
Experimental and Theoretical Study of O-Substituent Effect on the Fluorescence of 8-Hydroxyquinoline

 , ,
, ,
Abstract
:
1. Introduction
2. Results and Discussion
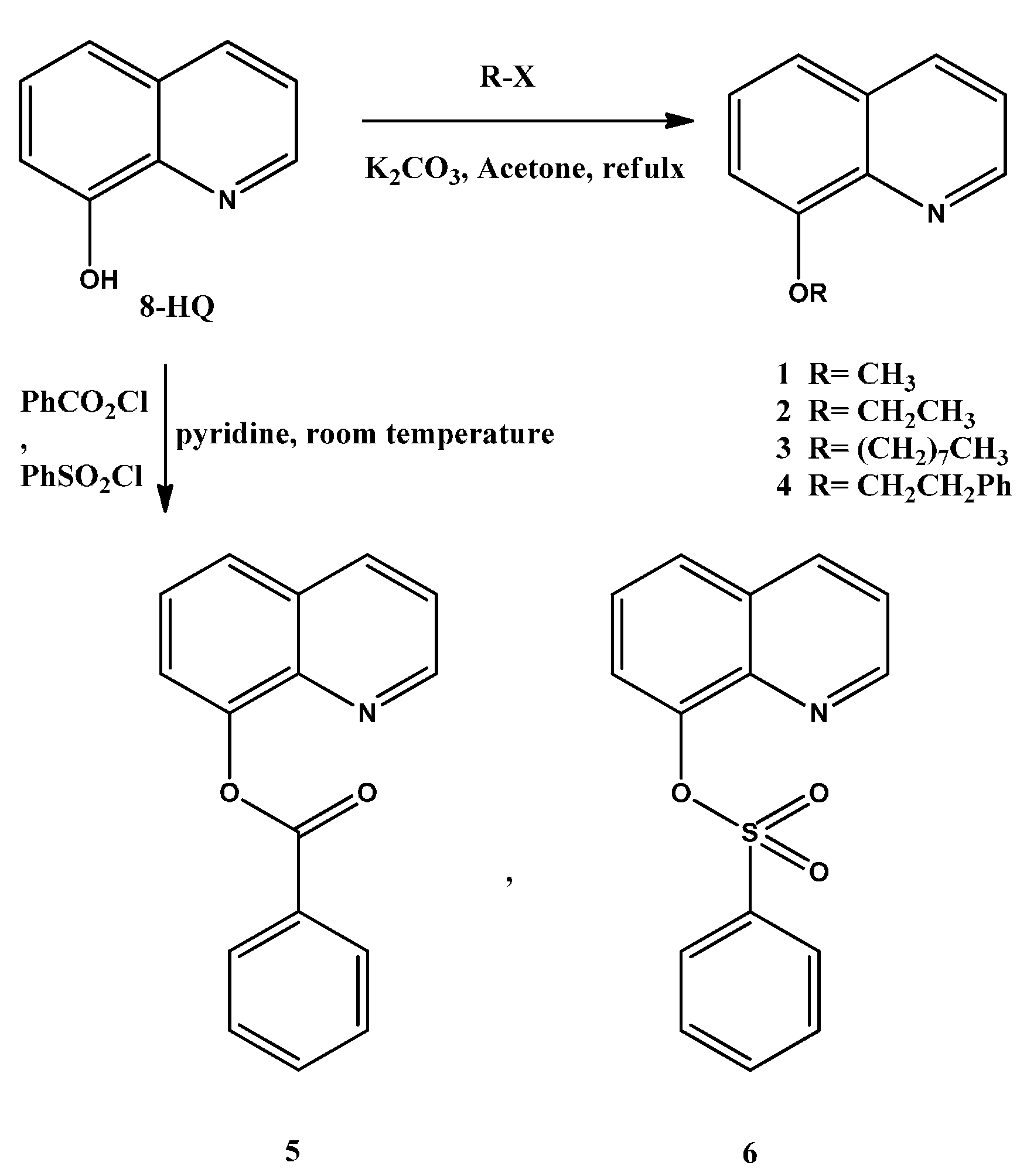
2.1. Synthesis

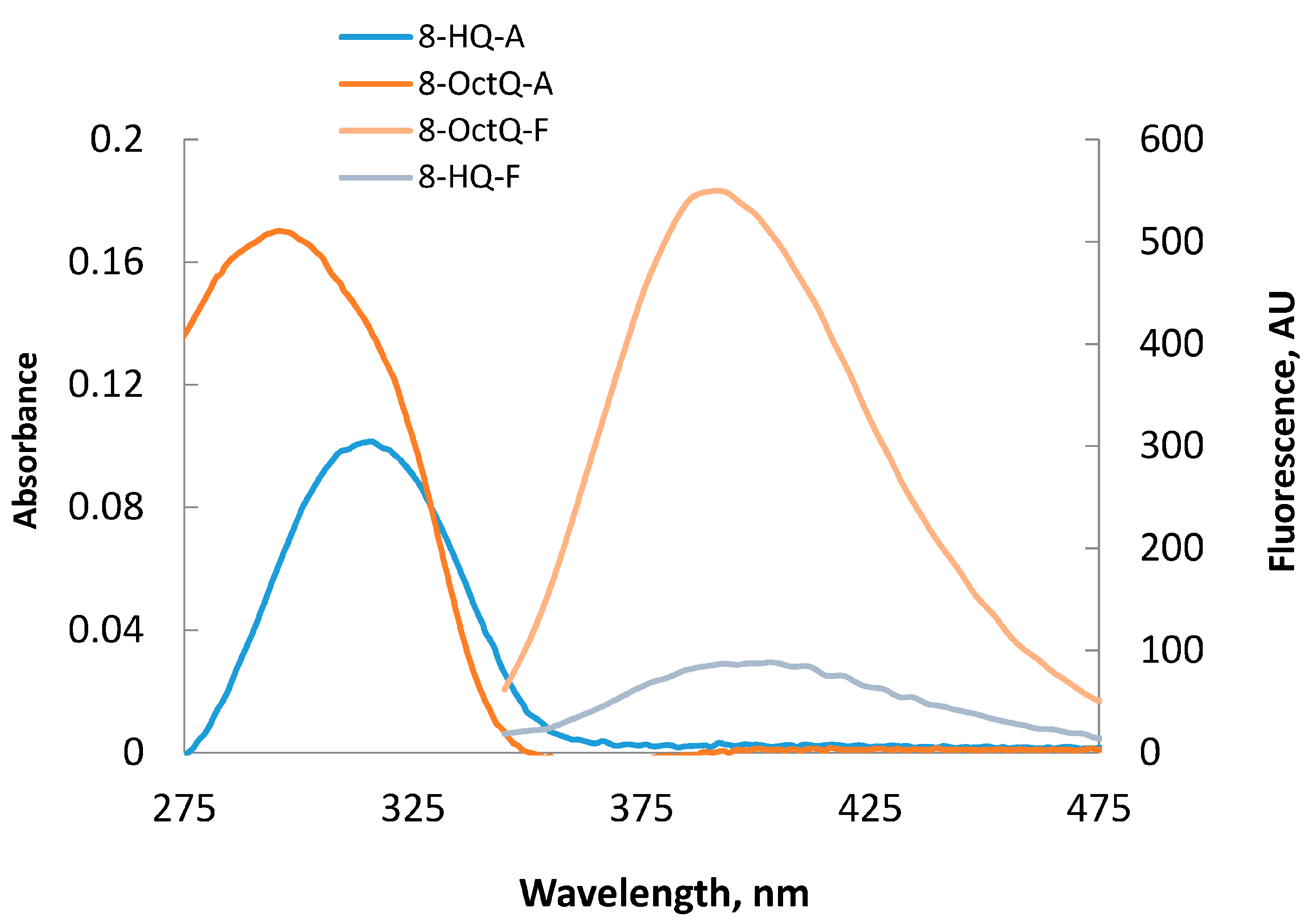
2.2. UV-Visible Spectra

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MeOH, KT(30) = 