
Ligand-Induced Dynamics of Neurotrophin Receptors Investigated by Single-Molecule Imaging Approaches

 ,
,
Abstract
:1. Introduction

2. Advanced Live-Cell Imaging Approaches for the Detection of Molecular Interactions and Single-Molecule Dynamics
3. Advanced Imaging Approaches Applied to the Study of NR Dynamics, Trafficking and Signaling

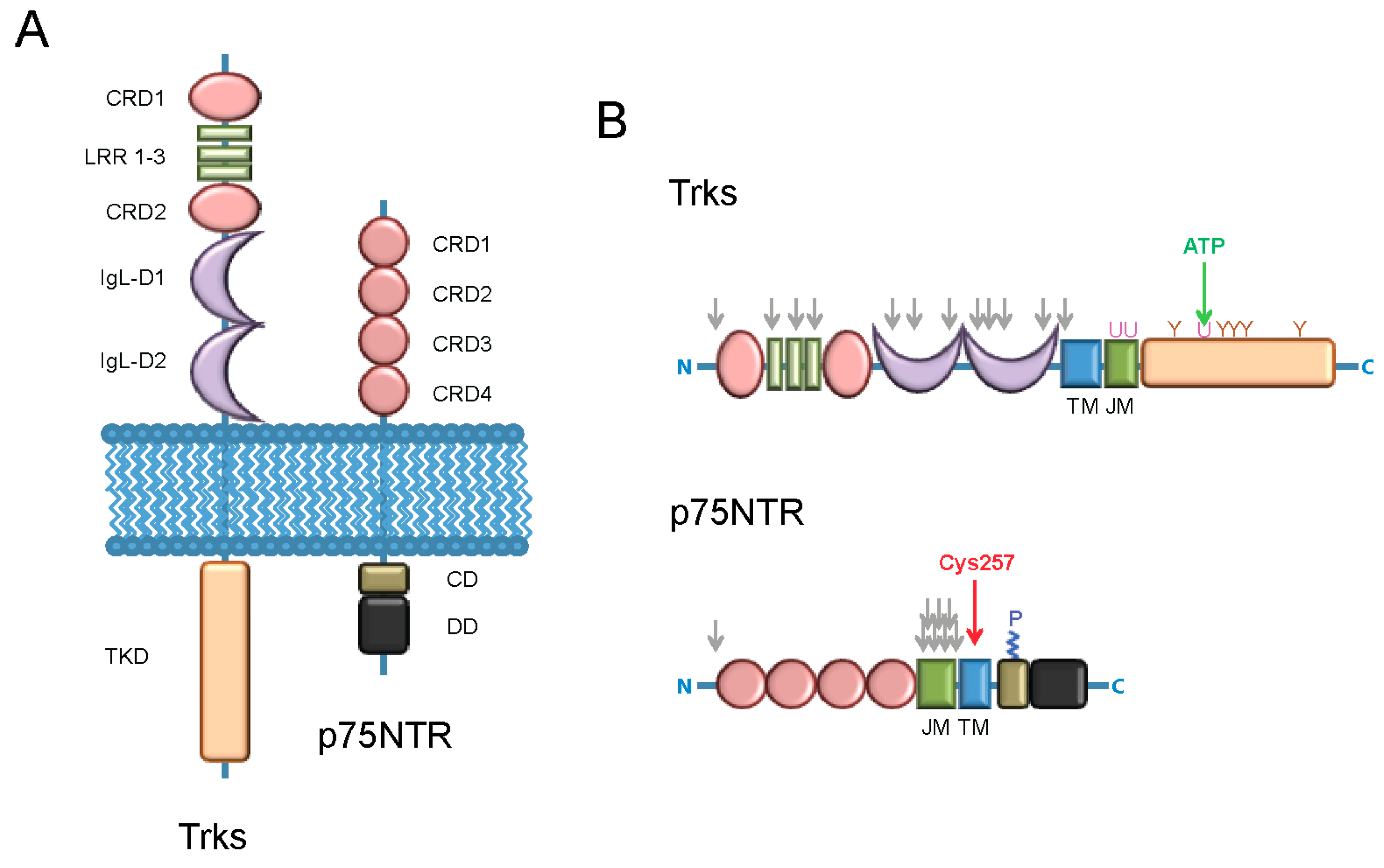{kind=link}
{kind=link}
| Membrane Protein | Host Cell | Labeling Strategy | Applied Trajectory Analysis | Biological Information Extracted | References |
|---|---|---|---|---|---|
| EGFR | CHO | Fab conjugated to Qdot | CSD-HMM | The EGFR monomer-dimer equilibrium is characterized: The receptor is found to exist in both states in resting conditions, with dimers primed for ligand binding and signaling. | [88] |
| CD36 | Macrophages | Fab conjugated to Cy3 or Qdot | MSD; Anisotropy of the scatter of particle positions; MSS | A subpopulation of receptors is identified, characterized by diffusion within linear confinement regions governed by cytoskeleton, whose peculiar geometry is found to promote receptor clustering as well as ligand-induced signaling and internalization. | [89] |
| FPR | CHO | Peptide ligand conjugated to Alexa594 | Analysis of spot intensities | The GPCR monomer-dimer equilibrium is characterized with quantitative details: the two-dimensional monomer-dimer equilibrium constant as well as the association and dissociation rate constants are computed. | [92] |
| AMPA receptors (with GluR2) | Primary hippocampal neurons from rat embryos | 0.5-μm latex beads coated with antibodies against GluR2 | MSD; TCZ; distance of trajectories from stained synaptic sites and endocytic pits | During maturation or when raising intracellular calcium, the equilibrium between fast diffusion and stationary behavior of AMPA receptors is more and more shifted to the second state. Stationary zones often correlates with synaptic sites but not with clathrin-coated pits targeted by Eps15. It is suggested that diffusion can rapidly regulate receptor numbers at synapses. | [108] |
| GlyR | Primary spinal cord neurons from Sprague Dawley rat embryos | Biotinylated Fab against GlyR 1 coupled to Streptavidin coated Qdots | MSD fitting for Brownian and confined trajectories; reconstruction of trajectories from Qdot-blinking-induced shorter ones | Cytoskeleton regulates the localization of GlyR and of gephyrin, the core scaffolding protein of inhibitory post-synaptic differentiation. Microtubules control GlyR lateral diffusion in the extra-synaptic membrane, actin at the synapses. | [109] |
| TrkA | SH-SY5Y | ACP chemical tag conjugated to Qdot or Atto633 | Analysis of combined distributions of parameters computed by MSD, MSS, and TCZ, and spot intensities analysis applied to trajectories and subtrajectories | The interplay between TrkA oligomerization states, local diffusivity and degree of anomalous diffusion is investigated as a function of the binding of different ligands, showing that each ligand promotes distinct TrkA trajectory patterns (“ligand fingerprinting effect”, see Section 3.1). | [104] |
| β1-AR, β2-AR, GABAB receptor | CHO | SNAP chemical tag conjugated to Alexa647 | MSD; Analysis of spot intensities | Three different GPCRs are found to exist at very different degrees of oligomerization (monomer-dimer for β1-AR and β2-AR, and dimer-tetramer for GABAB). The lifetime of such oligomeric states depend on receptor density but not on agonist stimulation. | [106] |
| CD59 | T24, Ptk2, NRK | Fab/IgG-gold, IgG-Cy3 or IgG-latex beads conjugates | TCZ | CD59 clusters were shown to undergo periods of actin-driven, stimulation-induced transient arrest of lateral diffusion (STALL), i.e., short-lived immobilization events throughout receptor trajectories. STALL events were correlated to the signaling of the receptor, via recruitment of activated PLCγ effector and subsequent IP3-Ca2+ spike under the STALL area. | [110] |
| AMPA receptors (with GluR2) | Primary hippocampal neurons from rat embryos | 0.5-μm latex beads coated with antibodies against GluR2 | MSD; TCZ; distance of trajectories from stained synaptic sites and endocytic pits | During maturation or when raising intracellular calcium, the equilibrium between fast diffusion and stationary behavior of AMPA receptors is more and more shifted to the second state. Stationary zones often correlates with synaptic sites but not with clathrin-coated pits targeted by Eps15. It is suggested that diffusion can rapidly regulate receptor numbers at synapses. | [108] |
3.1. Trk Receptors
| NGF Labeling Strategy | Ratio of Probes Per NGF Molecule | Cellular Model Testing NGF Activity | References |
|---|---|---|---|
| 125I-NGF | n.d. | Sympathetic neurons | [115] |
| Biotinylated NGF (NGF-b) | ~3 biotin molecules per NGF subunit | PC12 | [119] |
| NGF-b/Streptavidin Alexa647 | ~9 biotins per NGF molecule; 20 nM NGF-b is given to the cells and further detected with Streptavidin Alexa647 | PC12 and PC12nnr5 | [120] |
| NGF-b/streptavidin Qdot | ≤3 biotins per NGF subunit; NGF-b is conjugated to streptavidin-Qdot at a molar ratio of 1 NGF:1 Qdot | PC12 | [121,122] |
| ~1 biotin was bound per NGF molecule; 2nM NGF-b is given to the cells and further detected with 50–500 pM streptavidin-coated Qdot | Differentiated PC12 | [123] | |
