
Factor XIII B Subunit Polymorphisms and the Risk of Coronary Artery Disease

 ,
, 

Abstract
:1. Introduction
2. Results
2.1. Characterization of Study Population

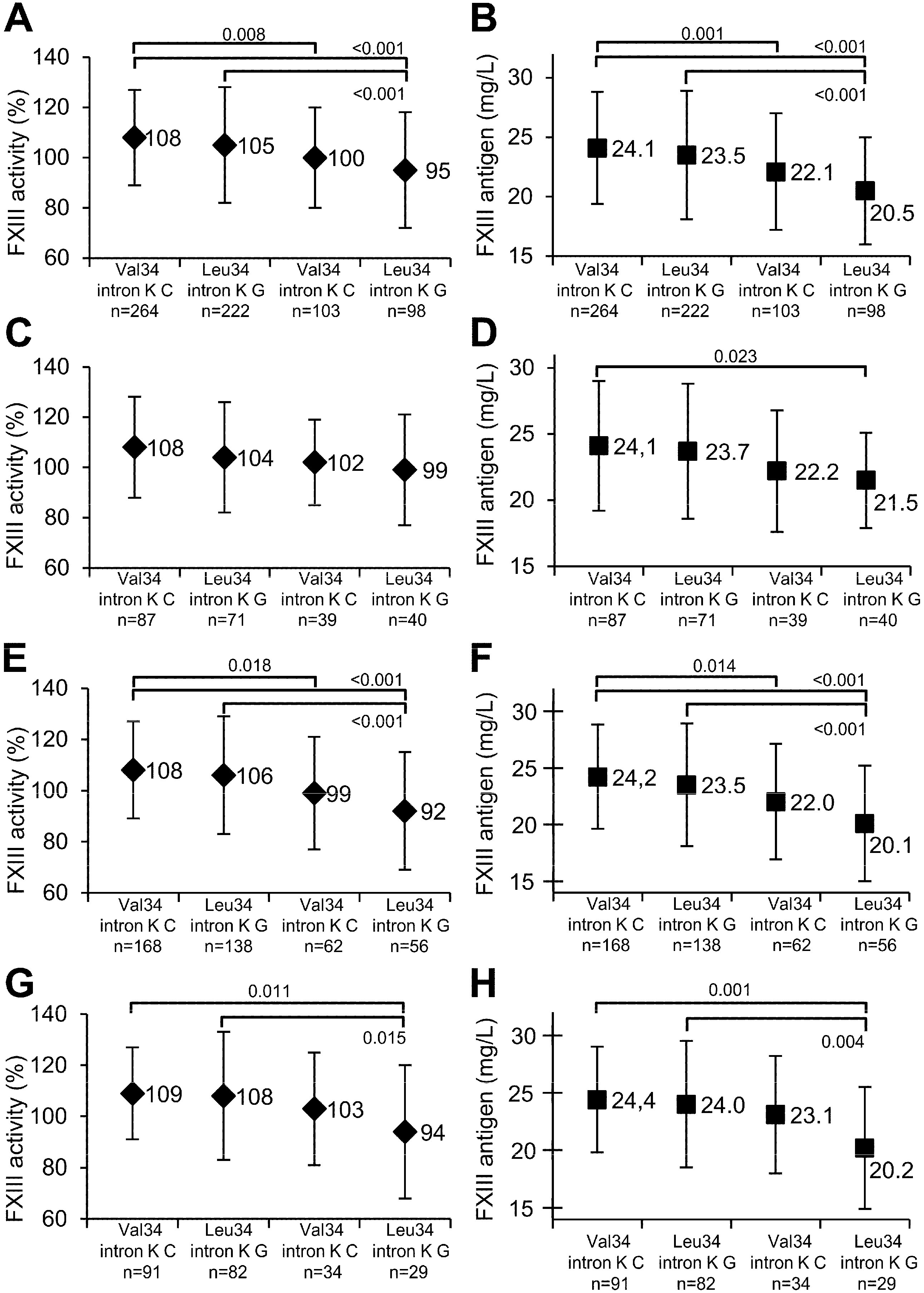{kind=link}
| Patients (n) | CAS−MI− (237) | CAS−MI+ (26) | CAS+MI− (214) | CAS+MI+ (210) |
|---|---|---|---|---|
| Gender (male/female) | 97/140 | 19/7 † | 144/70 ‡ | 164/46 ‡ |
| Age | 54 (48–64) | 56 (47–65) | 61 (54–70) † | 59 (51–68) † |
| Diabetes mellitus (−/+) | 218/19 | 20/6 * | 175/39 † | 161/49 ‡ |
| Current smoker (−/+) | 205/32 | 22/4 | 184/30 | 175/35 |
| Triglyceride (mmol/L) | 1.47 (1.03–2.19) | 1.46 (1.29–2.40) | 1.67 (1.25–2.26) * | 1.81 (1.35–2.48) ‡ |
| Cholesterol (mmol/L) | 5.60 ± 1.13 | 5.78 ± 1.17 | 5.71 ± 1.33 | 5.56 ± 1.12 |
| HDL-C (mmol/L) | 1.23 (1.01–1.49) | 1.13 (1.01–1.31) | 1.12 (0.96–1.33) † | 1.05 (0.90–1.25) ‡ |
| LDL-C (mmol/L) | 3.50 ± 0.98 | 3.70 ± 0.99 | 3.65 ± 1.15 | 3.48 ± 0.95 |
| ApoA-I (g/L) | 1.44 ± 0.27 | 1.34 ± 0.21 | 1.39 ± 0.30 | 1.32 ± 0.25 ‡ |
| ApoB (g/L) | 1.03 (0.90–1.18) | 1.15 (0.88–1.26) | 1.10 (0.95–1.27) * | 1.11 (0.96–1.29) † |
| Lp(a) (mg/L) | 126 (99–368) | 99 (99–300) | 126 (99–441) | 170 (99–642) * |
| Homocysteine (μmol/L) | 11.92 (9.68–14.75) | 13.38 (10.39–15.16) ‡ | 13.66 (10.98–16.28) ‡ | 13.62 (11.18–17.19) ‡ |
| Fibrinogen (g/L) | 3.78 (3.13–4.44) | 3.58 (2.95–5.11) | 3.88 (3.29–4.62) | 4.06 (3.23–5.03) * |
| FXIII activity (%) | 101 ± 20 | 103 ± 26 | 100 ± 22 | 101 ± 22 |
| FXIII antigen (mg/L) | 22.6 ± 4.8 | 23.4 ± 5.8 | 22.1 ± 5.2 | 22.4 ± 5.1 |
2.2. The Effect of the FXIII-B Polymorphisms on the Risk of Coronary Artery Disease
| Subjects | Population Controls n = 994 | CAS−MI− n = 237 | CAS+MI− n = 214 | CAS+MI+ n = 210 | CAS+ n = 424 | MI+ n = 236 |
|---|---|---|---|---|---|---|
| p.His95Arg | ||||||
| Wild-type n | 831 (83.6%) | 202 (85.2%) | 189 (88.3%) | 180 (85.7%) | 369 (87.0%) | 203 (86.0%) |
| Heterozygote n | 155 (15.6%) | 33 (14.0%) | 25 (11.7%) | 30 (14.3%) | 55 (13.0%) | 33 (14.0%) |
