
Mechanisms and Αpplications of Ιnterleukins in Cancer Immunotherapy

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction

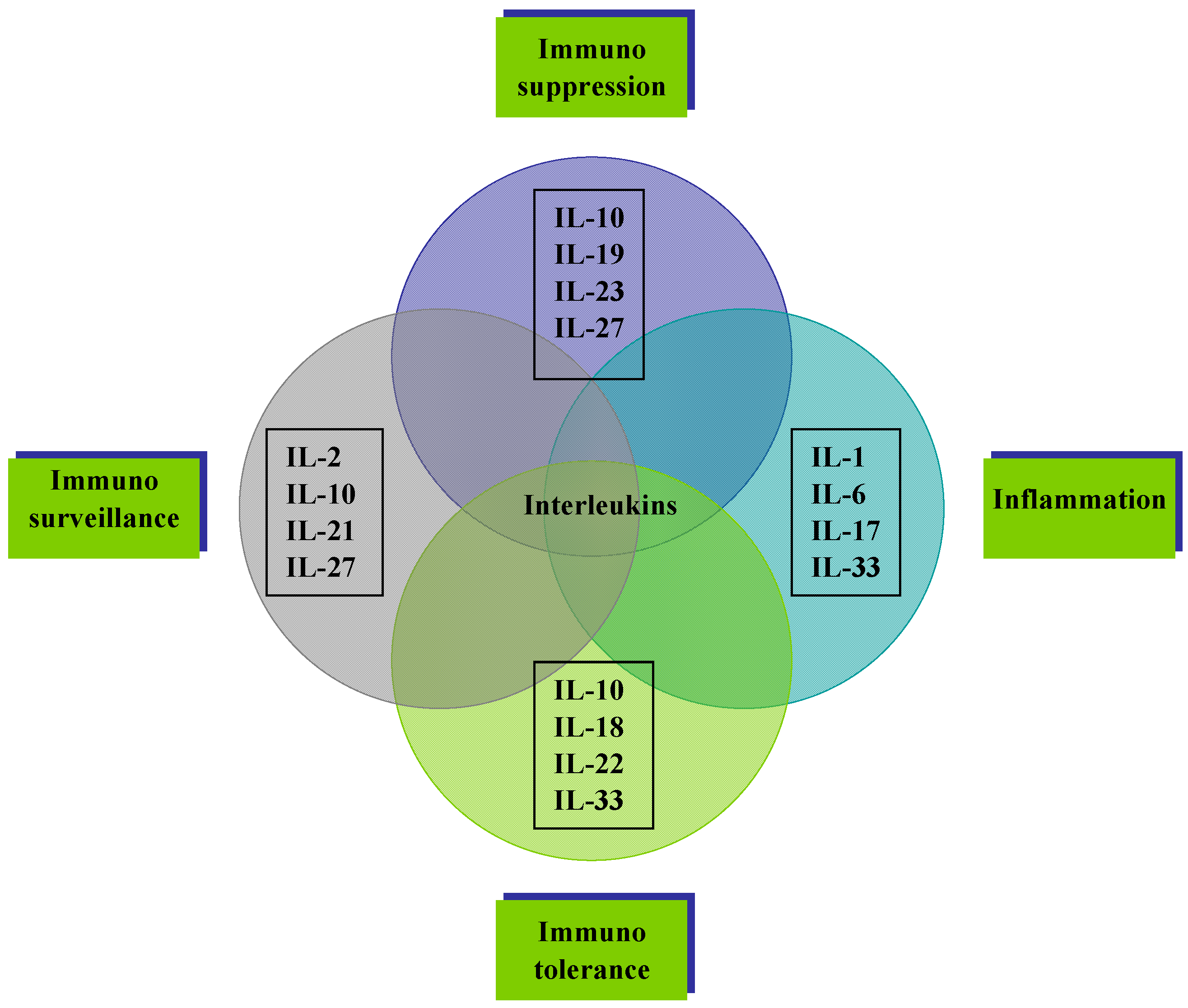
2. Interleukins
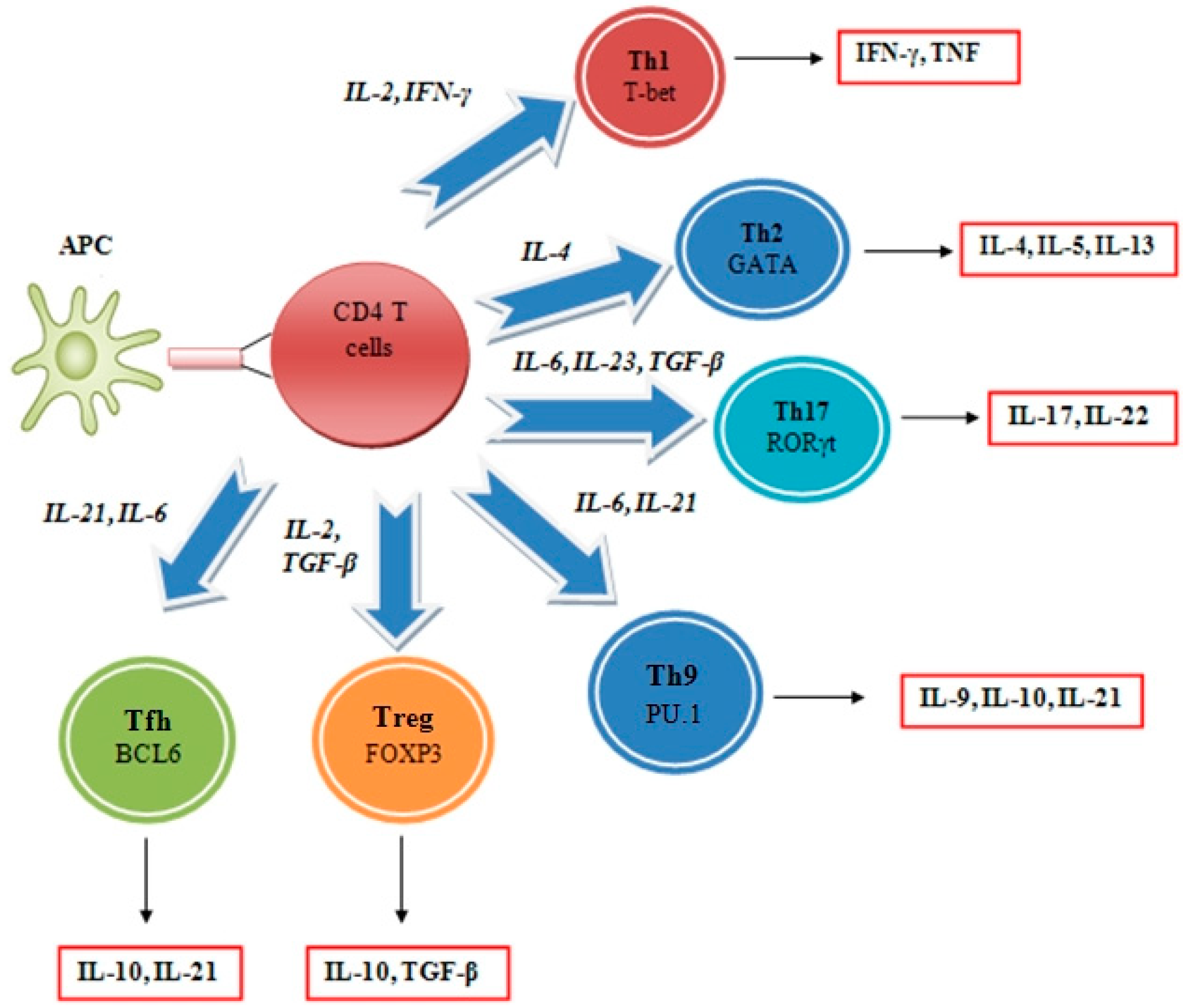
2.1. Function and Regulation

2.2. Interleukin Regulation of the Tumor Microenvironment
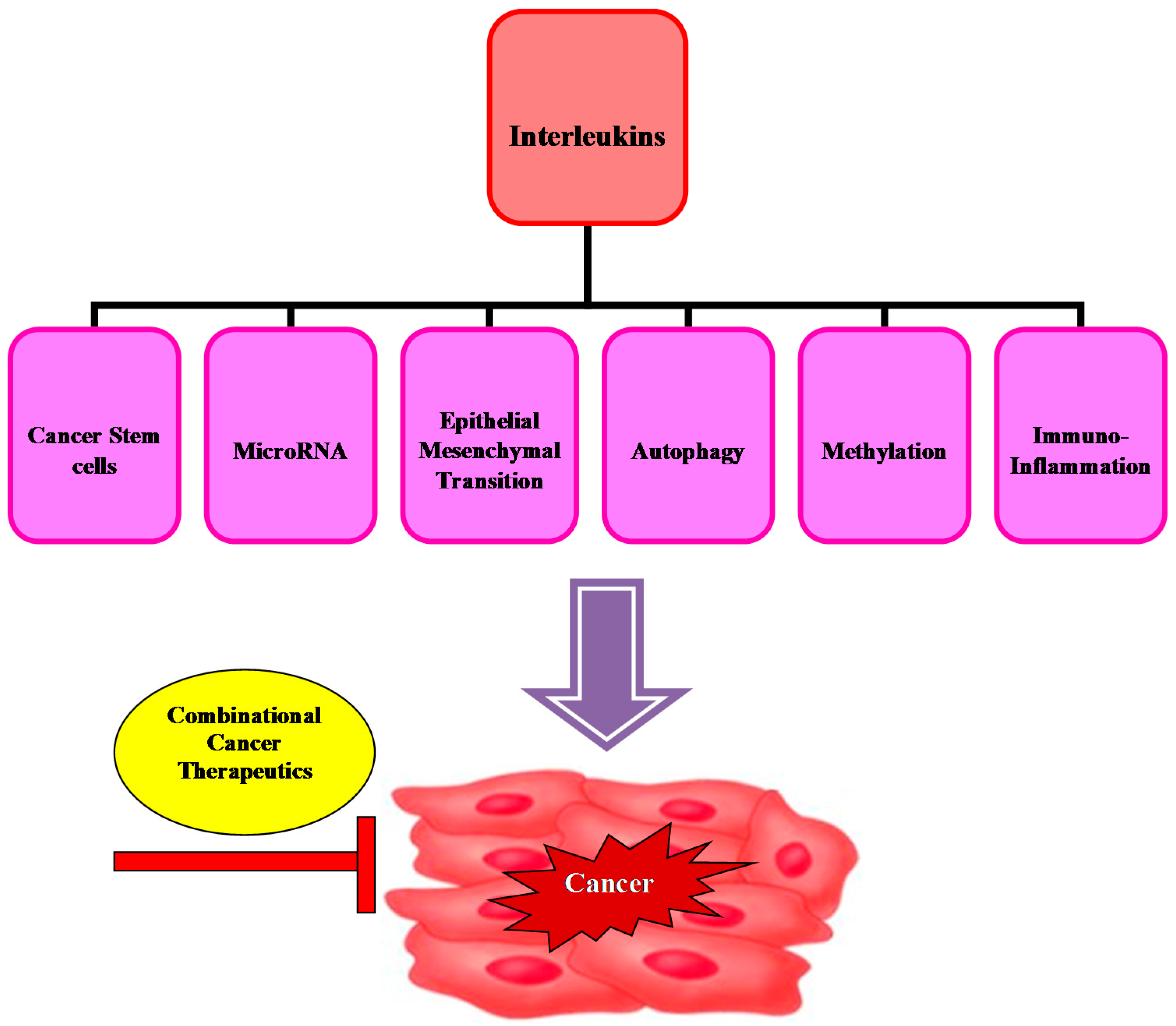
3. Interleukin Mechanisms Involved in Carcinogenesis
3.1. Crosstalk between Cancer Stem Cells and Cytokine Interleukins
3.2. Molecular Interplay of MicroRNA with Interleukins
3.3. Association of Epithelial Mesenchymal Transition with Interleukin Expression
3.4. Modulation of the Autophagic Machinery by Interleukins
3.5. Correlation between DNA Methylation and Interleukin Expression
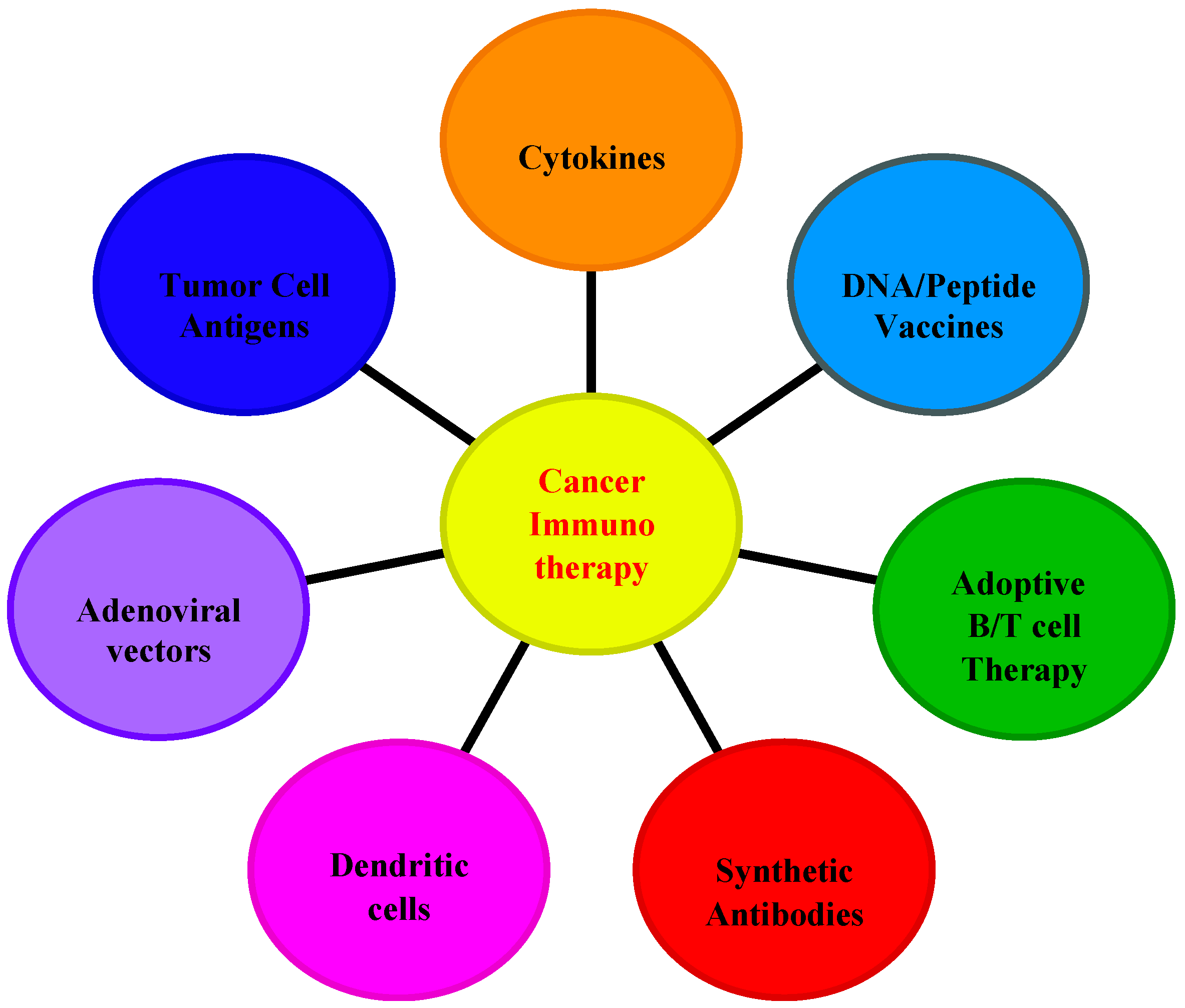
4. Applications of Interleukins in Cancer Immunotherapy
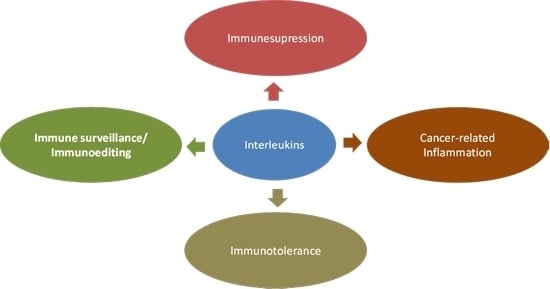
4.1. Interleukins in Cancer Immunoediting and Immunosurveillance

4.2. Linking Interleukin Expression and Inflammation-Driven Carcinogenesis
4.3. Strategies for IL-Based Cancer Immunotherapy
4.4. Combination Cancer Immunotherapy

5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yoshimoto, T.; Morishima, N.; Okumura, M.; Chiba, Y.; Xu, M.; Mizuguchi, J. Interleukins and cancer immunotherapy. Immunotherapy 2009, 5, 825–844. [Google Scholar] [CrossRef]
- Akdis, M.; Burgler, S.; Crameri, R.; Eiwegger, T.; Fujita, H.; Gomez, E.; Klunker, S.; Meyer, N.; O’Mahony, L.; Palomares, O.; et al. Interleukins, from 1 to 37, and interferon-γ: Receptors, functions, and roles in diseases. J. Allergy Clin. Immunol. 2011, 127, 701–721. [Google Scholar] [CrossRef] [PubMed]
- Tayal, V.; Kalra, B.S. Cytokines and anti-cytokines as therapeutics, an update. Eur. J. Pharmacol. 2008, 579, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Junttila, I.S.; Paul, W.E. Cytokine-induced cytokine production by conventional and innate lymphoid cells. Trends Immunol. 2012, 33, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, V.S. Interleukins: The search for an anticancer therapy. Semin. Oncol. Nurs. 1996, 12, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Rutz, S.; Ouyang, W. Regulation of interleukin-10 and interleukin-22 expression in T helper cells. Curr. Opin. Immunol. 2011, 23, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, A.; Im, S.H. Interleukin and interleukin receptor diversity: Role of alternative splicing. Int. Rev. Immunol. 2010, 29, 77–109. [Google Scholar] [CrossRef] [PubMed]
- Ngiow, S.F.; Teng, M.W.; Smyth, M.J. A balance of interleukin-12 and -23 in cancer. Trends Immunol. 2013, 34, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Yeung, Y.T.; McDonald, K.L.; Grewal, T.; Munoz, L. Interleukins in glioblastoma pathophysiology: Implications for therapy. Br. J. Pharmacol. 2013, 168, 591–606. [Google Scholar] [CrossRef] [PubMed]
