
Rare Titin (TTN) Variants in Diseases Associated with Sudden Cardiac Death

 ,
,
Abstract
:1. Introduction

2. Results

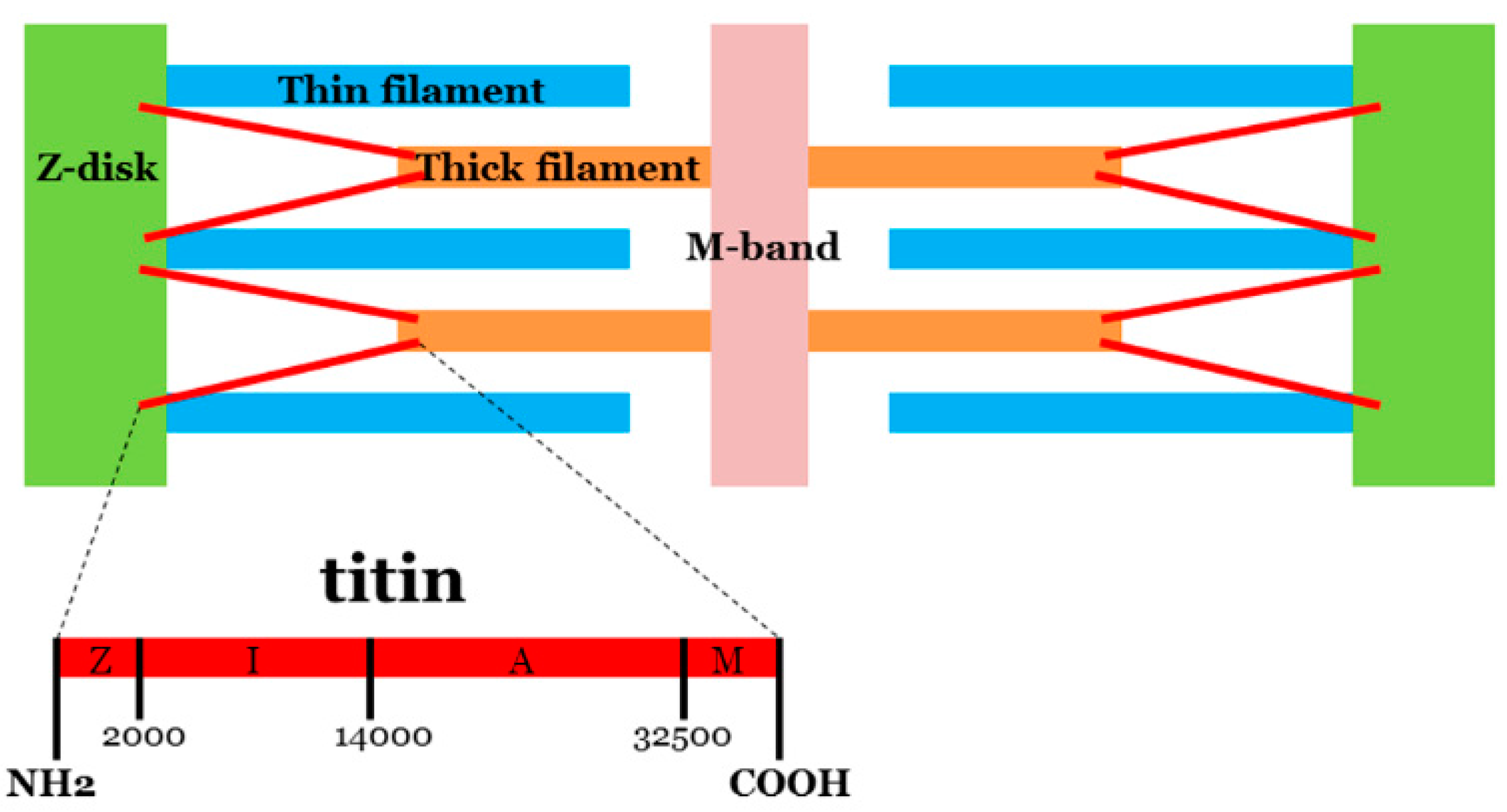{kind=link}
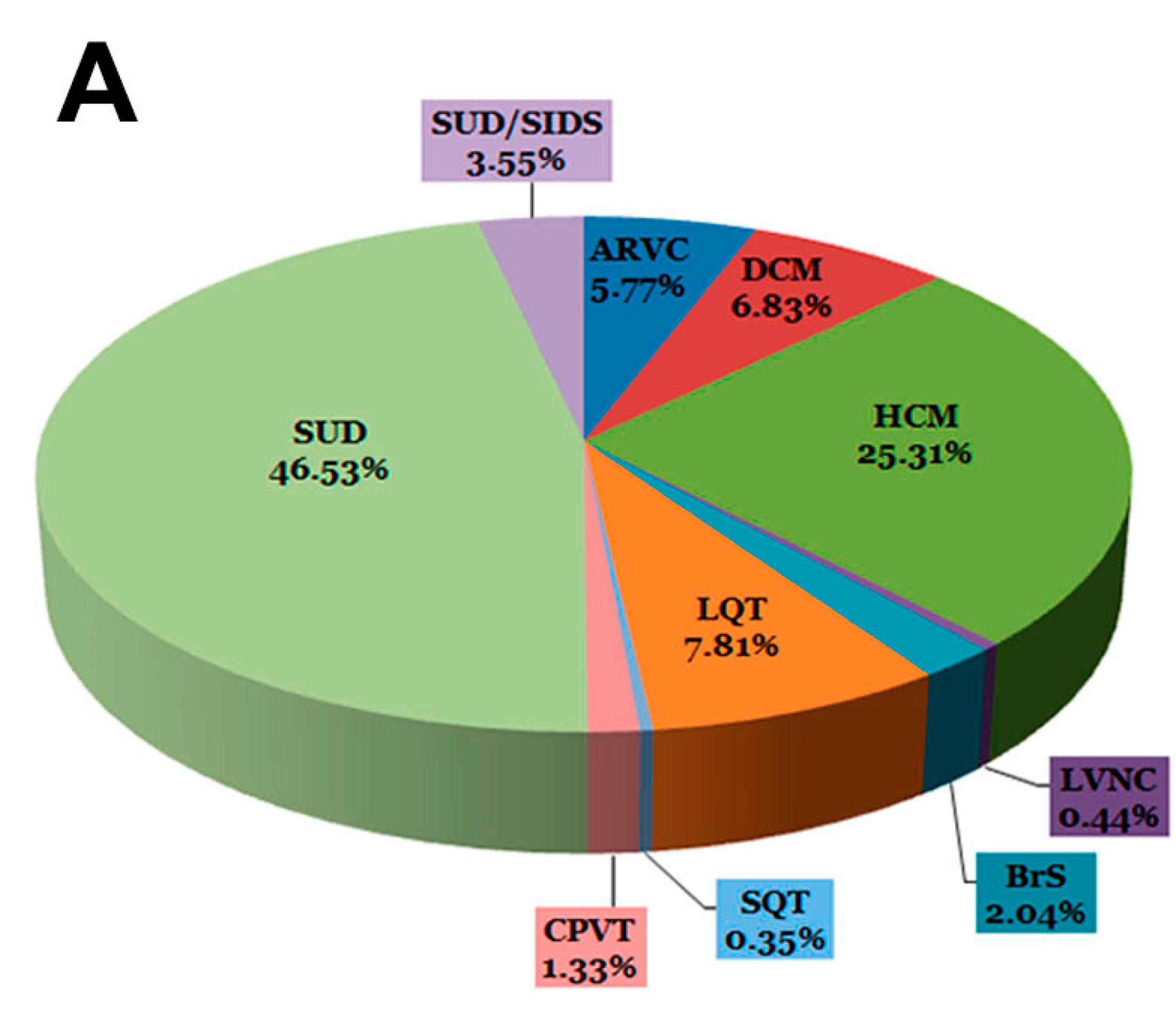{kind=link}
{kind=link}
| Disease | Samples | Total | |
|---|---|---|---|
| Channelopathies | BrS | 23 (2.04%) | 130 (11.55%) |
| LQT | 88 (7.81%) | ||
| SQT | 4 (0.35%) | ||
| CPVT | 15 (1.33%) | ||
| Cardiomyopathies | ARVC | 65 (5.77%) | 432 (38.36%) |
| HCM | 285 (25.31%) | ||
| DCM | 77 (6.83%) | ||
| LVNC | 5 (0.44%) | ||
| SUDs | SUD | 524 (46.53%) | 564 (50.09%) |
| SUD/SIDS | 40 (3.55%) | ||
| 1126 (100%) | 1126 (100%) |


2.1. Intronic Variants
| Variants | ||
|---|---|---|
| Intronic | 18 (3.24%) Novel: 8 (1.44%) | - |
| Exonic | 536 (96.75%) Novel: 274 (49.46%) | Missense 493 (88.99%) Novel: 233 (42.06%) |
| Nonsense 6 (1.08%) Novel: 5 (0.90%) | ||
| Indels 37 (6.68%) Novel: 36 (6.5%) | ||
| TOTAL | 554 (100%) Novel: 282 (50.90%) | - |
2.2. Exonic Variants
2.2.1. Missense
2.2.2. Indels
2.2.3. Nonsense
2.3. Cohorts
3. Discussion
| Total Samples 1126 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Channelopathies 130 | Cardiomyopathies 432 | Post-Mortem 564 | |||||||||
| BrS 23 | LQT 88 | SQT 4 | CPVT 15 | HCM 285 | DCM 77 | ARVC 65 | LVNC 5 | ||||
| Total Variants 554 | Post-Mortem 296 | - | - | - | - | - | - | - | - | - | 168 (56.75%) |
| Cardiomyopathies 325 | LVNC 5 | - | - | - | - | - | - | - | 2 (40%) | - | |
| ARVC 53 | - | - | - | - | - | - | 26 (49.05%) | - | - | ||
| DCM 64 | - | - | - | - | - | 32 (50%) | - | - | - | ||
| HCM 203 | - | - | - | - | 113 (55.65%) | - | - | - | - | ||
| Channelopathies 99 | CPVT 15 | - | - | - | 7 (46.66%) | - | - | - | - | - | |
| SQT 4 | - | - | - | - | - | - | - | - | - | ||
| LQT 57 | - | 29 (50.87%) | - | - | - | - | - | - | - | ||
| BrS 23 | 20 (86.95%) | - | - | - | - | - | - | - | - | ||
3.1. Clinical Implications
3.2. Limitations
4. Experimental Section
4.1. Cohort
4.2. Samples
4.3. Custom Sequencing Panel
4.4. Bioinformatics
4.5. Sanger Sequencing