| Homozygote n | 8 (0.8%) | 2 (0.8%) | – | – | – | – |
| Arg95 carrier frequency | 16.4% | 14.8% | 11.7% | 14.3% | 13.0% | 14.0% |
| Arg95 allele frequency | 8.6% | 7.8% | 5.8% | 7.1% | 6.5% | 7.0% |
| OR for Arg95 carriers non-adjusted | – | – | 0.76 (0.44, 1.32) | 0.96 (0.57, 1.63) | 0.86 (0.55, 1.36) | 0.94 (0.56, 1.57) |
| OR for Arg95 carriers adjusted | – | – | 0.76 (0.41, 1.39) | 1.18 (0.64, 2.17) | 0.95 (0.57, 1.59) | 1.11 (0.62, 2.01) |
| Intron K nt29756 C>G | ||||||
| Wild-type n | 712 (71.6%) | 158 (66.7%) | 155 (72.4%) | 151 (71.9%) | 306 (72.2%) | 173 (73.3%) |
| Heterozygote n | 259 (26.1%) | 74 (31.2%) | 52 (24.3%) | 55 (26.2%) | 107 (25.2%) | 59 (25.0%) |
| Homozygote n | 23 (2.3%) | 5 (2.1%) | 7 (3.3%) | 4 (1.9%) | 11 (2.6%) | 4 (1.7%) |
| G carrier frequency | 28.4% | 33.3% | 27.6% | 28.1% | 27.8% | 26.7% |
| G allele frequency | 15.3% | 17.7% | 15.4% | 15.0% | 15.2% | 14.2% |
| OR for G carriers non-adjusted | – | – | 0.76 (0.51,1.14) | 0.78 (0.52,1.17) | 0.77 (0.55, 1.09) | 0.73 (0.49, 1.08) |
| OR for G carriers adjusted | – | – | 0.82 (0.53,1.28) | 0.87 (0.55, 1.39) | 0.82 (0.56, 1.21) | 0.80 (0.51, 1.26) |
2.3. The Effect of the FXIII-B Polymorphisms on the Risk of Coronary Artery Disease in Individuals with Elevated Fibrinogen Concentration
| Subjects | CAS−MI− n = 63 | CAS+MI− n = 75 | CAS+MI+ n = 79 | CAS+ n = 154 | MI+ n = 88 |
|---|---|---|---|---|---|
| p.His95Arg | |||||
| Wild-type n | 55 (87.3%) | 69 (92.0%) | 67 (84.8%) | 136 (88.3%) | 74 (84.1%) |
| Heterozygote n | 8 (12.7%) | 6 (8.0%) | 12 (15.2%) | 18 (11.7%) | 14 (15.9%) |
| Homozygote n | – | – | – | – | – |
| Arg95 carrier frequency | 12.7% | 8.0% | 15.2% | 11.7% | 15.9% |
| Arg95 allele frequency | 6.3% | 4.0% | 7.6% | 5.8% | 8.0% |
| OR for Arg95 carriers non-adjusted | – | 0.6 (0.20, 1.83) | 1.23 (0.47, 3.23) | 0.91 (0.37, 2.22) | 1.3 (0.51, 3.32) |
| OR for Arg95 carriers adjusted | – | 0.81 (0.23, 2.92) | 1.55 (0.52, 4.65) | 1.07 (0.39, 2.96) | 1.5 (0.51, 4.38) |