- Christian, D.A.; Hunter, C.A. Particle-mediated delivery of cytokines for immunotherapy. Immunotherapy 2012, 4, 425–441. [Google Scholar] [CrossRef] [PubMed]
- Ramstead, A.G.; Jutila, M.A. Complex role of γδ T-cell-derived cytokines and growth factors in cancer. J. Interferon Cytokine Res. 2012, 32, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, A.; Kumar, Y. Cellular and molecular mechanisms in cancer immune escape: A comprehensive review. Expert Rev. Clin. Immunol. 2014, 10, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Rui, L.; Schmitz, R.; Ceribelli, M.; Staudt, L.M. Malignant pirates of the immune system. Nat. Immunol. 2011, 12, 933–934. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Poschke, I.; Kiessling, R. Tumour-induced immune suppression: Role of inflammatory mediators released by myelomonocytic cells. J. Intern. Med. 2014, 276, 154–170. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, A.A.; Watkins, S.K. Immune suppression in the tumor microenvironment: A role for dendritic cell-mediated tolerization of T cells. Cancer Immunol. Immunother. 2012, 61, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Stewart, T.J.; Smyth, M.J. Improving cancer immunotherapy by targeting tumor-induced immune suppression. Cancer Metastasis Rev. 2011, 30, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Arango, D.G.; Descoteaux, A. Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 2014, 7, 491. [Google Scholar]
- Brocker, C.; Thompson, D.; Matsumoto, A.; Nebert, D.W.; Vasiliou, V. Evolutionary divergence and functions of the human interleukin (IL) gene family. Hum. Genomics 2010, 5, 30–55. [Google Scholar] [CrossRef] [PubMed]
- Gadina, M.; Ferguson, P.R.; Johnston, J.A. New interleukins: Are there any more? Curr. Opin. Infect. Dis. 2003, 16, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Pascual, V.; O’Garra, A. From IL-2 to IL-37: The expanding spectrum of anti-inflammatory cytokines. Nat. Immunol. 2012, 13, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Pappu, R.; Ramirez-Carrozzi, V.; Sambandam, A. The interleukin-17 cytokine family: Critical players in host defence and inflammatory diseases. Immunology 2011, 134, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Ryungsa, K.; Manabu, E.; Kazuaki, T. Cancer immunoediting from immune surveillance to immune escape. Immunology 2007, 121, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, H.; Vesely, M.D.; Koboldt, D.C.; Rickert, C.G.; Uppaluri, R.; Magrini, V.J.; Arthur, C.D.; White, J.M.; Chen, Y.S.; Shea, L.K.; et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 2012, 482, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Gross, E.; Sunwoo, J.B.; Bui, J.D. Cancer immunosurveillance and immunoediting by natural killer cells. Cancer J. 2013, 19, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Oleinika, K.; Nibbs, R.J.; Graham, G.J.; Fraser, A.R. Suppression, subversion and escape: The role of regulatory T cells in cancer progression. Clin. Exp. Immunol. 