| ~3 biotins per NGF dimer; NGF-b is conjugated to streptavidin-Qdot at a molar ratio of 1 NGF:1 Qdot | Rat DRG neurons | [83] | |
| Cy3-NGF or Cy3.5-NGF | ~1.0–1.1 ratio between fluorophore and NGF | Chick embryonic DRG neurons; PC12 | [124,125,126] |
| Mono-biotinylated NGF via chemical tag | AVI-tag fused at NGF C-terminus; NGF-b is obtained transfecting the construct in HEK293FT cells together with the biotinylating BirA enzyme; NGF-b is conjugated to streptavidin-Qdot at a molar ratio of 1 NGF:1 QD | Rat DRG neurons | [127] |
| A4-tag fused at NGF C-terminus; NGF-b is obtained by labeling the purified protein with CoenzymeA-biotin substrates and PPTase enzyme | PC12 | [128] |

3.2. p75NTR
4. Conclusions and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lewin, G.R.; Carter, B.D. Neurotrophic Factors; Springer: Berlin-Heidelberg, Germany, 2014. [Google Scholar]
- Skaper, S.D. Neuarotrophic Factors: Methods and Protocols; Humana Press: Totowa, NJ, USA, 2012. [Google Scholar]
- Shooter, E.M. Early days of the nerve growth factor proteins. Annu. Rev. Neurosci. 2001, 24, 601–629. [Google Scholar] [CrossRef] [PubMed]
- Greene, L.A.; Shooter, E.M. The nerve growth factor: Biochemistry, synthesis, and mechanism of action. Annu. Rev. Neurosci. 1980, 3, 353–402. [Google Scholar] [CrossRef] [PubMed]
- Teng, K.K.; Felice, S.; Kim, T.; Hempstead, B.L. Understanding proneurotrophin actions: Recent advances and challenges. Dev. Neurobiol. 2012, 70, 350–359. [Google Scholar]
- Bruno, M.A.; Cuello, A.C. Activity-dependent release of precursor nerve growth factor, conversion to mature nerve growth factor, and its degradation by a protease cascade. Proc. Natl. Acad. Sci. USA 2006, 103, 6735–6740. [Google Scholar] [CrossRef] [PubMed]
- Skaper, S.D. The neurotrophin family of neurotrophic factors: An overview. Methods Mol. Biol. 2012, 846, 1–12. [Google Scholar] [PubMed]
- Ginty, D.D.; Segal, R.A. Retrograde neurotrophin signaling: Trk-ing along the axon. Curr. Opin. Neurobiol. 2002, 12, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Bothwell, M. Functional interactions of neurotrophins and neurotrophin receptors. Annu Rev. Neurosci. 1995, 18, 223–253. [Google Scholar] [CrossRef] [PubMed]
- Bronfman, F.C.; Lazo, O.M.; Flores, C.; Escudero, C.A. Spatiotemporal intracellular dynamics of neurotrophin and its receptors: Implications for neurotrophin signaling and neuronal function. Handb. Exp. Pharmacol. 2014, 220, 33–65. [Google Scholar] [PubMed]
- Matusica, D.; Coulson, E.J. Local versus long-range neurotrophin receptor signalling: Endosomes are not just carriers for axonal transport. Semin. Cell Dev. Biol. 2014, 31C, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Harrington, A.W.; Ginty, D.D. Long-distance retrograde neurotrophic factor signalling in neurons. Nat. Rev. Neurosci. 2013, 14, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Zweifel, L.S.; Kuruvilla, R.; Ginty, D.D. Functions and mechanisms of retrograde neurotrophin signalling. Nat. Rev. Neurosci. 2005, 6, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Yano, H.; Chao, M.V. Mechanisms of neurotrophin receptor vesicular transport. J. Neurobiol. 2004, 58, 244–257. [Google Scholar] [CrossRef] [PubMed]
- Chowdary, P.D.; Che, D.L.; Cui, B. Neurotrophin signaling via long-distance axonal transport. Annu. Rev. Phys. Chem. 2012, 63, 571–594. [Google Scholar] [CrossRef] [PubMed]
- Levi-Montalcini, R. The nerve growth factor 35 years later. Science 1987, 237, 1154–1162. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.; Cattaneo, A.; Mobley, W. Rita Levi-Montalcini: The story of an uncommon intellect and spirit. Neuroscience 2013, 252, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Hendry, I.A.; Stach, R.; Herrup, K. Characteristics of the retrograde axonal transport system for nerve growth factor in the sympathetic nervous system. Brain Res. 1974, 82, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Hendry, I.A.; Stockel, K.; Thoenen, H.; Iversen, L.L. The retrograde axonal transport of nerve growth factor. Brain Res. 1974, 68, 103–121. [Google Scholar] [CrossRef] [PubMed]
- Simi, A.; Ibanez, C.F. Assembly and activation of neurotrophic factor receptor complexes. Dev. Neurobiol. 2012, 70, 323–331. [Google Scholar]
- Schecterson, L.C.; Bothwell, M. Neurotrophin receptors: Old friends with new partners. Dev. Neurobiol. 2009, 70, 332–338. [Google Scholar]
- Reichardt, L.F. Neurotrophin-regulated signalling pathways. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2006, 361, 1545–1564. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.V. Neurotrophins and their receptors: A convergence point for many signalling pathways. Nat. Rev. Neurosci. 2003, 4, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Teng, K.K.; Hempstead, B.L. Neurotrophins and their receptors: Signaling trios in complex biological systems. Cell. Mol. Life Sci. 2004, 61, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Skeldal, S.; Matusica, D.; Nykjaer, A.; Coulson, E.J. Proteolytic processing of the p75 neurotrophin receptor: A prerequisite for signalling?: Neuronal life, growth and death signalling are crucially regulated by intra-membrane proteolysis and trafficking of p75(NTR). Bioessays 2011, 33, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Longo, F.M.; Massa, S.M. Small-molecule modulation of neurotrophin receptors: A strategy for the treatment of neurological disease. Nat. Rev. Drug Discov. 2013, 12, 507–525. [Google Scholar] [CrossRef] [PubMed]