| Intron K nt29756 C>G | |||||
| Wild-type n | 38 (60.3%) | 56 (74.7%) | 61 (77.2%) | 117 (76.0%) | 70 (79.5%) |
| Heterozygote n | 24 (38.1%) | 17 (22.7%) | 16 (20.3%) * | 33 (21.4%) * | 16 (18.2%) † |
| Homozygote n | 1 (1.6%) | 2 (2.6%) | 2 (2.5%) | 4 (2.6%) | 2 (2.3%) |
| G carrier frequency | 39.7% | 25.3% | 22.8% * | 24.0% * | 20.5% * |
| G allele frequency | 20.6% | 14.0% | 12.7% | 13.3% * | 11.4% * |
| OR for G carriers non-adjusted | – | 0.52 (0.25, 1.07) | 0.45 * (0.21–0.93) | 0.48 * (0.26, 0.90) | 0.39 * (0.19, 0.81) |
| OR for G carriers adjusted | – | 0.35 * (0.15, 0.83) | 0.42 * (0.19, 0.96) | 0.38 † (0.19, 0.79) | 0.37 * (0.17, 0.84) |
2.4. The Effect of Combined FXIII-A Val34Leu and FXIII-B Polymorphisms on the Risk of Coronary Artery Disease
| Subjects | CAS−MI− n = 63 | CAS+MI− n = 75 | CAS+MI+ n = 79 | CAS+ n = 154 | MI+ n = 88 |
|---|---|---|---|---|---|
| Val34 Homozygotes, Intron K C Homozygotes | |||||
| n | 19 | 30 | 33 | 63 | 37 |
| Leu34 carriers, Intron K C Homozygotes | |||||
| n | 19 | 26 | 28 | 54 | 33 |
| Unadjusted OR | – | 0.87 (0.38, 1.98) | 0.85 (0.38, 1.91) | 0.86 (0.41, 1.78) | 0.89 (0.41, 1.97) |
| Adjusted OR | – | 1.33 (0.51, 3.52) | 0.81 (0.31, 2.08) | 1.08 (0.48, 2.45) | 0.94 (0.38, 2.34) |
| Val34 homozygotes, Intron K G Carriers | |||||
| n | 10 | 10 | 15 | 25 | 15 |
| Unadjusted OR | – | 0.63 (0.22, 1.81) | 0.86 (0.32, 2.30) | 0.75 (0.31, 1.85) | 0.77 (0.29, 2.04) |
| Adjusted OR | – | 0.59 (0.18,1.96) | 0.92 (0.31, 2.76) | 0.76 (0.29, 2.02) | 0.86 (0.29, 2.52) |
| Leu34 Carriers, Intron K G Carriers | |||||
| n | 15 | 9 | 3 | 12 | 3 |
| Unadjusted OR | – | 0.38 (0.14, 1.04) | 0.12 (0.03, 0.45) † | 0.24 (0.10, 0.60) † | 0.10 (0.03, 0.40) ‡ |
| Adjusted OR | – | 0.30 (0.09, 0.96) * | 0.08 (0.02, 0.39) † | 0.19 (0.07, 0.55) † | 0.08 (0.02, 0.36) ‡ |
| Synergy factor unadjusted | – | 0.69 (0.23, 2.98) | 0.16 (0.03, 0.85) * | 0.37 (0.10, 1.35) | 0.15 (0.03, 0.80) * |
