
Lipid Metabolism, Apoptosis and Cancer Therapy

Division of Hematology/Oncology, Department of Internal Medicine, School of Medicine and Cancer Center, Saint Louis University, 3655 Vista Avenue, Saint Louis, MO 63110, USA
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2015, 16(1), 924-949; https://doi.org/10.3390/ijms16010924
Submission received: 8 July 2014
/
Accepted: 17 December 2014
/
Published: 2 January 2015
(This article belongs to the Collection Programmed Cell Death and Apoptosis)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Lipid metabolism is regulated by multiple signaling pathways, and generates a variety of bioactive lipid molecules. These bioactive lipid molecules known as signaling molecules, such as fatty acid, eicosanoids, diacylglycerol, phosphatidic acid, lysophophatidic acid, ceramide, sphingosine, sphingosine-1-phosphate, phosphatidylinositol-3 phosphate, and cholesterol, are involved in the activation or regulation of different signaling pathways. Lipid metabolism participates in the regulation of many cellular processes such as cell growth, proliferation, differentiation, survival, apoptosis, inflammation, motility, membrane homeostasis, chemotherapy response, and drug resistance. Bioactive lipid molecules promote apoptosis via the intrinsic pathway by modulating mitochondrial membrane permeability and activating different enzymes including caspases. In this review, we discuss recent data in the fields of lipid metabolism, lipid-mediated apoptosis, and cancer therapy. In conclusion, understanding the underlying molecular mechanism of lipid metabolism and the function of different lipid molecules could provide the basis for cancer cell death rationale, discover novel and potential targets, and develop new anticancer drugs for cancer therapy.
1. Introduction
Lipids are hydrophobic or amphipathic (hydrophilic and lipophilic) small molecules, and are not like proteins, nucleic acids, and polysaccharides which are large macromolecular polymers formed by the chemical linking of several small constituent molecules (these molecular building blocks are similar, or homologous, in structure) [1,2]. Thus far, there is still no widely accepted definition of lipids because lipids comprise an enormous number of chemically distinct molecules and are structurally quite diverse. Lipids are frequently defined as naturally occurring compounds that are insoluble in water but soluble in nonpolar solvents [1,2]. Amphipathic lipids form plasma membranes in which cells can maintain all biological events in an intracellular environment and respond to the changes of extracellular environment.
In all living cells, lipids are required to maintain cellular structure, provide energy and are involved in cell signaling. Lipid metabolism (anabolism and catabolism) generates a variety of biological intermediators. Many of these intermediators are bioactive lipid molecules (also known as signaling molecules or second messengers) which are produced by the activation of multiple signaling pathways and can also regulate multiple signaling pathways [3]. Lipid metabolism connects to signaling networks in the regulation of cell growth, proliferation, differentiation, survival, apoptosis, inflammation, motility, and membrane homeostasis [4,5,6]. Meanwhile, lipid metabolism can alter membrane composition and permeability which cause the development and progression of many diseases including a variety of cancers [7].
2. Lipid Metabolism
According to the International Lipid Classification and Nomenclature Committee, lipids are currently classified into eight categories: (1) fatty acids; (2) glycerolipids; (3) glycerophospholipids; (4) sphingolipids; (5) sterol lipids; (6) prenol lipids; (7) saccharolipids; and (8) polyketides [8]. In the cells, the structure of lipids determines their function and metabolic fate [1,2]. Lipids that are currently understood as most relevant to cancer development and chemotherapy are fatty acids, glycerolipids, glycerophospholipids, sphingolipids, and sterol lipids.
Fatty acids composed of a hydrocarbon chain with one terminal carboxyl group (COOH) are produced by fatty acid synthases from acetyl-CoA and malonyl-CoA precursors, by lipases in the degradation of glycerolipids or by phospholipase A1, A2 and B in the breakdown of glycerophospholipids. The degradation of fatty acids via β-oxidation leads to the release of energy (large quantities of ATP) and generates reactive oxygen species [9,10]. Glycerolipids, fatty acid esters of glycerol (mono-, di-, or tri-glyceride), are biosynthesized by the sn-glycerol-3-phosphate pathway which predominates in liver and adipose tissue and the monoacylglycerol pathway in the intestines [11].
Glycerophospholipids are the main component of biological membranes and contain at least one O-1-acyl, O-1-alkyl, or O-1-alkenyl residue attached to the glycerol moiety. The presence of an additional head group (such as choline, ethanolamine, serine, inositol, and glycerol) attached to the phosphate allows for many different glycerophospholipids. Both biosynthesis (CDP-DAG pathway and Kennedy pathway) and degradation (different phospholipases) of glycerophospholipids are regulated by different signaling pathways (Figure 1). Interestingly, many different bioactive lipid molecules, such as inositol trisphosphate, diacylglycerol, arachidonic acid, phosphatidic acid, and lysophosphatidic acid, are generated during glycerophospholipid metabolism, and these bioactive lipid molecules in turn regulate different signaling pathways in the cells [12,13]. The metabolism of glycerophospholipids is very complex and it is not fully understood how and when their substitutions and modifications occur.
Figure 1.
Glycerophospholipid metabolism. Left, glycerophospholipid synthesis; Right, glycerophospholipid degradation. The enzymes are choline kinase (ChoK), ethanolamine kinase (EthK), cytidine 5'-triphosphate (CTP)-phosphocholine (or phosphethanolamine) cytidylyltransferase (CCT), cholinephosphotransferase (CPT), ethanolaminephosphotransferase (EPT), phosphatidylethanolamine N-methyltransferase (PEMT), CDP-diacylglycerol synthase (CDS), phosphatidylglycerol synthase (PGS), phosphatidylserine synthase (PSS), phosphatidylserine decarboxylase (PSD), phosphatidylinositol synthase (PIS), phosphatidylinositol kinase (PIK), phosphatidylinositol phosphate kinase (PIPK), phospholipase A1 (PLA1), phospholipase A2 (PLA2), phospholipase B (PLB), phospholipase C (PLC), and phospholipase D (PLD). P-Cho, phosphocholine; P-Eth, phosphoethanolamine; CDP-Cho, CDP-choline; CDP-Eth, CDP-ethanolamine; PA, phosphatidic acid; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PG, phosphatidylglycerol; PS, phosphatidylserine; PI, phosphatidylinositol; PIP, phosphatidylinositol phosphate; PIP2, phosphatidylinositol bisphosphate; R1 and R2, acyl group; and Head groups are choline, inositol, serine, ethanolamine or glycerol.
Figure 1.
Glycerophospholipid metabolism. Left, glycerophospholipid synthesis; Right, glycerophospholipid degradation. The enzymes are choline kinase (ChoK), ethanolamine kinase (EthK), cytidine 5'-triphosphate (CTP)-phosphocholine (or phosphethanolamine) cytidylyltransferase (CCT), cholinephosphotransferase (CPT), ethanolaminephosphotransferase (EPT), phosphatidylethanolamine N-methyltransferase (PEMT), CDP-diacylglycerol synthase (CDS), phosphatidylglycerol synthase (PGS), phosphatidylserine synthase (PSS), phosphatidylserine decarboxylase (PSD), phosphatidylinositol synthase (PIS), phosphatidylinositol kinase (PIK), phosphatidylinositol phosphate kinase (PIPK), phospholipase A1 (PLA1), phospholipase A2 (PLA2), phospholipase B (PLB), phospholipase C (PLC), and phospholipase D (PLD). P-Cho, phosphocholine; P-Eth, phosphoethanolamine; CDP-Cho, CDP-choline; CDP-Eth, CDP-ethanolamine; PA, phosphatidic acid; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PG, phosphatidylglycerol; PS, phosphatidylserine; PI, phosphatidylinositol; PIP, phosphatidylinositol phosphate; PIP2, phosphatidylinositol bisphosphate; R1 and R2, acyl group; and Head groups are choline, inositol, serine, ethanolamine or glycerol.

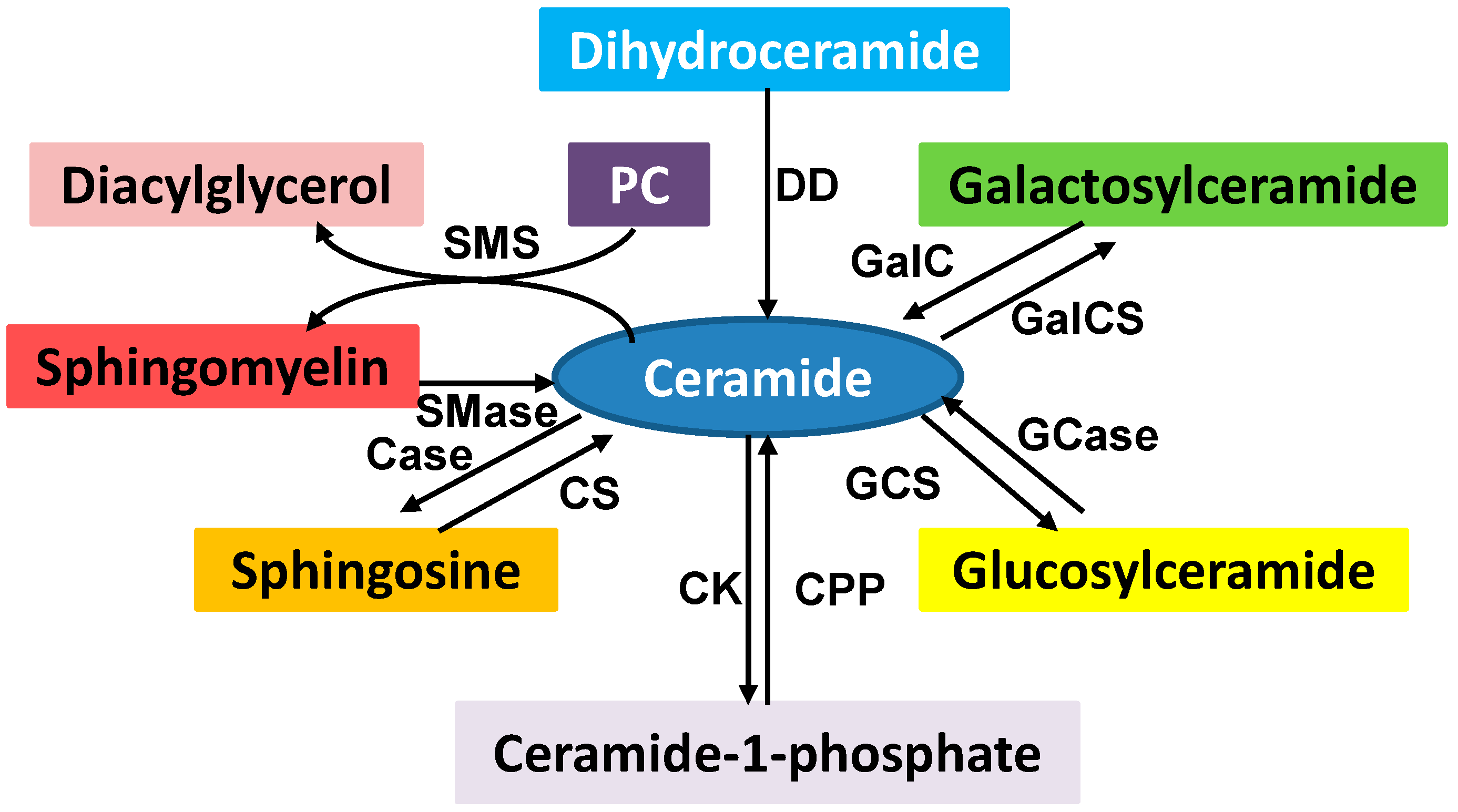
Sphingolipids, including the sphingomyelins and glycosphingolipids, are de novo synthesized in the endoplasmic reticulum (ER) from nonsphingolipid precursors [14]. Sphingomyelins can be hydrolyzed by sphingomyelinases to produce ceramides and phosphocholine. The conversion of sphingosine to sphingosine-1-phosphate, sphingosine to ceramide, ceramide to ceramide-1-phosphate, and ceramide to glucosylceramide is catalyzed by different enzymes (Figure 2). Sphingolipids are also structural components of cell membrane, and the products of sphingolipid metabolism such as ceramide, ceramide-1-phosphate, sphingosine, sphingosine-1-phosphate, and glucosylceramide act as bioactive lipid molecules in apoptotic and drug-resistant signaling.
Figure 2.
Sphingolipid metabolism. Ceramide is a key intermediator in sphingolipid metabolism. The enzymes involved in sphingolipid metabolism are ceramidase (Case), ceramide kinase (CK), ceramide-1-phophosphate phosphatase (CPP), ceramide synthase (CS), dihydroceramide desaturase (DD), galactosylceramide synthase (GalCS), galactocer (GalC), glucosylceramide synthase (GCS), glucosylceramidase (Gcase), sphingomyelinase (Smase), and sphingomyelin synthase (SMS). Many of these products play an important role in cell signaling which regulates a variety of cellular functions. SMS converts phosphatidylcholine (PC) and ceramide to sphingomyelin and diacylglycerol which brings two major classes of lipids in cell metabolism and signaling.
