
Exploring the Arabidopsis Proteome: Influence of Protein Solubilization Buffers on Proteome Coverage

 and
and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Proteome Analyses

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number Identified Proteins | NDSB | CHAPS | ND | SDS | TRIT |
|---|---|---|---|---|---|
| Average 2DE spots per gel | 1187 ± 21 | 1277 ± 41 | 1267 ± 22 | 1238 ± 17 | 1072 ± 29 |
| OFFGEL fractionated samples | 3383 | 3092 | 2906 | 3058 | 2990 |
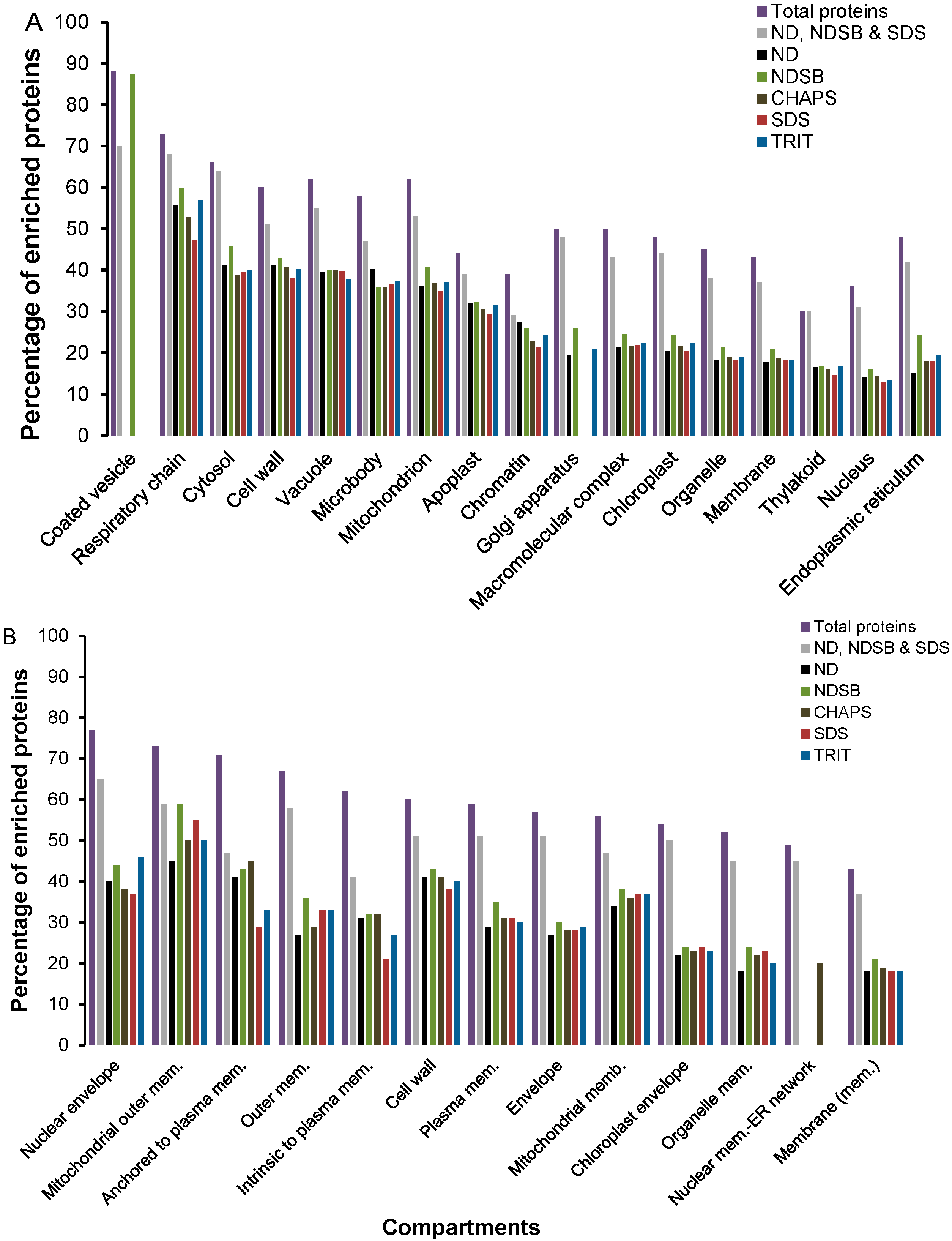
2.2. Functional Enrichment of Displayed Proteomes

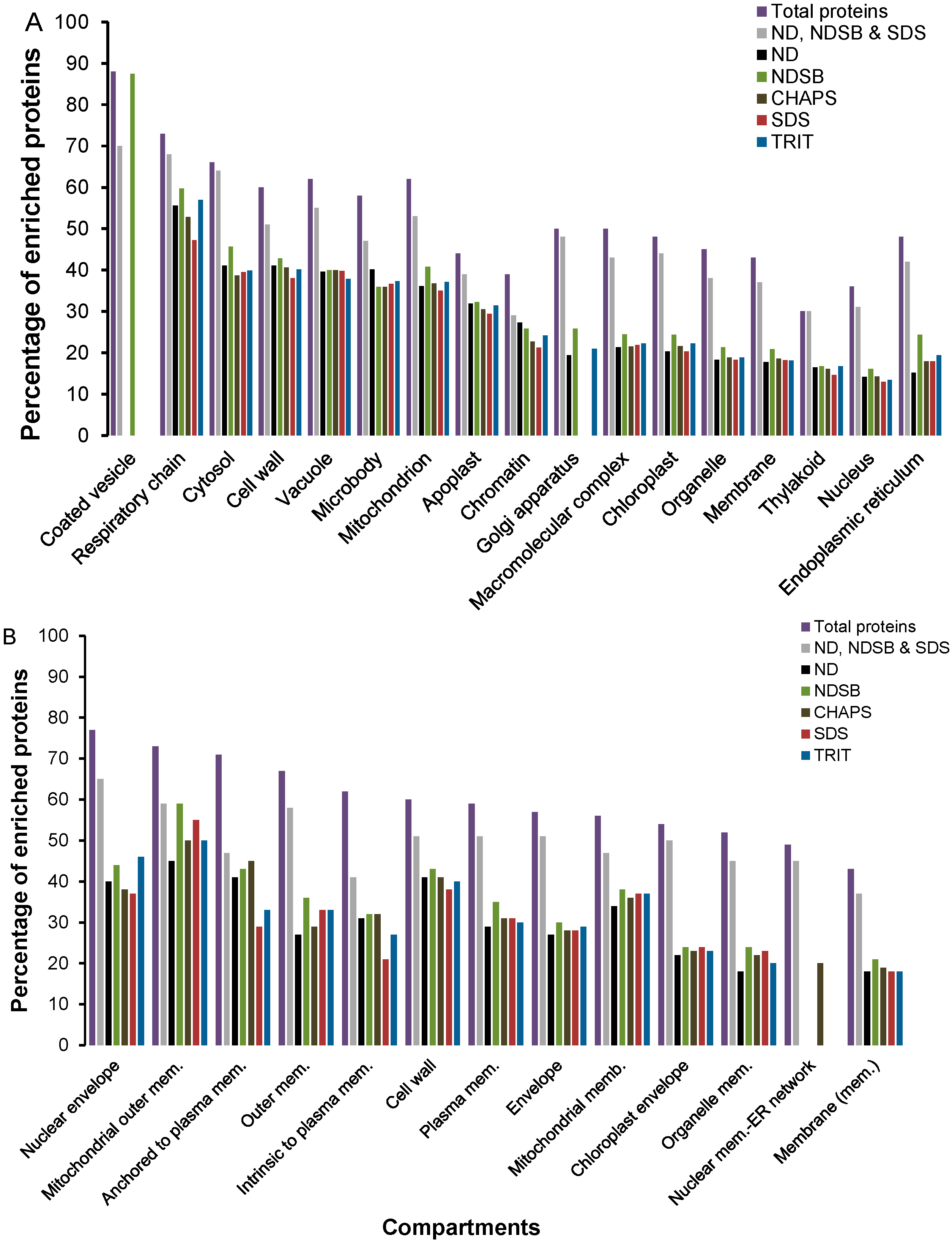
2.3. Physico-Chemical Properties of Buffer-Specific Proteins

2.4. Predictions of Post-Translational Modifications

3. Experimental Section
3.1. Arabidopsis Cell Suspension Culture and Protein Extraction
3.2. Comparative Two-Dimensional Gel Electrophoresis (2DE) of Arabidopsis Proteomes and In-Gel Digestion
3.3. Gel-Free Trypsin Digestion of Complex Protein Extracts
3.4. Peptide Fractionation by OFFGEL Fractionator
3.5. Protein Identification by LC–MS/MS
3.6. Computational Analysis of Identified Proteins
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sarnighausen, E.; Reski, R. Plant proteomics. Methods Mol. Biol. 2008, 484, 29–44. [Google Scholar] [PubMed]
- Gorg, A.; Obermaier, C.; Boguth, G.; Harder, A.; Scheibe, B.; Wildgruber, R.; Weiss, W. The current state of two-dimensional electrophoresis with immobilized pH gradients. Electrophoresis 2000, 21, 1037–1053. [Google Scholar] [CrossRef] [PubMed]
- Saravanan, R.S.; Rose, J.K.C. A critical evaluation of sample extraction techniques for enhanced proteomic analysis of recalcitrant plant tissues. Proteomics 2004, 4, 2522–2532. [Google Scholar] [CrossRef] [PubMed]
- Marondedze, C.; Thomas, L.A. Insights into fruit function from the proteome of the hypanthium. J. Plant Physiol. 2012, 169, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Rose, J.K.; Bashir, S.; Giovannoni, J.J.; Jahn, M.M.; Saravanan, R.S. Tackling the plant proteome: Practical approaches, hurdles and experimental tools. Plant J. 2004, 39, 715–733. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Scali, M.; Vignani, R.; Spadafora, A.; Sensi, E.; Mazzuca, S.; Cresti, M. Protein extraction for two-dimensional electrophoresis from olive leaf, a plant tissue containing high levels of interfering compounds. Electrophoresis 2003, 24, 2369–2375. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, S.C.; Witters, E.; Laukens, K.; Deckers, P.; Swennen, R.; Panis, B. Preparation of protein extracts from recalcitrant plant tissues: An evaluation of different methods for two-dimensional gel electrophoresis analysis. Proteomics 2005, 5, 2497–2507. [Google Scholar] [CrossRef] [PubMed]