| Synergy factor adjusted | – | 0.38 (0.23,1.62) | 0.11 (0.02, 0.61) * | 0.23 (0.06, 0.85) * | 0.10 (0.02, 0.53) † |
2.5. The Effect of FXIII-B Polymorphisms on FXIII Levels

| Subjects | Wild Type for the Mutation | Carriers of the Mutation | ||||
|---|---|---|---|---|---|---|
| n | FXIII Activity (%) | FXIII Antigen (mg/L) | n | FXIII Activity (%) | FXIII Antigen (mg/L) | |
| p.His95Arg Polymorphism | ||||||
| All | 594 | 103 ± 21 | 22.9 ± 5.0 | 93 | 109 ± 23 † | 24.0 ± 5.1 * |
| CAS−MI− | 202 | 103 ± 20 | 22.9 ± 4.7 | 35 | 112 ± 23 * | 24.3 ± 5.3 |
| CAS+MI− | 189 | 101 ± 22 | 22.6 ± 5.1 | 25 | 107 ± 25 | 23.7 ± 5.5 |
| CAS+MI+ | 180 | 106 ± 22 | 23.4 ± 5.3 | 30 | 107 ± 20 | 23.7 ± 4.3 |
| CAS+ | 369 | 103 ± 22 | 22.9 ± 5.2 | 55 | 107 ± 22 | 23.7 ± 4.9 |
| MI+ | 203 | 105 ± 23 | 23.4 ± 5.3 | 33 | 109 ± 21 | 24.0 ± 4.6 |
| Intron K nt29756 C>G Polymorphism | ||||||
| All | 486 | 106 ± 21 | 23.8 ± 5.0 | 201 | 97 ± 21 ‡ | 21.3 ± 4.7 ‡ |
| CAS−MI− | 158 | 106 ± 21 | 23.9 ± 5.0 | 79 | 100 ± 20 * | 21.8 ± 4.1 † |
| CAS+MI− | 155 | 106 ± 22 | 23.7 ± 5.1 | 59 | 94 ± 21‡ | 20.7 ± 4.9 ‡ |
| CAS+MI+ | 151 | 109 ± 20 | 24.2 ± 4.9 | 59 | 98 ± 24‡ | 21.6 ± 5.5 ‡ |
| CAS+ | 306 | 107 ± 21 | 23.9 ± 5.0 | 118 | 96 ± 22‡ | 21.1 ± 5.2 ‡ |
| MI+ | 173 | 108 ± 21 | 24.1 ± 5.0 | 63 | 99 ± 24† | 21.7 ± 5.3 † |
2.6. The Effect of Low FXIII Levels on the Risk of CAD
3. Discussion
| Subjects | CAS−MI− | CAS+MI− | CAS+MI+ | CAS+ | MI+ |
|---|---|---|---|---|---|
| FXIII activity upper tertile (n) | 21 | 33 | 40 | 73 | 47 |
| FXIII activity lower tertile (n) | 24 | 24 | 22 | 46 | 24 |
| Adjusted OR | – | 0.65 (0.25, 1.69) | 0.38 (0.15, 0.98) * | 0.52 (0.23, 1.17) | 0.39 (0.16, 0.96) * |
| FXIII antigen upper tertile (n) | 19 | 32 | 40 | 72 | 46 |
| FXIII antigen lower tertile (n) | 24 | 25 | 24 | 49 | 25 |
| Adjusted OR | – | 0.57 (0.23, 1.42) | 0.36 (0.14, 0.91) * | 0.49 (0.22, 1.08) | 0.35 (0.14, 0.86) * |
4. Experimental Section
4.1. Patients
4.2. Blood Sampling and Laboratory Methods
