; Xing et al., 2002 [151]; Unger et al., 2009 [152]; García-González et al., 2013 [153] | ||
| mouse | Bone marrow, GM-CSF, 5 days | - | in vivo | T cell priming; Maturation-resistant phenotype, IL10/IL12; Reduction of proinflammatory chemokines and cytokines | Immune-mediated diseases | Xing et al., 2002 [151] | |||
| mouse | Bone marrow, GM-CSF, 5 days | - | in vitro | T cell priming; Maturation-resistant phenotype, IL10/IL12 | Immune-mediated diseases | Moser et al., 1995 [154] | |||
| Dexamethasone plus monophosphoryl lipid A | human | Blood monocytes, GM-CSF and IL-4, 5–6 days | alloantigen | in vitro; pre-clinical | Stable phenotype and migratory capacity to lymphoid chemokines; T cell priming; Maturation-resistant phenotype, IL10/IL12 | Rheumatoid arthitis; Immune-mediated diseases; Prevention of graft rejection | García-González et al., 2013 [153] | ||
| Dexamethasone | human | Blood monocytes, GM-CSF and IL-4, 5–6 days | - | in vitro | Maturation-resistant phenotype, IL10/IL12; T cell priming | Immune-mediated diseases | Rea et al., 2000 [155] | ||
| Vitamin D3 | mouse | Bone marrow, GM-CSF, 5 days | - | in vivo | Reduce EAE severity; Maturation-resistant phenotype, IL10/IL12; Regulatory T cell induction | EAE; Autoimmunity | Farías et al., 2013 [156]; Unger et al., 2009 [152] | ||
| human | Blood monocytes, GM-CSF and IL-4, 5–6 days | myelin peptides | in vitro | Maturation-resistant phenotype, IL10/IL12; Reduce autoreactive T cell induction | MS; Autoimmunity | Raïch-Regué et al., 2012 [157] | |||
| Rapamycin | mouse | Bone marrow, GM-CSF, 5 days | alloantigen | in vitro | Maturation-resistant phenotype; Reduce T cell priming and allospecific T cell response | prevention of graft rejection | Turnquist et al., 2007 [143]; Taner et al., 2005 [144]; Hackstein et al., 2003 [158] | ||
| mouse | Bone marrow, GM-CSF, 5 days | alloantigen | in vivo | Reduce survival of alloantigen-specific CD8+ T cells in vivo | Prevention of graft rejection | Fischer et al., 2011 [145] | |||
| human | Blood monocytes, GM-CSF and IL-4, 5–6 days | alloantigen | in vitro | Maturation-resistant phenotype; Reduce T cell priming and allospecific T cell response | Immune-mediated diseases | Fedoric et al., 2008 [159] | |||
| Andrographolide | mouse | Bone marrow, GM-CSF, 5 days | MOG peptide | in vitro | Reduce T cell priming and antigen processing; NF-κB inhibition | Autoimmunity; EAE | Iruretagoyena et al., 2005 [146] | ||
| mouse | Bone marrow, GM-CSF, 5 days | MOG peptide | in vivo | Reduce EAE severity; NF-κB inhibition | Autoimmunity; EAE | Iruretagoyena et al., 2006 [149] | |||
| Aspirin | mouse | Bone marrow, GM-CSF, 5 days | alloantigen | in vitro | Maturation-resistant phenotype; IL10/IL12; Phagocytosis inhibition; Reduce T cell primi | Immune-mediated diseases | Hackstein et al., 2001 [160]; Buckland et al., 2006 [161]; Cai et al., 2011 [162] | ||
| Rosiglitazone | mouse | Bone marrow, GM-CSF, 5 days | MOG peptide | in vivo | Reduce T cell priming; Reduce EAE severity, NF-κB inhibition | Autoimmunity; EAE | Iruretagoyena et al., 2006 [149] | ||
| human | Blood monocytes, GM-CSF and IL-4, 5–6 days | - | in vitro | Reduce proinflammatory cytokine expression; Lipid accumulation appears to be diminished in these cells | Immune-mediated diseases | Szatmari et al., 2007 [147] | |||
| Troglitazone | human | Blood monocytes, GM-CSF and IL-4, 5–6 days | - | in vitro | Maturation-resistant phenotype, IL10/IL12 | Immune-mediated diseases | Volchenkov et al., 2013 [148] | ||
| Cobalt Protoporphyrin | human | Blood monocytes, GM-CSF and IL-4, 5–6 days | alloantigen | in vitro | Reduce T cell priming; Maturation-resistant phenotype, IL10/IL12; Reduce allospecific T cell response | Immune-mediated diseases; Prevention of graft rejection | Chauveau et al., 2005 [163] | ||
| Bay 11-7082 | mouse | Bone marrow, GM-CSF and IL-4, 5 days | methylated serum albumin | in vivo | Reduce disease severity; Reduce T cell response; NF-κB inhibition | CIA (Rheumatoid arthitis) | Martin et al., 2007 [164] | ||
| mouse | Bone marrow, GM-CSF, 5 days | - | in vitro | Maturation-resistant phenotype, IL10/IL12 | Immune-mediated diseases | Ade et al., 2007 [165] | |||
| Tacrolimus | mouse | Bone marrow, GM-CSF, 5 days | - | in vivo | - | CIA (Rheumatoid arthitis) | Ren et al., 2014 [166] | ||
| human | Blood monocytes, GM-CSF and IL-4, 5–6 days | - | in vitro | Maturation-resistant phenotype, IL10/IL12; Anti-inflammatory cytokine gene expression | Rheumatoid arthitis | Ren et al., 2014 [166] | |||
| IL-10 | human | Blood monocytes, GM-CSF and IL-4, 5–6 days | alloantigen; allergen | in vitro; pre-clinical | Maturation-resistant phenotype, IL10/IL12; Reduce T cell priming and allospecific T cell response | Systemic Lupus Erythematosus; Type 1 Diabetes; Immune-mediated diseases; Asthma and allergy | Sato et al., 1999 [167]; Knodler et al., 2008 [168]; Velten et al., 2004 [169]; Kubsch et al., 2003 [170]; Steinbrink et al., 2002 [171]; Li et al., 2010 [172]; Lopez et al., 2011 [173]; Crispin et al., 2012 [28] | ||
| mouse | Bone marrow, GM-CSF, 5 days | - | in vitro | Maturation-resistant phenotype | Immune-mediated diseases | Ruffner et al., 2009 [174] | |||
| rat | Bone marrow, GM-CSF, 5 days | - | in vivo | Maturation-resistant phenotype; Reduce T cell priming and allospecific T cell response | Prevention of graft rejection | Jiang et al., 2004 [175] | |||
| TGF-β | mouse | Bone marrow, GM-CSF, 5 days | insulin; allopeptides | in vivo | Long-term survival of the graft; Immune tolerance restoration | Prevention of graft rejection | Thomas et al., 2013 [176]; Yan et al, 2014 [177] | ||
| IL-10 and TGF-β | human | Blood monocytes, GM-CSF and IL-4, 5–6 days | insulin and GAD65; β2-glycoprotein I | in vitro; pre-clinical | Maturation-resistant phenotype, IL10/IL12; Reduced antigen specific T cell response | Antiphospholipid syndrome; Type 1 Diabetes | Segovia-Gamboa et al., 2014 [178]; Torres-Aguilar et al., 2012 [179] | ||
| Cholera toxin B | human | Blood monocytes, GM-CSF and IL-4, 5–6 days | - | in vitro | Maturation-resistant phenotype; Reduce T cell priming; regulatory T cell induction | Immune-mediated diseases | D’ambrosio et al., 2008 [180] | ||
| Gene therapy, IL-10 plus TGF-β | rat | Bone marrow, GM-CSF, 5 days | - | in vivo | Long-term survival of the graft; Maturation-resistant phenotype | Prevention of graft rejection | Chen et al., 2014 [181] | ||
| Gene therapy; silencing; IL-12/IL23/CD40/CD80/CD86/RelB | mouse | Bone marrow, GM-CSF or GM-CSF and IL-4, 5 days | collagen II; MOG petide; islet lysate | in vivo | Reduce disease severity and joint erosion; Reduce T cell priming; Reduced islet-specific T cell response; Reduce severity of Type 1 Diabetes | CIA (Rheumatoid arthitis); EAE; Type 1 Diabetes | Li et al., 2012 [182]; Zheng et al., 2010 [183]; Kalantari et al., 2014 [184]; Ma et al., 2003 [185]; Machen et al., 2004 [186] | ||
6.2. Pharmacologic Intervention
| Protocol for DC | Name | Targeted Disease | Results/Status | ClinicalTrials.gov Identifier | ||||
|---|---|---|---|---|---|---|---|---|
| Agent | Origin | Differentiation | Type of Study | Route | ||||
| Dexamethasone and Vitamin D3 | Blood monocytes | GM-CSF and IL-4, 5–6 days | Phase I; Proof of safety | Arthroscopically | AutoDECRA | Rheumatoid arthitis | No study results posted; Ongoing study | NCT01352858 |
| BAY11-7082 | Blood monocytes | GM-CSF and IL-4, 5–6 days | Phase I; Proof of safety | Intradermally | - | Rheumatoid arthitis (citrunillated peptides) | Safe and well tolerated; Ongoing study | - |
| Gene therapy; siRNA; CD40/CD80/CDD86 | Blood monocytes | GM-CSF and IL-4, 5–6 days | Phase I; Proof of safety | Intradermally | - | Type 1 Diabetes | Safe and well tolerated; Ongoing study | NCT00445913 |
| Low GM-CSF | Blood monocytes | low GM-CSF, 6 days | Phase I; feasibility study | Intravenous | The One Study | Kidney transplant | No study results posted; Ongoing study | - |
6.3. Biological Compounds
6.4. Gene Therapy
7. Conclusions
Acknowledgments
Abbreviations
| APCs | Antigen presenting cells |
| ANA | Anti-nuclear antibodies |
| BAFF | B cell activating factor |
| BLyS | B lymphocyte stimulator |
| cDCs | Conventional dendritic cells |
| CIA | Collagen induced arthritis |
| CoPP | Cobalt Protoporphyrin |
| DAMPs | Danger-associated molecular patterns |
| DCs | Dendritic cells |
| Dex | Dexamethasone |