5. Conclusions
Supplementary Materials
Acknowledgments
Authors Contributions
Conflicts of Interest
References
- Priori, S.G.; Aliot, E.; Blomstrom-Lundqvist, C.; Bossaert, L.; Breithardt, G.; Brugada, P.; Camm, J.A.; Cappato, R.; Cobbe, S.M.; di Mario, C.; et al. Task force on sudden cardiac death, european society of cardiology. Summary of recommendations. Ital. Heart J. Suppl. 2002, 3, 1051–1065. [Google Scholar]
- Arzamendi, D.; Benito, B.; Tizon-Marcos, H.; Flores, J.; Tanguay, J.F.; Ly, H.; Doucet, S.; Leduc, L.; Leung, T.K.; Campuzano, O.; et al. Increase in sudden death from coronary artery disease in young adults. Am. Heart J. 2011, 161, 574–580. [Google Scholar] [PubMed]
- Campuzano, O.; Allegue, C.; Partemi, S.; Iglesias, A.; Oliva, A.; Brugada, R. Negative autopsy and sudden cardiac death. Int. J. Legal Med. 2014, 128, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, O.; Beltran-Alvarez, P.; Iglesias, A.; Scornik, F.; Perez, G.; Brugada, R. Genetics and cardiac channelopathies. Genet. Med. 2010, 12, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Cerrone, M.; Delmar, M. Desmosomes and the sodium channel complex: Implications for arrhythmogenic cardiomyopathy and brugada syndrome. Trends Cardiovasc. Med. 2014, 24, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Cerrone, M.; Lin, X.; Zhang, M.; Agullo-Pascual, E.; Pfenniger, A.; Chkourko Gusky, H.; Novelli, V.; Kim, C.; Tirasawadichai, T.; Judge, D.P.; et al. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a brugada syndrome phenotype. Circulation 2014, 129, 1092–1103. [Google Scholar] [CrossRef] [PubMed]
- Amin, A.S. SCN5A-related dilated cardiomyopathy: What do we know? Heart Rhythm 2014, 11, 1454–1455. [Google Scholar] [CrossRef] [PubMed]
- Herman, D.S.; Lam, L.; Taylor, M.R.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Van Spaendonck-Zwarts, K.Y.; Posafalvi, A.; van den Berg, M.P.; Hilfiker-Kleiner, D.; Bollen, I.A.; Sliwa, K.; Alders, M.; Almomani, R.; van Langen, I.M.; van der Meer, P.; et al. Titin gene mutations are common in families with both peripartum cardiomyopathy and dilated cardiomyopathy. Eur. Heart J. 2014, 21, 2165–2173. [Google Scholar] [CrossRef] [PubMed]