- Casaletto, J.B.; McClatchey, A.I. Spatial regulation of receptor tyrosine kinases in development and cancer. Nat. Rev. Cancer 2012, 12, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Roux, P.P.; Barker, P.A. Neurotrophin signaling through the p75 neurotrophin receptor. Prog. Neurobiol. 2002, 67, 203–233. [Google Scholar] [CrossRef] [PubMed]
- Underwood, C.K.; Coulson, E.J. The p75 neurotrophin receptor. Int. J. Biochem. Cell Biol. 2008, 40, 1664–1668. [Google Scholar] [CrossRef] [PubMed]
- Enokido, Y.; Wyatt, S.; Davies, A.M. Developmental changes in the response of trigeminal neurons to neurotrophins: Influence of birthdate and the ganglion environment. Development 1999, 126, 4365–4373. [Google Scholar] [PubMed]
- Gavazzi, I.; Kumar, R.D.; McMahon, S.B.; Cohen, J. Growth responses of different subpopulations of adult sensory neurons to neurotrophic factors in vitro. Eur. J. Neurosci. 1999, 11, 3405–3414. [Google Scholar] [CrossRef] [PubMed]
- Vilar, M.; Charalampopoulos, I.; Kenchappa, R.S.; Reversi, A.; Klos-Applequist, J.M.; Karaca, E.; Simi, A.; Spuch, C.; Choi, S.; Friedman, W.J.; et al. Ligand-independent signaling by disulfide-crosslinked dimers of the p75 neurotrophin receptor. J. Cell Sci. 2009, 122, 3351–3357. [Google Scholar] [CrossRef] [PubMed]
- Vilar, M.; Charalampopoulos, I.; Kenchappa, R.S.; Simi, A.; Karaca, E.; Reversi, A.; Choi, S.; Bothwell, M.; Mingarro, I.; Friedman, W.J.; et al. Activation of the p75 neurotrophin receptor through conformational rearrangement of disulphide-linked receptor dimers. Neuron 2009, 62, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Arevalo, J.C.; Waite, J.; Rajagopal, R.; Beyna, M.; Chen, Z.Y.; Lee, F.S.; Chao, M.V. Cell survival through Trk neurotrophin receptors is differentially regulated by ubiquitination. Neuron 2006, 50, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Geetha, T.; Jiang, J.; Wooten, M.W. Lysine 63 polyubiquitination of the nerve growth factor receptor TrkA directs internalization and signaling. Mol. Cell 2005, 20, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Makkerh, J.P.; Ceni, C.; Auld, D.S.; Vaillancourt, F.; Dorval, G.; Barker, P.A. p75 neurotrophin receptor reduces ligand-induced Trk receptor ubiquitination and delays Trk receptor internalization and degradation. EMBO Rep. 2005, 6, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Kiris, E.; Wang, T.; Yanpallewar, S.; Dorsey, S.G.; Becker, J.; Bavari, S.; Palko, M.E.; Coppola, V.; Tessarollo, L. TrkA in vivo function is negatively regulated by ubiquitination. J. Neurosci. 2014, 34, 4090–4098. [Google Scholar] [CrossRef] [PubMed]
- Underwood, C.K.; Reid, K.; May, L.M.; Bartlett, P.F.; Coulson, E.J. Palmitoylation of the C-terminal fragment of p75(NTR) regulates death signaling and is required for subsequent cleavage by γ-secretase. Mol. Cell. Neurosci. 2008, 37, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Hempstead, B.L.; Martin-Zanca, D.; Kaplan, D.R.; Parada, L.F.; Chao, M.V. High-affinity NGF binding requires coexpression of the Trk proto-oncogene and the low-affinity NGF receptor. Nature 1991, 350, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Verdi, J.M.; Birren, S.J.; Ibanez, C.F.; Persson, H.; Kaplan, D.R.; Benedetti, M.; Chao, M.V.; Anderson, D.J. p75NGFR regulates Trk signal transduction and NGF-induced neuronal differentiation in MAH cells. Neuron 1994, 12, 733–745. [Google Scholar] [CrossRef] [PubMed]
- Frade, J.M.; Rodriguez-Tebar, A.; Barde, Y.A. Induction of cell death by endogenous nerve growth factor through its p75 receptor. Nature 1996, 383, 166–168. [Google Scholar] [CrossRef] [PubMed]
- Casaccia-Bonnefil, P.; Carter, B.D.; Dobrowsky, R.T.; Chao, M.V. Death of oligodendrocytes mediated by the interaction of nerve growth factor with its receptor p75. Nature 1996, 383, 716–719. [Google Scholar] [CrossRef] [PubMed]
- Barrett, G.L.; Bartlett, P.F. The p75 nerve growth factor receptor mediates survival or death depending on the stage of sensory neuron development. Proc. Natl. Acad. Sci. USA 1994, 91, 6501–6505. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, S.; Davies, A.M. Regulation of expression of mRNAs encoding the nerve growth factor receptors p75 and TrkA in developing sensory neurons. Development 1993, 119, 635–648. [Google Scholar] [PubMed]
- Yoon, S.O.; Casaccia-Bonnefil, P.; Carter, B.; Chao, M.V. Competitive signaling between TrkA and p75 nerve growth factor receptors determines cell survival. J. Neurosci. 1998, 18, 3273–3281. [Google Scholar] [PubMed]
- Esposito, D.; Patel, P.; Stephens, R.M.; Perez, P.; Chao, M.V.; Kaplan, D.R.; Hempstead, B.L. The cytoplasmic and transmembrane domains of the p75 and TrkA receptors regulate high affinity binding to nerve growth factor. J. Biol. Chem. 2001, 276, 32687–32695. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Cao, P.; Yu, H.J.; Jiang, T. Crystal structure of the neurotrophin-3 and p75NTR symmetrical complex. Nature 2008, 454, 789–793. [Google Scholar] [PubMed]