| EAE | Experimental Autoimmune Encephalitis |
| HO-1 | Hemeoxygenase 1 |
| IC | Immune complex |
| ILT | Immunoglobulin-like Transcript |
| MS | Multiple sclerosis |
| mTOR | Mammalian target of rapamycin |
| NF-κB | Nuclear factor kappa B |
| PAMPs | Pathogen-associated molecular patterns |
| PD-1 | Programmed death 1 |
| pDCs | Plasmacytoid dendritic cells |
| PPAR | Peroxysome proliferator-activated receptor |
| RA | Rheumatoid Arthritis |
| RAPA | Rapamycin |
| RNAi | interference RNA |
| SLE | Systemic Lupus Erythematosus |
| SS | Sjögren’ |
| s syndrome; T1D | Type 1 Diabetes |
| Th | T helper |
| TLRs | Toll Like Receptors |
| Treg | Regulatory T cells |
| VD3 | 1α,25-dihydroxyvitamin D3 |
Conflicts of Interest
References
- Gratz, I.K.; Rosenblum, M.D.; Maurano, M.M.; Paw, J.S.; Truong, H.A.; Marshak-Rothstein, A.; Abbas, A.K. Cutting edge: Self-antigen controls the balance between effector and regulatory T cells in peripheral tissues. J. Immunol. 2014, 192, 1351–1355. [Google Scholar]
- Luo, X.; Yang, W.; Ye, D.-Q.; Cui, H.; Zhang, Y.; Hirankarn, N.; Qian, X.; Tang, Y.; Lau, Y.L.; de Vries, N.; et al. A functional variant in microrna-146a promoter modulates its expression and confers disease risk for systemic lupus erythematosus. PLoS Genet. 2011, 7, e1002128. [Google Scholar]
- Ginzler, E.M.; Dooley, M.A.; Aranow, C.; Kim, M.Y.; Buyon, J.; Merrill, J.T.; Petri, M.; Gilkeson, G.S.; Wallace, D.J.; Weisman, M.H.; et al. Mycophenolate mofetil or intravenous cyclophosphamide for lupus nephritis. N. Engl. J. Med. 2005, 353, 2219–2228. [Google Scholar] [CrossRef]
- Perez, V.L.; van Parijs, L.; Biuckians, A.; Zheng, X.X.; Strom, T.B.; Abbas, A.K. Induction of peripheral T cell tolerance in vivo requires ctla-4 engagement. Immunity 1997, 6, 411–417. [Google Scholar]
- Bernatsky, S.; Boivin, J.F.; Joseph, L.; Manzi, S.; Ginzler, E.; Gladman, D.D.; Urowitz, M.; Fortin, P.R.; Petri, M.; Barr, S.; et al. Mortality in systemic lupus erythematosus. Arthritis Rheumatol. 2006, 54, 2550–2557. [Google Scholar] [CrossRef]
- Carreño, L.J.; Pacheco, R.; Gutierrez, M.A.; Jacobelli, S.; Kalergis, A.M. Disease activity in systemic lupus erythematosus is associated with an altered expression of low-affinity fcγ receptors and costimulatory molecules on dendritic cells. Immunology 2009, 128, 334–341. [Google Scholar]
- Crispin, J.C.; Kyttaris, V.C.; Terhorst, C.; Tsokos, G.C. T cells as therapeutic targets in sle. Nat. Rev. Rheumatol. 2010, 6, 317–325. [Google Scholar]
- Gaipl, U.S.; Munoz, L.E.; Grossmayer, G.; Lauber, K.; Franz, S.; Sarter, K.; Voll, R.E.; Winkler, T.; Kuhn, A.; Kalden, J.; et al. Clearance deficiency and systemic lupus erythematosus (SLE). J. Autoimmun. 2007, 28, 114–121. [Google Scholar] [CrossRef]
- Gerl, V.; Lischka, A.; Panne, D.; Großmann, P.; Berthold, R.; Hoyer, B.F.; Biesen, R.; Bruns, A.; Alexander, T.; Jacobi, A.; et al. Blood dendritic cells in systemic lupus erythematosus exhibit altered activation state and chemokine receptor function. Ann. Rheum. Dis. 2010, 69, 1370–1377. [Google Scholar]
- Ding, D.; Mehta, H.; McCune, W.J.; Kaplan, M.J. Aberrant phenotype and function of myeloid dendritic cells in systemic lupus erythematosus. J. Immunol. 2006, 177, 5878–5889. [Google Scholar]
- Schwarz, E.; Ritchlin, C. Clinical development of anti-rankl therapy. Arthritis Res. Ther. 2007, 9, S7. [Google Scholar]
- Orban, T.; Bundy, B.; Becker, D.J.; DiMeglio, L.A.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Greenbaum, C.J.; Marks, J.B.; Monzavi, R.; et al. Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: A randomised, double-blind, placebo-controlled trial. Lancet 2011, 378, 412–419. [Google Scholar] [CrossRef]
- Houssiau, F.; Ginzler, E. Current treatment of lupus nephritis. Lupus 2008, 17, 426–430. [Google Scholar]
- Hahn, B.H. Belimumab for systemic lupus erythematosus. N. Engl. J. Med. 2013, 368, 1528–1535. [Google Scholar] [CrossRef]
- Navarra, S.V.; Guzmán, R.M.; Gallacher, A.E.; Hall, S.; Levy, R.A.; Jimenez, R.E.; Li, E.K.M.; Thomas, M.; Kim, H.-Y.; León, M.G.; et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: A randomised, placebo-controlled, phase 3 trial. Lancet 2010, 377, 721–731. [Google Scholar]
- Furie, R.; Petri, M.; Zamani, O.; Cervera, R.; Wallace, D.J.; Tegzova, D.; Sanchez-Guerrero, J.; Schwarting, A.; Merrill, J.T.; Chatham, W.W.; et al. A phase III, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheumatol. 2011, 63, 3918–3930. [Google Scholar] [CrossRef]
- Lee, J.-H.; Kim, T.-H.; Park, H.E.; Lee, E.G.; Jung, N.-C.; Song, J.-Y.; Seo, H.G.; Seung, K.-B.; Chang, K.; Lim, D.-S. Myosin-primed tolerogenic dendritic cells ameliorate experimental autoimmune myocarditis. Cardiovasc. Res. 2014, 101, 203–210. [Google Scholar]
- Mukhopadhaya, A.; Hanafusa, T.; Jarchum, I.; Chen, Y.-G.; Iwai, Y.; Serreze, D.V.; Steinman, R.M.; Tarbell, K.V.; DiLorenzo, T.P. Selective delivery of β cell antigen to dendritic cells in vivo leads to deletion and tolerance of autoreactive CD8+ T cells in nod mice. Proc. Natl. Acad. Sci. USA 2008, 105, 6374–6379. [Google Scholar]
- Dufait, I.; Liechtenstein, T.; Lanna, A.; Bricogne, C.; Laranga, R.; Padella, A.; Breckpot, K.; Escors, D. Retroviral and lentiviral vectors for the induction of immunological tolerance. Scientifica 2012, 2012. [Google Scholar] [CrossRef]
- Anderson, M.S.; Venanzi, E.S.; Chen, Z.; Berzins, S.P.; Benoist, C.; Mathis, D. The cellular mechanism of aire control of T cell tolerance. Immunity 2005, 23, 227–239. [Google Scholar]
- Carreno, L.J.; Gonzalez, P.A.; Kalergis, A.M. Modulation of T cell function by TCR/pMHC binding kinetics. Immunobiology 2006, 211, 47–64. [Google Scholar] [CrossRef]
- Iruretagoyena, M.I.; Wiesendanger, M.; Kalergis, A.M. The dendritic cell-T cell synapse as a determinant of autoimmune pathogenesis. Curr. Pharm. Des. 2006, 12, 131–147. [Google Scholar]
- Kalergis, A.M. Modulation of T cell immunity by TCR/pMHC dwell time and activating/inhibitory receptor pairs on the antigen-presenting cell. Curr. Pharm. Des. 2003, 9, 233–244. [Google Scholar]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef]
- Grewal, I.S.; Foellmer, H.G.; Grewal, K.D.; Xu, J.; Hardardottir, F.; Baron, J.L.; Janeway, C.A., Jr.; Flavell, R.A. Requirement for CD40 ligand in costimulation induction, T cell activation, and experimental allergic encephalomyelitis. Science 1996, 273, 1864–1867. [Google Scholar]
- Linsley, P.S.; Brady, W.; Grosmaire, L.; Aruffo, A.; Damle, N.K.; Ledbetter, J.A. Binding of the B cell activation antigen b7 to CD28 costimulates T cell proliferation and interleukin 2 mrna accumulation. J. Exp. Med. 1991, 173, 721–730. [Google Scholar] [CrossRef]
- Hubo, M.; Trinschek, B.; Kryczanowsky, F.; Tüttenberg, A.; Steinbrink, K.; Jonuleit, H. Costimulatory molecules on immunogenic versus tolerogenic human dendritic cells. Front. Immunol. 2013, 4, 82. [Google Scholar]
- Crispin, J.C.; Vargas-Rojas, M.I.; Monsivais-Urenda, A.; Alcocer-Varela, J. Phenotype and function of dendritic cells of patients with systemic lupus erythematosus. Clin. Immunol. 2012, 143, 45–50. [Google Scholar]
- Ito, T.; Wang, Y.-H.; Duramad, O.; Hori, T.; Delespesse, G.J.; Watanabe, N.; Qin, F.X.-F.; Yao, Z.; Cao, W.; Liu, Y.-J. TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J. Exp. Med. 2005, 202, 1213–1223. [Google Scholar] [CrossRef]
- M.Gaspal, F.; Withers, D.; Saini, M.; Bekiaris, V.; McConnell, F.M.; White, A.; Khan, M.; Yagita, H.; Walker, L.S.K.; Anderson, G.; et al. Abrogation of CD30 and OX40 signals prevents autoimmune disease in foxp3-deficient mice. J. Exp. Med. 2011, 208, 1579–1584. [Google Scholar]
- Gerritse, K.; Laman, J.D.; Noelle, R.J.; Aruffo, A.; Ledbetter, J.A.; Boersma, W.J.; Claassen, E. CD40-CD40 ligand interactions in experimental allergic encephalomyelitis and multiple sclerosis. Proc. Natl. Acad. Sci. USA 1996, 93, 2499–2504. [Google Scholar]
- Bagenstose, L.M.; Agarwal, R.K.; Silver, P.B.; Harlan, D.M.; Hoffmann, S.C.; Kampen, R.L.; Chan, C.-C.; Caspi, R.R. Disruption of CD40/CD40-ligand interactions in a retinal autoimmunity model results in protection without tolerance. J. Immunol. 2005, 175, 124–130. [Google Scholar]
- Witsch, E.J.; Peiser, M.; Hutloff, A.; Büchner, K.; Dorner, B.G.; Jonuleit, H.; Mages, H.W.; Kroczek, R.A. ICOS and CD28 reversely regulate IL-10 on re-activation of human effector T cells with mature dendritic cells. Eur. J. Immunol. 2002, 32, 2680–2686. [Google Scholar]