- Peled, Y.; Gramlich, M.; Yoskovitz, G.; Feinberg, M.S.; Afek, A.; Polak-Charcon, S.; Pras, E.; Sela, B.A.; Konen, E.; Weissbrod, O.; et al. Titin mutation in familial restrictive cardiomyopathy. Int. J. Cardiol. 2014, 171, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Brun, F.; Barnes, C.V.; Sinagra, G.; Slavov, D.; Barbati, G.; Zhu, X.; Graw, S.L.; Spezzacatene, A.; Pinamonti, B.; Merlo, M.; et al. Titin and desmosomal genes in the natural history of arrhythmogenic right ventricular cardiomyopathy. J. Med. Genet. 2014, 51, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Dowling, J.J. Titin and centronuclear myopathy: The tip of the iceberg for TTN-ic mutations? Neurology 2013, 81, 1189–1190. [Google Scholar] [CrossRef] [PubMed]
- LeWinter, M.M.; Granzier, H.L. Titin is a major human disease gene. Circulation 2013, 127, 938–944. [Google Scholar] [CrossRef] [PubMed]
- LeWinter, M.M.; Granzier, H.L. Cardiac titin and heart disease. J. Cardiovasc. Pharmacol. 2014, 63, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Golbus, J.R.; Puckelwartz, M.J.; Fahrenbach, J.P.; Dellefave-Castillo, L.M.; Wolfgeher, D.; McNally, E.M. Population-based variation in cardiomyopathy genes. Circ. Cardiovasc. Genet. 2012, 5, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Buermans, H.P.; den Dunnen, J.T. Next generation sequencing technology: Advances and applications. Biochim. Biophys. Acta 2014, 1842, 1932–1941. [Google Scholar] [CrossRef] [PubMed]
- Polychronakos, C.; Seng, K.C. Exome diagnostics: Already a reality? J. Med. Genet. 2011, 48. [Google Scholar] [CrossRef] [PubMed]