- He, X.L.; Garcia, K.C. Structure of nerve growth factor complexed with the shared neurotrophin receptor p75. Science 2004, 304, 870–875. [Google Scholar] [CrossRef] [PubMed]
- Wehrman, T.; He, X.; Raab, B.; Dukipatti, A.; Blau, H.; Garcia, K.C. Structural and mechanistic insights into nerve growth factor interactions with the TrkA and p75 receptors. Neuron 2007, 53, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Wiesmann, C.; Ultsch, M.H.; Bass, S.H.; de Vos, A.M. Crystal structure of nerve growth factor in complex with the ligand-binding domain of the TrkA receptor. Nature 1999, 401, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Barker, P.A. High affinity not in the vicinity? Neuron 2007, 53, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Matusica, D.; Skeldal, S.; Sykes, A.M.; Palstra, N.; Sharma, A.; Coulson, E.J. An intracellular domain fragment of the p75 neurotrophin receptor (p75(NTR)) enhances tropomyosin receptor kinase A (TrkA) receptor function. J. Biol. Chem. 2013, 288, 11144–11154. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.; Kermani, P.; Teng, K.K.; Hempstead, B.L. Regulation of cell survival by secreted proneurotrophins. Science 2001, 294, 1945–1948. [Google Scholar] [CrossRef] [PubMed]
- Nykjaer, A.; Lee, R.; Teng, K.K.; Jansen, P.; Madsen, P.; Nielsen, M.S.; Jacobsen, C.; Kliemannel, M.; Schwarz, E.; Willnow, T.E.; et al. Sortilin is essential for proNGF-induced neuronal cell death. Nature 2004, 427, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Kim, T.; Ozkan, E.; Light, M.; Torkin, R.; Teng, K.K.; Hempstead, B.L.; Garcia, K.C. Molecular and structural insight into proNGF engagement of p75NTR and sortilin. J. Mol. Biol. 2010, 396, 967–984. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Cornish, V.W.; Min, W. Chemical tags: Inspiration for advanced imaging techniques. Curr. Opin. Chem. Biol. 2013, 17, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Suarez, M.; Ting, A.Y. Fluorescent probes for super-resolution imaging in living cells. Nat. Rev. Mol. Cell Biol. 2008, 9, 929–943. [Google Scholar] [CrossRef] [PubMed]
- Hinner, M.J.; Johnsson, K. How to obtain labeled proteins and what to do with them. Curr. Opin. Biotechnol. 2010, 21, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Pierobon, P.; Cappello, G. Quantum dots to tail single bio-molecules inside living cells. Adv. Drug Deliv. Rev. 2012, 64, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Tsien, R.Y. Building and breeding molecules to spy on cells and tumors. FEBS Lett. 2005, 579, 927–932. [Google Scholar] [CrossRef] [PubMed]
- Wombacher, R.; Cornish, V.W. Chemical tags: Applications in live cell fluorescence imaging. J. Biophotonics 2011, 4, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Cella Zanacchi, F.; Lavagnino, Z.; Perrone Donnorso, M.; del Bue, A.; Furia, L.; Faretta, M.; Diaspro, A. Live-cell 3D super-resolution imaging in thick biological samples. Nat. Methods 2011, 8, 1047–1049. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Saez, A.J.; Schwille, P. Single molecule techniques for the study of membrane proteins. Appl. Microbiol. Biotechnol. 2007, 76, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Hell, S.W. Toward fluorescence nanoscopy. Nat. Biotechnol. 2003, 21, 1347–1355. [Google Scholar] [CrossRef] [PubMed]
- Kusumi, A.; Tsunoyama, T.A.; Hirosawa, K.M.; Kasai, R.S.; Fujiwara, T.K. Tracking single molecules at work in living cells. Nat. Chem. Biol. 2014, 10, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Levi, V.; Gratton, E. Exploring dynamics in living cells by tracking single particles. Cell Biochem. Biophys. 2007, 48, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ntziachristos, V. Fluorescence molecular imaging. Annu. Rev. Biomed. Eng. 2006, 8, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Elangovan, M.; Day, R.N.; Periasamy, A. Nanosecond fluorescence resonance energy transfer-fluorescence lifetime imaging microscopy to localize the protein interactions in a single living cell. J. Microsc. 2002, 205, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa-Ankerhold, H.C.; Ankerhold, R.; Drummen, G.P. Advanced fluorescence microscopy techniques–FRAP, FLIP, FLAP, FRET and FLIM. Molecules 2012, 17, 4047–4132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekar, R.B.; Periasamy, A. Fluorescence resonance energy transfer (FRET) microscopy imaging of live cell protein localizations. J. Cell Biol. 2003, 160, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Wallrabe, H.; Periasamy, A. Imaging protein molecules using FRET and FLIM microscopy. Curr. Opin. Biotechnol. 2005, 16, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Albertazzi, L.; Arosio, D.; Marchetti, L.; Ricci, F.; Beltram, F. Quantitative FRET analysis with the EGFP-mCherry fluorescent protein pair. Photochem. Photobiol. 2009, 85, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, L.; Comelli, L.; D’Innocenzo, B.; Puzzi, L.; Luin, S.; Arosio, D.; Calvello, M.; Mendoza-Maldonado, R.; Peverali, F.; Trovato, F.; et al. Homeotic proteins participate in the function of human-DNA replication origins. Nucleic Acids Res. 2010, 38, 8105–8119. [Google Scholar] [CrossRef] [PubMed]