- Yoshinaga, S.K.; Whoriskey, J.S.; Khare, S.D.; Sarmiento, U.; Guo, J.; Horan, T.; Shih, G.; Zhang, M.; Coccia, M.A.; Kohno, T.; et al. T-cell co-stimulation through B7RP-1 and ICOS. Nature 1999, 402, 827–832. [Google Scholar] [CrossRef]
- Kopf, M.; Coyle, A.J.; Schmitz, N.; Barner, M.; Oxenius, A.; Gallimore, A.; Gutierrez-Ramos, J.-C.; Bachmann, M.F. Inducible costimulator protein (ICOS) controls T helper cell subset polarization after virus and parasite infection. J. Exp. Med. 2000, 192, 53–62. [Google Scholar]
- Sim, G.C.; Martin-Orozco, N.; Jin, L.; Yang, Y.; Wu, S.; Washington, E.; Sanders, D.; Lacey, C.; Wang, Y.; Vence, L.; et al. IL-2 therapy promotes suppressive ICOS+ Treg expansion in melanoma patients. J. Clin. Investig. 2014, 124, 99–110. [Google Scholar]
- Prevot, N.; Briet, C.; Lassmann, H.; Tardivel, I.; Roy, E.; Morin, J.; Mak, T.W.; Tafuri, A.; Boitard, C. Abrogation of ICOS/ICOS ligand costimulation in nod mice results in autoimmune deviation toward the neuromuscular system. Eur. J. Immunol. 2010, 40, 2267–2276. [Google Scholar]
- Waterhouse, P.; Penninger, J.M.; Timms, E.; Wakeham, A.; Shahinian, A.; Lee, K.P.; Thompson, C.B.; Griesser, H.; Mak, T.W. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 1995, 270, 985–988. [Google Scholar]
- Podojil, J.; Miller, S. Targeting the B7 family of co-stimulatory molecules. BioDrugs 2013, 27, 1–13. [Google Scholar] [CrossRef]
- Fife, B.T.; Griffin, M.D.; Abbas, A.K.; Locksley, R.M.; Bluestone, J.A. Inhibition of T cell activation and autoimmune diabetes using a B cell surface-linked CTLA-4 agonist. J. Clin. Investig. 2006, 116, 2252–2261. [Google Scholar] [CrossRef]
- Picchianti Diamanti, A.; Rosado, M.M.; Scarsella, M.; Germano, V.; Giorda, E.; Cascioli, S.; Laganà, B.; D’Amelio, R.; Carsetti, R. Abatacept (CTLA4-Ig) improves B cell function and treg inhibitory capacity in rheumatoid arthritis patients non responding to anti-tnf-alpha agents. Clin. Exp. Immunol. 2014. [Google Scholar] [CrossRef]
- Jin, Y.; Qu, A.; Wang, G.M.; Hao, J.; Gao, X.; Xie, S. Simultaneous stimulation of fas-mediated apoptosis and blockade of costimulation prevent autoimmune diabetes in mice induced by multiple low-dose streptozotocin. Gene Ther. 2004, 11, 982–991. [Google Scholar]
- Feng, Y.G.; Jin, Y.Z.; Zhang, Q.Y.; Hao, J.; Wang, G.M.; Xie, S.S. CTLA4-FAS ligand gene transfer mediated by adenovirus induce long-time survival of murine cardiac allografts. Transplant. Proc. 2005, 37, 2379–2381. [Google Scholar] [CrossRef]
- Ikawa, K.; Araki, H.; Tsujino, Y.; Hayashi, Y.; Igarashi, K.; Hatada, Y.; Hagihara, H.; Ozawa, T.; Ozaki, K.; Kobayashi, T.; et al. Hyperexpression of the gene for a bacillus alpha-amylase in bacillus subtilis cells: Enzymatic properties and crystallization of the recombinant enzyme. Biosci. Biotechnol. Biochem. 1998, 62, 1720–1725. [Google Scholar]
- Yamazaki, T.; Akiba, H.; Iwai, H.; Matsuda, H.; Aoki, M.; Tanno, Y.; Shin, T.; Tsuchiya, H.; Pardoll, D.M.; Okumura, K.; et al. Expression of programmed death 1 ligands by murine T cells and apc. J. Immunol. 2002, 169, 5538–5545. [Google Scholar]
- Keir, M.E.; Francisco, L.M.; Sharpe, A.H. PD-1 and its ligands in T-cell immunity. Curr. Opin. Immunol. 2007, 19, 309–314. [Google Scholar] [CrossRef]
- Dilek, N.; Poirier, N.; Hulin, P.; Coulon, F.; Mary, C.; Ville, S.; Vie, H.; Clémenceau, B.; Blancho, G.; Vanhove, B. Targeting CD28, CTLA-4 and PD-L1 costimulation differentially controls immune synapses and function of human regulatory and conventional T-cells. PLoS One 2013, 8, e83139. [Google Scholar]
- Francisco, L.M.; Salinas, V.H.; Brown, K.E.; Vanguri, V.K.; Freeman, G.J.; Kuchroo, V.K.; Sharpe, A.H. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 2009, 206, 3015–3029. [Google Scholar] [CrossRef]
- Yogev, N.; Frommer, F.; Lukas, D.; Kautz-Neu, K.; Karram, K.; Ielo, D.; von Stebut, E.; Probst, H.-C.; van den Broek, M.; Riethmacher, D.; et al. Dendritic cells ameliorate autoimmunity in the cns by controlling the homeostasis of PD-1 receptor+ regulatory T cells. Immunity 2012, 37, 264–275. [Google Scholar] [CrossRef] [Green Version]
- Schreiner, B.; Bailey, S.L.; Shin, T.; Chen, L.; Miller, S.D. PD-1 ligands expressed on myeloid-derived apc in the CNS regulate T-cell responses in eae. Eur. J. Immunol. 2008, 38, 2706–2717. [Google Scholar]
- Latchman, Y.E.; Liang, S.C.; Wu, Y.; Chernova, T.; Sobel, R.A.; Klemm, M.; Kuchroo, V.K.; Freeman, G.J.; Sharpe, A.H. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc. Natl. Acad. Sci. USA 2004, 101, 10691–10696. [Google Scholar]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an itim motif-carrying immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef]
- Watanabe, N.; Gavrieli, M.; Sedy, J.R.; Yang, J.; Fallarino, F.; Loftin, S.K.; Hurchla, M.A.; Zimmerman, N.; Sim, J.; Zang, X.; et al. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat. Immunol. 2003, 4, 670–679. [Google Scholar]
- Oya, Y.; Watanabe, N.; Owada, T.; Oki, M.; Hirose, K.; Suto, A.; Kagami, S.-I.; Nakajima, H.; Kishimoto, T.; Iwamoto, I.; et al. Development of autoimmune hepatitis-like disease and production of autoantibodies to nuclear antigens in mice lacking B and T lymphocyte attenuator. Arthritis Rheumatol. 2008, 58, 2498–2510. [Google Scholar]
- Hitoshi, Y.; Lorens, J.; Kitada, S.-I.; Fisher, J.; LaBarge, M.; Ring, H.Z.; Francke, U.; Reed, J.C.; Kinoshita, S.; Nolan, G.P. Toso, a cell surface, specific regulator of FAS-induced apoptosis in T cells. Immunity 1998, 8, 461–471. [Google Scholar]
- Brenner, D.; Brüstle, A.; Lin, G.H.Y.; Lang, P.A.; Duncan, G.S.; Knobbe-Thomsen, C.B.; St. Paul, M.; Reardon, C.; Tusche, M.W.; Snow, B.; et al. Toso controls encephalitogenic immune responses by dendritic cells and regulatory T cells. Proc. Natl. Acad. Sci. USA 2014, 111, 1060–1065. [Google Scholar] [CrossRef]
- Vinay, D.S.; Choi, J.H.; Kim, J.D.; Choi, B.K.; Kwon, B.S. Role of endogenous 4-1BB in the development of systemic lupus erythematosus. Immunology 2007, 122, 394–400. [Google Scholar]
- Sun, Y.; Chen, H.M.; Subudhi, S.K.; Chen, J.; Koka, R.; Chen, L.; Fu, Y.-X. Costimulatory molecule-targeted antibody therapy of a spontaneous autoimmune disease. Nat. Med. 2002, 8, 1405–1413. [Google Scholar] [CrossRef]
- Wakkach, A.; Cottrez, F.; Groux, H. Differentiation of regulatory T cells 1 is induced by cd2 costimulation. J. Immunol. 2001, 167, 3107–3113. [Google Scholar]
- Pasare, C.; Medzhitov, R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science 2003, 299, 1033–1036. [Google Scholar]
- Wan, S.; Xia, C.; Morel, L. Il-6 produced by dendritic cells from lupus-prone mice inhibits CD4+CD25+ T cell regulatory functions. J. Immunol. 2007, 178, 271–279. [Google Scholar] [CrossRef]
- Bondanza, A.; Zimmermann, V.S.; dell’Antonio, G.; Dal Cin, E.; Capobianco, A.; Sabbadini, M.G.; Manfredi, A.A.; Rovere-Querini, P. Cutting edge: Dissociation between autoimmune response and clinical disease after vaccination with dendritic cells. J. Immunol. 2003, 170, 24–27. [Google Scholar]
- Ma, L.; Chan, K.W.; Trendell-Smith, N.J.; Wu, A.; Tian, L.; Lam, A.C.; Chan, A.K.; Lo, C.K.; Chik, S.; Ko, K.H.; et al. Systemic autoimmune disease induced by dendritic cells that have captured necrotic but not apoptotic cells in susceptible mouse strains. Eur. J. Immunol. 2005, 35, 3364–3375. [Google Scholar] [Green Version]
- Georgiev, M.; Agle, L.M.A.; Chu, J.L.; Elkon, K.B.; Ashany, D. Mature dendritic cells readily break tolerance in normal mice but do not lead to disease expression. Arthritis Rheumatol. 2005, 52, 225–238. [Google Scholar]
- Mackay, F.; Woodcock, S.A.; Lawton, P.; Ambrose, C.; Baetscher, M.; Schneider, P.; Tschopp, J.; Browning, J.L. Mice transgenic for baff develop lymphocytic disorders along with autoimmune manifestations. J. Exp. Med. 1999, 190, 1697–1710. [Google Scholar]
- Vincent, F.; Northcott, M.; Hoi, A.; Mackay, F.; Morand, E. Association of serum B cell activating factor from the tumour necrosis factor family (baff) and a proliferation-inducing ligand (april) with central nervous system and renal disease in systemic lupus erythematosus. Lupus 2013, 22, 873–884. [Google Scholar] [CrossRef]
- Kim, J.Y.; Yang, Y.; Moon, J.-S.; Lee, E.Y.; So, S.H.; Lee, H.-S.; Park, K.D.; Choi, Y.-C. Serum baff expression in patients with myasthenia gravis. J. Neuroimmunol. 2008, 199, 151–154. [Google Scholar]