2013, 171, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Amedei, A.; Prisco, D.; D’Elios, M.M. The use of cytokines and chemokines in the cancer immunotherapy. Recent Pat. Anticancer Drug Discov. 2013, 8, 126–142. [Google Scholar] [CrossRef] [PubMed]
- Introna, M.; Golay, J.; Rambaldi, A. Cytokine induced killer (CIK) cells for the treatment of haematological neoplasms. Immunol. Lett. 2013, 155, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kared, H.; Camous, X.; Larbi, A. T cells and their cytokines in persistent stimulation of the immune system. Curr. Opin. Immunol. 2014, 29, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.M.; Verma, N.D.; Tran, G.T.; Hodgkinson, S.J. Distinct regulatory CD4+ T cell subsets; Differences between naïve and antigen specific T regulatory cells. Curr. Opin. Immunol. 2011, 23, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Végran, F.; Berger, H.; Boidot, R.; Mignot, G.; Bruchard, M.; Dosset, M.; Chalmin, F.; Rébé, C.; Dérangère, V.; Ryffel, B.; et al. The transcription factor IRF1 dictates the IL-21-dependent anticancer functions of TH9 cells. Nat. Immunol. 2014, 15, 758–766. [Google Scholar] [CrossRef] [PubMed]
- Shale, M.; Schiering, C.; Powrie, F. CD4+ T-cell subsets in intestinal inflammation. Immunol. Rev. 2013, 252, 164–182. [Google Scholar] [CrossRef] [PubMed]
- Heikamp, E.B.; Powell, J.D. Sensing the immune microenvironment to coordinate T cell metabolism, differentiation and function. Semin. Immunol. 2012, 24, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Zarogoulidis, P.; Lampaki, S.; Yarmus, L.; Kioumis, I.; Pitsiou, G.; Katsikogiannis, N.; Hohenforst-Schmidt, W.; Li, Q.; Huang, H.; Sakkas, A.; et al. Interleukin-7 and interleukin-15 for cancer. J. Cancer 2014, 5, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Voronov, E.; Carmi, Y.; Apte, R.N. The role IL-1 in tumor-mediated angiogenesis. Front. Physiol. 2014, 5, 114. [Google Scholar] [CrossRef] [PubMed]
- Drexler, S.K.; Yazdi, A.S. Complex roles of inflammasomes in carcinogenesis. Cancer J. 2013, 19, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Mo, H.Y.; Xiong, G.; Zhang, L.; He, J.; Huang, Z.F.; Liu, Z.W.; Chen, Q.Y.; Du, Z.M.; Zheng, L.M.; et al. Tumor microenvironment macrophage inhibitory factor directs the accumulation of interleukin-17-producing tumor-infiltrating lymphocytes and predicts favorable survival in nasopharyngeal carcinoma patients. J. Biol. Chem. 2012, 287, 35484–35495. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Fan, P.; Yin, T.; Chen, Q.; Shi, H.; Liu, S.; Li, H.; Jing, Q.; Yan, Y.; Zhang, H.; et al. Local delivery of recombinant adenovirus expressing hepatitis B virus X protein and interleukin-12 results in antitumor effects via inhibition of hepatoma cell growth and intervention of tumor microenvironment. Int. J. Mol. Med. 2012, 30, 599–605. [Google Scholar] [PubMed]