- Digman, M.A.; Dalal, R.; Horwitz, A.F.; Gratton, E. Mapping the number of molecules and brightness in the laser scanning microscope. Biophys. J. 2008, 94, 2320–2332. [Google Scholar] [CrossRef] [PubMed]
- Di Rienzo, C.; Jacchetti, E.; Cardarelli, F.; Bizzarri, R.; Beltram, F.; Cecchini, M. Unveiling LOX-1 receptor interplay with nanotopography: Mechanotransduction and atherosclerosis onset. Sci. Rep. 2013, 3, 1141. [Google Scholar] [CrossRef] [PubMed]
- James, N.G.; Digman, M.A.; Gratton, E.; Barylko, B.; Ding, X.; Albanesi, J.P.; Goldberg, M.S.; Jameson, D.M. Number and brightness analysis of LRRK2 oligomerization in live cells. Biophys. J. 2012, 102, L41–L43. [Google Scholar] [CrossRef] [PubMed]
- Ossato, G.; Digman, M.A.; Aiken, C.; Lukacsovich, T.; Marsh, J.L.; Gratton, E. A two-step path to inclusion formation of huntingtin peptides revealed by number and brightness analysis. Biophys. J. 2010, 98, 3078–3085. [Google Scholar] [CrossRef] [PubMed]
- Storti, B.; Bizzarri, R.; Cardarelli, F.; Beltram, F. Intact microtubules preserve transient receptor potential vanilloid 1 (TRPV1) functionality through receptor binding. J. Biol. Chem. 2012, 287, 7803–7811. [Google Scholar] [CrossRef] [PubMed]
- Youker, R.T.; Bruns, J.R.; Costa, S.A.; Rbaibi, Y.; Lanni, F.; Kashlan, O.B.; Teng, H.; Weisz, O.A. Multiple motifs regulate apical sorting of p75 via a mechanism that involves dimerization and higher-order oligomerization. Mol. Biol. Cell 2013, 24, 1996–2007. [Google Scholar] [CrossRef] [PubMed]
- Reits, E.A.; Neefjes, J.J. From fixed to FRAP: Measuring protein mobility and activity in living cells. Nat. Cell Biol. 2001, 3, E145–E147. [Google Scholar] [CrossRef] [PubMed]
- Klonis, N.; Rug, M.; Harper, I.; Wickham, M.; Cowman, A.; Tilley, L. Fluorescence photobleaching analysis for the study of cellular dynamics. Eur. Biophys. J. 2002, 31, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Saxton, M.J.; Jacobson, K. Single-particle tracking: Applications to membrane dynamics. Annu. Rev. Biophys. Biomol. Struct. 1997, 26, 373–399. [Google Scholar] [CrossRef] [PubMed]
- Cui, B.; Wu, C.; Chen, L.; Ramirez, A.; Bearer, E.L.; Li, W.P.; Mobley, W.C.; Chu, S. One at a time, live tracking of NGF axonal transport using quantum dots. Proc. Natl. Acad. Sci. USA 2007, 104, 13666–13671. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, B.; Zhuang, X. Virus trafficking-learning from single-virus tracking. Nat. Rev. Microbiol. 2007, 5, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Akhmanova, A.; Steinmetz, M.O. Tracking the ends: A dynamic protein network controls the fate of microtubule tips. Nat. Rev. Mol. Cell Biol. 2008, 9, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Morisaki, T.; Muller, W.G.; Golob, N.; Mazza, D.; McNally, J.G. Single-molecule analysis of transcription factor binding at transcription sites in live cells. Nat. Commun. 2014, 5, 4456. [Google Scholar] [PubMed]
- Hibino, K.; Hiroshima, M.; Takahashi, M.; Sako, Y. Single-molecule imaging of fluorescent proteins expressed in living cells. Methods Mol. Biol. 2009, 544, 451–460. [Google Scholar] [PubMed]
- Chung, I.; Akita, R.; Vandlen, R.; Toomre, D.; Schlessinger, J.; Mellman, I. Spatial control of EGF receptor activation by reversible dimerization on living cells. Nature 2010, 464, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Jaqaman, K.; Kuwata, H.; Touret, N.; Collins, R.; Trimble, W.S.; Danuser, G.; Grinstein, S. Cytoskeletal control of CD36 diffusion promotes its receptor and signaling function. Cell 2011, 146, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Opazo, P.; Labrecque, S.; Tigaret, C.M.; Frouin, A.; Wiseman, P.W.; de Koninck, P.; Choquet, D. CaMKII triggers the diffusional trapping of surface AMPARs through phosphorylation of stargazin. Neuron 2010, 67, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Triller, A.; Choquet, D. Surface trafficking of receptors between synaptic and extrasynaptic membranes: And yet they do move! Trends Neurosci. 2005, 28, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Kasai, R.S.; Suzuki, K.G.; Prossnitz, E.R.; Koyama-Honda, I.; Nakada, C.; Fujiwara, T.K.; Kusumi, A. Full characterization of GPCR monomer-dimer dynamic equilibrium by single molecule imaging. J. Cell Biol. 2011, 192, 463–480. [Google Scholar] [CrossRef] [PubMed]
- Piguet, J.; Schreiter, C.; Segura, J.M.; Vogel, H.; Hovius, R. Acetylcholine receptor organization in membrane domains in muscle cells: Evidence for rapsyn-independent and rapsyn-dependent mechanisms. J. Biol. Chem. 2011, 286, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Winter, P.W.; van Orden, A.K.; Roess, D.A.; Barisas, B.G. Actin-dependent clustering of insulin receptors in membrane microdomains. Biochim. Biophys. Acta 2011, 1818, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Johnsson, N.; George, N.; Johnsson, K. Protein chemistry on the surface of living cells. Chembiochem 2005, 6, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.G.; Kasai, R.S.; Fujiwara, T.K.; Kusumi, A. Single-molecule imaging of receptor-receptor interactions. Methods Cell Biol. 2013, 117, 373–390. [Google Scholar] [PubMed]
- Meijering, E.; Dzyubachyk, O.; Smal, I. Methods for cell and particle tracking. Methods Enzymol. 2012, 504, 183–200. [Google Scholar] [PubMed]