- Vannucchi, G.; Covelli, D.; Curro, N.; Dazzi, D.; Maffini, A.; Campi, I.; Bonara, P.; Guastella, C.; Pignataro, L.; Ratiglia, R.; et al. Serum baff concentrations in patients with graves’ disease and orbitopathy before and after immunosuppressive therapy. J. Clin. Endocrinol. Metab. 2012, 97, E755–E759. [Google Scholar]
- Nagai, M.; Hirayama, K.; Ebihara, I.; Shimohata, H.; Kobayashi, M.; Koyama, A. Serum levels of baff and april in myeloperoxidase anti-neutrophil cytoplasmic autoantibody-associated renal vasculitis: Association with disease activity. Nephron Clin. Pract. 2011, 118, c339–c345. [Google Scholar] [CrossRef]
- Xin, G.; Cui, Z.; Su, Y.; Xu, L.-X.; Zhao, M.-H.; Li, K.-S. Serum baff and april might be associated with disease activity and kidney damage in patients with anti-glomerular basement membrane disease. Nephrology 2013, 18, 209–214. [Google Scholar]
- Gross, J.A.; Dillon, S.R.; Mudri, S.; Johnston, J.; Littau, A.; Roque, R.; Rixon, M.; Schou, O.; Foley, K.P.; Haugen, H.; et al. TACI-Ig neutralizes molecules critical for B cell development and autoimmune disease: Impaired B cell maturation in mice lacking BLYS. Immunity 2001, 15, 289–302. [Google Scholar]
- Gross, J.A.; Johnston, J.; Mudri, S.; Enselman, R.; Dillon, S.R.; Madden, K.; Xu, W.; Parrish-Novak, J.; Foster, D.; Lofton-Day, C.; et al. Taci and bcma are receptors for a tnf homologue implicated in B-cell autoimmune disease. Nature 2000, 404, 995–999. [Google Scholar] [CrossRef]
- Ginzler, E.; Wax, S.; Rajeswaran, A.; Copt, S.; Hillson, J.; Ramos, E.; Singer, N. Atacicept in combination with mmf and corticosteroids in lupus nephritis: Results of a prematurely terminated trial. Arthritis Res. Ther. 2012, 14, R33. [Google Scholar]
- Gorelik, L.; Gilbride, K.; Dobles, M.; Kalled, S.L.; Zandman, D.; Scott, M.L. Normal B cell homeostasis requires B cell activation factor production by radiation-resistant cells. J. Exp. Med. 2003, 198, 937–945. [Google Scholar]
- Jego, G.; Palucka, A.K.; Blanck, J.-P.; Chalouni, C.; Pascual, V.; Banchereau, J. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity 2003, 19, 225–234. [Google Scholar]
- Nishimoto, N.; Kishimoto, T. Interleukin 6: From bench to bedside. Nat. Clin. Pract. Rheum. 2006, 2, 619–626. [Google Scholar]
- Yokota, S.; Imagawa, T.; Mori, M.; Miyamae, T.; Takei, S.; Iwata, N.; Umebayashi, H.; Murata, T.; Miyoshi, M.; Tomiita, M.; et al. Longterm safety and effectiveness of the anti-interleukin 6 receptor monoclonal antibody tocilizumab in patients with systemic juvenile idiopathic arthritis in Japan. J. Rheumatol. 2014, 41, 759–767. [Google Scholar] [CrossRef]
- Conaghan, P.G.; Peterfy, C.; Olech, E.; Kaine, J.; Ridley, D.; DiCarlo, J.; Friedman, J.; Devenport, J.; Troum, O. The effects of tocilizumab on osteitis, synovitis and erosion progression in rheumatoid arthritis: Results from the ACT-RAY MRI substudy. Ann. Rheum. Dis. 2014, 73, 810–816. [Google Scholar]
- Yao, X.; Huang, J.; Zhong, H.; Shen, N.; Faggioni, R.; Fung, M.; Yao, Y. Targeting interleukin-6 in inflammatory autoimmune diseases and cancers. Pharmacol. Ther. 2014, 141, 125–139. [Google Scholar]
- Harigai, M.; Kawamoto, M.; Hara, M.; Kubota, T.; Kamatani, N.; Miyasaka, N. Excessive production of IFN-γ in patients with systemic lupus erythematosus and its contribution to induction of b lymphocyte stimulator/B cell-activating factor/TNF ligand superfamily-13b. J. Immunol. 2008, 181, 2211–2219. [Google Scholar] [CrossRef]
- Litinskiy, M.B.; Nardelli, B.; Hilbert, D.M.; He, B.; Schaffer, A.; Casali, P.; Cerutti, A. DCs induce CD40-independent immunoglobulin class switching through BLYS and april. Nat. Immunol. 2002, 3, 822–829. [Google Scholar]
- Yao, Y.; Richman, L.; Higgs, B.W.; Morehouse, C.A.; de los Reyes, M.; Brohawn, P.; Zhang, J.; White, B.; Coyle, A.J.; Kiener, P.A.; et al. Neutralization of interferon-α/β-inducible genes and downstream effect in a phase I trial of an anti-interferon-α monoclonal antibody in systemic lupus erythematosus. Arthritis Rheumatol. 2009, 60, 1785–1796. [Google Scholar]
- Panchanathan, R.; Choubey, D. Murine baff expression is up-regulated by estrogen and interferons: Implications for sex bias in the development of autoimmunity. Mol. Immunol. 2013, 53, 15–23. [Google Scholar] [CrossRef]
- Batista, F.D.; Harwood, N.E. The who, how and where of antigen presentation to B cells. Nat. Rev. Immunol. 2009, 9, 15–27. [Google Scholar]
- Fayette, J.; Dubois, B.; Vandenabeele, S.; Bridon, J.-M.; Vanbervliet, B.; Durand, I.; Banchereau, J.; Caux, C.; Brière, F. Human dendritic cells skew isotype switching of CD40-activated naive B cells towards IgA1 and IgA2. J. Exp. Med. 1997, 185, 1909–1918. [Google Scholar]
- Wan, S.; Zhou, Z.; Duan, B.; Morel, L. Direct B cell stimulation by dendritic cells in a mouse model of lupus. Arthritis Rheumatol. 2008, 58, 1741–1750. [Google Scholar] [CrossRef]
- Poeck, H.; Wagner, M.; Battiany, J.; Rothenfusser, S.; Wellisch, D.; Hornung, V.; Jahrsdorfer, B.; Giese, T.; Endres, S.; Hartmann, G. Plasmacytoid dendritic cells, antigen, and CpG-C license human B cells for plasma cell differentiation and immunoglobulin production in the absence of T-cell help. Blood 2004, 103, 3058–3064. [Google Scholar]
- Morva, A.; Lemoine, S.; Achour, A.; Pers, J.O.; Youinou, P.; Jamin, C. Maturation and function of human dendritic cells are regulated by B lymphocytes. Blood 2012, 119, 106–114. [Google Scholar]
- Shaw, J.; Wang, Y.H.; Ito, T.; Arima, K.; Liu, Y.J. Plasmacytoid dendritic cells regulate B-cell growth and differentiation via CD70. Blood 2010, 115, 3051–3057. [Google Scholar]
- Arens, R.; Nolte, M.A.; Tesselaar, K.; Heemskerk, B.; Reedquist, K.A.; van Lier, R.A.W.; van Oers, M.H.J. Signaling through CD70 regulates B cell activation and IgG production. J. Immunol. 2004, 173, 3901–3908. [Google Scholar] [CrossRef]
- Teichmann, L.L.; Ols, M.L.; Kashgarian, M.; Reizis, B.; Kaplan, D.H.; Shlomchik, M.J. Dendritic cells in lupus are not required for activation of T and B cells but promote their expansion, resulting in tissue damage. Immunity 2010, 33, 967–978. [Google Scholar] [CrossRef]
- Qian, L.; Qian, C.; Chen, Y.; Bai, Y.; Bao, Y.; Lu, L.; Cao, X. Regulatory dendritic cells program B cells to differentiate into CD19hiFcγiibhi regulatory B cells through IFN-beta and CD40l. Blood 2012, 120, 581–591. [Google Scholar] [CrossRef]
- Berggren, O.; Hagberg, N.; Weber, G.; Alm, G.V.; Rönnblom, L.; Eloranta, M.-L. B lymphocytes enhance interferon-α production by plasmacytoid dendritic cells. Arthritis Rheumatol. 2012, 64, 3409–3419. [Google Scholar]
- Karni, A.; Abraham, M.; Monsonego, A.; Cai, G.; Freeman, G.J.; Hafler, D.; Khoury, S.J.; Weiner, H.L. Innate immunity in multiple sclerosis: Myeloid dendritic cells in secondary progressive multiple sclerosis are activated and drive a proinflammatory immune response. J. Immunol. 2006, 177, 4196–4202. [Google Scholar]
- Tucci, M.; Calvani, N.; Richards, H.B.; Quatraro, C.; Silvestris, F. The interplay of chemokines and dendritic cells in the pathogenesis of lupus nephritis. Ann. N. Y. Acad. Sci. 2005, 1051, 421–432. [Google Scholar] [CrossRef]
- Decker, P.; Kotter, I.; Klein, R.; Berner, B.; Rammensee, H.G. Monocyte-derived dendritic cells over-express CD86 in patients with systemic lupus erythematosus. Rheumatology (Oxf.) 2006, 45, 1087–1095. [Google Scholar]
- Jin, O.; Kavikondala, S.; Sun, L.; Fu, R.; Mok, M.-Y.; Chan, A.; Yeung, J.; Lau, C.-S. Systemic lupus erythematosus patients have increased number of circulating plasmacytoid dendritic cells, but decreased myeloid dendritic cells with deficient cd83 expression. Lupus 2008, 17, 654–662. [Google Scholar] [CrossRef]
- Kanakoudi-Tsakalidou, F.; Farmaki, E.; Tzimouli, V.; Taparkou, A.; Paterakis, G.; Trachana, M.; Pratsidou-Gertsi, P.; Nalbanti, P.; Papachristou, F. Simultaneous changes in serum HMGB1 and IFN-α levels and in LAIR-1 expression on plasmatoid dendritic cells of patients with juvenile SLE. New therapeutic options? Lupus 2014, 23, 305–312. [Google Scholar]
- Hilliard, B.; Zizzo, G.; Ulas, M.; Linan, M.; Schreiter, J.; Cohen, P. Increased expression of mer tyrosine kinase in circulating dendritic cells and monocytes of lupus patients: Correlations with plasma interferon activity and steroid therapy. Arthritis Res. Ther. 2014, 16, R76. [Google Scholar]