4.3. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Komaromi, I.; Bagoly, Z.; Muszbek, L. Factor XIII: Novel structural and functional aspects. J. Thromb. Haemost. 2011, 9, 9–20. [Google Scholar] [CrossRef]
- Muszbek, L.; Bereczky, Z.; Bagoly, Z.; Komaromi, I.; Katona, E. Factor XIII: A coagulation factor with multiple plasmatic and cellular functions. Physiol. Rev. 2011, 91, 931–972. [Google Scholar] [CrossRef]
- Schroeder, V.; Kohler, H.P. New developments in the area of factor XIII. J. Thromb. Haemost. 2013, 11, 234–244. [Google Scholar] [CrossRef] [Green Version]
- Muszbek, L.; Bereczky, Z.; Bagoly, Z.; Shemirani, A.H.; Katona, E. Factor XIII and atherothrombotic diseases. Semin. Thromb. Hemost. 2010, 36, 18–33. [Google Scholar] [CrossRef]
- Bereczky, Z.; Balogh, E.; Katona, E.; Czuriga, I.; Edes, I.; Muszbek, L. Elevated factor XIII level and the risk of myocardial infarction in women. Haematologica 2007, 92, 287–288. [Google Scholar] [CrossRef]
- Shemirani, A.H.; Szomjak, E.; Csiki, Z.; Katona, E.; Bereczky, Z.; Muszbek, L. Elevated factor XIII level and the risk of peripheral artery disease. Haematologica 2008, 93, 1430–1432. [Google Scholar] [CrossRef]
- Ariens, R.A.; Philippou, H.; Nagaswami, C.; Weisel, J.W.; Lane, D.A.; Grant, P.J. The factor XIII V34L polymorphism accelerates thrombin activation of factor XIII and affects cross-linked fibrin structure. Blood 2000, 96, 988–995. [Google Scholar]
- Balogh, I.; Szoke, G.; Karpati, L.; Wartiovaara, U.; Katona, E.; Komaromi, I.; Haramura, G.; Pfliegler, G.; Mikkola, H.; Muszbek, L. Val34leu polymorphism of plasma factor XIII: Biochemistry and epidemiology in familial thrombophilia. Blood 2000, 96, 2479–2486. [Google Scholar]
- Wartiovaara, U.; Mikkola, H.; Szoke, G.; Haramura, G.; Karpati, L.; Balogh, I.; Lassila, R.; Muszbek, L.; Palotie, A. Effect of val34leu polymorphism on the activation of the coagulation factor XIII-A. Thromb. Haemost. 2000, 84, 595–600. [Google Scholar]