- Qian, X.; Gu, L.; Ning, H.; Zhang, Y.; Hsueh, E.C.; Fu, M.; Hu, X.; Wei, L.; Hoft, D.F.; Liu, J. Increased Th17 cells in the tumor microenvironment is mediated by IL-23 via tumor-secreted prostaglandin E2. J. Immunol. 2013, 190, 5894–5902. [Google Scholar] [CrossRef] [PubMed]
- Hodge, L.S.; Ziesmer, S.C.; Yang, Z.Z.; Secreto, F.J.; Gertz, M.A.; Novak, A.J.; Ansell, S.M. IL-21 in the bone marrow microenvironment contributes to IgM secretion and proliferation of malignant cells in Waldenstrom macroglobulinemia. Blood 2012, 120, 3774–3782. [Google Scholar] [CrossRef] [PubMed]
- Kryczek, I.; Lin, Y.; Nagarsheth, N.; Peng, D.; Zhao, L.; Zhao, E.; Vatan, L.; Szeliga, W.; Dou, Y.; Owens, S.; et al. IL-22+CD4+ T cells promote colorectal cancer stemness via STAT3 transcription factor activation and induction of the methyltransferase DOT1L. Immunity 2014, 40, 772–784. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Yao, Q.; Liu, Y.; Du, S.; Liu, A.; Guo, Z.; Sun, A.; Ruan, J.; Chen, L.; Ye, C.; et al. IL-6-induced epithelial-mesenchymal transition promotes the generation of breast cancer stemlike cells analogous to mammosphere cultures. Int. J. Oncol. 2012, 40, 1171–1179. [Google Scholar] [PubMed]
- Krishnamurthy, S.; Warner, K.A.; Dong, Z.; Imai, A.; Nör, C.; Ward, B.B.; Helman, J.I.; Taichman, R.S.; Bellile, E.L.; McCauley, L.K.; et al. Endothelial interleukin-6 defines the tumorigenic potential of primary human cancer stem cells. Stem Cells 2014, 32, 2845–2857. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, L.; Pappan, L.; Galliher-Beckley, A.; Shi, J. IL-1β promotes stemness and invasiveness of colon cancer cells through Zeb1 activation. Mol. Cancer 2012, 11, 87. [Google Scholar] [CrossRef] [PubMed]
- Xiang, T.; Long, H.; He, L.; Han, X.; Lin, K.; Liang, Z.; Zhuo, W.; Xie, R.; Zhu, B. Interleukin-17 produced by tumor microenvironment promotes self-renewal of CD133+ cancer stem-like cells in ovarian cancer. Oncogene 2013. [CrossRef]
- Van Wolfswinkel, J.C.; Ketting, R.F. The role of small non-coding RNAs in genome stability and chromatin organization. J. Cell Sci. 2010, 123, 1825–1839. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, K.; Sivasankar, V. MicroRNAs-biology and clinical applications. J. Oral Maxillofac. Pathol. 2014, 18, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Jasinski-Bergner, S.; Mandelboim, O.; Seliger, B. The role of microRNAs in the control of innate immune response in cancer. J. Natl. Cancer Inst. 2014, 106, dju257. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Takeda, S. Regulation of bone metastasis by microRNAs. Clin. Calcium 2014, 24, 1209–1215. [Google Scholar] [PubMed]