- Chenouard, N.; Smal, I.; de Chaumont, F.; Maska, M.; Sbalzarini, I.F.; Gong, Y.; Cardinale, J.; Carthel, C.; Coraluppi, S.; Winter, M.; et al. Objective comparison of particle tracking methods. Nat. Methods 2014, 11, 281–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalet, X. Mean square displacement analysis of single-particle trajectories with localization error: Brownian motion in an isotropic medium. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2010, 82, 041914. [Google Scholar] [CrossRef] [PubMed]
- Callegari, A.; Luin, S.; Marchetti, L.; Duci, A.; Cattaneo, A.; Beltram, F. Single particle tracking of acyl carrier protein (ACP)-tagged TrkA receptors in PC12nnr5 cells. J. Neurosci. Methods 2012, 204, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Pinaud, F.; Michalet, X.; Iyer, G.; Margeat, E.; Moore, H.P.; Weiss, S. Dynamic partitioning of a glycosyl-phosphatidylinositol-anchored protein in glycosphingolipid-rich microdomains imaged by single-quantum dot tracking. Traffic 2009, 10, 691–712. [Google Scholar] [CrossRef] [PubMed]
- Ewers, H.; Smith, A.E.; Sbalzarini, I.F.; Lilie, H.; Koumoutsakos, P.; Helenius, A. Single-particle tracking of murine polyoma virus-like particles on live cells and artificial membranes. Proc. Natl. Acad. Sci. USA 2005, 102, 15110–15115. [Google Scholar] [CrossRef] [PubMed]
- Simson, R.; Sheets, E.D.; Jacobson, K. Detection of temporary lateral confinement of membrane proteins using single-particle tracking analysis. Biophys. J. 1995, 69, 989–993. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, L.; Callegari, A.; Luin, S.; Signore, G.; Viegi, A.; Beltram, F.; Cattaneo, A. Ligand signature in the membrane dynamics of single TrkA receptor molecules. J. Cell Sci. 2013, 126, 4445–4456. [Google Scholar] [CrossRef] [PubMed]
- Huet, S.; Karatekin, E.; Tran, V.S.; Fanget, I.; Cribier, S.; Henry, J.P. Analysis of transient behavior in complex trajectories: Application to secretory vesicle dynamics. Biophys. J. 2006, 91, 3542–3559. [Google Scholar] [CrossRef] [PubMed]
- Calebiro, D.; Rieken, F.; Wagner, J.; Sungkaworn, T.; Zabel, U.; Borzi, A.; Cocucci, E.; Zurn, A.; Lohse, M.J. Single-molecule analysis of fluorescently labeled G-protein-coupled receptors reveals complexes with distinct dynamics and organization. Proc. Natl. Acad. Sci. USA 2013, 110, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Das, S.K.; Darshi, M.; Cheley, S.; Wallace, M.I.; Bayley, H. Membrane protein stoichiometry determined from the step-wise photobleaching of dye-labelled subunits. Chembiochem 2007, 8, 994–999. [Google Scholar] [CrossRef] [PubMed]
- Borgdorff, A.J.; Choquet, D. Regulation of AMPA receptor lateral movements. Nature 2002, 417, 649–653. [Google Scholar] [CrossRef] [PubMed]
- Charrier, C.; Ehrensperger, M.V.; Dahan, M.; Levi, S.; Triller, A. Cytoskeleton regulation of glycine receptor number at synapses and diffusion in the plasma membrane. J. Neurosci. 2006, 26, 8502–8511. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.G.; Fujiwara, T.K.; Edidin, M.; Kusumi, A. Dynamic recruitment of phospholipase C gamma at transiently immobilized GPI-anchored receptor clusters induces IP3-Ca2+ signaling: Single-molecule tracking study 2. J. Cell Biol. 2007, 177, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Watson, F.L.; Heerssen, H.M.; Moheban, D.B.; Lin, M.Z.; Sauvageot, C.M.; Bhattacharyya, A.; Pomeroy, S.L.; Segal, R.A. Rapid nuclear responses to target-derived neurotrophins require retrograde transport of ligand-receptor complex. J. Neurosci. 1999, 19, 7889–7900. [Google Scholar] [PubMed]
- Egan, M.F.; Kojima, M.; Callicott, J.H.; Goldberg, T.E.; Kolachana, B.S.; Bertolino, A.; Zaitsev, E.; Gold, B.; Goldman, D.; Dean, M.; et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 2003, 112, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Kohara, K.; Kitamura, A.; Adachi, N.; Nishida, M.; Itami, C.; Nakamura, S.; Tsumoto, T. Inhibitory but not excitatory cortical neurons require presynaptic brain-derived neurotrophic factor for dendritic development, as revealed by chimera cell culture. J. Neurosci. 2003, 23, 6123–6131. [Google Scholar] [PubMed]
- Jullien, J.; Guili, V.; Derrington, E.A.; Darlix, J.L.; Reichardt, L.F.; Rudkin, B.B. Trafficking of TrkA-green fluorescent protein chimerae during nerve growth factor-induced differentiation. J. Biol. Chem. 2003, 278, 8706–8716. [Google Scholar] [CrossRef] [PubMed]
- Claude, P.; Hawrot, E.; Dunis, D.A.; Campenot, R.B. Binding, internalization, and retrograde transport of 125I-nerve growth factor in cultured rat sympathetic neurons. J. Neurosci. 1982, 2, 431–442. [Google Scholar] [PubMed]
- Seefeldt, B.; Kasper, R.; Seidel, T.; Tinnefeld, P.; Dietz, K.J.; Heilemann, M.; Sauer, M. Fluorescent proteins for single-molecule fluorescence applications. J. Biophotonics 2008, 1, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Clary, D.O.; Weskamp, G.; Austin, L.R.; Reichardt, L.F. TrkA cross-linking mimics neuronal responses to nerve growth factor. Mol. Biol. Cell 1994, 5, 549–563. [Google Scholar] [CrossRef] [PubMed]