- Lim, B.C.; Ariens, R.A.; Carter, A.M.; Weisel, J.W.; Grant, P.J. Genetic regulation of fibrin structure and function: Complex gene-environment interactions may modulate vascular risk. Lancet 2003, 361, 1424–1431. [Google Scholar] [CrossRef]
- Kohler, H.P.; Stickland, M.H.; Ossei-Gerning, N.; Carter, A.; Mikkola, H.; Grant, P.J. Association of a common polymorphism in the factor XIII gene with myocardial infarction. Thromb. Haemost. 1998, 79, 8–13. [Google Scholar]
- Bereczky, Z.; Balogh, E.; Katona, E.; Pocsai, Z.; Czuriga, I.; Szeles, G.; Karpati, L.; Adany, R.; Edes, I.; Muszbek, L. Modulation of the risk of coronary sclerosis/myocardial infarction by the interaction between factor XIII subunit A Val34Leu polymorphism and fibrinogen concentration in the high risk hungarian population. Thromb. Res. 2007, 120, 567–573. [Google Scholar] [CrossRef]
- Voko, Z.; Bereczky, Z.; Katona, E.; Adany, R.; Muszbek, L. Factor XIII Val34Leu variant protects against coronary artery disease. A meta-analysis. Thromb. Haemost. 2007, 97, 458–463. [Google Scholar]
- Wells, P.S.; Anderson, J.L.; Scarvelis, D.K.; Doucette, S.P.; Gagnon, F. Factor XIII Val34Leu variant is protective against venous thromboembolism: A huge review and meta-analysis. Am. J. Epidemiol. 2006, 164, 101–109. [Google Scholar] [CrossRef]
- Board, P.G. Genetic polymorphism of the B subunit of human coagulation factor XIII. Am. J. Hum. Genet. 1980, 32, 348–353. [Google Scholar]
- Leifheit, H.J.; Cleve, H. Analysis of the genetic polymorphism of coagulation factor XIIIB (FXIIIB) by isoelectric focusing. Electrophoresis 1988, 9, 426–429. [Google Scholar] [CrossRef]
- Komanasin, N.; Catto, A.J.; Futers, T.S.; van Hylckama Vlieg, A.; Rosendaal, F.R.; Ariens, R.A. A novel polymorphism in the factor XIII B-subunit (His95Arg): Relationship to subunit dissociation and venous thrombosis. J. Thromb. Haemost. 2005, 3, 2487–2496. [Google Scholar] [CrossRef]
- International Hapmap Project. Available online: http://hapmap.ncbi.nlm.nih.gov/ (accessed on 15 October 2014).