Figure 2.
Sphingolipid metabolism. Ceramide is a key intermediator in sphingolipid metabolism. The enzymes involved in sphingolipid metabolism are ceramidase (Case), ceramide kinase (CK), ceramide-1-phophosphate phosphatase (CPP), ceramide synthase (CS), dihydroceramide desaturase (DD), galactosylceramide synthase (GalCS), galactocer (GalC), glucosylceramide synthase (GCS), glucosylceramidase (Gcase), sphingomyelinase (Smase), and sphingomyelin synthase (SMS). Many of these products play an important role in cell signaling which regulates a variety of cellular functions. SMS converts phosphatidylcholine (PC) and ceramide to sphingomyelin and diacylglycerol which brings two major classes of lipids in cell metabolism and signaling.

Sterol lipids, such as cholesterol, are biosynthesized in a highly complex series of at least thirty different enzymatic reactions to form four linked hydrocarbon rings (hexagons and pentagon) and are not readily biodegradable. Cholesterol is an integral component of cellular membranes, determines membrane rigidity and fluidity, and plays a crucial role in membrane organization, dynamics, and function [15]. Some steroid lipids, such as vitamins, testosterone, estrogen, and cortisone are ligands and can regulate cell signaling to control a myriad of bodily functions. Recently, more and more data supports that the levels of cellular cholesterol are significantly increased in cancer cells and tissues, and cholesterol promotes cell proliferation, tumor progression, and drug resistance [16,17].
Prenol lipids, Saccharolipids and Polyketides are produced mainly in bacteria, fungi and plants [18,19,20]. Some of these lipids and their derivatives are vitamins or antimicrobial, antiparasitic, and anticancer agents. For instance, vitamins A, E, and K belong to prenol lipids, lipopolysaccharides are one of the most familiar saccharolipids [19], and erythromycins, tetracyclines, avermectins, and epothilones are polyketides or their derivatives [21].
Lipoproteins are not classified as lipids but are a group of biochemical assemblies that contains both proteins and lipids, covalently or non-covalently bound to the proteins, which allow fats to move through the water inside and outside of the cells. The proteins serve to emulsify lipid molecules. Most importantly, lipoproteins can be enzymes, transporters, structural proteins, antigens, adhesins, or toxins that can regulate cellular functions including apoptosis [22].
3. Regulation of Lipid Metabolism
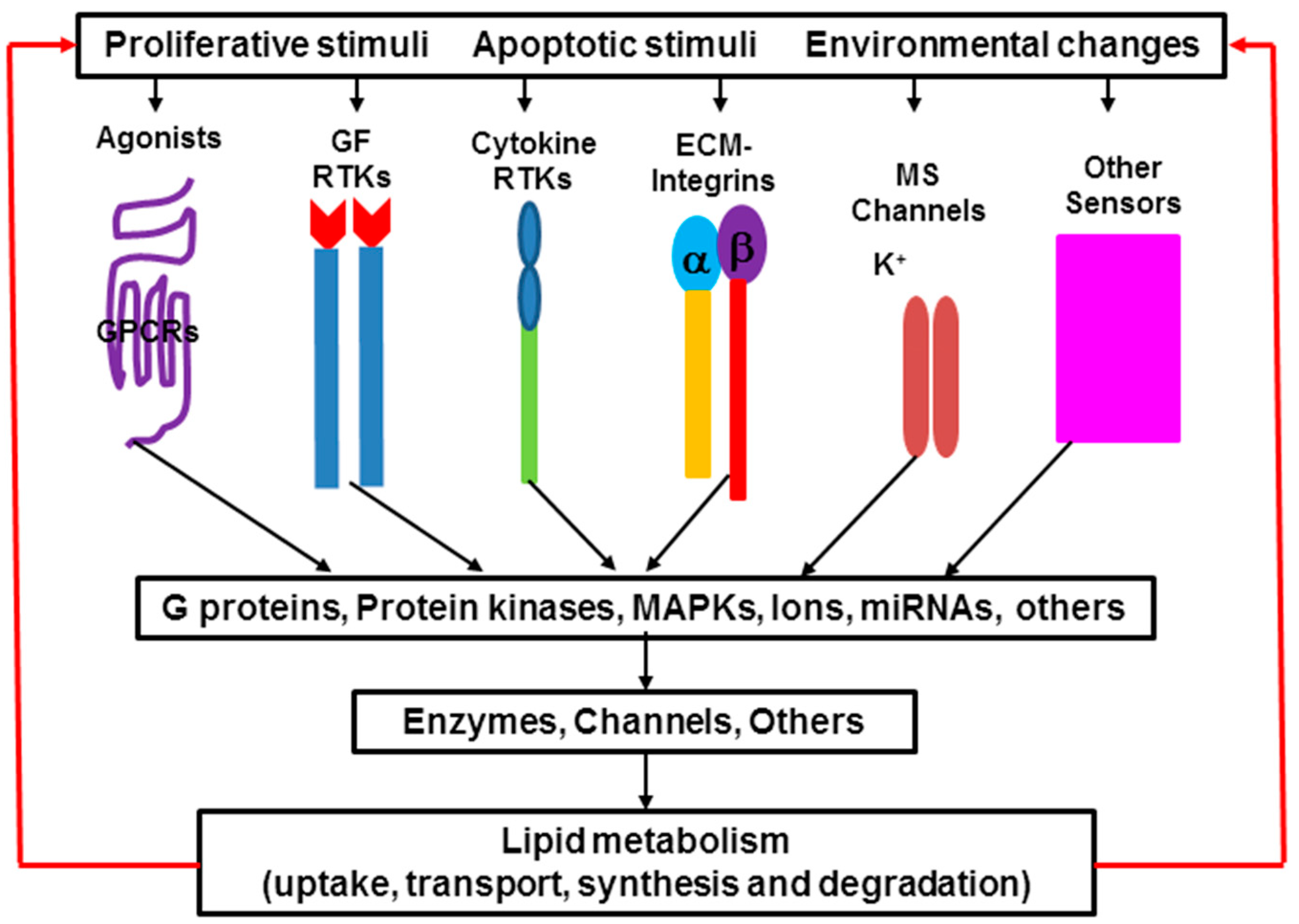
Lipid metabolism, including lipid uptake, transport, synthesis, and degradation, is a complex process. Biosynthesis and degradation of different lipids are regulated by different signaling pathways, and the same lipid can be regulated by different signaling pathways in different tissues and cells as well as under physiological, pathophysiological or therapeutic conditions [1,2]. Either activation or inhibition of these signaling pathways is based on cell needs and responds to environmental changes (Figure 3). There are more than one hundred enzymes regulating lipid metabolism in the cells. Recent studies show that expression of many of these enzymes is regulated by microRNA (miRNA). It indicates that miRNA also plays an important role in lipid metabolism. Alteration of lipid metabolism leads to the changes of membrane compositions, protein distribution and function, gene expression, and cellular functions, and further causes the development and progression of many diseases such as inflammation, hypertension, diabetes, liver disease, heart disease, renal disease, neurological disorder, cystic fibrosis, and cancer [3,23]. On the other hand, manipulation of lipid metabolism can lead cancer cells to apoptosis.
Figure 3.
Signaling transduction in lipid metabolism. Extracellular signals induce different pathways that regulate lipid metabolism in the cells. Protein kinases include protein kinase A, B and C; mitogen-activated protein kinases (MAPK) includes extracellular signal-regulated kinases (ERK), p38 kinase and c-Jun N-terminal kinases (JNK); Enzymes that are involved in lipid uptake, transport, synthesis and degradation; Ions: Ca2+, K+, Na+, H+. Some bioactive lipid molecules are ligands, and in turn induce different signaling pathways. MS, mechanical stress.
Figure 3.
Signaling transduction in lipid metabolism. Extracellular signals induce different pathways that regulate lipid metabolism in the cells. Protein kinases include protein kinase A, B and C; mitogen-activated protein kinases (MAPK) includes extracellular signal-regulated kinases (ERK), p38 kinase and c-Jun N-terminal kinases (JNK); Enzymes that are involved in lipid uptake, transport, synthesis and degradation; Ions: Ca2+, K+, Na+, H+. Some bioactive lipid molecules are ligands, and in turn induce different signaling pathways. MS, mechanical stress.

3.1. Signaling Pathways in Lipid Metabolism
G protein-coupled receptors (GPCRs): GPCRs are a superfamily of receptors that are vital in a wide array of physiological processes and are the most important class of membrane proteins in clinical medicine accounting for approximately 40% of all current therapeutics [24,25]. Allosteric ligands bind to GPCRs leading to the activation of G protein and downstream enzymes involved in lipid uptake, transport, synthesis, and degradation. Over the past three decades, research has been not only centered on the identification of GPCR-signaling that regulates lipid metabolism but has also demonstrated that some bioactive lipid molecules, such as lysophosphatidic acid, sphingosine-1 phosphate, free fatty acids, and platelet activating factors, are ligands for activating GPCR-signaling [26,27,28].
Tyrosine kinases: Cytokines and growth factors exert their biological effects by binding to specific cell surface receptors on target cells. Most of these receptors have a tyrosine kinase activity domain that is localized at the cytoplasmic region of the molecule [29,30]. The interaction of the cytokines and growth factors with the receptors induces the kinase activity of the receptor, and further activates downstream effectors such as protein phosphorylation and enzyme activation. Cytokine signaling pathways respond to innate immunity and inflammation which can induce phospholipases (A2, C, and D), sphingomyelinases, and the enzymes that regulate cholesterol metabolism [31,32]. Mitogenic signaling carried out by growth factors regulates cell growth and proliferation which is involved in the activation of many lipid-metabolism-related enzymes [29,32]. Recently, a large body of evidence has indicated that agonists of some GPCRs can activate growth factor receptor tyrosine kinases in the absence of growth factor [33,34]. The transactivation by GPCRs also links to cellular lipid synthesis and degradation.
Integrin signaling: Integrin signaling governs cellular adhesion and transmits signals leading to the activation of intracellular signaling pathways aimed to prevent apoptosis. This regulation is associated with lipid metabolism. Integrin-associated Lyn kinase can promote cell survival by suppressing acid sphingomyelinase activity [35]. In bovine pulmonary artery endothelial cells integrin signaling causes arachidonic acid release by membrane translocation and phosphorylation of cytosolic phospholipase A2 as well as tyrosine phosphorylation of the mitogen-activated protein kinase (MAPK) [36]. Tyrosine kinase Btk regulates E-selectin-mediated integrin activation and neutrophil recruitment by controlling phospholipase C γ2 and phosphatidylinositol-3 Kinase γ pathways [37]. Integrin signaling also links large integrin-associated intracellular protein complexes, which act as anchors for the cytoskeleton and as signaling hotspots, and regulates trafficking of cholesterol-enriched membrane microdomains known as lipid rafts [38,39].
Ion-channel signaling: Cells commonly use membrane lipids to modulate the function of ion channels. The interaction of ion channels with membrane lipids can be highly specific and is often important for full functional and structural integrity of ion channels [40]. Recent studies indicate mechanistic insights into how lipid modification (for example palmitoylation) controls large conductance calcium- and voltage-activated potassium channel trafficking and cross-talk with phosphorylation-dependent signaling pathways [41]. In addition, a constantly growing body of literature reveals that some agonists, despite their direct effect on ion channels, may also influence functions of ion channels via cellular lipid metabolism. For instance, the conversion of sphingomyelin to ceramide or phosphatidylinositol bisphosphate to diacylglycerol not only contributes to the membrane surface potential, but also affects the functional properties of some channels (from the opened state to the closed state of the channels) [42,43].
Other signaling: Signaling pathways that respond to environmental changes such as pH changes, oxidative stress, and mechanical stress also play an important role in lipid metabolism. The pH-dependent group II phospholipase A2 enhances membrane phospholipid degradation which contributes to chemical hypoxic and ischemic injury of rat hepatocytes [44], and pH also activates phospholipase C during intracellular infection [45]. Oxidative stress induces arachidonate release from human lung cells [46] and redox-active antioxidant modulation of lipid signaling in vascular endothelial cells by the regulation of phospholipase D, phospholipase A2, lipoxygenase, and cyclooxygenase [47]. Mechanical forces generated from blood pressure and blood flow are responsible for lipid metabolism through the activation of phospholipases A2, C, and D, phosphatidylinositol 3-kinase, sphingomyelinase, and phosphatidylcholine biosynthesis via a variety of signaling pathways [48]. Sterols, bile acids, and fatty acids are the endogenous ligands of many nuclear receptors such as orphan receptor, farnesoid X receptor, peroxisome proliferator-activated receptor, vitamin D receptor, constitutive androstane receptor, and pregnane X receptor [49]. These receptors coordinately regulate lipid, glucose, energy, and drug metabolism [50]. Many non-enzymatic proteins, such as p53, caveolin, and cell-death-inducing DNA fragmentation factor 45-like effector, are also involved in lipid metabolism [51,52]. The signaling network of lipid metabolism can activate different kinases and a variety of other enzymes that regulate different cell functions.