- Kim, J.S.; Yu, S.K.; Lee, M.H.; Park, M.G.; Park, E.; Kim, S.G.; Lee, S.Y.; Kim, C.S.; Kim, H.J.; Chun, H.S.; et al. MicroRNA-205 directly regulates the tumor suppressor, interleukin-24, in human KB oral cancer cells. Mol. Cells 2013, 35, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Majid, S.; Dar, A.A.; Saini, S.; Yamamura, S.; Hirata, H.; Tanaka, Y.; Deng, G.; Dahiya, R. MicroRNA-205-directed transcriptional activation of tumor suppressor genes in prostate cancer. Cancer 2010, 116, 5637–5649. [Google Scholar] [CrossRef] [PubMed]
- Pollari, S.; Leivonen, S.K.; Perälä, M.; Fey, V.; Käkönen, S.M.; Kallioniemi, O. Identification of microRNAs inhibiting TGF-β-induced IL-11 production in bone metastatic breast cancer cells. PLoS One 2012, 7, e37361. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Liu, J.; Wang, Z.; Gu, X.; Fan, Y.; Zhang, W.; Xu, L.; Zhang, J.; Cai, D. NF-κB dependent microRNA-425 up-regulation promotes gastric cancer cell growth by targeting PTEN upon IL-1β induction. Mol. Cancer 2014, 26, 13–40. [Google Scholar]
- Park, S.J.; Cheon, E.J.; Lee, M.H.; Kim, H.A. MicroRNA-127–5p regulates matrix metalloproteinase 13 expression and interleukin-1β-induced catabolic effects in human chondrocytes. Arthritis Rheumatol. 2013, 65, 3141–3152. [Google Scholar] [CrossRef]
- Steinestel, K.; Eder, S.; Jan Schrader, A.; Steinestel, J. Clinical significance of epithelialmesenchymal transition. Clin. Transl. Med. 2014, 3, 17. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Li, J.; Xu, L.; Wang, W.; Luo, M.; Luo, S.; Ma, L.; Li, K.; Gong, S.; He, L.; et al. IL4 and IL-17A provide a Th2/Th17-polarized inflammatory milieu in favor of TGF-β1 to induce bronchial epithelial-mesenchymal transition (EMT). Int. J. Clin. Exp. Pathol. 2013, 6, 1481–1492. [Google Scholar] [PubMed]
- Desai, S.; Laskar, S.; Pandey, B.N. Autocrine IL-8 and VEGF mediate epithelial-mesenchymal transition and invasiveness via p38/JNK-ATF-2 signalling in A549 lung cancer cells. Cell Signal. 2013, 25, 1780–1791. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.W.; Liu, L.J.; Huang, J. Interleukin-6-induced epithelial-mesenchymal transition through signal transducer and activator of transcription 3 in human cervical carcinoma. Int. J. Oncol. 2014, 45, 165–176. [Google Scholar] [PubMed]
- Yadav, A.; Kumar, B.; Datta, J.; Teknos, T,N.; Kumar, P. IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Mol. Cancer Res. 2011, 9, 1658–1667. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Choi, S.Y.; Lee, J.H.; Lee, M.; Nam, E.S.; Jeong, A.L.; Lee, S.; Han, S.; Lee, M.S.; Lim, J.S.; et al. Interleukin-32β stimulates migration of MDA-MB-231 and MCF-7cells via the VEGF-STAT3 signaling pathway. Cell Oncol. 2013, 36, 493–503. [Google Scholar] [CrossRef]