- Norton, N.; Li, D.; Rampersaud, E.; Morales, A.; Martin, E.R.; Zuchner, S.; Guo, S.; Gonzalez, M.; Hedges, D.J.; Robertson, P.D.; et al. Exome sequencing and genome-wide linkage analysis in 17 families illustrate the complex contribution of TTN truncating variants to dilated cardiomyopathy. Circ. Cardiovasc. Genet. 2013, 6, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Tennessen, J.A.; Bigham, A.W.; O’Connor, T.D.; Fu, W.; Kenny, E.E.; Gravel, S.; McGee, S.; Do, R.; Liu, X.; Jun, G.; et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 2012, 337, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.; Graw, S.; Sinagra, G.; Barnes, C.; Slavov, D.; Brun, F.; Pinamonti, B.; Salcedo, E.E.; Sauer, W.; Pyxaras, S.; et al. Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy-overlap syndromes. Circulation 2011, 124, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Lopes, L.R.; Zekavati, A.; Syrris, P.; Hubank, M.; Giambartolomei, C.; Dalageorgou, C.; Jenkins, S.; McKenna, W.; Plagnol, V.; Elliott, P.M. Genetic complexity in hypertrophic cardiomyopathy revealed by high-throughput sequencing. J. Med. Genet. 2013, 50, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Brion, M.; Allegue, C.; Santori, M.; Gil, R.; Blanco-Verea, A.; Haas, C.; Bartsch, C.; Poster, S.; Madea, B.; Campuzano, O.; et al. Sarcomeric gene mutations in sudden infant death syndrome (SIDS). Forensic Sci. Int. 2012, 219, 278–281. [Google Scholar] [CrossRef] [PubMed]
- Kruger, M.; Linke, W.A. The giant protein titin: A regulatory node that integrates myocyte signaling pathways. J. Biol. Chem. 2011, 286, 9905–9912. [Google Scholar] [CrossRef] [PubMed]
- Giudicessi, J.R.; Kapplinger, J.D.; Tester, D.J.; Alders, M.; Salisbury, B.A.; Wilde, A.A.; Ackerman, M.J. Phylogenetic and physicochemical analyses enhance the classification of rare nonsynonymous single nucleotide variants in type 1 and 2 long-QT syndrome. Circ. Cardiovasc. Genet. 2012, 5, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Riuro, H.; Campuzano, O.; Berne, P.; Arbelo, E.; Iglesias, A.; Perez-Serra, A.; Coll-Vidal, M.; Partemi, S.; Mademont-Soler, I.; Pico, F.; et al. Genetic analysis, in silico prediction, and family segregation in long QT syndrome. Eur. J. Hum. Genet. 