- Giavalisco, P.; Nordhoff, E.; Lehrach, H.; Gobom, J.; Klose, J. Extraction of proteins from plant tissues for two-dimensional electrophoresis analysis. Electrophoresis 2003, 24, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Sheoran, I.S.; Ross, A.R.S.; Olson, D.J.H.; Sawhney, V.K. Compatibility of plant protein extraction methods with mass spectrometry for proteome analysis. Plant Sci. 2009, 176, 99–104. [Google Scholar] [CrossRef]
- Barry, R.; Soloviev, M. Quantitative protein profiling using antibody arrays. Proteomics 2004, 4, 3717–3726. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.R.; Griffin, T.J. Gel-free mass spectrometry-based high throughput proteomics: Tools for studying biological response of proteins and proteomes. Proteomics 2006, 6, 4678–4687. [Google Scholar] [CrossRef] [PubMed]
- Hurkman, W.J.; Tanaka, C.K. Solubilization of plant membrane-proteins for analysis by two-dimensional gel-electrophoresis. Plant Physiol. 1986, 81, 802–806. [Google Scholar] [CrossRef] [PubMed]
- Isaacson, T.; Damasceno, C.M.B.; Saravanan, R.S.; He, Y.; Catala, C.; Saladie, M.; Rose, J.K.C. Sample extraction techniques for enhanced proteomic analysis of plant tissues. Nat. Protoc. 2006, 1, 769–774. [Google Scholar] [CrossRef] [PubMed]
- Ashoub, A.; Berberich, T.; Beckhaus, T.; Bruggemann, W. A competent extraction method of plant proteins for 2-D gel electrophoresis. Electrophoresis 2011, 32, 2975–2978. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Vignani, R.; Scali, M.; Cresti, M. A universal and rapid protocol for protein extraction from recalcitrant plant tissues for proteomic analysis. Electrophoresis 2006, 27, 2782–2786. [Google Scholar] [CrossRef] [PubMed]
- Marondedze, C.; Turek, I.; Parrott, B.; Thomas, L.; Jankovic, B.; Lilley, K.S.; Gehring, C. Structural and functional characteristics of cGMP-dependent methionine oxidation in Arabidopsis thaliana proteins. Cell Commun. Signal 2013, 11, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Pridmore, A.M.; Devine, D.A.; Bonass, W.A.; Silley, P. Influence of sample preparation technique on two-dimensional gel electrophoresis of proteins from porphyromonas gingivalis. Lett. Appl. Microbiol. 