- Balanescu, A.; Radu, E.; Nat, R.; Regalia, T.; Bojinca, V.; Predescu, V.; Predeteanu, D. Co-stimulatory and adhesion molecules of dendritic cells in rheumatoid arthritis. J. Cell. Mol. Med. 2002, 6, 415–425. [Google Scholar] [CrossRef]
- Nieminen, J.K.; Vakkila, J.; Salo, H.M.; Ekström, N.; Härkönen, T.; Ilonen, J.; Knip, M.; Vaarala, O. Altered phenotype of peripheral blood dendritic cells in pediatric type 1 diabetes. Diabetes Care 2012, 35, 2303–2310. [Google Scholar]
- Liu, Y.J. Ipc: Professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu. Rev. Immunol. 2005, 23, 275–306. [Google Scholar] [CrossRef]
- Izaguirre, A.; Barnes, B.J.; Amrute, S.; Yeow, W.S.; Megjugorac, N.; Dai, J.; Feng, D.; Chung, E.; Pitha, P.M.; Fitzgerald-Bocarsly, P. Comparative analysis of IRF and IFN-alpha expression in human plasmacytoid and monocyte-derived dendritic cells. J. Leukoc. Biol. 2003, 74, 1125–1138. [Google Scholar]
- Ito, T.; Kanzler, H.; Duramad, O.; Cao, W.; Liu, Y.J. Specialization, kinetics, and repertoire of type 1 interferon responses by human plasmacytoid predendritic cells. Blood 2006, 107, 2423–2431. [Google Scholar] [CrossRef]
- Gottenberg, J.-E.; Cagnard, N.; Lucchesi, C.; Letourneur, F.; Mistou, S.; Lazure, T.; Jacques, S.; Ba, N.; Ittah, M.; Lepajolec, C.; et al. Activation of ifn pathways and plasmacytoid dendritic cell recruitment in target organs of primary sjögren’s syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 2770–2775. [Google Scholar] [CrossRef]
- Ronnblom, L.E.; Alm, G.V.; Oberg, K. Autoimmune phenomena in patients with malignant carcinoid tumors during interferon-alpha treatment. Acta Oncol. 1991, 30, 537–540. [Google Scholar]
- Wilson, L.E.; Widman, D.; Dikman, S.H.; Gorevic, P.D. Autoimmune disease complicating antiviral therapy for hepatitis C virus infection. Semin. Arthritis Rheum. 2002, 32, 163–173. [Google Scholar] [CrossRef]
- Kalkner, K.M.; Ronnblom, L.; Karlsson Parra, A.K.; Bengtsson, M.; Olsson, Y.; Oberg, K. Antibodies against double-stranded DNA and development of polymyositis during treatment with interferon. QJM 1998, 91, 393–399. [Google Scholar] [CrossRef]
- Bennett, L.; Palucka, A.K.; Arce, E.; Cantrell, V.; Borvak, J.; Banchereau, J.; Pascual, V. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 2003, 197, 711–723. [Google Scholar] [CrossRef]
- Piccinini, A.M.; Midwood, K.S. Dampening inflammation by modulating TLR signalling. Med. Inflamm. 2010, 2010. [Google Scholar]
- Wen, Z.; Xu, L.; Chen, X.; Xu, W.; Yin, Z.; Gao, X.; Xiong, S. Autoantibody induction by DNA-containing immune complexes requires hmgb1 with the TLR2/microrna-155 pathway. J. Immunol. 2013, 190, 5411–5422. [Google Scholar] [CrossRef]
- Leadbetter, E.A.; Rifkin, I.R.; Hohlbaum, A.M.; Beaudette, B.C.; Shlomchik, M.J.; Marshak-Rothstein, A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and toll-like receptors. Nature 2002, 416, 603–607. [Google Scholar]
- Doring, Y.; Manthey, H.D.; Drechsler, M.; Lievens, D.; Megens, R.T.; Soehnlein, O.; Busch, M.; Manca, M.; Koenen, R.R.; Pelisek, J.; et al. Auto-antigenic protein-DNA complexes stimulate plasmacytoid dendritic cells to promote atherosclerosis. Circulation 2012, 125, 1673–1683. [Google Scholar] [CrossRef]
- Sandgren, S.; Wittrup, A.; Cheng, F.; Jonsson, M.; Eklund, E.; Busch, S.; Belting, M. The human antimicrobial peptide LL-37 transfers extracellular DNA plasmid to the nuclear compartment of mammalian cells via lipid rafts and proteoglycan-dependent endocytosis. J. Biol. Chem. 2004, 279, 17951–17956. [Google Scholar]
- Lande, R.; Gregorio, J.; Facchinetti, V.; Chatterjee, B.; Wang, Y.H.; Homey, B.; Cao, W.; Su, B.; Nestle, F.O.; Zal, T.; et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature 2007, 449, 564–569. [Google Scholar] [CrossRef]
- Ganguly, D.; Chamilos, G.; Lande, R.; Gregorio, J.; Meller, S.; Facchinetti, V.; Homey, B.; Barrat, F.J.; Zal, T.; Gilliet, M. Self-RNA–antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J. Exp. Med. 2009, 206, 1983–1994. [Google Scholar]
- Dombrowski, Y.; Schauber, J. Cathelicidin LL-37: A defense molecule with a potential role in psoriasis pathogenesis. Exp. Dermatol. 2012, 21, 327–330. [Google Scholar]
- Gilliet, M.; Lande, R. Antimicrobial peptides and self-DNA in autoimmune skin inflammation. Curr. Opin. Immunol. 2008, 20, 401–407. [Google Scholar] [CrossRef]
- Sacre, K.; Criswell, L.A.; McCune, J.M. Hydroxychloroquine is associated with impaired interferon-alpha and tumor necrosis factor-alpha production by plasmacytoid dendritic cells in systemic lupus erythematosus. Arthritis Res. Ther. 2012, 14, R155. [Google Scholar] [CrossRef]
- Ruiz-Irastorza, G.; Ramos-Casals, M.; Brito-Zeron, P.; Khamashta, M.A. Clinical efficacy and side effects of antimalarials in systemic lupus erythematosus: A systematic review. Ann. Rheum. Dis. 2010, 69, 20–28. [Google Scholar] [CrossRef]
- Means, T.K.; Latz, E.; Hayashi, F.; Murali, M.R.; Golenbock, D.T.; Luster, A.D. Human lupus autoantibody-DNA complexes activate dcs through cooperation of CD32 and TLR9. J. Clin. Investig. 2005, 115, 407–417. [Google Scholar]
- Bave, U.; Magnusson, M.; Eloranta, M.L.; Perers, A.; Alm, G.V.; Ronnblom, L. Fc gamma RIIa is expressed on natural IFN-alpha-producing cells (plasmacytoid dendritic cells) and is required for the IFN-alpha production induced by apoptotic cells combined with lupus IgG. J. Immunol. 2003, 171, 3296–3302. [Google Scholar]
- Lovgren, T.; Eloranta, M.L.; Kastner, B.; Wahren-Herlenius, M.; Alm, G.V.; Ronnblom, L. Induction of interferon-alpha by immune complexes or liposomes containing systemic lupus erythematosus autoantigen- and sjogren’s syndrome autoantigen-associated rna. Arthritis Rheumatol. 2006, 54, 1917–1927. [Google Scholar]
- Mold, C.; Clos, T.W.D. C-reactive protein inhibits plasmacytoid dendritic cell interferon responses to autoantibody immune complexes. Arthritis Rheumatol. 2013, 65, 1891–1901. [Google Scholar]
- Chen, D.Y.; Chen, Y.M.; Wen, M.C.; Hsieh, T.Y.; Hung, W.T.; Lan, J.L. The potential role of Th17 cells and Th17-related cytokines in the pathogenesis of lupus nephritis. Lupus 2012, 21, 1385–1396. [Google Scholar] [CrossRef]
- Rana, A.; Minz, R.W.; Aggarwal, R.; Anand, S.; Pasricha, N.; Singh, S. Gene expression of cytokines (TNF-alpha, IFN-gamma), serum profiles of IL-17 and IL-23 in paediatric systemic lupus erythematosus. Lupus 2012, 21, 1105–1112. [Google Scholar] [CrossRef]
- Wong, C.K.; Lit, L.C.; Tam, L.S.; Li, E.K.; Wong, P.T.; Lam, C.W. Hyperproduction of IL-23 and IL-17 in patients with systemic lupus erythematosus: Implications for Th17-mediated inflammation in auto-immunity. Clin. Immunol. 2008, 127, 385–393. [Google Scholar] [CrossRef]
- Pan, W.C.; Chen, R.M.; Shen, Y.C.; Chen, C.C.; Ueng, Y.F. Suppressive effect of tobacco smoke extracts on oral p-glycoprotein function and its impact in smoke-induced insult to oral epidermal cells. Toxicol. Lett. 2009, 185, 116–123. [Google Scholar]
- Steinman, R.M.; Hawiger, D.; Nussenzweig, M.C. Tolerogenic dendritic cells. Annu. Rev. Immunol. 2003, 21, 685–711. [Google Scholar]
- Guerder, S.; Joncker, N.; Mahiddine, K.; Serre, L. Dendritic cells in tolerance and autoimmune diabetes. Curr. Opin. Immunol. 2013, 25, 670–675. [Google Scholar] [CrossRef]
- Gordon, J.R.; Ma, Y.; Churchman, L.; Gordon, S.A.; Dawicki, W. Regulatory dendritic cells for immunotherapy in immunologic diseases. Front. Immunol. 2014, 5, 7. [Google Scholar]
- Van Brussel, I.; Lee, W.P.; Rombouts, M.; Nuyts, A.H.; Heylen, M.; de Winter, B.Y.; Cools, N.; Schrijvers, D.M. Tolerogenic dendritic cell vaccines to treat autoimmune diseases: Can the unattainable dream turn into reality? Autoimmun. Rev. 2014, 13, 138–150. [Google Scholar]
- Raïch-Regué, D.; Glancy, M.; Thomson, A.W. Regulatory dendritic cell therapy: From rodents to clinical application. Immunol. Lett. 2014, 161, 216–221. [Google Scholar] [CrossRef]
- Inaba, K.; Inaba, M.; Romani, N.; Aya, H.; Deguchi, M.; Ikehara, S.; Muramatsu, S.; Steinman, R.M. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 1992, 176, 1693–1702. [Google Scholar]
- Romani, N.; Gruner, S.; Brang, D.; Kämpgen, E.; Lenz, A.; Trockenbacher, B.; Konwalinka, G.; Fritsch, P.O.; Steinman, R.M.; Schuler, G. Proliferating dendritic cell progenitors in human blood. J. Exp. Med. 1994, 180, 83–93. [Google Scholar]