- Sundara Rajan, S.; Vu, T.Q. Quantum dots monitor TrkA receptor dynamics in the interior of neural PC12 cells. Nano Lett. 2006, 6, 2049–2059. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, M.B.; Hawrot, E.; Breakefield, X.O. Receptor binding activities of biotinylated derivatives of β-nerve growth factor. J. Neurochem. 1986, 46, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Bronfman, F.C.; Tcherpakov, M.; Jovin, T.M.; Fainzilber, M. Ligand-induced internalization of the p75 neurotrophin receptor: A slow route to the signaling endosome. J. Neurosci. 2003, 23, 3209–3220. [Google Scholar] [PubMed]
- Rajan, S.S.; Liu, H.Y.; Vu, T.Q. Ligand-bound quantum dot probes for studying the molecular scale dynamics of receptor endocytic trafficking in live cells. ACS Nano 2008, 2, 1153–1166. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.Q.; Maddipati, R.; Blute, T.A.; Nehilla, B.J.; Nusblat, L.; Desai, T.A. Peptide-conjugated quantum dots activate neuronal receptors and initiate downstream signaling of neurite growth. Nano Lett. 2005, 5, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Echarte, M.M.; Bruno, L.; Arndt-Jovin, D.J.; Jovin, T.M.; Pietrasanta, L.I. Quantitative single particle tracking of NGF-receptor complexes: Transport is bidirectional but biased by longer retrograde run lengths. FEBS Lett. 2007, 581, 2905–2913. [Google Scholar] [CrossRef] [PubMed]
- Tani, T.; Miyamoto, Y.; Fujimori, K.E.; Taguchi, T.; Yanagida, T.; Sako, Y.; Harada, Y. Trafficking of a ligand-receptor complex on the growth cones as an essential step for the uptake of nerve growth factor at the distal end of the axon: A single-molecule analysis. J. Neurosci. 2005, 25, 2181–2191. [Google Scholar] [CrossRef] [PubMed]
- Shibata, S.C.; Hibino, K.; Mashimo, T.; Yanagida, T.; Sako, Y. Formation of signal transduction complexes during immobile phase of NGFR movements. Biochem. Biophys. Res. Commun. 2006, 342, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Nomura, M.; Nagai, T.; Harada, Y.; Tani, T. Facilitated intracellular transport of TrkA by an interaction with nerve growth factor. Dev. Neurobiol. 2011, 71, 634–649. [Google Scholar] [CrossRef] [PubMed]
- Sung, K.; Maloney, M.T.; Yang, J.; Wu, C. A novel method for producing mono-biotinylated, biologically active neurotrophic factors: An essential reagent for single molecule study of axonal transport. J. Neurosci. Methods 2011, 200, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, L.; de Nadai, T.; Bonsignore, F.; Calvello, M.; Signore, G.; Viegi, A.; Beltram, F.; Luin, S.; Cattaneo, A. Site-specific labeling of neurotrophins and their receptors via short and versatile peptide tags. PLoS One 2014, 9, e113708. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Zhang, K.; Cui, B. Functional characterization and axonal transport of quantum dot labeled BDNF. Integr. Biol. (Camb.) 2012, 4, 953–960. [Google Scholar] [CrossRef]
- Vermehren-Schmaedick, A.; Krueger, W.; Jacob, T.; Ramunno-Johnson, D.; Balkowiec, A.; Lidke, K.A.; Vu, T.Q. Heterogeneous intracellular trafficking dynamics of brain-derived neurotrophic factor complexes in the neuronal soma revealed by single quantum dot tracking. PLoS One 2014, 9, e95113. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Cironi, P.; Lin, A.J.; Xu, Y.; Hrvatin, S.; Golan, D.E.; Silver, P.A.; Walsh, C.T.; Yin, J. Genetically encoded short peptide tags for orthogonal protein labeling by Sfp and AcpS phosphopantetheinyl transferases. ACS Chem. Biol. 2007, 2, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Capsoni, S.; Covaceuszach, S.; Marinelli, S.; Ceci, M.; Bernardo, A.; Minghetti, L.; Ugolini, G.; Pavone, F.; Cattaneo, A. Taking pain out of NGF: A “painless” NGF mutant, linked to hereditary sensory autonomic neuropathy type V, with full neurotrophic activity. PLoS One 2011, 6, e17321. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, F.; Covaceuszach, S.; Konarev, P.V.; Gonfloni, S.; Malerba, F.; Schwarz, E.; Svergun, D.I.; Cattaneo, A.; Lamba, D. Intrinsic structural disorder of mouse proNGF. Proteins 2009, 75, 990–1009. [Google Scholar] [CrossRef] [PubMed]
- Covaceuszach, S.; Capsoni, S.; Marinelli, S.; Pavone, F.; Ceci, M.; Ugolini, G.; Vignone, D.; Amato, G.; Paoletti, F.; Lamba, D.; et al. In vitro receptor binding properties of a “painless” NGF mutein, linked to hereditary sensory autonomic neuropathy type V. Biochem. Biophys. Res. Commun. 2010, 391, 824–829. [Google Scholar] [CrossRef] [PubMed]
- Ivanisevic, L.; Zheng, W.; Woo, S.B.; Neet, K.E.; Saragovi, H.U. TrkA receptor “hot spots” for binding of NT-3 as a heterologous ligand. J. Biol. Chem. 2007, 282, 16754–16763. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Maruyama, I.N. Nerve growth factor receptor TrkA exists as a preformed, yet inactive, dimer in living cells. FEBS Lett. 2011, 585, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Maruyama, I.N. Brain-derived neurotrophic factor receptor TrkB exists as a preformed dimer in living cells. J. Mol. Signal. 2012, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.; Claus, J.; Jovin, T.M.; Arndt-Jovin, D.J. Distribution of resting and ligand-bound ErbB1 and ErbB2 receptor tyrosine kinases in living cells using number and brightness analysis. Proc. Natl. Acad. Sci. USA 2010, 107, 16524–16529. [Google Scholar] [CrossRef] [PubMed]