3.2. Enzymes in Lipid Metabolism
Fatty acid synthase, choline kinase, ceramide synthase, phosphatidylinositol-3 or -4 kinases, and the enzymes that catalyze the generation of fatty acids, phospholipids, and cholesterol all play integral roles in lipid biosynthesis which can alter lipid compositions in cellular membranes and cell fates by regulating cellular functions. Fatty acid synthase and choline kinase are overexpressed in many cancer cell lines and tumors [53,54]. Understanding the regulation of these enzymes in cancer cells not only to explore how they contribute to cancer cell growth, proliferation and tumor progression, but also to evaluate whether their specific inhibitors can be used for cancer chemotherapy [55]. The levels of cellular cholesterol are much higher in cancer cell lines and tissues than their normal compartments [56,57,58,59]. The data indicate that overexpression of enzymes in cholesterol synthesis or activation of these enzymes occurs in cancer cells. Cholesterol is capable of promoting cell proliferation, migration, tumor progression, and chemotherapy resistance [16,17]. Cholesterol-lowering agents could induce cancer cell apoptosis and exhibit important antitumor activity [60,61,62,63,64,65].
Phospholipases A2, C, and D, sphingomyelinases and ceramidase are key enzymes in the regulation of lipid degradation during cell response to stimuli. Phospholipase A2-mediated phospholipid degradation generates free fatty acids and accompanies with the generation of lysophospholipids which are emerging as a novel class of inflammatory lipids [1,2]. Phospholipase C hydrolyzes glycerophospholipids to generate diacylglycerol which uniquely functions as a basic component of membranes, an intermediate in lipid metabolism, and a key element in lipid-mediated signaling [66]. Inositol trisphosphate, one of the products of phospholipase C in the hydrolysis of polyphosphoinositides, binds to inositol-trisphosphate receptor and evokes intracellular Ca2+ signaling for normal cell survival and is also actively involved in apoptosis induction and progression [67]. Phospholipase D catalyzes the hydrolysis of the terminal diester bond of glycerophospholipids with the formation of phosphatidic acid plus head groups. Phosphatidic acid regulates the activity of small GTPases or directly binds some small GTPases to membranes [68]. Recent studies show that phosphatidic acid directly interacts with mammalian target of rapamycin (mTOR) in a manner that is competitive with rapamycin and is required for the stability and kinase activity of both mTOR complexes (mTORC1 and mTORC2) [69,70]. Phospholipase d-phosphatidic acid-mTOR signaling leads to podocyte hypertrophy and apoptosis [71]. Sphingomyelinases hydrolyze sphingomyelin to produce ceramide which can also be synthesized de novo by ceramide synthase. Ceramide was the first lipid molecule linked to cell death signaling about two decades ago and has received considerable attention as a key regulator of programmed cell death [72,73].
Many enzymes, such as cyclooxygenase, lipoxygenase, prostaglandins synthase, diacylglycerol kinase, phosphatidate phosphatase, and glucosylceramide synthase, are also very important in the cells because they can convert one lipid molecule to another in the cells (Figure 1 and Figure 2) [1,2,3,4,5]. For instance, ceramide can be converted to sphingosine, ceramide-1-phosphate, and glucosylceramide by ceramidase, ceramide kinase, or glucosylceramide synthase [74,75]. These converted derivatives are also important signaling molecules but play different roles in the regulation of cellular functions [72,73,74,75].
3.3. MicroRNAs (MiRNAs) in Lipid Metabolism
MiRNAs are small, evolutionarily conserved, and non-coding RNA molecules (containing about 22 nucleotides) that can regulate gene expression at the posttranscriptional level. To date, more than 30 miRNAs, including miRNA-122, miRNA-33, and miRNA-370, have been discovered to play important roles in the regulation of lipid metabolism such as fatty acid oxidation, cholesterol efflux, and the biosynthesis of fatty acids, cholesterol, and triacylglycerol [76,77,78,79].
MiRNA-122 was initially identified as a highly abundant miRNA (~70% of total miRNA) in the liver [80]. This conserved, liver-specific miRNA has been associated with the regulation of liver metabolism, including the biosynthesis of fatty acids, triacylglycerol, and cholesterol, which can stimulate the production of endoplasmic reticulum-associated lipid droplets and cholesterol-rich membrane domains (lipid rafts or caveolae) [81,82]. MiRNA-122 also promotes the propagation of hepatitis C virus by multiple mechanisms, has been shown to be down-regulated in hepatocellular carcinoma, and plays a role in fatty liver disease [81,83]. MiRNA-33 is highly conserved across species and can be found in numerous cell types, including macrophages, hepatocytes, and endothelial cells. It has two isoforms (a and b) and is an intronic miRNA located in a non-coding region of sterol-regulatory binding factor (SRBF) genes which are involved in cholesterol uptake and synthesis [84]. MiRNA-33, which targets ATP-binding cassette transporter subfamily A1 (ABCA) gene controlling the movement of cholesterol out of the cell, is also a key posttranscriptional regulator of cellular cholesterol homeostasis [85]. MiRNA-33a and miRNA-33b also contribute to the regulation of fatty acid metabolism (β-oxidation) by modulating the expression of carnitine palmitoyltransferase1A, carnitine O-octanyltransferase, and hydroxylacyl-CoA dehydrogenase-3-ketoacyl-CoA thiolase [86]. MiRNA-33 is responsive to alterations in cholesterol levels associated with diet or medication, making miRNA potential biomarkers of response to environmental stimuli and targets of therapeutic interventions. Recent studies showed that miRNA-370 has similar effects on lipid metabolism as miRNA-122. MiRNA-370 controls the expression of miRNA-122, targets and regulates lipid metabolism by up-regulating multiple genes coding for SRBF 1c, diacylglycerol O-acyltransferase 2, fatty acid synthase, and acyl-CoA carboxylase 1 [87]. MiRNA-370 also targets carnitine palmitoyl transferase which mediates the transport of long-chain fatty acids across the membrane and fatty acid oxidation [76]. MiRNA-378, another intronic miRNA located within the genomic sequence of peroxisome proliferator-activated receptor γ coactivator-1α (a master regulator of energy metabolism), also plays important roles in regulating lipid metabolism by targeting estrogen-related receptors and GA-binding protein α in adipocyte differentiation and lipid synthesis [88]. MiRNA-106, -144 and -758 regulate ABCA1 expression involved in cholesterol metabolism. MiRNA-96, -125, -185, -223 and -455 have been recently described to regulate SRBF 1 expression and HDL (high-density lipoprotein) uptake [76,77]. MiRNA-1, -143, -206 and -371 can promote adipogenesis [78]. In contrast, miRNA-27, -130, -206, and -369 negatively regulate adipocyte differentiation [76,77,78,79].
4. Lipid and Apoptotic Signaling
Cell death that maintains organismal and cellular homeostasis has been defined as an irreversible loss of plasma membrane integrity which is associated with changes in membrane lipid metabolism. Three types of cell death can be distinguished in mammalian cells according to morphological criteria: autophagy, necrosis and apoptosis [89]. Cell death, either progressive or acute, is also a hallmark characteristic of cancer treatment and various diseases, including cardiac disease, brain injury, and renal failure [90].
4.1. Cell Death and Regulation
Autophagy is an evolutionarily conserved catabolic pathway that allows cells to degrade and recycle cellular components. This process mainly maintains a balance between the manufacture of cellular components and the breakdown of damaged or unnecessary organelles and cellular constituents. Disruption of autophagy is involved in diverse human diseases including cancer. Autophagy is death receptor-independent, and target of rapamycin (TOR) acts as an efficient gatekeeper, on which it exerts an inhibitory effect [91]. Necrosis is an accidental and uncontrolled form of cell death lacking underlying signaling events. The factors that cause necrosis are external to the cells or tissues, such as physical damage (mechanical stress and detergent-induced cytolysis), infection, toxins, or trauma, which can lead to cell injury and result in the unregulated digestion of cell components and the premature death of cells. Necrosis is often associated with pathological conditions such as injury of organs. There are two main necrotic pathways: the death receptor pathway which is stimulated by tumor necrosis factor (TNF) α, Fas ligand, and TNF-related apoptosis-inducing ligand (TRAIL) and the mitochondrial pathway that leads to the generation of reactive oxygen species, ATP depletion, the accumulation of H+ and acidosis, and mitochondrial dysfunction [92]. A relatively new form of necrosis, termed necroptosis or programmed necrosis, has been identified. It exhibits the features of necrosis and apoptosis, and is caspase independent but receptor interaction protein kinase dependent [93].
Apoptosis, or so-called programmed cell death, is a type of cell death that is not involved in an inflammatory response and occurs in a tightly controlled manner. In contrast to autophagy and necrosis, apoptotic signaling is triggered either by death receptor (extrinsic pathway) or by mitochondria (intrinsic pathway) [94,95]. The extrinsic pathway is that death receptor activated by TNF-α, Fas ligand (CD95/APO1) or TRAIL leads to the assembly of a death-inducing signaling complex formed by the death receptors, adapter proteins, and caspases (cysteinyl aspartate specific proteases) such as caspase-8 and caspase-10. Caspase-8 directly triggers caspase-3 activation or can interact with the intrinsic apoptotic pathway by cleaving Bid (a pro-apoptotic member of the Bcl-2 (B-cell lymphoma 2) family) to form the truncated Bid (tBid) which translocates to the mitochondria and results in the release of cytochrome c. The intrinsic pathway can be induced by a variety of upstream receptor-independent stimuli, such as anticancer drugs, toxins, and radiation, and causes the alteration of mitochondrial membrane property and function. The permeabilization of mitochondrial outer membrane results in the release of cytochrome c. Cytochrome c interacts with apoptotic protease activating factor 1 (Apaf-1) and pro-caspase-9 to form a caspase activation complex, the apoptosome. The response to death receptor and the permeabilization of mitochondrial membrane are directly associated with lipid metabolism.
4.2. Bioactive Lipid Molecules and Apoptosis
As described above, many bioactive lipid molecules play an important role in the regulation of many different cell functions. Here we discuss the bioactive lipid molecules that are produced in lipid metabolism and are associated with apoptosis.
4.2.1. Fatty Acids
Fatty acids can induce apoptosis in different cell types [96,97,98,99]. Short chain fatty acids (C2–5) inhibit histone deacetylases, resulting in a hyperacetylation of core histone proteins [53]. Hyperacetylation of histones is associated with transcriptional regulation and growth inhibition in colonic epithelial cells [54]. Long-chain fatty acids induce ER stress which in turn activates c-jun N-terminal kinase (JNK) and CEBP (CCAAT/enhancer binding protein)-homologous protein (CHOP). JNK leads to the up-regulation of the pro-apoptotic BH3 (Bcl-2 homology domain 3) only proteins p53 (tumor protein p53)-upregulated modulator of apoptosis (PUMA). CHOP enhances the expression of the proapoptotic BH3-only protein Bim, contributes to PUMA up-regulation, and mediates the generation of reactive oxygen species (ROS). Bim, in cooperation with PUMA, induces the activation of the multi-domain executioner proapoptotic protein Bax. Bax activation results in mitochondrial membrane permeabilization, activation of the caspase cascade and leads to cell death [100].
Oxidation of fatty acids is the source of increased production of mitochondrial ROS [10]. At low levels ROS is a signaling molecule, while at high levels it can damage organelles, particularly the mitochondria. The toxicity of fatty acid oxidation is related to both the chain length and the degree of unsaturation. The longer the chain is and the more unsaturated the species, the more toxic it is [101]. 4-Hydroxy-2-nonenal (HNE), a major α,β-unsaturated aldehyde product of n-6 fatty acid oxidation, is a highly toxic and most abundant stable end product of lipid peroxidation, and has been considered as an oxidative stress marker [102]. ROS can cause unwanted stress that can activate stress-activated protein kinase or JNK [102]. Oxidative damage and the associated mitochondrial dysfunction result in energy depletion, accumulation of cytotoxic mediators, modulation ligand-independent signaling by Fas (CD95) receptor, and caspase activation which lead to apoptosis. Fatty acids also activate AMP (adenosine monophosphate)-activated protein kinase [103], extracellular signal-regulated kinase (ERK) [104], GPCR signaling [105], Toll-like receptor 4/NF-κB [106], Src-JNK [107], and protein kinase C [108] signaling as well as sphingomyelinase-ceramide signaling [109] in the regulation of apoptosis. These data clearly demonstrate that oxidation of fatty acids has been implicated in the tissue damage, dysfunction, and injury associated with aging and other pathological states such as cancer, metabolic diseases, neurodegenerative diseases, cardiovascular and inflammatory complications [102,110].