- Chang, P.C.; Wang, T.Y.; Chang, Y.T.; Chu, C.Y.; Lee, C.L.; Hsu, H.W.; Zhou, T.A.; Wu, Z.; Kim, R.H.; Desai, S.J.; et al. Autophagy pathway is required for IL-6 induced neuroendocrine differentiation and chemoresistance of prostate cancer LNCaP cells. PLoS One 2014, 9, e88556. [Google Scholar] [CrossRef] [PubMed]
- Buchser, W.J.; Laskow, T.C.; Pavlik, P.J.; Lin, H.M.; Lotze, M.T. Cell-mediated autophagy promotes cancer cell survival. Cancer Res. 2012, 72, 2970–2979. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Tong, Y.; Ni, W.; Liu, J.; Xu, W.; Li, L.; Liu, X.; Meng, H.; Qian, W. Inhibition of autophagy induced by overexpression of mda-7/interleukin-24 strongly augments the antileukemia activity in vitro and in vivo. Cancer Gene Ther. 2010, 17, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Bai, B.; Sha, S.; Yu, P.; An, Y.; Wang, S.; Kong, X.; Liu, C.; Wei, N.; Feng, Q.; et al. Interleukin-1β induces autophagy by affecting calcium homeostasis and trypsinogen activation in pancreatic acinar cells. Int. J. Clin. Exp. Pathol. 2014, 7, 3620–3623. [Google Scholar] [PubMed]
- Liang, X.; de Vera, M.E.; Buchser, W.J.; Romo de Vivar Chavez, A.; Loughran, P.; Beer Stolz, D.; Basse, P.; Wang, T.; van Houten, B.; Zeh, H.J.; et al. Inhibiting systemic autophagy during interleukin 2 immunotherapy promotes long-term tumor regression. Cancer Res. 2012, 72, 2791–2801. [Google Scholar] [CrossRef] [PubMed]
- Tekpli, X.; Landvik, N.E.; Anmarkud, K.H.; Skaug, V.; Haugen, A.; Zienolddiny, S. DNA methylation at promoter regions of interleukin 1B, interleukin 6, and interleukin 8 in non-small cell lung cancer. Cancer Immunol. Immunother. 2013, 62, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Gasche, J.A.; Hoffmann, J.; Boland, C.R.; Goel, A. Interleukin-6 promotes tumorigenesis by altering DNA methylation in oral cancer cells. Int. J. Cancer 2011, 129, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.A.; Bhambra, U.; Charalambous, M.P.; David, R.M.; Edwards, R.J.; Lightfoot, T.; Boobis, A.R.; Gooderham, N.J. Interleukin-6 mediated up-regulation of CYP1B1 and CYP2E1 in colorectal cancer involves DNA methylation, miR27b and STAT3. Br. J. Cancer 2014, 111, 2287–2296. [Google Scholar] [CrossRef] [PubMed]
- Baird, A.M.; Gray, S.G.; O’Byrne, K.J. IL-20 is epigenetically regulated in NSCLC and down regulates the expression of VEGF. Eur. J. Cancer 2011, 47, 1908–1918. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The three Es of cancer immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, T.; Saddawi-Konefka, R.; Vermi, W.; Koebel, C.M.; Arthur, C.; White, J.M.; Uppaluri, R.; Andrews, D.M.; Ngiow, S.F.; Teng, M.W.; et al. Cancer immunoediting by the innate immune system in the absence of adaptive immunity. J. Exp. Med. 2012, 209, 1869–1882. [Google Scholar] [CrossRef] [PubMed]