- Endres, N.F.; Das, R.; Smith, A.W.; Arkhipov, A.; Kovacs, E.; Huang, Y.; Pelton, J.G.; Shan, Y.; Shaw, D.E.; Wemmer, D.E.; et al. Conformational coupling across the plasma membrane in activation of the EGF receptor. Cell 2013, 152, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Teramura, Y.; Ichinose, J.; Takagi, H.; Nishida, K.; Yanagida, T.; Sako, Y. Single-molecule analysis of epidermal growth factor binding on the surface of living cells. EMBO J. 2006, 25, 4215–4222. [Google Scholar] [CrossRef] [PubMed]
- Arkhipov, A.; Shan, Y.; Das, R.; Endres, N.F.; Eastwood, M.P.; Wemmer, D.E.; Kuriyan, J.; Shaw, D.E. Architecture and membrane interactions of the EGF receptor. Cell 2013, 152, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Deinhardt, K.; Reversi, A.; Berninghausen, O.; Hopkins, C.R.; Schiavo, G. Neurotrophins Redirect p75NTR from a clathrin-independent to a clathrin-dependent endocytic pathway coupled to axonal transport. Traffic 2007, 8, 1736–1749. [Google Scholar] [CrossRef] [PubMed]
- Escudero, C.A.; Lazo, O.M.; Galleguillos, C.; Parraguez, J.I.; Lopez-Verrilli, M.A.; Cabeza, C.; Leon, L.; Saeed, U.; Retamal, C.; Gonzalez, A.; et al. The p75 neurotrophin receptor evades the endolysosomal route in neuronal cells, favouring multivesicular bodies specialised for exosomal release. J. Cell Sci. 2014, 127, 1966–1979. [Google Scholar] [CrossRef] [PubMed]
- Formaggio, E.; Cantu, C.; Chiamulera, C.; Fumagalli, G.F. p75 neurotrophin receptor distribution and transport in cultured neurons. Neurosci. Res. 2008, 62, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Los, G.V.; Encell, L.P.; McDougall, M.G.; Hartzell, D.D.; Karassina, N.; Zimprich, C.; Wood, M.G.; Learish, R.; Ohana, R.F.; Urh, M.; et al. HaloTag: A novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. 2008, 3, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Mok, S.A.; Lund, K.; Lapointe, P.; Campenot, R.B. A Halo Tag® method for assessing the retrograde axonal transport of the p75 neurotrophin receptor and other proteins in compartmented cultures of rat sympathetic neurons. J. Neurosci. Methods 2013, 214, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Kanning, K.C.; Hudson, M.; Amieux, P.S.; Wiley, J.C.; Bothwell, M.; Schecterson, L.C. Proteolytic processing of the p75 neurotrophin receptor and two homologs generates C-terminal fragments with signaling capability. J. Neurosci. 2003, 23, 5425–5436. [Google Scholar] [PubMed]
- Jung, K.M.; Tan, S.; Landman, N.; Petrova, K.; Murray, S.; Lewis, R.; Kim, P.K.; Kim, D.S.; Ryu, S.H.; Chao, M.V.; et al. Regulated intramembrane proteolysis of the p75 neurotrophin receptor modulates its association with the TrkA receptor. J. Biol. Chem. 2003, 278, 42161–42169. [Google Scholar] [CrossRef] [PubMed]
- Sykes, A.M.; Palstra, N.; Abankwa, D.; Hill, J.M.; Skeldal, S.; Matusica, D.; Venkatraman, P.; Hancock, J.F.; Coulson, E.J. The effects of transmembrane sequence and dimerization on cleavage of the p75 neurotrophin receptor by γ-secretase. J. Biol. Chem. 2012, 287, 43810–43824. [Google Scholar] [CrossRef] [PubMed]
- Kenchappa, R.S.; Zampieri, N.; Chao, M.V.; Barker, P.A.; Teng, H.K.; Hempstead, B.L.; Carter, B.D. Ligand-dependent cleavage of the P75 neurotrophin receptor is necessary for NRIF nuclear translocation and apoptosis in sympathetic neurons. Neuron 2006, 50, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Skeldal, S.; Sykes, A.M.; Glerup, S.; Matusica, D.; Palstra, N.; Autio, H.; Boskovic, Z.; Madsen, P.; Castren, E.; Nykjaer, A.; et al. Mapping of the interaction site between sortilin and the p75 neurotrophin receptor reveals a regulatory role for the sortilin intracellular domain in p75 neurotrophin receptor shedding and apoptosis. J. Biol. Chem. 2012, 287, 43798–43809. [Google Scholar] [CrossRef] [PubMed]
- Jacquier, V.; Prummer, M.; Segura, J.M.; Pick, H.; Vogel, H. Visualizing odorant receptor trafficking in living cells down to the single-molecule level. Proc. Natl. Acad. Sci. USA 2006, 103, 14325–14330. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, E.; Marion, S.; Mishra, B.B.; John, M.; Kratzke, R.; Ahmad, S.F.; Holzer, D.; Anand, P.K.; Weiss, D.G.; Griffiths, G.; Kuznetsov, S.A. Initial receptor-ligand interactions modulate gene expression and phagosomal properties during both early and late stages of phagocytosis. Eur. J. Cell Biol. 2010, 89, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marchetti, L.; Luin, S.; Bonsignore, F.; De Nadai, T.; Beltram, F.; Cattaneo, A. Ligand-Induced Dynamics of Neurotrophin Receptors Investigated by Single-Molecule Imaging Approaches. Int. J. Mol. Sci. 2015, 16, 1949-1979. https://doi.org/10.3390/ijms16011949
Marchetti L, Luin S, Bonsignore F, De Nadai T, Beltram F, Cattaneo A. Ligand-Induced Dynamics of Neurotrophin Receptors Investigated by Single-Molecule Imaging Approaches. International Journal of Molecular Sciences. 2015; 16(1):1949-1979. https://doi.org/10.3390/ijms16011949
Chicago/Turabian StyleMarchetti, Laura, Stefano Luin, Fulvio Bonsignore, Teresa De Nadai, Fabio Beltram, and Antonino Cattaneo. 2015. "Ligand-Induced Dynamics of Neurotrophin Receptors Investigated by Single-Molecule Imaging Approaches" International Journal of Molecular Sciences 16, no. 1: 1949-1979. https://doi.org/10.3390/ijms16011949