- Moreau, A.; Varey, E.; Bouchet-Delbos, L.; Cuturi, M.-C. Cell therapy using tolerogenic dendritic cells in transplantation. Transplant. Res. 2012, 1, 13. [Google Scholar] [Green Version]
- Klechevsky, E.; Banchereau, J. Human dendritic cells subsets as targets and vectors for therapy. Ann. N. Y. Acad. Sci. 2013, 1284, 24–30. [Google Scholar] [CrossRef]
- Everts, B.; Pearce, E.J. Metabolic control of dendritic cell activation and function: Recent advances and clinical implications. Front. Immunol. 2014, 5, 203. [Google Scholar]
- Lewis, J.S.; Roche, C.; Zhang, Y.; Brusko, T.M.; Wasserfall, C.H.; Atkinson, M.; Clare-Salzler, M.J.; Keselowsky, B.G. Combinatorial delivery of immunosuppressive factors to dendritic cells using dual-sized microspheres. J. Mater. Chem. B Mater. Biol. Med. 2014, 2, 2562–2574. [Google Scholar]
- Krawczyk, C.M.; Holowka, T.; Sun, J.; Blagih, J.; Amiel, E.; DeBerardinis, R.J.; Cross, J.R.; Jung, E.; Thompson, C.B.; Jones, R.G.; et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 2010, 115, 4742–4749. [Google Scholar] [CrossRef]
- Ferreira, G.B.; Kleijwegt, F.S.; Waelkens, E.; Lage, K.; Nikolic, T.; Hansen, D.A.; Workman, C.T.; Roep, B.O.; Overbergh, L.; Mathieu, C. Differential protein pathways in 1,25-dihydroxyvitamin D3 and dexamethasone modulated tolerogenic human dendritic cells. J. Proteome Res. 2011, 11, 941–971. [Google Scholar]
- Wobben, R.; Hüsecken, Y.; Lodewick, C.; Gibbert, K.; Fandrey, J.; Winning, S. Role of hypoxia inducible factor-1α for interferon synthesis in mouse dendritic cells. Biol. Chem. 2013, 394, 495. [Google Scholar]
- Turnquist, H.R.; Raimondi, G.; Zahorchak, A.F.; Fischer, R.T.; Wang, Z.; Thomson, A.W. Rapamycin-conditioned dendritic cells are poor stimulators of allogeneic CD4+ T cells, but enrich for antigen-specific Foxp3+ T regulatory cells and promote organ transplant tolerance. J. Immunol. 2007, 178, 7018–7031. [Google Scholar]
- Taner, T.; Hackstein, H.; Wang, Z.; Morelli, A.E.; Thomson, A.W. Rapamycin-treated, alloantigen-pulsed host dendritic cells induce Ag-specific T cell regulation and prolong graft survival. Am. J. Transplant. 2005, 5, 228–236. [Google Scholar]
- Fischer, R.T.; Turnquist, H.R.; Wang, Z.; Beer-Stolz, D.; Thomson, A.W. Rapamycin-conditioned, alloantigen-pulsed myeloid dendritic cells present donor MHC class I/peptide via the semi-direct pathway and inhibit survival of antigen-specific CD8+ T cells in vitro and in vivo. Transplant. Immunol. 2011, 25, 20–26. [Google Scholar] [CrossRef]
- Iruretagoyena, M.I.; Tobar, J.A.; Gonzalez, P.A.; Sepulveda, S.E.; Figueroa, C.A.; Burgos, R.A.; Hancke, J.L.; Kalergis, A.M. Andrographolide interferes with T cell activation and reduces experimental autoimmune encephalomyelitis in the mouse. J. Pharmacol. Exp. Ther. 2005, 312, 366–372. [Google Scholar]
- Szatmari, I.; Torocsik, D.; Agostini, M.; Nagy, T.; Gurnell, M.; Barta, E.; Chatterjee, K.; Nagy, L. Ppargamma regulates the function of human dendritic cells primarily by altering lipid metabolism. Blood 2007, 110, 3271–3280. [Google Scholar]
- Volchenkov, R.; Karlsen, M.; Jonsson, R.; Appel, S. Type 1 regulatory T cells and regulatory B cells induced by tolerogenic dendritic cells. Scand. J. Immunol. 2013, 77, 246–254. [Google Scholar]
- Iruretagoyena, M.I.; Sepúlveda, S.E.; Lezana, J.P.; Hermoso, M.; Bronfman, M.; Gutiérrez, M.A.; Jacobelli, S.H.; Kalergis, A.M. Inhibition of nuclear factor-κB enhances the capacity of immature dendritic cells to induce antigen-specific tolerance in experimental autoimmune encephalomyelitis. J. Pharmacol. Exp. Ther. 2006, 318, 59–67. [Google Scholar]
- Volchenkov, R.; Brun, J.; Jonsson, R.; Appel, S. In vitro suppression of immune responses using monocyte-derived tolerogenic dendritic cells from patients with primary sjogren’s syndrome. Arthritis Res. Ther. 2013, 15, R114. [Google Scholar]
- Xing, N.; ML, L.M.; Bachman, L.A.; McKean, D.J.; Kumar, R.; Griffin, M.D. Distinctive dendritic cell modulation by vitamin D(3) and glucocorticoid pathways. Biochem. Biophys. Res. Commun 2002, 297, 645–652. [Google Scholar] [CrossRef]
- Unger, W.W.J.; Laban, S.; Kleijwegt, F.S.; van der Slik, A.R.; Roep, B.O. Induction of treg by monocyte-derived dc modulated by vitamin D3 or dexamethasone: Differential role for PD-L1. Eur. J. Immunol. 2009, 39, 3147–3159. [Google Scholar]
- Garcia-Gonzalez, P.; Morales, R.; Hoyos, L.; Maggi, J.; Campos, J.; Pesce, B.; Garate, D.; Larrondo, M.; Gonzalez, R.; Soto, L.; et al. A short protocol using dexamethasone and monophosphoryl lipid a generates tolerogenic dendritic cells that display a potent migratory capacity to lymphoid chemokines. J. Transl. Med. 2013, 11, 128. [Google Scholar] [CrossRef]
- Moser, M.; de Smedt, T.; Sornasse, T.; Tielemans, F.; Chentoufi, A.A.; Muraille, E.; van Mechelen, M.; Urbain, J.; Leo, O. Glucocorticoids down-regulate dendritic cell function in vitro and in vivo. Eur. J. Immunol. 1995, 25, 2818–2824. [Google Scholar]
- Rea, D.; van Kooten, C.; van Meijgaarden, K.E.; Ottenhoff, T.H.; Melief, C.J.; Offringa, R. Glucocorticoids transform CD40-triggering of dendritic cells into an alternative activation pathway resulting in antigen-presenting cells that secrete IL-10. Blood 2000, 95, 3162–3167. [Google Scholar]
- Farias, A.S.; Spagnol, G.S.; Bordeaux-Rego, P.; Oliveira, C.O.F.; Fontana, A.G.M.; de Paula, R.F.O.; Santos, M.P.A.; Pradella, F.; Moraes, A.S.; Oliveira, E.C.; et al. Vitamin D3 induces IDO+ tolerogenic DCs and enhances Treg, reducing the severity of EAE. CNS Neurosci. Ther. 2013, 19, 269–277. [Google Scholar] [CrossRef]
- Raϊch-Regué, D.; Grau-López, L.; Naranjo-Gómez, M.; Ramo-Tello, C.; Pujol-Borrell, R.; Martínez-Cáceres, E.; Borràs, F.E. Stable antigen-specific T-cell hyporesponsiveness induced by tolerogenic dendritic cells from multiple sclerosis patients. Eur. J. Immunol. 2012, 42, 771–782. [Google Scholar]
- Hackstein, H.; Taner, T.; Zahorchak, A.F.; Morelli, A.E.; Logar, A.J.; Gessner, A.; Thomson, A.W. Rapamycin inhibits IL-4—Induced dendritic cell maturation in vitro and dendritic cell mobilization and function in vivo. Blood 2003, 101, 4457–4463. [Google Scholar]
- Fedoric, B.; Krishnan, R. Rapamycin downregulates the inhibitory receptors ILT2, ILT3, ILT4 on human dendritic cells and yet induces T cell hyporesponsiveness independent of Foxp3 induction. Immunol. Lett. 2008, 120, 49–56. [Google Scholar]
- Hackstein, H.; Morelli, A.E.; Larregina, A.T.; Ganster, R.W.; Papworth, G.D.; Logar, A.J.; Watkins, S.C.; Falo, L.D.; Thomson, A.W. Aspirin inhibits in vitro maturation and in vivo immunostimulatory function of murine myeloid dendritic cells. J. Immunol. 2001, 166, 7053–7062. [Google Scholar] [CrossRef]
- Buckland, M.; Jago, C.; Fazekesova, H.; George, A.; Lechler, R.; Lombardi, G. Aspirin modified dendritic cells are potent inducers of allo-specific regulatory T-cells. Int. Immunopharmacol. 2006, 6, 1895–1901. [Google Scholar] [CrossRef]
- Cai, D.T.; Ho, Y.H.S.; Chiow, K.H.; Wee, S.H.; Han, Y.; Peh, M.T.; Wong, S.H. Aspirin regulates snare protein expression and phagocytosis in dendritic cells. Mol. Membr. Biol. 2011, 28, 90–102. [Google Scholar] [CrossRef]
- Chauveau, C.; Remy, S.; Royer, P.J.; Hill, M.; Tanguy-Royer, S.; Hubert, F.X.; Tesson, L.; Brion, R.; Beriou, G.; Gregoire, M.; et al. Heme oxygenase-1 expression inhibits dendritic cell maturation and proinflammatory function but conserves IL-10 expression. Blood 2005, 106, 1694–1702. [Google Scholar]
- Martin, E.; Capini, C.; Duggan, E.; Lutzky, V.P.; Stumbles, P.; Pettit, A.R.; O’Sullivan, B.; Thomas, R. Antigen-specific suppression of established arthritis in mice by dendritic cells deficient in NF-κB. Arthritis Rheumatol. 2007, 56, 2255–2266. [Google Scholar]
- Ade, N.; Antonios, D.; Kerdine-Romer, S.; Boisleve, F.; Rousset, F.; Pallardy, M. NF-κB plays a major role in the maturation of human dendritic cells induced by NiSO4 but not by DNCB. Toxicol. Sci. 2007, 99, 488–501. [Google Scholar] [CrossRef]
- Ren, Y.; Yang, Y.; Yang, J.; Xie, R.; Fan, H. Tolerogenic dendritic cells modified by tacrolimus suppress CD4+ T-cell proliferation and inhibit collagen-induced arthritis in mice. Int. Immunopharmacol. 2014, 21, 247–254. [Google Scholar] [CrossRef]
- Sato, K.; Nagayama, H.; Tadokoro, K.; Juji, T.; Takahashi, T.A. Extracellular signal-regulated kinase, stress-activated protein kinase/c-Jun N-terminal kinase, and p38mapk are involved in IL-10-mediated selective repression of TNF-α-induced activation and maturation of human peripheral blood monocyte-derived dendritic cells. J. Immunol. 1999, 162, 3865–3872. [Google Scholar]