4.2.2. Phosphatidic Acid
Phosphatidic acid is a key intermediate in glycerophospholipid metabolism. Earlier studies showed that phosphatidic acid is a signaling molecule with growth factor-like properties, inducing DNA synthesis and cell proliferation [111] and stimulating phospholipase C activation and calcium release [112] in the cultured cells. Later, phosphatidic acid, as a second messenger, can activate NADPH (nicotinamide adenine dinucleotide phosphate) oxidase in human polymorph nuclear leukocytes [113], which could link to apoptotic signaling. Recent evidence supports the involvement of phosphatidic acid in apoptotic signaling. For instance, cerium activates phospholipase D and produces phosphatidic acid which induces the biphasic burst of superoxide anions and regulates MAPK (mitogen-activated protein kinases)-mediated apoptosis [114]. Increasing levels of production of phosphatidic acid on the mitochondrial surface results in mitochondrial aggregation and facilitates the fusion process [115]. Phosphatidic acid, one of the major acidic phospholipids found in lysosome membrane, is essential for tBid-induced lysosomal membrane permeabilization and links to the lysosomal-mitochondrial mediated apoptotic pathway [69]. Galectin-8, a potent pro-apoptotic agent, induces phospholipase D/phosphatidic acid signaling pathway that enhances ERK-mediated apoptosis in Jurkat T cells [116]. We recently found that phosphatidic acid, produced by shear stress-induced phospholipase D activation, stimulates mTOR signaling, and causes podocyte hypertrophy and apoptosis [71]. Taken together, phosphatidic acid plays an important role in the regulation of apoptotic signaling.
4.2.3. Ceramide
Ceramide is capable of triggering apoptosis in almost any cell, including tumor cells. Ceramide can be generated by a de novo pathway (ceramide synthase) or by sphingomyelinases in response to various stress stimuli, such as cytokines, heat shock, growth factors, vitamin D, TNF-α, CD95/Fas, chemotherapeutic agents, toxin, irradiation, UV-light, and infection by different signaling pathways [72]. Elevation of cellular ceramide levels directly or indirectly regulates the activities of a number of enzymes and signaling components, including MAP kinases, ceramide-activated kinase, ceramide-activating serine/threonine phosphatases such as protein phosphatase 1A and 2A, protein kinase C ζ, phospholipases such as phospholipase A2 or D, CPP32-like caspases, cathepsin D, transcription factors such as NF-κB, and kinase suppressor ras [117,118,119,120,121,122]. These enzymes and signaling components play an important role in the regulation of apoptotic signaling. On the other hand, an irreversible step in apoptotic processing is mitochondrial outer membrane permeabilization which releases critical proteins such as cytochrome c. The channels for protein release are controlled by Bcl-2 family proteins based on cell physiological function: anti-apoptotic proteins (Bcl-x, Bcl-w, and others) destabilize the channels whereas pro-apoptotic proteins (Bax, BAD, Bak, Bok, and others) act synergistically with ceramide to increase membrane permeability [123]. Ceramide can self-assemble in the mitochondrial outer membrane to form large stable channels capable of releasing cytochrome c [124]. Cytochrome c further interacts with Apaf-1, activates several caspases and forces cell to undergo apoptosis. The role of ceramide in apoptosis indicates that ceramide could be a potential anticancer drug.
4.2.4. Cholesterol
Cholesterol modulates cell signaling through the cholesterol–protein interaction, cholesterol–phospholipid interaction, and membrane dynamics. Increasing cholesterol levels promotes cell proliferation, tumor progression, and chemotherapy resistance [16,17]. However, recent studies show that cholesterol is also involved in apoptotic signaling. A significant fraction of cholesterol that accumulates in atherosclerotic lesions is oxidized to yield a number of derivatives, called oxysterols. Cholesterol oxidation produces 7α-hydroxy-, 7β-hydroxy-, 7-keto-, 20-hydroxy-, and 25-hydroxycholesterol, and can also attack the Δ5 double bond of cholesterol, forming cholesterol-5,6-epoxide which could react spontaneously with nucleophiles and behave like alkylating agents with direct carcinogenic properties [125]. Oxysterols increase intracellular levels of ROS, induce modification of cellular proteins (pro- and anti-apoptotic molecules), and alter gene expression and mitochondrial membrane properties. It is clear that accumulation of oxysterols may strongly stimulate the mitochondrial pathway of apoptosis [126]. On the other hand, toxic amyloid beta peptides (Aβ) are overproduced and accumulate in mitochondrial matrix of experimental models of Alzheimer’s disease. Specific mitochondrial cholesterol pool sensitizes to Aβ-induced oxidant cell death and caspase-independent apoptosis by cholesterol-mediated perturbation of mitochondrial membrane dynamics [127]. Recent investigations have shown that dendrogenin A is a selective inhibitor of cholesterol epoxide hydrolase and it triggered tumor re-differentiation and growth control in mice and improved animal survival [128]; cholesterol-5,6-epoxide metabolites, moreover, contribute to the anticancer pharmacology of Tamoxifen [129]. Although cholesterol promotes cell proliferation, oxidized cholesterol can lead cells to apoptosis.
4.2.5. Apolipoproteins
Some apolipoprotein Ls (ApoL1 and ApoL6) share structural and functional similarities with Bcl-2 family proteins that play crucial roles in regulating apoptosis. ApoL1 is inducible by p53 in p53-induced cell death [130], and overexpression of ApoL6 induces the release of cytochrome c and Smac/DIABLO from mitochondria and activation of caspase-9 via a mitochondria-mediated pathway [131]. The levels of apolipoprotein E (ApoE) mRNA and protein are up-regulated during staurosporine-induced apoptosis and are also correlated with increased caspase-3 activity and apoptotic morphological changes [132]. ApoE synthesis induced by neuronal damage or stress indicates its neurotoxic effect and is associated with apoptotic signaling [133]. The genesis of atherosclerosis is also associated with lipoprotein oxidation [126]. The oxidized low-density lipoprotein could enhance arterial apoptosis via mitochondrial and death receptor pathways [134]. The oxidative stress has also been implicated in the cardiovascular complications in chronic renal failure patients [133].
4.2.6. Intracellular Calcium
Ca2+ is not a lipid but has strong correlations with lipid metabolism and cell death. Intracellular calcium homeostasis is crucial for healthy cells, and the disruption of intracellular calcium homeostasis will cause cell damage, and even death [135,136]. In mammalian cells, the endoplasmic reticulum (ER) forms the main intracellular Ca2+ reservoir. Intracellular Ca2+ can be mobilized from ER by inositol trisphosphate (IP3). IP3 is produced by phospholipase C-hydrolyzed phosphatidylinositol bisphosphate and can bind to IP3 receptor on the ER [137]. Extracellular Ca2+ enters to the cells controlled by membrane transporter such as plasma membrane calcium ATPases and channels such as Trp. These transporters and channels are regulated by lipids [126]. Apoptotic cells rely on increased intracellular Ca2+ concentrations [135], mediated by the release from ER and by capacitive Ca2+ influx through transporters or channels [137,138,139]. Mitochondrial uptake of Ca2+ causes ATP production, mitochondria outer membrane permeabilization and release of cytochrome c [135]. Recently, intracellular organelles coordinate complex molecular mechanisms in the regulation of Ca2+ signaling and lipid metabolism. A number of experimental evidence support that cell apoptosis regulated by alteration of intracellular Ca2+ homeostasis is associated with the tight interplay between ER and mitochondria known as the mitochondria-associated membrane (MAM) [140,141,142,143]. In mammalian cells, the formation of these contact sites appear to be required for key cellular events including rapid transmission of calcium from the ER to mitochondria, the import of phosphatidylserine into mitochondria from the ER for decarboxylation to phosphatidylethanolamine, the formation of autophagosomes, and the regulation of the morphology, dynamics and functions of mitochondria, and cell survival [144]. In a mouse model of the human lysosomal storage disease, GM1-ganglioside is accumulated in the glycosphingolipid-enriched microdomain (GEM) fractions of MAMs [145]. Meanwhile, the MAM fractions from rat liver contain highly active sphingolipid-specific glycosyltransferases [146].
5. Lipid Metabolism, Cancer Treatment and Drug Resistance
Cancer is characterized by uncontrolled cell growth with increased proliferation and decreased apoptosis and enhances migrating behavior of cells by promoting their ability to invade adjacent tissues and/or metastasize to non-adjacent organs and tissues. Cell proliferation requires duplication of all macromolecular components during each cell division. Aberrant lipid metabolism is now recognized as one of the key features of cancer cells because cell proliferation requires increased lipid biosynthesis, and lipid catabolism produces bioactive molecules which act as signal molecules to regulate cancer metastasis [1,2,7].
5.1. Lipid Metabolism in Cancer
Three classical lipids, fatty acids, phospholipids and cholesterol, are dramatically increased and actively biosynthesized in cancer cells and tumors. At first, evidence shows that expression and activity of fatty acid synthase are extremely low in nearly all nonmalignant adult tissues, whereas it is significantly up-regulated in a number of solid and aggressive cancers [147]. Fatty acids are also building blocks for glycerolipids, glycerophospholipids, and other lipids. Secondly, expression of choline kinase, a key enzyme in biosynthesis of phosphatidylcholine, is up-regulated in a variety of cancer cell lines and tumors, and choline kinase can be activated by different growth factors and oncogene-coding proteins such as ras [148]. Thirdly, active sterol biosynthesis remains an essential metabolic component of cell proliferation. Up-regulating cholesterol biosynthesis and cholesterol efflux are only discovered in proliferating normal tissues and tumors [15,16,17]. Transcriptional profiling by microarray has demonstrated that refractory cancers exhibit significant overexpression of a number of genes in cholesterol biosynthetic pathway [56]. Cholesterol biosynthesis happens much earlier than DNA synthesis, and inhibiting cholesterol biosynthesis slows cell growth, suggesting a linkage between the cholesterol and DNA synthetic pathways [149]. Lipid metabolism in cancer cells remains largely unknown. Recently, lipidomics (also called lipid profiling) has provided more details of lipid metabolism by comparing lipid profiles of normal and cancer cells or tissues [150,151], which could be useful for identifying clinical biomarkers for earlier diagnosis, and allow evaluation of determining the efficacy of cancer therapy.
5.2. Anticancer Drugs, Lipid Metabolism and Apoptosis
A number of anti-cancer drugs are lipid-based or effective in terms of their ability to regulate lipid metabolism. Many anticancer drugs, such as cytarabine, daunorubicin, doxorubicin, etoposide, fludarabine, irinotecan, paclitaxel, tamoxifen, taxol, vinblastine, and vincristine, can impact ceramide accumulation by inducing ceramide synthase to catalyze de novo ceramide synthesis or by activating sphingmyelinase to catalyze sphingomyelin degradation [117,152]. Based on the structure of ceramide, ceramide analogs such as ceramidoids, 4, 6-diene-ceramide, and C16-serinol are also used as anticancer drugs [117]. Some anticancer drugs target fatty acid synthesis by inhibiting fatty acid synthase [153], phospholipase A2 [154], and lipases [155]. Some anticancer drugs can significantly reduce the levels of cellular cholesterol by blocking different steps of cholesterol biosynthesis [16,17,156]. Some anticancer drugs are developed based on blocking the conversion of lipid products [75]. In each case, anticancer drugs cause the alternations of lipid metabolism in cancer cells and the result leads to cancer cells to growth arrest and/or apoptosis.