- Pruissen, D.M.; Rosendaal, F.R.; Frijns, C.J.; Kappelle, L.J.; Vos, H.L.; Algra, A. Prothrombotic gene variants and mortality after cerebral ischemia of arterial origin. Neuroepidemiology 2011, 37, 109–113. [Google Scholar] [CrossRef]
- Reiner, A.P.; Heckbert, S.R.; Vos, H.L.; Ariens, R.A.; Lemaitre, R.N.; Smith, N.L.; Lumley, T.; Rea, T.D.; Hindorff, L.A.; Schellenbaum, G.D.; et al. Genetic variants of coagulation factor XIII, postmenopausal estrogen therapy, and risk of nonfatal myocardial infarction. Blood 2003, 102, 25–30. [Google Scholar] [CrossRef]
- Iwata, H.; Kitano, T.; Umetsu, K.; Yuasa, I.; Yamazaki, K.; Kemkes-Matthes, B.; Ichinose, A. Distinct C-terminus of the B subunit of factor XIII in a population-associated major phenotype: The first case of complete allele-specific alternative splicing products in the coagulation and fibrinolytic systems. J. Thromb. Haemost. 2009, 7, 1084–1091. [Google Scholar] [CrossRef]
- Ryan, A.W.; Hughes, D.A.; Tang, K.; Kelleher, D.P.; Ryan, T.; McManus, R.; Stoneking, M. Natural selection and the molecular basis of electrophoretic variation at the coagulation F13B locus. Eur. J. Hum. Genet. 2009, 17, 219–227. [Google Scholar] [CrossRef]
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Simoons, M.L.; Chaitman, B.R.; White, H.D.; Katus, H.A.; Apple, F.S.; Lindahl, B.; Morrow, D.A.; et al. Third universal definition of myocardial infarction. J. Am. Coll. Cardiol. 2012, 60, 1581–1598. [Google Scholar] [CrossRef]
- Katona, E.; Penzes, K.; Csapo, A.; Fazakas, F.; Udvardy, M.L.; Bagoly, Z.; Orosz, Z.Z.; Muszbek, L. Interaction of factor XIII subunits. Blood 2014, 123, 1757–1763. [Google Scholar] [CrossRef] [Green Version]
- Girolami, A.; Burul, A.; Fabris, F.; Cappellato, G.; Betterle, C. Studies on factor XIII antigen in congenital factor XIII deficiency. A tentative classification of the disease in two groups. Folia Haematol. 1978, 105, 131–141. [Google Scholar]
- Capellato, M.G.; Lazzaro, A.R.; Marafioti, F.; Polato, G.; Girolami, A. A new family with congenital factor XIII deficiency showing a deficit of both subunit A and B. Type I factor XIII deficiency. Haematologia 1987, 20, 179–187. [Google Scholar]
- Saito, M.; Asakura, H.; Yoshida, T.; Ito, K.; Okafuji, K.; Yoshida, T.; Matsuda, T. A familial factor XIII subunit B deficiency. Br. J. Haematol. 1990, 74, 290–294. [Google Scholar] [CrossRef]
- Hashiguchi, T.; Saito, M.; Morishita, E.; Matsuda, T.; Ichinose, A. Two genetic defects in a patient with complete deficiency of the B-subunit for coagulation factor XIII. Blood 1993, 82, 145–150. [Google Scholar]
- Rodeghiero, F.; Tosetto, A. Factor XIII subunit B deficiency. Br. J. Haematol. 1990, 76, 317. [Google Scholar] [CrossRef]
- Rodeghiero, F.; Tosetto, A.; di Bona, E.; Castaman, G. Clinical pharmacokinetics of a placenta-derived factor XIII concentrate in type I and type II factor XIII deficiency. Am. J. Hematol. 1991, 36, 30–34. [Google Scholar] [CrossRef]
- Souri, M.; Koseki-Kuno, S.; Takeda, N.; Degen, J.L.; Ichinose, A. Administration of factor XIII B subunit increased plasma factor XIII A subunit levels in factor XIII B subunit knock-out mice. Int. J. Hematol. 2008, 87, 60–68. [Google Scholar] [CrossRef]
- Polgar, J.; Hidasi, V.; Muszbek, L. Non-proteolytic activation of cellular protransglutaminase (placenta macrophage factor XIII). Biochem. J. 1990, 267, 557–560. [Google Scholar]
- Souri, M.; Kaetsu, H.; Ichinose, A. Sushi domains in the B subunit of factor XIII responsible for oligomer assembly. Biochemistry 2008, 47, 8656–8664. [Google Scholar] [CrossRef]