- Knodler, A.; Schmidt, S.M.; Bringmann, A.; Weck, M.M.; Brauer, K.M.; Holderried, T.A.W.; Heine, A.K.; Grunebach, F.; Brossart, P. Post-transcriptional regulation of adapter molecules by IL-10 inhibits TLR-mediated activation of antigen-presenting cells. Leukemia 2008, 23, 535–544. [Google Scholar]
- Velten, F.W.; Duperrier, K.; Bohlender, J.; Metharom, P.; Goerdt, S. A gene signature of inhibitory MHC receptors identifies a BDCA3+ subset of IL-10-induced dendritic cells with reduced allostimulatory capacity in vitro. Eur. J. Immunol. 2004, 34, 2800–2811. [Google Scholar]
- Kubsch, S.; Graulich, E.; Knop, J.; Steinbrink, K. Suppressor activity of anergic T cells induced by IL-10-treated human dendritic cells: Association with IL-2- and CTLA-4-dependent G1 arrest of the cell cycle regulated by p27Kip1. Eur. J. Immunol. 2003, 33, 1988–1997. [Google Scholar] [CrossRef]
- Steinbrink, K.; Graulich, E.; Kubsch, S.; Knop, J.; Enk, A.H. CD4+ and CD8+ anergic T cells induced by interleukin-10-treated human dendritic cells display antigen-specific suppressor activity. Blood 2002, 99, 2468–2476. [Google Scholar] [CrossRef]
- Li, X.; Yang, A.; Huang, H.; Zhang, X.; Town, J.; Davis, B.; Cockcroft, D.W.; Gordon, J.R. Induction of type 2 T helper cell allergen tolerance by il-10-differentiated regulatory dendritic cells. Am. J. Respir. Cell Mol. Biol. 2010, 42, 190–199. [Google Scholar] [CrossRef]
- Lopez, M.N.; Pesce, B.; Kurte, M.; Perez, C.; Segal, G.; Roa, J.; Aguillon, J.C.; Mendoza-Naranjo, A.; Gesser, B.; Larsen, C.; et al. A synthetic peptide homologous to IL-10 functional domain induces monocyte differentiation to TGF-beta+ tolerogenic dendritic cells. Immunobiology 2011, 216, 1117–1126. [Google Scholar]
- Ruffner, M.A.; Kim, S.H.; Bianco, N.R.; Francisco, L.M.; Sharpe, A.H.; Robbins, P.D. B7-1/2, but not PD-L1/2 molecules, are required on IL-10-treated tolerogenic DC and DC-derived exosomes for in vivo function. Eur. J. Immunol. 2009, 39, 3084–3090. [Google Scholar] [CrossRef]
- Jiang, H.; Hou, L.; Qiao, H.; Pan, S.; Zhou, B.; Liu, C.; Sun, X. Administration of tolerogenic dendritic cells induced by interleukin-10 prolongs rat splenic allograft survival. Transplant. Proc. 2004, 36, 3255–3259. [Google Scholar]
- Thomas, D.C.; Wong, F.S.; Zaccone, P.; Green, E.A.; Wållberg, M. Protection of islet grafts through transforming growth factor-β-induced tolerogenic dendritic cells. Diabetes 2013, 62, 3132–3142. [Google Scholar]
- Yan, F.; Cai, L.; Hui, Y.; Chen, S.; Meng, H.; Huang, Z. Tolerogenic dendritic cells suppress murine corneal allograft rejection by modulating CD28/CTLA-4 expression on regulatory T cells. Cell Biol. Int. 2014, 38, 835–848. [Google Scholar]
- Segovia-Gamboa, N.; Rodríguez-Arellano, M.E.; Rangel-Cruz, R.; Sánchez-Díaz, M.; Ramírez-Reyes, J.C.; Faradji, R.; González-Domínguez, É.; Sánchez-Torres, C. Tolerogenic dendritic cells induce antigen-specific hyporesponsiveness in insulin- and glutamic acid decarboxylase 65-autoreactive T lymphocytes from type 1 diabetic patients. Clin. Immunol. 2014, 154, 72–83. [Google Scholar]
- Torres-Aguilar, H.; Blank, M.; Kivity, S.; Misgav, M.; Luboshitz, J.; Pierangeli, S.S.; Shoenfeld, Y. Tolerogenic dendritic cells inhibit antiphospholipid syndrome derived effector/memory CD4+ T cell response to Β2GPI. Ann. Rheum. Dis. 2012, 71, 120–128. [Google Scholar]
- D’Ambrosio, A.; Colucci, M.; Pugliese, O.; Quintieri, F.; Boirivant, M. Cholera toxin b subunit promotes the induction of regulatory T cells by preventing human dendritic cell maturation. J. Leukoc. Biol. 2008, 84, 661–668. [Google Scholar] [CrossRef]
- Chen, L.; Zheng, L.; He, W.; Qiu, M.; Gao, L.; Liu, J.; Huang, A. Cotransfection with IL-10 and TGF-beta1 into immature dendritic cells enhances immune tolerance in a rat liver transplantation model. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G575–G581. [Google Scholar] [CrossRef]
- Li, R.; Zheng, X.; Popov, I.; Zhang, X.; Wang, H.; Suzuki, M.; Necochea-Campion, R.D.; French, P.W.; Chen, D.; Siu, L.; et al. Gene silencing of IL-12 in dendritic cells inhibits autoimmune arthritis. J. Transl. Med. 2012, 10, 19. [Google Scholar]
- Zheng, X.; Suzuki, M.; Ichim, T.E.; Zhang, X.; Sun, H.; Zhu, F.; Shunnar, A.; Garcia, B.; Inman, R.D.; Min, W. Treatment of autoimmune arthritis using RNA interference-modulated dendritic cells. J. Immunol. 2010, 184, 6457–6464. [Google Scholar] [CrossRef]
- Kalantari, T.; Karimi, M.H.; Ciric, B.; Yan, Y.; Rostami, A.; Kamali-Sarvestani, E. Tolerogenic dendritic cells produced by lentiviral-mediated CD40- and IL-23p19-specific shRNA can ameliorate experimental autoimmune encephalomyelitis by suppressing Th17 cells. Clin. Exp. Immunol. 2014, 176, 180–189. [Google Scholar] [CrossRef]
- Ma, L.; Qian, S.; Liang, X.; Wang, L.; Woodward, J.E.; Giannoukakis, N.; Robbins, P.D.; Bertera, S.; Trucco, M.; Fung, J.J.; et al. Prevention of diabetes in nod mice by administration of dendritic cells deficient in nuclear transcription factor-κB activity. Diabetes 2003, 52, 1976–1985. [Google Scholar]
- Machen, J.; Harnaha, J.; Lakomy, R.; Styche, A.; Trucco, M.; Giannoukakis, N. Antisense oligonucleotides down-regulating costimulation confer diabetes-preventive properties to nonobese diabetic mouse dendritic cells. J. Immunol. 2004, 173, 4331–4341. [Google Scholar]
- Hackstein, H.; Taner, T.; Logar, A.J.; Thomson, A.W. Rapamycin inhibits macropinocytosis and mannose receptor-mediated endocytosis by bone marrow-derived dendritic cells. Blood 2002, 100, 1084–1087. [Google Scholar] [CrossRef]
- Leung, D.Y.M.; Bloom, J.W. Update on glucocorticoid action and resistance. J. Allergy Clin. Immunol. 2003, 111, 3–22. [Google Scholar] [CrossRef]
- Harry, R.A.; Anderson, A.E.; Isaacs, J.D.; Hilkens, C.M.U. Generation and characterisation of therapeutic tolerogenic dendritic cells for rheumatoid arthritis. Ann. Rheum. Dis. 2010, 69, 2042–2050. [Google Scholar]
- Chiurchiù, V.; Cencioni, M.T.; Bisicchia, E.; de Bardi, M.; Gasperini, C.; Borsellino, G.; Centonze, D.; Battistini, L.; Maccarrone, M. Distinct modulation of human myeloid and plasmacytoid dendritic cells by anandamide in multiple sclerosis. Ann. Neurol. 2013, 73, 626–636. [Google Scholar] [CrossRef]
- Gross, C.C.; Jonuleit, H.; Wiendl, H. Fulfilling the dream: Tolerogenic dendritic cells to treat multiple sclerosis. Eur. J. Immunol. 2012, 42, 569–572. [Google Scholar] [CrossRef]
- Švajger, U.; Vidmar, A.; Jeras, M. Niflumic acid renders dendritic cells tolerogenic and up-regulates inhibitory molecules ilt3 and ilt4. Int. Immunopharmacol. 2008, 8, 997–1005. [Google Scholar]
- Stallone, G.; Pontrelli, P.; Infante, B.; Gigante, M.; Netti, G.S.; Ranieri, E.; Grandaliano, G.; Gesualdo, L. Rapamycin induces ILT3highILT4high dendritic cells promoting a new immunoregulatory pathway. Kidney Int. 2014, 85, 888–897. [Google Scholar]
- Ko, H.; Hambly, B.D.; Eris, J.M.; Levidiotis, V.; Wyburn, K.; Wu, H.; Chadban, S.J.; Yin, J.L. Dentritic cell derived IL-18 production is inhibited by rapamycin and sanglifehrin A, but not cyclosporine A. Transplant. Immunol. 2008, 20, 99–105. [Google Scholar]
- Tardif, V.; Riquelme, S.A.; Remy, S.; Carreño, L.J.; Cortés, C.M.; Simon, T.; Hill, M.; Louvet, C.; Riedel, C.A.; Blancou, P.; et al. Carbon monoxide decreases endosome-lysosome fusion and inhibits soluble antigen presentation by dendritic cells to T cells. Eur. J. Immunol. 2013, 43, 2832–2844. [Google Scholar]
- Simon, T.; Pogu, S.; Tardif, V.; Rigaud, K.; Rémy, S.; Piaggio, E.; Bach, J.-M.; Anegon, I.; Blancou, P. Carbon monoxide-treated dendritic cells decrease β1-integrin induction on cd8+ T cells and protect from type 1 diabetes. Eur. J. Immunol. 2013, 43, 209–218. [Google Scholar]
- O’Neill, L.A.J. Toll-like receptor signal transduction and the tailoring of innate immunity: A role for mal? Trends Immunol. 2002, 23, 296–300. [Google Scholar] [CrossRef]
- Thomas, R.; Street, S.; Ramnoruth, N.; Pahau, H.; Law, S.; Brunck, M.; Hyde, C.; O’Sullivan, B.; Capini, C.; Tran, A.; et al. Feasibility, safety and clinical effects of single intradermal administration of autologous tolerising dendritic cells exposed to citrullinated peptides in patients with rheumatoid arthritis. Arthritis Rheumatol. 2011, 63, 2430. [Google Scholar]