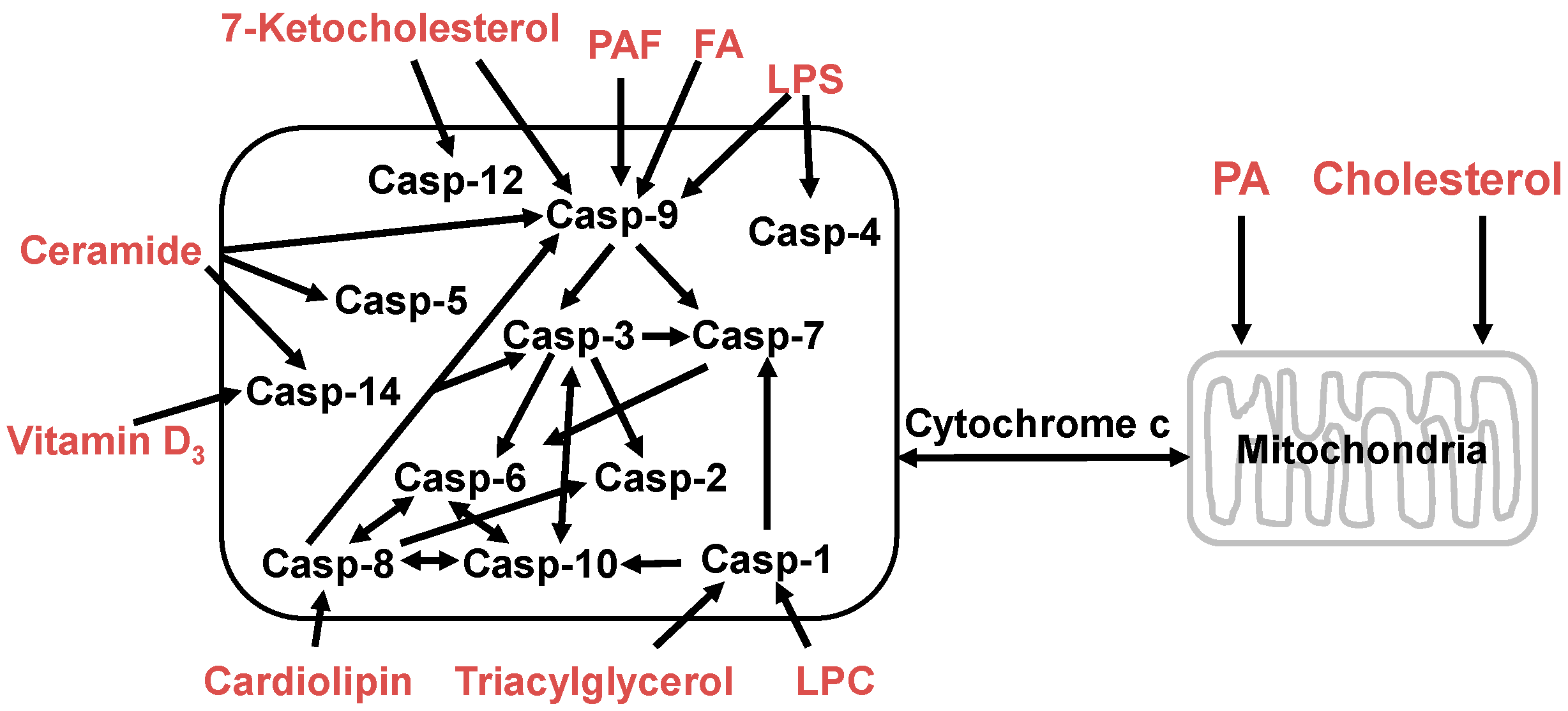
Some lipids directly induce caspase activation leading to programmed cell death. For example, triglyceride [157], lysophosphatidylcholine [158], lipopolysaccharide [159], and cholesterol [160] can induce or trigger caspase-1 activation. Apoptosis induced by fatty acids and their derivatives is associated with significant activation of caspase-2, -3, -6, -7, -8 and -9 [161,162]. Cardiolipin is a mitochondria-specific phospholipid and provides an essential activating platform for caspase-8 on mitochondria [163]. The 7-Ketocholesterol activates caspases-3, -7, -8, and -12 in human microvascular endothelial cells in vitro [164]. In rat small intestine platelet-activating factor promotes mucosal apoptosis via Fas ligand-mediating caspase-9 active pathway [165]. Vitamin D3 induces caspase-14 expression and enhances caspase-14 activation in processing organotypic skin cultures [166]. Ceramide stimulates caspase-3, -5, -7, -8, -9 and -14 activities leading to apoptosis in many cells and tissues [166,167,168,169,170,171]. These lipids can be used or developed as anticancer drugs (Figure 4).
5.3. Lipid Metabolism and Drug Resistance
In spite of many significant progresses in cancer therapy, cancer is still a major disease that causes more than 8 million deaths, or about 15% of all human deaths around the world every year because most cancer patients eventually develop drug resistance [172]. Drug resistance of cancer cells represents a serious barrier to successful clinic treatment, and inherent drug resistance of cancer cells is caused by multiple mechanisms. The molecular mechanisms of drug resistance can be caused by gene mutations which can enzymatically deactivate the drug, alter the drug-specific binding site, decrease drug permeability and/or increase active efflux (pumping out) of the drugs across plasma membrane, and/or change of the metabolic pathway to yield different non-cytotoxic products. Many of these processes are associated with the alteration of lipid metabolism. Ceramide is a center of sphingolipid metabolism, and more than eleven different enzymes use ceramide as a substrate (ceramidase, ceramide kinase, glycosylceramide synthase, galactosylceramide synthase, and sphingomyelin synthase) or directly convert other molecules to ceramide (dihydroceramide desaturase, sphingomyelinase, ceramide-1-phosphate phosphatase, glucocerebrosidase, galactocerebrosidase, and ceramide synthase) [75,163]. One of the best examples of how changing metabolic pathways may lead cancer cell to drug resistance is that ceramide-generating cancer chemotherapeutic drugs impact the accumulation of ceramide [163] and the increased levels of cellular ceramide drives cancer cell death [73]. Accumulation of cellular ceramide also activates glucosylceramide synthase which converts ceramide to glucosylceramide, thereby reducing ceramide levels in the cells [75]. Glucosylceramide has been demonstrated to stimulate cell growth and DNA synthesis which drive cancer cell resistance to chemotherapy [173]. Other ceramide derivatives such as ceramide-1-phosphate and sphingosine-1-phosphate also regulate cell survival and proliferation pathways, and could lead to drug resistance, as well [161].
Figure 4.
Lipids regulate apoptotic signaling in the cells. Phosphatidic acid (PA) and cholesterol can modulate mitochondrial membrane permeability and triggers apoptosis via the lysosomal-mitochondrial pathway; and the bioactive lipid molecules, such as platelet-activating factor (PAF), fatty acids (FA), lysophosphatidylcholine (LPC), lipopolysaccharide (LPS), 7-ketocholesterol, ceramide, vitamin D3, cardiolipin, and triacylglycerol, can induce apoptotic signaling pathways by activating different caspases. The released cytochrome c in mitochondrial pathway interacts with Apaf-1 and pro-caspase-9 to form a caspase (Casp) activation complex, the apoptosome, or caspase activation can cleave Bid (a pro-apoptotic member of the Bcl-2 family) to form the truncated Bid (tBid) which translocates to the mitochondria and results in the release of cytochrome c.
Figure 4.
Lipids regulate apoptotic signaling in the cells. Phosphatidic acid (PA) and cholesterol can modulate mitochondrial membrane permeability and triggers apoptosis via the lysosomal-mitochondrial pathway; and the bioactive lipid molecules, such as platelet-activating factor (PAF), fatty acids (FA), lysophosphatidylcholine (LPC), lipopolysaccharide (LPS), 7-ketocholesterol, ceramide, vitamin D3, cardiolipin, and triacylglycerol, can induce apoptotic signaling pathways by activating different caspases. The released cytochrome c in mitochondrial pathway interacts with Apaf-1 and pro-caspase-9 to form a caspase (Casp) activation complex, the apoptosome, or caspase activation can cleave Bid (a pro-apoptotic member of the Bcl-2 family) to form the truncated Bid (tBid) which translocates to the mitochondria and results in the release of cytochrome c.

6. Conclusions
Lipid metabolism is very complex and is regulated by a complex signaling network in the cells. The same lipid molecule, via different signaling pathways or under different conditions, can generate different metabolites. Understanding and defining signaling pathways of lipid metabolism in cancer cells can provide rational targets for therapy, and determining the function of different lipid molecules could develop new anticancer drugs for clinical evaluation. Clearly, better understanding of lipid metabolism in cancer therapy and apoptosis requires further elucidation and investigation to develop new and better cancer treatments for future cancer patients.
Abbreviations
| ABCA | ATP-binding cassette transporter subfamily A1 |
| Aβ | amyloid beta peptides |
| Apaf | apoptotic protease activating factor |
| ApoL | apolipoprotein L |
| ApoE | apolipoprotein E |
| CHOP | C/EBP-homologous protein |
| ERK | Extracellular signal-regulated kinase |
| GPCR | G protein-coupled receptor |
| HDL | high-density lipoproteins |
| HMG-CoA | 3-Hydroxy-3-methyl-glutaryl-CoA |
| JNK | c-jun N-terminal kinase |
| LDL | low-density lipoproteins |
| MAM | mitochondria associated membrane |
| MAPK | mitogen-activated protein kinase |
| miRNA | microRNA |
| mTOR | mammalian target of rapamycin |
| PA | phosphatidic acid |
| PUMA | proteins p53-upregulated modulator of apoptosis |
| ROS | reactive oxygen species |
| SRBF | sterol-regulatory binding factor |
| TNF | tumor necrosis factor |
| RAIL | TNF-related apoptosis-inducing ligand |
Acknowledgments
This work was partially completed at the Ellis Fischel Cancer Center, Department of Medicine, University of Missouri, Columbia.