1999, 28, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Kamo, M.; Tsugita, A. N-terminal amino acid sequencing of 2-de spots. In 2-D Proteome Analysis Protocols; Link, A.J., Ed.; Humana Press: Totowa, NJ, USA, 1999; Volume 112, pp. 461–466. [Google Scholar]
- Porubleva, L.; Vander Velden, K.; Kothari, S.; Oliver, D.J.; Chitnis, P.R. The proteome of maize leaves: Use of gene sequences and expressed sequence tag data for identification of proteins with peptide mass fingerprints. Electrophoresis 2001, 22, 1724–1738. [Google Scholar] [CrossRef] [PubMed]
- Tanca, A.; Biosa, G.; Pagnozzi, D.; Addis, M.F.; Uzzau, S. Comparison of detergent-based sample preparation workflows for ltq-orbitrap analysis of the Escherichia coli proteome. Proteomics 2013, 13, 2597–2607. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-J.; Chen, W.-Y.; Wu, J.-H. Total protein extraction for metaproteomics analysis of methane producing biofilm: The effects of detergents. Int. J. Mol. Sci. 2014, 15, 10169–10184. [Google Scholar] [CrossRef] [PubMed]
- Adessi, C.; Miege, C.; Albrieux, C.; Rabilloud, T. Two-dimensional electrophoresis of membrane proteins: A current challenge for immobilized pH gradients. Electrophoresis 1997, 18, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Urquhart, B.L.; Atsalos, T.E.; Roach, D.; Basseal, D.J.; Bjellqvist, B.; Britton, W.L.; Humphery-Smith, I. “proteomic contigs” of mycobacterium tuberculosis and mycobacterium bovis (BCG) using novel immobilised ph gradients. Electrophoresis 1997, 18, 1384–1392. [Google Scholar] [CrossRef] [PubMed]
- Chevallet, M.; Santoni, V.; Poinas, A.; Rouquié, D.; Fuchs, A.; Kieffer, S.; Lunardi, J.; Garin, J.; Rabilloud, T. New zwitterionic detergents improve the analysis of membrane proteins by two-dimensional electrophoresis. Electrophoresis 1998, 19, 1901–1909. [Google Scholar] [CrossRef] [PubMed]
- Ito, J.; Parsons, H.T.; Heazlewood, J.L. The Arabidopsis cytosolic proteome: The metabolic heart of the cell. Front. Plant Sci. 2014, 5, 21. [Google Scholar] [CrossRef] [PubMed]