- O'Sullivan, T.; Saddawi-Konefka, R.; Gross, E.; Tran, M.; Mayfield, S.P.; Ikeda, H.; Bui, J.D. Interleukin-17D mediates tumor rejection through recruitment of natural killer cells. Cell Rep. 2014, 7, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Jauch, D.; Martin, M.; Schiechl, G.; Kesselring, R.; Schlitt, H.J.; Geissler, E.K.; Fichtner-Feigl, S. Interleukin 21 controls tumour growth and tumour immunosurveillance in colitis-associated tumorigenesis in mice. Gut 2011, 60, 1678–1686. [Google Scholar] [CrossRef] [PubMed]
- Volonté, A.; di Tomaso, T.; Spinelli, M.; Todaro, M.; Sanvito, F.; Albarello, L.; Bissolati, M.; Ghirardelli, L.; Orsenigo, E.; Ferrone, S.; et al. Cancer-initiating cells from colorectal cancer patients escape from T cell-mediated immunosurveillance in vitro through membrane-bound IL-4. J. Immunol. 2014, 192, 523–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, H.-C.; Velichko, S.; Hung, L.-Y.; Wu, R. IL-17A and Th17 cells in lung inflammation: An update on the role of Th17 cell differentiation and IL-17R signaling in host defense against infection. Clin. Dev. Immunol. 2013, 2013, 267971. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Kang, H.; Fung, A.; Zhao, H.; Wang, T.; Ma, D. The role of interleukin 17 in tumour proliferation, angiogenesis, and metastasis. Mediat. Inflamm. 2014, 2014, 623759. [Google Scholar]
- Hayata, K.; Iwahashi, M.; Ojima, T.; Katsuda, M.; Iida, T.; Nakamori, M.; Ueda, K.; Nakamura, M.; Miyazawa, M.; Tsuji, T.; et al. Inhibition of IL-17A in tumor microenvironment augments cytotoxicity of tumor-infiltrating lymphocytes in tumor-bearing mice. PLoS One 2013, 8, e53131. [Google Scholar] [CrossRef] [PubMed]
- Chae, W.J.; Bothwell, A.L. IL-17F deficiency inhibits small intestinal tumorigenesis in ApcMin/+ mice. Biochem. Biophys. Res. Commun. 2011, 414, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Girardin, A.; McCall, J.; Black, M.A.; Edwards, F.; Phillips, V.; Taylor, E.S.; Reeve, A.E.; Kemp, R.A. Inflammatory and regulatory T cells contribute to a unique immune microenvironment in tumor tissue of colorectal cancer patients. Int. J. Cancer 2013, 132, 1842–1850. [Google Scholar] [CrossRef] [PubMed]
- Petanidis, S.; Anestakis, D.; Argyraki, M.; Hadzopoulou-Cladaras, M.; Salifoglou, A. Differential expression of IL-17, 22 and 23 in the progression of colorectal cancer in patients with K-Ras mutation: Ras signal inhibition and crosstalk with GM-CSF and IFN-γ. PLoS One 2013, 8, e73616. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, L.; Xu, W.; Qian, H.; Ye, S.; Zhu, W.; Cao, H.; Yan, Y.; Li, W.; Wang, M.; et al. Experimental therapy for lung cancer: Umbilical cord-derived mesenchymal stem cell-mediated interleukin-24 delivery. Curr. Cancer Drug Targets 2013, 13, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Oshima, T.; Imada, T.; Masuda, M.; Debnath, B.; Grande, F.; Garofalo, A.; Neamati, N. Stabilization of MDA-7/IL-24 for colon cancer therapy. Cancer Lett. 2013, 335, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.L.; Yu, Y.H.; Tian, H.; Ren, G.P.; Wang, H.; Zhou, B.; Han, X.H.; Yu, Q.Z.; Li, D.S. Genetically engineered Newcastle disease virus expressing interleukin-2 and TNF-related apoptosis-inducing ligand for cancer therapy. Cancer Biol. Ther. 2014, 15, 1226–1238. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.; Kim, C.S.; Kim, S.B.; Kim, Y.M.; Kwon, S.W.; Kim, Y.; Kim, H.; Lee, H. Combination therapy of renal cell carcinoma or breast cancer patients with dendritic cell vaccine and IL-2: Results from a phase I/II trial. J. Transl. Med. 2011, 9, 178. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Zhao, H.; Zhang, L.; Liu, X. The efficacy of combination therapy using adenoassociated virus-mediated co-expression of apoptin and interleukin-24 on hepatocellular carcinoma. Tumour Biol. 2013, 34, 3027–3034. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Li, X.; He, X.; Qiu, Y.; Zhu, L.; Qi, F. Interleukin-15 gene therapy and the mammalian target of rapamycin inhibitor everolimus inhibit the growth of metastatic breast cancer. J. Gene Med. 2013, 15, 366–367. [Google Scholar] [CrossRef] [PubMed]
- Denies, S.; Cicchelero, L.; van Audenhove, I.; Sanders, N.N. Combination of interleukin-12 gene therapy, metronomic cyclophosphamide and DNA cancer vaccination directs all arms of the immune system towards tumor eradication. J. Control. Release 2014, 187, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Newman, R.G.; Dee, M.J.; Malek, T.R.; Podack, ER.; Levy, R.B. Heat shock protein vaccination and directed IL-2 therapy amplify tumor immunity rapidly following bone marrow transplantation in mice. Blood 2014, 123, 3045–3055. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anestakis, D.; Petanidis, S.; Kalyvas, S.; Nday, C.M.; Tsave, O.; Kioseoglou, E.; Salifoglou, A. Mechanisms and Αpplications of Ιnterleukins in Cancer Immunotherapy. Int. J. Mol. Sci. 2015, 16, 1691-1710. https://doi.org/10.3390/ijms16011691
Anestakis D, Petanidis S, Kalyvas S, Nday CM, Tsave O, Kioseoglou E, Salifoglou A. Mechanisms and Αpplications of Ιnterleukins in Cancer Immunotherapy. International Journal of Molecular Sciences. 2015; 16(1):1691-1710. https://doi.org/10.3390/ijms16011691
Chicago/Turabian StyleAnestakis, Doxakis, Savvas Petanidis, Spyridon Kalyvas, Christiane M. Nday, Olga Tsave, Efrosini Kioseoglou, and Athanasios Salifoglou. 2015. "Mechanisms and Αpplications of Ιnterleukins in Cancer Immunotherapy" International Journal of Molecular Sciences 16, no. 1: 1691-1710. https://doi.org/10.3390/ijms16011691