2015, 23, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.M.; Ware, J.S.; Herman, D.S.; Schafer, S.; Baksi, J.; Bick, A.G.; Buchan, R.J.; Walsh, R.; John, S.; Wilkinson, S.; et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci. Transl. Med. 2015, 7, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Rehm, H.L.; Bale, S.J.; Bayrak-Toydemir, P.; Berg, J.S.; Brown, K.K.; Deignan, J.L.; Friez, M.J.; Funke, B.H.; Hegde, M.R.; Lyon, E. Acmg clinical laboratory standards for next-generation sequencing. Genet. Med. 2013, 15, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Tester, D.J.; Ackerman, M.J. Genetic testing for potentially lethal, highly treatable inherited cardiomyopathies/channelopathies in clinical practice. Circulation 2011, 123, 1021–1037. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, O.; Sanchez-Molero, O.; Allegue, C.; Coll, M.; Mademont-Soler, I.; Selga, E.; Ferrer-Costa, C.; Mates, J.; Iglesias, A.; Sarquella-Brugada, G.; et al. Post-mortem genetic analysis in juvenile cases of sudden cardiac death. Forensic Sci. Int. 2014, 245C, 30–37. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campuzano, O.; Sanchez-Molero, O.; Mademont-Soler, I.; Riuró, H.; Allegue, C.; Coll, M.; Pérez-Serra, A.; Mates, J.; Picó, F.; Iglesias, A.; et al. Rare Titin (TTN) Variants in Diseases Associated with Sudden Cardiac Death. Int. J. Mol. Sci. 2015, 16, 25773-25787. https://doi.org/10.3390/ijms161025773
Campuzano O, Sanchez-Molero O, Mademont-Soler I, Riuró H, Allegue C, Coll M, Pérez-Serra A, Mates J, Picó F, Iglesias A, et al. Rare Titin (TTN) Variants in Diseases Associated with Sudden Cardiac Death. International Journal of Molecular Sciences. 2015; 16(10):25773-25787. https://doi.org/10.3390/ijms161025773
Chicago/Turabian StyleCampuzano, Oscar, Olallo Sanchez-Molero, Irene Mademont-Soler, Helena Riuró, Catarina Allegue, Monica Coll, Alexandra Pérez-Serra, Jesus Mates, Ferran Picó, Anna Iglesias, and et al. 2015. "Rare Titin (TTN) Variants in Diseases Associated with Sudden Cardiac Death" International Journal of Molecular Sciences 16, no. 10: 25773-25787. https://doi.org/10.3390/ijms161025773