- Gamborg, O.L.; Miller, R.A.; Ojima, K. Nutrient requirements of suspension cultures of soybean root cells. Exp. Cell Res. 1968, 50, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.; Marondedze, C.; Ederli, L.; Pasqualini, S.; Gehring, C. Proteomic signatures implicate camp in light and temperature responses in Arabidopsis thaliana. J. Proteomics 2013, 83, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Groen, A.; Thomas, L.; Lilley, K.S.; Marondedze, C. Identification and quantitation of signal molecule-dependent protein phosphorylation. In Cyclic Nucleotide Signaling in Plants: Methods and Protocols; Gehring, C., Ed.; Springer Science: New York, NY, USA, 2013; pp. 121–137. [Google Scholar]
- BINGO. Available online: http://cytoscapeweb.cytoscape.org/ (accessed on 24 June 2014).
- Tusnady, G.E.; Simon, I. Principles governing amino acid composition of integral membrane proteins: Application to topology prediction. J. Mol. Biol. 1998, 283, 489–506. [Google Scholar] [CrossRef] [PubMed]
- HMMTOP Server. Available online: http://www.enzim.hu/hmmtop (accessed on 1 June 2014).
- GRAVY Calculator. Available online: http://www.gravy-calculator.de (accessed on 1 June 2014).
- Beausoleil, S.A.; Villen, J.; Gerber, S.A.; Rush, J.; Gygi, S.P. A probability-based approach for high-throughput protein phosphorylation analysis. Nat. Biotechnol. 2006, 24, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Big-PI Plant Predictor. Available online: http://www.mendel.imp.univie.ac.at/gpi/plant_server.html (accessed on 21 April 2013).
- NMT–MYR predictor. Available online: http://www.mendel.imp.univie.ac.at/myristate/SUPLpredictor.htm (accessed on 4 May 2013).
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marondedze, C.; Wong, A.; Groen, A.; Serrano, N.; Jankovic, B.; Lilley, K.; Gehring, C.; Thomas, L. Exploring the Arabidopsis Proteome: Influence of Protein Solubilization Buffers on Proteome Coverage. Int. J. Mol. Sci. 2015, 16, 857-870. https://doi.org/10.3390/ijms16010857
Marondedze C, Wong A, Groen A, Serrano N, Jankovic B, Lilley K, Gehring C, Thomas L. Exploring the Arabidopsis Proteome: Influence of Protein Solubilization Buffers on Proteome Coverage. International Journal of Molecular Sciences. 2015; 16(1):857-870. https://doi.org/10.3390/ijms16010857
Chicago/Turabian StyleMarondedze, Claudius, Aloysius Wong, Arnoud Groen, Natalia Serrano, Boris Jankovic, Kathryn Lilley, Christoph Gehring, and Ludivine Thomas. 2015. "Exploring the Arabidopsis Proteome: Influence of Protein Solubilization Buffers on Proteome Coverage" International Journal of Molecular Sciences 16, no. 1: 857-870. https://doi.org/10.3390/ijms16010857