- Tuut, M.; Hense, H.W. Smoking, other risk factors and fibrinogen levels. Evidence of effect modification. Ann. Epidemiol. 2001, 11, 232–238. [Google Scholar] [CrossRef]
- Lanktree, M.B.; Hegele, R.A. Gene-gene and gene-environment interactions: New insights into the prevention, detection and management of coronary artery disease. Genome Med. 2009, 1, 28. [Google Scholar] [CrossRef]
- Bereczky, Z.; Balogh, E.; Katona, E.; Czuriga, I.; Karpati, L.; Shemirani, A.H.; Edes, I.; Muszbek, L. Decreased factor XIII levels in factor XIII A subunit Leu34 homozygous patients with coronary artery disease. Thromb. Res. 2008, 121, 469–476. [Google Scholar] [CrossRef]
- Montagnana, M.; Danese, E.; Lippi, G. Genetic risk factors of atherothrombosis. Pol. Arch. Med. Wewn. 2014, 124, 474–482. [Google Scholar]
- Kunicki, T.J.; Williams, S.A.; Nugent, D.J. Genetic variants that affect platelet function. Curr. Opin. Hematol. 2012, 19, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Fedele, F.; Mancone, M.; Chilian, W.M.; Severino, P.; Canali, E.; Logan, S.; de Marchis, M.L.; Volterrani, M.; Palmirotta, R.; Guadagni, F. Role of genetic polymorphisms of ion channels in the pathophysiology of coronary microvascular dysfunction and ischemic heart disease. Basic Res. Cardiol. 2013, 108, 387. [Google Scholar] [CrossRef]
- Stephen, B.H.; Steven, R.C.; Warren, S.B.; Deborah, G.G.; Thomas, B.N. Designing Clinical Research, 3rd ed.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2007. [Google Scholar]
- Széles, G.; Voko, Z.; Jenei, T.; Kardos, L.; Pocsai, Z.; Bajtay, A.; Papp, E.; Pasti, G.; Kosa, Z.; Molnar, I.; et al. A preliminary evaluation of a health monitoring programme in Hungary. Eur. J. Public Health 2005, 15, 26–32. [Google Scholar] [CrossRef]
- Shemirani, A.H.; Muszbek, L. Rapid detection of the factor XIII Val34Leu (163 G→T) polymorphism by real-time pcr using fluorescence resonance energy transfer detection and melting curve analysis. Clin. Chem. Lab. Med. 2004, 42, 877–879. [Google Scholar]
- Karpati, L.; Penke, B.; Katona, E.; Balogh, I.; Vamosi, G.; Muszbek, L. A modified, optimized kinetic photometric assay for the determination of blood coagulation factor XIII activity in plasma. Clin. Chem. 2000, 46, 1946–1955. [Google Scholar]
- Katona, E.; Haramura, G.; Karpati, L.; Fachet, J.; Muszbek, L. A simple, quick one-step ELISA assay for the determination of complex plasma factor XIII (A2B2). Thromb. Haemost. 2000, 83, 268–273. [Google Scholar]
- Cortina-Borja, M.; Smith, A.D.; Combarros, O.; Lehmann, D.J. The synergy factor: A statistic to measure interactions in complex diseases. BMC Res. Notes 2009, 2, 105. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mezei, Z.A.; Bereczky, Z.; Katona, É.; Gindele, R.; Balogh, E.; Fiatal, S.; Balogh, L.; Czuriga, I.; Ádány, R.; Édes, I.; et al. Factor XIII B Subunit Polymorphisms and the Risk of Coronary Artery Disease. Int. J. Mol. Sci. 2015, 16, 1143-1159. https://doi.org/10.3390/ijms16011143
Mezei ZA, Bereczky Z, Katona É, Gindele R, Balogh E, Fiatal S, Balogh L, Czuriga I, Ádány R, Édes I, et al. Factor XIII B Subunit Polymorphisms and the Risk of Coronary Artery Disease. International Journal of Molecular Sciences. 2015; 16(1):1143-1159. https://doi.org/10.3390/ijms16011143
Chicago/Turabian StyleMezei, Zoltán A., Zsuzsanna Bereczky, Éva Katona, Réka Gindele, Emília Balogh, Szilvia Fiatal, László Balogh, István Czuriga, Róza Ádány, István Édes, and et al. 2015. "Factor XIII B Subunit Polymorphisms and the Risk of Coronary Artery Disease" International Journal of Molecular Sciences 16, no. 1: 1143-1159. https://doi.org/10.3390/ijms16011143