Author Contributions
This manuscript was conceived jointly by both authors. Chunfa Huang wrote the first draft of the manuscript and both authors have approved the final manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Vance, J.E.; Vance, D. Biochemistry of Lipids, Lipoproteins and Membranes, 5th ed.; Elsevier: Amsterdam, The Netherlands, 2008. [Google Scholar]
- Prasad, S.K. Biochemistry of Lipids; Discovery Publishing House: New Delhi, DEL, India, 2010. [Google Scholar]
- Mattes, R.D. Fat taste and lipid metabolism in humans. Physiol. Behav. 2005, 86, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Zechner, R.; Zimmermann, R.; Eichmann, T.O.; Kohlwein, S.D.; Haemmerle, G.; Lass, A.; Madeo, F. Fat signals—Lipases and lipolysis in lipid metabolism and signaling. Cell Metab. 2012, 15, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Krycer, J.R.; Sharpe, L.J.; Luu, W.; Brown, A.J. The Akt-SREBP nexus: Cell signaling meets lipid metabolism. Trends Endocrinol. Metab. 2010, 21, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.R.; Schulze, A. Lipid metabolism in cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef] [PubMed]
- Fahy, E.; Subramaniam, S.; Murphy, R.; Nishijima, M.; Raetz, C.; Shimizu, T.; Spener, F.; van Meer, G.; Wakelam, M.; Dennis, E.A.; et al. Update of the LIPID MAPS comprehensive classification system for lipids. J. Lipid Res. 2009, 50, S9–S14. [Google Scholar] [CrossRef] [PubMed]
- Petho, G.; Reeh, P.W. Sensory and signaling mechanisms of bradykinin, eicosanoids, platelet-activating factor, and nitric oxide in peripheral nociceptors. Physiol. Rev. 2012, 92, 1699–1775. [Google Scholar] [CrossRef] [PubMed]
- Rosca, M.G.; Vazquez, E.J.; Chen, Q.; Kerner, J.; Kern, T.S.; Hoppel, C.L. Oxidation of fatty acids is the source of increased mitochondrial reactive oxygen species production in kidney cortical tubules in early diabetes. Diabetes 2012, 61, 2074–2083. [Google Scholar] [CrossRef] [PubMed]
- Athenstaedt, K.; Daum, G. The life cycle of neutral lipids: Synthesis, storage and degradation. Cell. Mol. Life Sci. 2006, 63, 1355–1369. [Google Scholar] [CrossRef] [PubMed]
- Tappia, P.S.; Singal, T. Phospholipid-mediated signaling and heart disease. Subcell. Biochem. 2008, 49, 299–324. [Google Scholar] [PubMed]
- Oude Weernink, P.A.; Han, L.; Jakobs, K.H.; Schmidt, M. Dynamic phospholipid signaling by G protein-coupled receptors. Biochim. Biophys. Acta 2007, 1768, 888–900. [Google Scholar] [CrossRef] [PubMed]
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An overview of sphingolipid metabolism: From synthesis to breakdown. Adv. Exp. Med. Biol. 2010, 688, 1–23. [Google Scholar] [PubMed]
- Maxfield, F.R.; Tabas, I. Role of cholesterol and lipid organization in disease. Nature 2005, 438, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Luu, W.; Sharpe, L.J.; Gelissen, I.C.; Brown, A.J. The role of signalling in cellular cholesterol homeostasis. IUBMB Life 2013, 65, 675–684. [Google Scholar] [CrossRef]
- Gorin, A.; Gabitova, L.; Astsaturov, I. Regulation of cholesterol biosynthesis and cancer signaling. Curr. Opin. Pharmacol. 2012, 12, 710–716. [Google Scholar] [CrossRef] [PubMed]
- Kuzuyama, T.; Seto, H. Diversity of the biosynthesis of the isoprene units. Nat. Prod. Rep. 2003, 20, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Raetz, C.R.; Garrett, T.A.; Reynolds, C.M.; Shaw, W.A.; Moore, J.D.; Smith, D.C., Jr.; Ribeiro, A.A.; Murphy, R.C.; Ulevitch, R.J.; Fearns, C.; et al. Kdo2-Lipid A of Escherichia coli, a defined endotoxin that activates macrophages via TLR-4. J. Lipid Res. 2006, 47, 1097–1111. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.T. Polyketide and nonribosomal peptide antibiotics: Modularity and versatility. Science 2004, 303, 1805–1810. [Google Scholar] [CrossRef] [PubMed]
- Minto, R.E.; Blacklock, B.J. Biosynthesis and function of polyacetylenes and allied natural products. Prog. Lipid Res. 2008, 47, 233–306. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.J. Physiological aspects of low-density lipoprotein oxidation. Curr. Opin. Lipidol. 2000, 11, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Lammert, E.; Zee, M. Metabolism of Human Diseases: Organ Physiology and Pathophysiology; Springer: Berlin, Germany, 2014. [Google Scholar]
- Wootten, D.; Christopoulos, A.; Sexton, P.M. Emerging paradigms in GPCR allostery: Implications for drug discovery. Nat. Rev. Drug Discov. 2013, 12, 630–644. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.I.; Lewis, R.J. Emerging opportunities for allosteric modulation of G-protein coupled receptors. Biochem. Pharmacol. 2013, 85, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Archbold, J.K.; Martin, J.L.; Sweet, M.J. Towards selective lysophospholipid GPCR modulators. Trends Pharmacol. Sci. 2014, 35, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Ishii, S.; Shimizu, T. Platelet-activating factor (PAF) receptor and genetically engineered PAF receptor mutant mice. Prog. Lipid Res. 2000, 39, 41–82. [Google Scholar] [CrossRef] [PubMed]
- Rayasam, G.V.; Tulasi, V.K.; Davis, J.A.; Bansal, V.S. Fatty acid receptors as new therapeutic targets for diabetes. Expert Opin. Ther. Targets. 2007, 11, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Perona, R. Cell signalling: Growth factors and tyrosine kinase receptors. Clin. Transl. Oncol. 2006, 8, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Greenhalgh, C.J.; Hilton, D.J. Negative regulation of cytokine signaling. J. Leukoc. Biol. 2001, 70, 348–356. [Google Scholar] [PubMed]
- Lebman, D.A.; Spiegel, S. Cross-talk at the crossroads of sphingosine-1-phosphate, growth factors, and cytokine signaling. J. Lipid Res. 2008, 49, 1388–1394. [Google Scholar] [CrossRef] [PubMed]
- McLaren, J.E.; Michael, D.R.; Ashlin, T.G.; Ramji, D.P. Cytokines, macrophage lipid metabolism and foam cells: Implications for cardiovascular disease therapy. Prog. Lipid Res. 2011, 50, 331–347. [Google Scholar] [CrossRef] [PubMed]
- Pyne, N.J.; Pyne, S. Receptor tyrosine kinase-G-protein-coupled receptor signalling platforms: Out of the shadow? Trends Pharmacol. Sci. 2011, 32, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Delcourt, N.; Bockaert, J.; Marin, P. GPCR-jacking: From a new route in RTK signalling to a new concept in GPCR activation. Trends Pharmacol. Sci. 2007, 28, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Chudakova, D.A.; Zeidan, Y.H.; Wheeler, B.W.; Yu, J.; Novgorodov, S.A.; Kindy, M.S.; Hannun, Y.A.; Gudz, T.I. Integrin-associated Lyn kinase promotes cell survival by suppressing acid sphingomyelinase activity. J. Biol. Chem. 2008, 283, 28806–28816. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Patel, R.; Sen, N.; Quadri, S.; Parthasarathi, K.; Bhattacharya, J. Dual signaling by the αvβ3-integrin activates cytosolic PLA2 in bovine pulmonary artery endothelial cells. Am. J. Physiol. 2001, 280, L1049–L1056. [Google Scholar]
- Mueller, H.; Stadtmann, A.; van Aken, H.; Hirsch, E.; Wang, D.; Ley, K.; Zarbock, A. Tyrosine kinase Btk regulates E-selectin-mediated integrin activation and neutrophil recruitment by controlling phospholipase C (PLC) γ2 and PI3Kγ pathways. Blood 2010, 115, 3118–3127. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Balcioglu, H.E.; Danen, E.H. Integrin signaling in control of tumor growth and progression. Int. J. Biochem. Cell Biol. 2013, 45, 1012–1015. [Google Scholar] [CrossRef] [PubMed]
- Salanueva, I.J.; Cerezo, A.; Guadamillas, M.C.; del Pozo, M.A. Integrin regulation of caveolin function. J. Cell. Mol. Med. 2007, 11, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Hunte, C.; Richers, S. Lipids and membrane protein structures. Curr. Opin. Struct. Biol. 2008, 18, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Shipston, M.J. Regulation of large conductance calcium- and voltage-activated potassium (BK) channels by S-palmitoylation. Biochem. Soc. Trans. 2013, 41, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Kasimova, M.A.; Tarek, M.; Shaytan, A.K.; Shaitan, K.V.; Delemotte, L. Voltage-gated ion channel modulation by lipids: Insights from molecular dynamics simulations. Biochim. Biophys. Acta 2014, 1838, 1322–1331. [Google Scholar] [CrossRef] [PubMed]
- Minke, B.; Cook, B. TRP channel proteins and signal transduction. Physiol. Rev. 2002, 82, 429–472. [Google Scholar] [PubMed]
- Wang, H.; Harrison-Shostak, D.C.; Lemasters, J.J.; Herman, B. Contribution of pH-dependent group II phospholipase A2 to chemical hypoxic injury in rat hepatocytes. FASEB J. 1996, 10, 1319–1325. [Google Scholar] [PubMed]
- Marquis, H.; Hager, E.J. pH-regulated activation and release of a bacteria-associated phospholipase C during intracellular infection by Listeria monocytogenes. Mol. Microbiol. 2000, 35, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Pawliczak, R.; Huang, X.L.; Nanavaty, U.B.; Lawrence, M.; Madara, P.; Shelhamer, J.H. Oxidative stress induces arachidonate release from human lung cells through the epithelial growth factor receptor pathway. Am. J. Respir. Cell Mol. Biol. 2002, 27, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Steinhour, E.; Sherwani, S.I.; Mazerik, J.N.; Ciapala, V.; O’Connor Butler, E.; Cruff, J.P.; Magalang, U.; Parthasarathy, S.; Sen, C.K.; Marsh, C.B.; et al. Redox-active antioxidant modulation of lipid signaling in vascular endothelial cells: Vitamin C induces activation of phospholipase D through phospholipase A2, lipoxygenase, and cyclooxygenase. Mol. Cell. Biochem. 2008, 315, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Huang, C. Hypertension, mechanical force, and renal disease. Ann. Clin. Exp. Hypertens. 2014, 2, 1009. [Google Scholar]
- Chiang, J.Y. Nuclear receptor regulation of lipid metabolism: Potential therapeutics for dyslipidemia, diabetes, and chronic heart and liver diseases. Curr. Opin. Investig. Drugs. 2005, 6, 994–1001. [Google Scholar] [PubMed]
- Papacleovoulou, G.; Abu-Hayyeh, S.; Williamson, C. Nuclear receptor-driven alterations in bile acid and lipid metabolic pathways during gestation. Biochim. Biophys. Acta 2011, 1812, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, I.; Rotter, V. Regulation of lipid metabolism by p53—Fighting two villains with one sword. Trends Endocrinol. Metab. 2012, 23, 567–575. [Google Scholar] [CrossRef]
- Xu., L.; Zhou, L.; Li, P. CIDE proteins and lipid metabolism. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1094–1098. [Google Scholar] [CrossRef] [PubMed]
- Boffa, L.C.; Lupton, J.R.; Mariani, M.R. Modulation of colonic epithelial cell proliferation, histone acetylation, and luminal short chain fatty acids by variation of dietary fiber in rats. Cancer Res. 1992, 52, 5906–5912. [Google Scholar] [PubMed]
- Matthews, G.M.; Howarth, G.S.; Butler, R.N. Short-chain fatty acid modulation of apoptosis in the Kato III human gastric carcinoma cell line. Cancer Biol. Ther. 2007, 6, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Little, J.L.; Kridel, S.J. Fatty acid synthase activity in tumor cells. Subcell. Biochem. 2008, 49, 169–194. [Google Scholar] [PubMed]
- Krycer, J.R.; Brown, A.J. Cholesterol accumulation in prostate cancer: A classic observation from a modern perspective. Biochim. Biophys. Acta 2013, 1835, 219–229. [Google Scholar] [PubMed]
- Hilvo, M.; Denkert, C.; Lehtinen, L.; Muller, B.; Brockmoller, S.; Seppanen-Laakso, T.; Budczies, J.; Bucher, E.; Yetukuri, L.; Castillo, S.; et al. Novel theranostic opportunities offered by characterization of altered membrane lipid metabolism in breast cancer progression. Cancer Res. 2011, 71, 3236–3245. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.A.; Iliopoulos, D.; Joshi, A.; Zhang, Y.; Jaeger, S.A.; Bulyk, M.; Tsichlis, P.N.; Liu, S.X.; Struhl, K. A transcriptional signature and common gene networks link cancer with lipid metabolism and diverse human diseases. Cancer Cell 2010, 17, 348–361. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Fernandez Matilla, M.; Dass, S.; Buch, M.H.; Rawstron, A.C.; Vital, E.M.; Emery, P. Statins do not influence clinical response and B cell depletion after rituximab treatment in rheumatoid arthritis. Ann. Rheum. Dis. 2013, 72, 463–464. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, B.; Bhendwal, S.; Halmos, B.; Moss, S.F.; Ramey, W.G.; Holt, P.R. Lovastatin augments apoptosis induced by chemotherapeutic agents in colon cancer cells. Clin. Cancer Res. 1999, 5, 2223–2229. [Google Scholar] [PubMed]