- Hackstein, H.; Thomson, A.W. Dendritic cells: Emerging pharmacological targets of immunosuppressive drugs. Nat. Rev. Immunol. 2004, 4, 24–35. [Google Scholar] [CrossRef]
- Llanos, C.; Mackern-Oberti, J.P.; Vega, F.; Jacobelli, S.H.; Kalergis, A.M. Tolerogenic dendritic cells as a therapy for treating lupus. Clin. Immunol. 2013, 148, 237–245. [Google Scholar] [CrossRef]
- Ding, Y.; Chen, D.; Tarcsafalvi, A.; Su, R.; Qin, L.; Bromberg, J.S. Suppressor of cytokine signaling 1 inhibits IL-10-mediated immune responses. J. Immunol. 2003, 170, 1383–1391. [Google Scholar]
- Murray, P.J. Understanding and exploiting the endogenous interleukin-10/STAT3-mediated anti-inflammatory response. Curr. Opin. Pharmacol. 2006, 6, 379–386. [Google Scholar]
- Figueroa-Vega, N.; Galindo-Rodríguez, G.; Bajaña, S.; Portales-Pérez, D.; Abud-Mendoza, C.; Sánchez-Torres, C.; González-Amaro, R. Phenotypic analysis of IL-10-treated, monocyte-derived dendritic cells in patients with systemic lupus erythematosus. Scand. J. Immunol. 2006, 64, 668–676. [Google Scholar]
- Carreno, L.J.; Riedel, C.A.; Kalergis, A.M. Induction of tolerogenic dendritic cells by NF-kappaB blockade and FCγ receptor modulation. Methods Mol. Biol. 2010, 677, 339–353. [Google Scholar]
- Zhou, Z.; Li, W.; Song, Y.; Wang, L.; Zhang, K.; Yang, J.; Zhang, W.; Su, H.; Zhang, Y. Growth differentiation factor-15 suppresses maturation and function of dendritic cells and inhibits tumor-specific immune response. PLoS One 2013, 8, e78618. [Google Scholar]
- Bimczok, D.; Rau, H.; Wundrack, N.; Naumann, M.; Rothkötter, H.-J.; McCullough, K.; Summerfield, A. Cholera toxin promotes the generation of semi-mature porcine monocyte-derived dendritic cells that are unable to stimulate T cells. Vet. Res. 2007, 38, 597–612. [Google Scholar]
- Pauley, K.; Cha, S. Rnai therapeutics in autoimmune disease. Pharmaceuticals 2013, 6, 287–294. [Google Scholar]
- Apparailly, F.; Jorgensen, C. SiRNA-based therapeutic approaches for rheumatic diseases. Nat. Rev. Rheumatol. 2013, 9, 56–62. [Google Scholar]
- Mathieu, P.; Chauveau, C.; Bouchet, D.; Guillot, C.; Tesson, L.; Anegon, I. Genetic engineering in allotransplantation of vascularized organs. Curr. Gene Ther. 2002, 2, 9–21. [Google Scholar]
- Zheng, X.; Suzuki, M.; Zhang, X.; Ichim, T.; Zhu, F.; Ling, H.; Shunnar, A.; Wang, M.; Garcia, B.; Inman, R.; et al. RNAi-mediated CD40-CD154 interruption promotes tolerance in autoimmune arthritis. Arthritis Res. Ther. 2010, 12, R13. [Google Scholar]
- Ferenbach, D.A.; Ramdas, V.; Spencer, N.; Marson, L.; Anegon, I.; Hughes, J.; Kluth, D.C. Macrophages expressing heme oxygenase-1 improve renal function in ischemia/reperfusion injury. Mol. Ther. 2010, 18, 1706–1713. [Google Scholar]
- Qiu, T.; Zhu, H.-C.; Liu, X.-H.; Dong, W.-C.; Weng, X.-D.; Hu, C.-H.; Kuang, Y.-L.; Gao, R.-H.; Dan, C.; Tao, T. Lentiviral-mediated shrna against RelB induces the generation of tolerogenic dendritic cells. Int. Immunopharmacol. 2012, 12, 501–509. [Google Scholar]
- Giannoukakis, N.; Phillips, B.; Finegold, D.; Harnaha, J.; Trucco, M. Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care 2011, 34, 2026–2032. [Google Scholar]
- Giannoukakis, N. Interview: Immunoregulatory dendritic cells to treat autoimmunity are ready for the clinic. Immunotherapy 2013, 5, 919–921. [Google Scholar]
- Johnson, L.; Jackson, D. Control of dendritic cell trafficking in lymphatics by chemokines. Angiogenesis 2014, 17, 335–345. [Google Scholar]
- Scheinecker, C.; McHugh, R.; Shevach, E.M.; Germain, R.N. Constitutive presentation of a natural tissue autoantigen exclusively by dendritic cells in the draining lymph node. J. Exp. Med. 2002, 196, 1079–1090. [Google Scholar]
- Tang, Q.; Adams, J.Y.; Tooley, A.J.; Bi, M.; Fife, B.T.; Serra, P.; Santamaria, P.; Locksley, R.M.; Krummel, M.F.; Bluestone, J.A. Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat. Immunol. 2006, 7, 83–92. [Google Scholar]
- Llanos, C.; Carreño, L.J.; Kalergis, A.M. Contribution of dendritic cell/T cell interactions to triggering and maintaining autoimmunity. Biol. Res. 2011, 44, 53–61. [Google Scholar]
- Fife, B.T.; Pauken, K.E.; Eagar, T.N.; Obu, T.; Wu, J.; Tang, Q.; Azuma, M.; Krummel, M.F.; Bluestone, J.A. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat. Immunol. 2009, 10, 1185–1192. [Google Scholar]
- Sercarz, E.E.; Lehmann, P.V.; Ametani, A.; Benichou, G.; Miller, A.; Moudgil, K. Dominance and crypticity of T cell antigenic determinants. Ann. Rev. Immunol. 1993, 11, 729–766. [Google Scholar]
- Al-Hashimi, H.; Bhowmik, A. Generalised lymphadenopathy as the first manifestation of lupus nephritis. BMJ Case Rep. 2010, 2010. [Google Scholar] [CrossRef]
- Healy, L.J.; Collins, H.L.; Thompson, S.J. Systemic administration of tolerogenic dendritic cells ameliorates murine inflammatory arthritis. Open Rheumatol. J. 2008, 2, 71–80. [Google Scholar]
- Ferreira, G.B.; Gysemans, C.A.; Demengeot, J.; da Cunha, J.P.M.C.M.; Vanherwegen, A.-S.; Overbergh, L.; van Belle, T.L.; Pauwels, F.; Verstuyf, A.; Korf, H.; et al. 1,25-dihydroxyvitamin d3 promotes tolerogenic dendritic cells with functional migratory properties in nod mice. J. Immunol. 2014, 192, 4210–4220. [Google Scholar]
- Ahrens, E.T.; Flores, R.; Xu, H.; Morel, P.A. In vivo imaging platform for tracking immunotherapeutic cells. Nat. Biotechnol. 2005, 23, 983–987. [Google Scholar]
- Kim, S.H.; Kim, S.; Evans, C.H.; Ghivizzani, S.C.; Oligino, T.; Robbins, P.D. Effective treatment of established murine collagen-induced arthritis by systemic administration of dendritic cells genetically modified to express IL-4. J. Immunol. 2001, 166, 3499–3505. [Google Scholar]
- Anderson, A.E.; Swan, D.J.; Sayers, B.L.; Harry, R.A.; Patterson, A.M.; von Delwig, A.; Robinson, J.H.; Isaacs, J.D.; Hilkens, C.M.U. LPS activation is required for migratory activity and antigen presentation by tolerogenic dendritic cells. J. Leukoc. Biol. 2009, 85, 243–250. [Google Scholar]
- Cozzani, E.; Drosera, M.; Gasparini, G.; Parodi, A. Serology of lupus erythematosus: Correlation between immunopathological features and clinical aspects. Autoimmun. Dis. 2014, 2014, 13. [Google Scholar]
- Caux, C.; Dezutter-Dambuyant, C.; Schmitt, D.; Banchereau, J. GM-CSF and TNF-alpha cooperate in the generation of dendritic langerhans cells. Nature 1992, 360, 258–261. [Google Scholar]
- Hayden, H.; Friedl, J.; Dettke, M.; Sachet, M.; Hassler, M.; Dubsky, P.; Bachleitner-Hofmann, T.; Gnant, M.; Stift, A. Cryopreservation of monocytes is superior to cryopreservation of immature or semi-mature dendritic cells for dendritic cell-based immunotherapy. J. Immunother. 2009, 32, 638–654. [Google Scholar]
- Strasser, E.F.; Eckstein, R. Optimization of leukocyte collection and monocyte isolation for dendritic cell culture. Transfus. Med. Rev. 2010, 24, 130–139. [Google Scholar]
- Silveira, G.F.; Wowk, P.F.; Machado, A.M.B.; dos Santos, C.N.D.; Bordignon, J. Immature dendritic cells generated from cryopreserved human monocytes show impaired ability to respond to LPS and to induce allogeneic lymphocyte proliferation. PLoS One 2013, 8, e71291. [Google Scholar]
- Galipeau, J. The mesenchymal stromal cells dilemma—Does a negative phase III trial of random donor mesenchymal stromal cells in steroid-resistant graft-versus-host disease represent a death knell or a bump in the road? Cytotherapy 2013, 15, 2–8. [Google Scholar]
- Giancola, R.; Bonfini, T.; Iacone, A. Cell therapy: cGMP facilities and manufacturing. Muscles Ligaments Tendons J. 2012, 2, 243–247. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mackern-Oberti, J.P.; Vega, F.; Llanos, C.; Bueno, S.M.; Kalergis, A.M. Targeting Dendritic Cell Function during Systemic Autoimmunity to Restore Tolerance. Int. J. Mol. Sci. 2014, 15, 16381-16417. https://doi.org/10.3390/ijms150916381
Mackern-Oberti JP, Vega F, Llanos C, Bueno SM, Kalergis AM. Targeting Dendritic Cell Function during Systemic Autoimmunity to Restore Tolerance. International Journal of Molecular Sciences. 2014; 15(9):16381-16417. https://doi.org/10.3390/ijms150916381
Chicago/Turabian StyleMackern-Oberti, Juan P., Fabián Vega, Carolina Llanos, Susan M. Bueno, and Alexis M. Kalergis. 2014. "Targeting Dendritic Cell Function during Systemic Autoimmunity to Restore Tolerance" International Journal of Molecular Sciences 15, no. 9: 16381-16417. https://doi.org/10.3390/ijms150916381