- Cho, S.J.; Kim, J.S.; Kim, J.M.; Lee, J.Y.; Jung, H.C.; Song, I.S. Simvastatin induces apoptosis in human colon cancer cells and in tumor xenografts, and attenuates colitis-associated colon cancer in mice. Int. J. Cancer 2008, 123, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Zhang, Q.; Lin, Y.; Reddy, B.S.; Yang, C.S. Combination of atorvastatin and celecoxib synergistically induces cell cycle arrest and apoptosis in colon cancer cells. Int. J. Cancer 2008, 122, 2115–2124. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.K.; Oza, A.M.; Siu, L.L. The statins as anticancer agents. Clin. Cancer Res. 2003, 9, 10–19. [Google Scholar] [PubMed]
- Sassano, A.; Platanias, L.C. Statins in tumor suppression. Cancer Lett. 2008, 260, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Bardou, M.; Barkun, A.; Martel, M. Effect of statin therapy on colorectal cancer. Gut 2010, 59, 1572–1585. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, S.; Mérida, I. Diacylglycerol, when simplicity becomes complex. Trends Biochem. Sci. 2007, 32, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Decrock, E.; de Bock, M.; Wang, N.; Gadicherla, A.K.; Bol, M.; Delvaeye, T.; Vandenabeele, P.; Vinken, M.; Bultynck, G.; Krysko, D.V.; et al. IP3, a small molecule with a powerful message. Biochim. Biophys. Acta 2013, 1833, 1772–1786. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Du, G. Phosphatidic acid signaling regulation of Ras superfamily of small guanosine triphosphatases. Biochim. Biophys. Acta 2009, 1791, 850–855. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.Y.; Frohman, M.A. Mitochondria: Signaling with phosphatidic acid. Int. J. Biochem. Cell Biol. 2012, 44, 1346–1350. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.A. Phosphatidic acid signaling to mTOR: Signals for the survival of human cancer cells. Biochim. Biophys. Acta 2009, 1791, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Bruggeman, L.A.; Hydo, L.M.; Miller, R.T. Shear stress induces cell apoptosis via a c-Src-phospholipase D-mTOR signaling pathway in cultured podocytes. Exp. Cell Res. 2012, 318, 1075–1785. [Google Scholar] [CrossRef] [PubMed]
- Mullen, T.D.; Obeid, L.M. Ceramide and apoptosis: Exploring the enigmatic connections between sphingolipid metabolism and programmed cell death. Anticancer Agents Med. Chem. 2012, 12, 340–363. [Google Scholar] [CrossRef] [PubMed]
- Obeid, L.M.; Linardic, C.M.; Karolak, L.A.; Hannun, Y.A. Programmed cell death induced by ceramide. Science 1993, 259, 1769–1771. [Google Scholar] [CrossRef] [PubMed]
- Kihara, A.; Mitsutake, S.; Mizutani, Y.; Igarashi, Y. Metabolism and biological functions of two phosphorylated sphingolipids, sphingosine 1-phosphate and ceramide 1-phosphate. Prog. Lipid Res. 2007, 46, 126–144. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Y.; Hill, R.A.; Li, Y.T. Ceramide glycosylation catalyzed by glucosylceramide synthase and cancer drug resistance. Adv. Cancer Res. 2013, 117, 59–89. [Google Scholar] [PubMed]
- Aranda, J.F.; Madrigal-Matute, J.; Rotllan, N.; Fernández-Hernando, C. MicroRNA modulation of lipid metabolism and oxidative stress in cardiometabolic diseases. Free Radic. Biol. Med. 2013, 64, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Flowers, E.; Froelicher, E.S.; Aouizerat, B.E. MicroRNA regulation of lipid metabolism. Metabolism 2013, 62, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Rottiers, V.; Näär, A.M. MicroRNAs in metabolism and metabolic disorders. Nat. Rev. Mol. Cell Biol. 2012, 13, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.Q.; Cao, J.; Zhou, Y.J.; Liang, X.; Du, Y.L.; Wan, Y.J.; Li, Y.Y. The effect of miRNA-122 in regulating fat deposition in a cell line model. J. Cell. Biochem. 2014, 115, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.C.; Hsu, S.D.; Hsu, C.S.; Lai, T.C.; Chen, S.J.; Shen, R.; Huang, Y.; Chen, H.C.; Lee, C.H.; Tsai, T.F.; et al. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J. Clin. Investig. 2012, 122, 2884–2897. [Google Scholar] [CrossRef] [PubMed]
- Esau, C.; Davis, S.; Murray, S.F.; Yu, X.X.; Pandey, S.K.; Pear, M.; Watts, L.; Booten, S.L.; Graham, M.; McKay, R.; et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006, 3, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Krützfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of microRNAs in vivo with “antagomirs”. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Lanford, R.E.; Hildebrandt-Eriksen, E.S.; Petri, A.; Persson, R.; Lindow, M.; Munk, M.E.; Kauppinen, S.; Ørum, H. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 2010, 327, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Najafi-Shoushtari, S.H.; Kristo, F.; Li, Y.; Shioda, T.; Cohen, D.E.; Gerszten, R.E.; Näär, A.M. MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science 2010, 328, 1566–1569. [Google Scholar] [CrossRef] [PubMed]
- Marquart, T.J.; Allen, R.M.; Ory, D.S.; Baldán, A. miR-33 links SREBP-2 induction to repression of sterol transporters. Proc. Natl. Acad. Sci. USA 2010, 107, 12228–12232. [Google Scholar] [CrossRef] [PubMed]
- Rayner, K.J.; Suárez, Y.; Dávalos, A.; Parathath, S.; Fitzgerald, M.L.; Tamehiro, N.; Fisher, E.A.; Moore, K.J.; Fernández-Hernando, C. MiR-33 contributes to the regulation of cholesterol homeostasis. Science 2010, 328, 1570–1573. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Drosatos, K.; Hiyama, Y.; Goldberg, I.J.; Zannis, V.I. MicroRNA-370 controls the expression of microRNA-122 and Cpt1alpha and affects lipid metabolism. J. Lipid Res. 2010, 51, 1513–1523. [Google Scholar] [CrossRef] [PubMed]
- Gerin, I.; Bommer, G.T.; McCoin, C.S.; Sousa, K.M.; Krishnan, V.; MacDougald, O.A. Roles for miRNA-378/378∗ in adipocyte gene expression and lipogenesis. Am. J. Physiol. 2010, 299, E198–E206. [Google Scholar]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of cell death: Recom mendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009, 16, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, Y.; Steller, H. Programmed cell death in animal development and disease. Cell 2011, 147, 742–758. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Levine, B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell 2014, 157, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 2013, 1833, 3448–3459. [Google Scholar] [CrossRef] [PubMed]
- Christofferson, D.E.; Yuan, J. Necroptosis as an alternative form of programmed cell death. Curr. Opin. Cell Biol. 2010, 22, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Golstein, P.; Kroemer, G. Cell death by necrosis: Towards a molecular definition. Trends Biochem. Sci. 2007, 32, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Pinedo, C. Signaling pathways that regulate life and cell death: Evolution of apoptosis in the context of self-defense. Adv. Exp. Med. Biol. 2012, 738, 124–143. [Google Scholar] [PubMed]
- Shimabukuro, M.; Zhou, Y.T.; Levi, M.; Unger, R.H. Fatty acid-induced beta cell apoptosis: A link between obesity and diabetes. Proc. Natl. Acad. Sci. USA 1998, 95, 2498–2502. [Google Scholar] [CrossRef] [PubMed]
- Karaskov, E.; Scott, C.; Zhang, L.; Teodoro, T.; Ravazzola, M.; Volchuk, A. Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic beta-cell apoptosis. Endocrinology 2006, 147, 3398–3407. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.Y.; Rabkin, S.W. Palmitate-induced apoptosis in cardiomyocytes is mediated through alterations in mitochondria: Prevention by cyclosporin A. Biochim. Biophys. Acta 2000, 1485, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Chai, W.; Liu, Z. p38 mitogen-activated protein kinase mediates palmitate-induced apoptosis but not inhibitor of nuclear factor-κB degradation in human coronary artery endothelial cells. Endocrinology 2007, 148, 1622–1628. [Google Scholar] [CrossRef] [PubMed]
- Malhi, H.; Gores, G.J. Molecular mechanisms of lipotoxicity in nonalcoholic fatty liver disease. Semin. Liver Dis. 2008, 28, 360–309. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.C.; Kehrer, J.P. Free radicals and apoptosis: Relationships with glutathione, thioredoxin and the Bcl family of proteins. Front. Biosci. 2005, 10, 1727–1738. [Google Scholar] [CrossRef] [PubMed]
- Dalleau, S.; Baradat, M.; Guéraud, F.; Huc, L. Cell death and diseases related to oxidative stress: 4-hydroxynonenal (HNE) in the balance. Cell Death Differ. 2013, 20, 1615–1630. [Google Scholar] [CrossRef] [PubMed]
- Elamin, E.E.; Masclee, A.A.; Dekker, J.; Pieters, H.J.; Jonkers, D.M. Short-chain fatty acids activate AMP-activated protein kinase and ameliorate ethanol-induced intestinal barrier dysfunction in Caco-2 cell monolayers. J. Nutr. 2013, 143, 1872–1881. [Google Scholar] [CrossRef] [PubMed]
- Bi, L.; Chiang, J.Y.; Ding, W.X.; Dunn, W.; Roberts, B.; Li, T. Saturated fatty acids activate ERK signaling to downregulate hepatic sortilin 1 in obese and diabetic mice. J. Lipid Res. 2013, 54, 2754–2762. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.H.; Kang, S.G.; Park, J.H.; Yanagisawa, M.; Kim, C.H. Short-chain fatty acids activate GPR41 and GPR43 on intestinal epithelial cells to promote inflammatory responses in mice. Gastroenterology 2013, 145, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Rutkowsky, J.M.; Snodgrass, R.G.; Ono-Moore, K.D.; Schneider, D.A.; Newman, J.W.; Adams, S.H.; Hwang, D.H. Saturated fatty acids activate TLR-mediated proinflammatory signaling pathways. J. Lipid Res. 2012, 53, 2002–2013. [Google Scholar] [CrossRef] [PubMed]
- Holzer, R.G.; Park, E.J.; Li, N.; Tran, H.; Chen, M.; Choi, C.; Solinas, G.; Karin, M. Saturated fatty acids induce c-Src clustering within membrane subdomains, leading to JNK activation. Cell 2011, 147, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, H.; Bao, Y.; Zhang, X.; Yu, Y. Free fatty acids induce endothelial dysfunction and activate protein kinase C and nuclear factor-κB pathway in rat aorta. Int. J. Cardiol. 2011, 152, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.H.; Kohli, R.; Gores, G.J. Mechanisms of lipotoxicity in NAFLD and clinical implications. J. Pediatr. Gastroenterol. Nutr. 2011, 53, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, Y.C.; Sharma, R.; Sharma, A.; Yadav, S.; Singhal, S.S.; Chaudhary, P.; Awasthi, S. Self-regulatory role of 4-hydroxynonenal in signaling for stress-induced programmed cell death. Free Radic. Biol. Med. 2008, 45, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Knauss, T.C.; Jaffer, F.E.; Abboud, H.E. Phosphatidic acid modulates DNA synthesis, phospholipase C, and platelet-derived growth factor mRNAs in cultured mesangial cells. Role of protein kinase C. J. Biol. Chem. 1990, 265, 14457–14463. [Google Scholar] [PubMed]
- Moolenaar, W.H.; Kruijer, W.; Tilly, B.C.; Verlaan, I.; Bierman, A.J.; de Laat, S.W. Growth factor-like action of phosphatidic acid. Nature 1986, 323, 171–173. [Google Scholar] [CrossRef] [PubMed]
- Agwu, D.E.; McPhail, L.C.; Sozzani, S.; Bass, D.A.; McCall, C.E. Phosphatidic acid as a second messenger in human polymorphonuclear leukocytes. Effects on activation of NADPH oxidase. J. Clin. Investig. 1991, 88, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Lu, S.H.; Yuan, Y.J. Cerium elicitor-induced phosphatidic acid triggers apoptotic signaling development in Taxus cuspidata cell suspension cultures. Chem. Phys. Lipids 2009, 159, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Zhou, H.; Zhao, X.; Wolff, D.W.; Tu, Y.; Liu, H.; Wei, T.; Yang, F. Phosphatidic acid mediates the targeting of tBid to induce lysosomal membrane permeabilization and apoptosis. J. Lipid Res. 2012, 53, 2102–2114. [Google Scholar] [CrossRef] [PubMed]
- Norambuena, A.; Metz, C.; Vicuña, L.; Silva, A.; Pardo, E.; Oyanadel, C.; Massardo, L.; González, A.; Soza, A. Galectin-8 induces apoptosis in Jurkat T cells by phosphatidic acid-mediated ERK1/2 activation supported by protein kinase A down-regulation. J. Biol. Chem. 2009, 284, 12670–12679. [Google Scholar] [CrossRef] [PubMed]
- Saddoughi, S.A.; Ogretmen, B. Diverse functions of ceramide in cancer cell death and proliferation. Adv. Cancer Res. 2013, 117, 37–58. [Google Scholar] [PubMed]
- Uchida, Y. Ceramide signaling in mammalian epidermis. Biochim. Biophys. Acta 2014, 1841, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Chalfant, C.E.; Szulc, Z.; Roddy, P.; Bielawska, A.; Hannun, Y.A. The structural requirements for ceramide activation of serine–threonine protein phosphatases. J. Lipid Res. 2004, 45, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Lozano, J.; Berra, E.; Municio, M.M.; Diaz-Meco, M.T.; Dominguez, I.; Sanz, L.; Moscat, J. Protein kinase C zeta isoformis critical for kappa B-dependent promoter activation by sphingomyelinase. J. Biol. Chem. 1994, 269, 19200–19202. [Google Scholar] [PubMed]
- Heinrich, M.; Wickel, M.; Schneider-Brachert, W.; Sandberg, C.; Gahr, J.; Schwandner, R.; Weber, T.; Saftig, P.; Peters, C.J.; Brunner, J.; et al. Cathepsin D targeted by acid sphingomyelinase-derived ceramide. EMBO J. 1999, 18, 5252–5263. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yao, B.; Delikat, S.; Bayoumy, S.; Lin, X.H.; Basu, S.; McGinley, M.; Chan-Hui, P.-Y.; Lichenstein, H.; Kolesnick, R.; et al. Kinase suppressor of Ras is ceramide-activated protein kinase. Cell 1997, 89, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Colombini, M. Membrane channels formed by ceramide. Handb. Exp. Pharmacol. 2013, 2013, 109–126. [Google Scholar]
- Siskind, L.J.; Kolesnick, R.N.; Colombini, M. Ceramide channels increase the permeability of the mitochondrial outer membrane to small proteins. J. Biol. Chem. 2002, 277, 26796–26803. [Google Scholar] [CrossRef] [PubMed]
- Sottero, B.; Gamba, P.; Gargiulo, S.; Leonarduzzi, G.; Poli, G. Cholesterol oxidation products and disease: An emerging topic of interest in medicinal chemistry. Curr. Med. Chem. 2009, 16, 685–705. [Google Scholar] [CrossRef] [PubMed]
- Schroepfer, G.J., Jr. Oxysterols: Modulators of cholesterol metabolism and other processes. Physiol. Rev. 2000, 80, 361–554. [Google Scholar] [PubMed]
- Colell, A.; Fernández, A.; Fernández-Checa, J.C. Mitochondria, cholesterol and amyloid beta peptide: A dangerous trio in Alzheimer disease. J. Bioenerg. Biomembr. 2009, 41, 417–423. [Google Scholar] [CrossRef] [PubMed]
- De Medina, P.; Paillasse, M.R.; Segala, G.; Voisin, M.; Mhamdi, L.; Dalenc, F.; Lacroix-Triki, M.; Filleron, T.; Pont, F.; Saati, T.A.; et al. Dendrogenin A arises from cholesterol and histamine metabolism and shows cell differentiation and anti-tumour properties. Nat. Commun. 2013, 4, 1840. [Google Scholar] [CrossRef] [PubMed]
- Segala, G.; de Medina, P.; Iuliano, L.; Zerbinati, C.; Paillasse, M.R.; Noguer, E.; Dalenc, F.; Payré, B.; Jordan, V.C.; Record, M.; et al. 5,6-Epoxy-cholesterols contribute to the anticancer pharmacology of tamoxifen in breast cancer cells. Biochem. Pharmacol. 2013, 86, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Wan, G.; Zhaorigetu, S.; Liu, Z.; Kaini, R.; Jiang, Z.; Hu, C.A. Apolipoprotein L1, a novel Bcl-2 homology domain 3-only lipid-binding protein, induces autophagic cell death. J. Biol. Chem. 2008, 283, 21540–21549. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Lu, H.; Jiang, Z.; Pastuszyn, A.; Hu, C.A. Apolipoprotein L6, a novel proapoptotic Bcl-2 homology 3-only protein, induces mitochondria-mediated apoptosis in cancer cells. Mol. Cancer Res. 2005, 3, 21–31. [Google Scholar] [PubMed]
- Quinn, C.M.; Kågedal, K.; Terman, A.; Stroikin, U.; Brunk, U.T.; Jessup, W.; Garner, B. Induction of fibroblast apolipoprotein E expression during apoptosis, starvation-induced growth arrest and mitosis. Biochem. J. 2004, 378, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W.; Huang, Y. Apolipoprotein e sets the stage: Response to injury triggers neuropathology. Neuron 2012, 76, 871–885. [Google Scholar] [CrossRef] [PubMed]
- Salvayre, R.; Auge, N.; Benoist, H.; Negre-Salvayre, A. Oxidized low-density lipoprotein-induced apoptosis. Biochim. Biophys. Acta 2002, 1585, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Chan, S.L. Calcium orchestrates apoptosis. Nat. Cell Biol. 2003, 5, 1041–1043. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Prieto, C.; Riaz Ahmed, K.B.; Chen, Z.; Zhou, Y.; Hammoudi, N.; Kang, Y.; Lou, C.; Mei, Y.; Jin, Z.; Huang, P.; et al. Effective killing of leukemia cells by the natural product OSW-1 through disruption of cellular calcium homeostasis. J. Biol. Chem. 2013, 288, 3240–3250. [Google Scholar] [CrossRef] [PubMed]
- Kiviluoto, S.; Vervliet, T.; Ivanova, H.; Decuypere, J.P.; de Smedt, H.; Missiaen, L.; Bultynck, G.; Parys, J.B. Regulation of inositol 1,4,5-trisphosphate receptors during endoplasmic reticulum stress. Biochim. Biophys. Acta 2013, 1833, 1612–1624. [Google Scholar] [CrossRef] [PubMed]
- Murgia, M.; Giorgi, C.; Pinton, P.; Rizzuto, R. Controlling metabolism and cell death: At the heart of mitochondrial calcium signalling. J. Mol. Cell. Cardiol. 2009, 46, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Boczek, T.; Lisek, M.; Kowalski, A.; Pikula, S.; Niewiarowska, J.; Wiktorska, M.; Zylinska, L. Downregulation of PMCA2 or PMCA3 reorganizes Ca2+ handling systems in differentiating PC12 cells. Cell. Calcium 2012, 52, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, A.R.; Verfaillie, T.; Agostinis, P. New functions of mitochondria associated membranes in cellular signaling. Biochim. Biophys. Acta 2014, 1843, 2253–2262. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Rizzuto, R.; Hajnoczky, G.; Su, T.P. MAM: More than just a housekeeper. Trends Cell Biol. 2009, 19, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Grimm, S. The ER-mitochondria interface: The social network of cell death. Biochim. Biophys. Acta 2012, 1823, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Patergnani, S.; Pinton, P. The endoplasmic reticulum-mitochondria connection: One touch, multiple functions. Biochim. Biophys. Acta 2014, 1837, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Raturi, A.; Simmen, T. Where the endoplasmic reticulum and the mitochondrion tie the knot: The mitochondria-associated membrane (MAM). Biochim. Biophys. Acta 2013, 1833, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E. MAM (mitochondria-associated membranes) in mammalian cells: Lipids and beyond. Biochim. Biophys. Acta 2014, 1841, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Ardail, D.; Popa, I.; Bodennec, J.; Louisot, P.; Schmitt, D.; Portoukalian, J. The mitochondria-associated endoplasmic-reticulum subcompartment (MAM fraction) of rat liver contains highly active sphingolipid-specific glycosyltransferases. Biochem. J. 2003, 371, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Lupu, R.; Menendez, J.A. Pharmacological inhibitors of Fatty Acid Synthase (FASN) catalyzed endogenous fatty acid biogenesis: A new family of anti-cancer agents? Curr. Pharm. Biotechnol. 2006, 7, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Janardhan, S.; Srivani, P.; Sastry, G.N. Choline kinase: An important target for cancer. Curr. Med. Chem. 2006, 13, 1169–1186. [Google Scholar] [CrossRef] [PubMed]
- Danilo, C.; Frank, P.G. Cholesterol and breast cancer development. Curr. Opin. Pharmacol. 2012, 12, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Wenk, M.R. The emerging field of lipidomics. Nat. Rev. Drug Discov. 2005, 4, 594–610. [Google Scholar] [CrossRef] [PubMed]
- Postle, A.D. Lipidomics. Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 127–133. [Google Scholar] [PubMed]
- Senchenkov, A.; Litvak, D.A.; Cabot, M.C. Targeting ceramide metabolism—A strategy for overcoming drug resistance. J. Natl. Cancer Inst. 2001, 93, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Kridel, S.J.; Lowther, W.T.; Pemble, C.W. 4th Fatty acid synthase inhibitors: New directions for oncology. Expert Opin. Investig. Drugs 2007, 16, 1817–1829. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Mukae, S.; Murakami, M.; Ishikawa, Y.; Ishii, T.; Ohki, H.; Matsumoto, M.; Komiyama, K. Butyrate inhibits oral cancer cell proliferation and regulates expression of secretory phospholipase A2-X and COX-2. Anticancer Res. 2007, 27, 1493–1502. [Google Scholar] [PubMed]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Benakanakere, I.; Johnson, T.; Sleightholm, R.; Villeda, V.; Arya, M.; Bobba, R.; Freter, C.; Huang, C. Targeting cholesterol synthesis increases chemoimmuno-sensitivity in chronic lymphocytic leukemia cells. Exp. Hematol. Oncol. 2014, 3, 24. [Google Scholar] [CrossRef] [PubMed]
- Kotas, M.E.; Jurczak, M.J.; Annicelli, C.; Gillum, M.P.; Cline, W.; Shulman, G.I.; Medzhitov, R. Role of caspase-1 in regulation of triglyceride metabolism. Proc. Natl. Acad. Sci. USA 2013, 110, 4810–4815. [Google Scholar] [CrossRef] [PubMed]
- Schilling, T.; Eder, C. Sodium dependence of lysophosphatidylcholine-induced caspase-1 activity and reactive oxygen species generation. Immunobiology 2011, 216, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Schumann, R.R.; Belka, C.; Reuter, D.; Lamping, N.; Kirschning, C.J.; Weber, J.R.; Pfeil, D. Lipopolysaccharide activates caspase-1 (interleukin-1-converting enzyme) in cultured monocytic and endothelial cells. Blood 1998, 91, 577–584. [Google Scholar] [PubMed]
- Schroeder, G.N.; Hilbi, H. Cholesterol is required to trigger caspase-1 activation and macrophage apoptosis after phagosomal escape of Shigella. Cell. Microbiol. 2007, 9, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Kroemer, G. Caspase-3 and prostaglandins signal for tumor regrowth in cancer therapy. Oncogene 2012, 31, 2805–2808. [Google Scholar] [CrossRef] [PubMed]
- Němcová-Fürstová, V.; James, R.F.; Kovář, J. Inhibitory effect of unsaturated fatty acids on saturated fatty acid-induced apoptosis in human pancreatic β-cells: Activation of caspases and ER stress induction. Cell. Physiol. Biochem. 2011, 27, 525–538. [Google Scholar] [CrossRef] [PubMed]
- Gonzalvez, F.; Schug, Z.T.; Houtkooper, R.H.; MacKenzie, E.D.; Brooks, D.G.; Wanders, R.J.; Petit, P.X.; Vaz, F.M.; Gottlieb, E. Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J. Cell Biol. 2008, 183, 681–696. [Google Scholar] [CrossRef] [PubMed]
- Luthra, S.; Dong, J.; Gramajo, A.L.; Chwa, M.; Kim, D.W.; Neekhra, A.; Kuppermann, B.D.; Kenney, M.C. 7-Ketocholesterol activates caspases-3/7, -8, and -12 in human microvascular endothelial cells in vitro. Microvasc. Res. 2008, 75, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Iwakiri, R.; Ootani, A.; Fujise, T.; Tsunada, S.; Fujimoto, K. Platelet-activating factor promotes mucosal apoptosis via FasL-mediating caspase-9 active pathway in rat small intestine after ischemia-reperfusion. FASEB J. 2003, 17, 1156–1158. [Google Scholar] [PubMed]
- Jiang, Y.J.; Kim, P.; Uchida, Y.; Elias, P.M.; Bikle, D.D.; Grunfeld, C.; Feingold, K.R. Ceramides stimulate caspase-14 expression in human keratinocytes. Exp. Dermatol. 2013, 22, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Lippens, S.; Kockx, M.; Denecker, G.; Knaapen, M.; Verheyen, A.; Christiaen, R.; Tschachler, E.; Vandenabeele, P.; Declercq, W. Vitamin D3 induces caspase-14 expression in psoriatic lesions and enhances caspase-14 processing in organotypic skin cultures. Am. J. Pathol. 2004, 165, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Edelmann, B.; Bertsch, U.; Tchikov, V.; Winoto-Morbach, S.; Perrotta, C.; Jakob, M.; Adam-Klages, S.; Kabelitz, D.; Schütze, S. Caspase-8 and caspase-7 sequentially mediate proteolytic activation of acid sphingomyelinase in TNF-R1 receptosomes. EMBO J. 2011, 30, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Lafont, E.; Dupont, R.; Andrieu-Abadie, N.; Okazaki, T.; Schulze-Osthoff, K.; Levade, T.; Benoist, H.; Ségui, B. Ordering of ceramide formation and caspase-9 activation in CD95L-induced Jurkat leukemia T cell apoptosis. Biochim. Biophys. Acta 2012, 1821, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.; Neumeyer, J.; Jakob, M.; Hallas, C.; Tchikov, V.; Winoto-Morbach, S.; Wickel, M.; Schneider-Brachert, W.; Trauzold, A.; Hethke, A.; et al. Cathepsin D links TNF-induced acid sphingomyelinase to Bid-mediated caspase-9 and -3 activation. Cell Death Differ. 2004, 11, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Lin, L.; Cheng, Q.; Xu, Q.; Zhang, J.; Tomlinson, S.; Jin, J.; Chen, X.; He, S. The role of acid sphingomyelinase and caspase 5 in hypoxia-induced HuR cleavage and subsequent apoptosis in hepatocytes. Biochim. Biophys. Acta 2012, 1821, 1453–1461. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2013. CA Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.C.; Radin, N.S. Stimulation of liver growth and DNA synthesis by glucosylceramide. Lipids 1988, 23, 508–510. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Huang, C.; Freter, C. Lipid Metabolism, Apoptosis and Cancer Therapy. Int. J. Mol. Sci. 2015, 16, 924-949. https://doi.org/10.3390/ijms16010924
AMA Style
Huang C, Freter C. Lipid Metabolism, Apoptosis and Cancer Therapy. International Journal of Molecular Sciences. 2015; 16(1):924-949. https://doi.org/10.3390/ijms16010924
Chicago/Turabian StyleHuang, Chunfa, and Carl Freter. 2015. "Lipid Metabolism, Apoptosis and Cancer Therapy" International Journal of Molecular Sciences 16, no. 1: 924-949. https://doi.org/10.3390/ijms16010924