55.4 | CH3CN, KT(30) = 45.6 | CHCl3, KT(30) = 37.7 | ||||
|---|---|---|---|---|---|---|---|
| λmax, nm A | λmax, nm F | λmax, nm A | Molar Absorbivity, M−1·cm−1 | λmax, nm F | λmax, nm A | λmax, nm F | |
| 8-HQ | 315 | - | 316 | 10,200 | 400 | 311 | - |
| 8-MeQ | 305 | 407 | 305 | 10,700 | 388 | 300 | 390 |
| 8-EtQ | 306 | 405 | 302 | 13,900 | 392 | 307 | 392 |
| 8-PhetQ | 312 | 406 | 304 | 31,700 | 387 | 305 | 387 |
| 8-OctQ | 306 | 405 | 296 | 17,000 | 392 | 307 | 392 |
| 8-OateQ | 283 | - | 276 | 22,400 | 385 | 279 | 393 |
| 8-SulfonateQ | 274 | 359 | 273 | 19,200 | 392 | 275 | 397 |
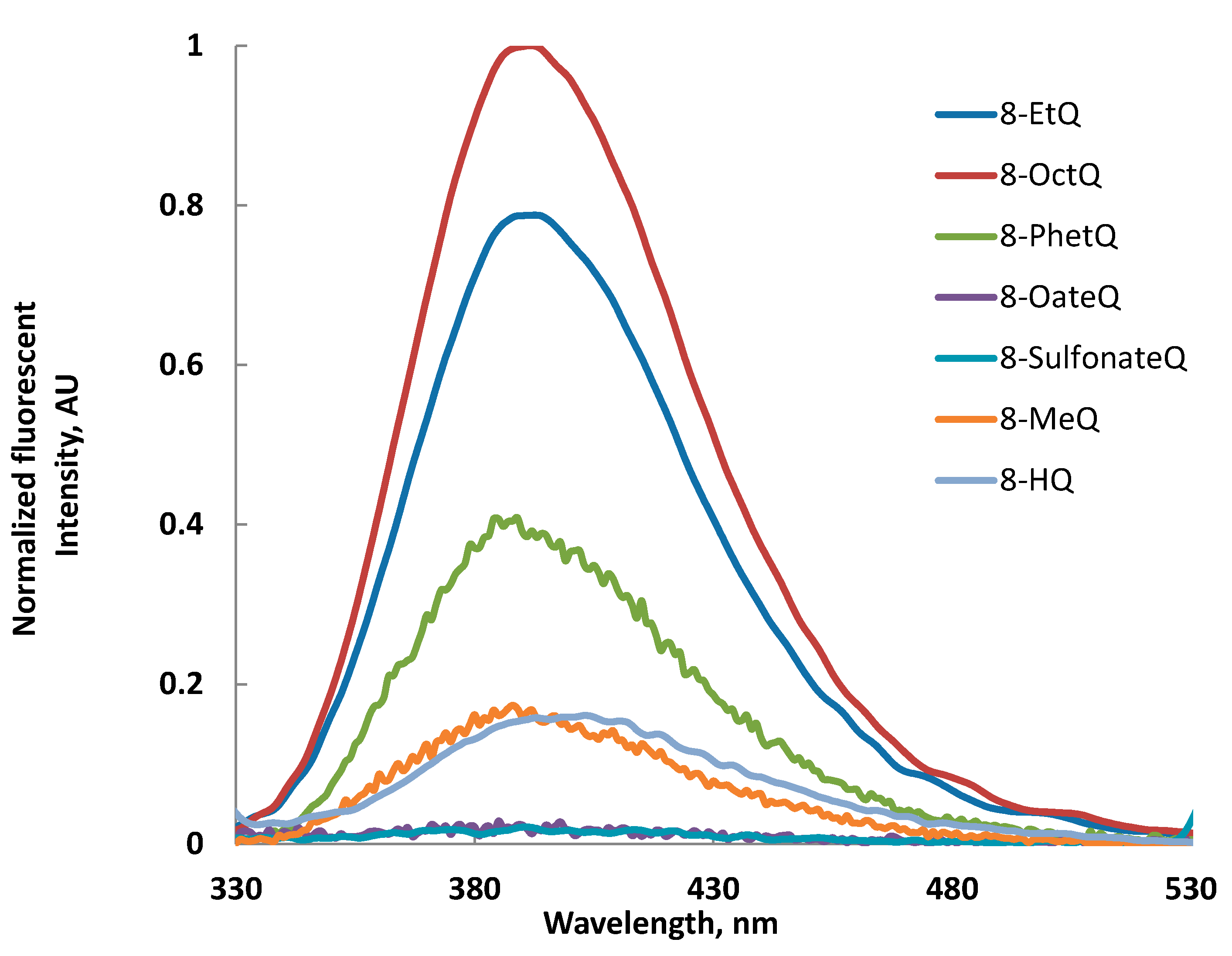
2.3. Fluorescence Spectra


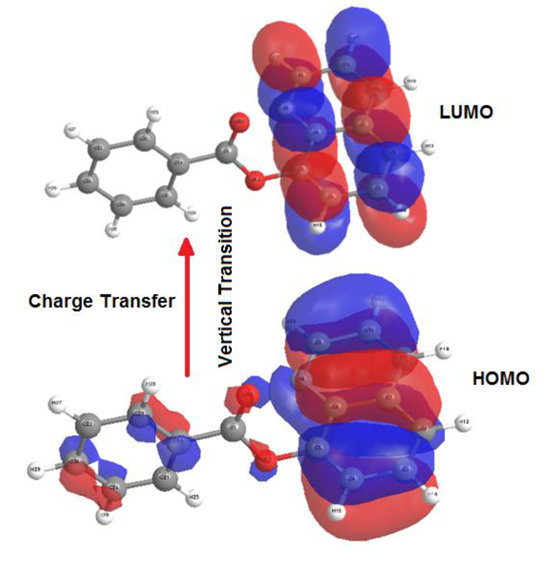
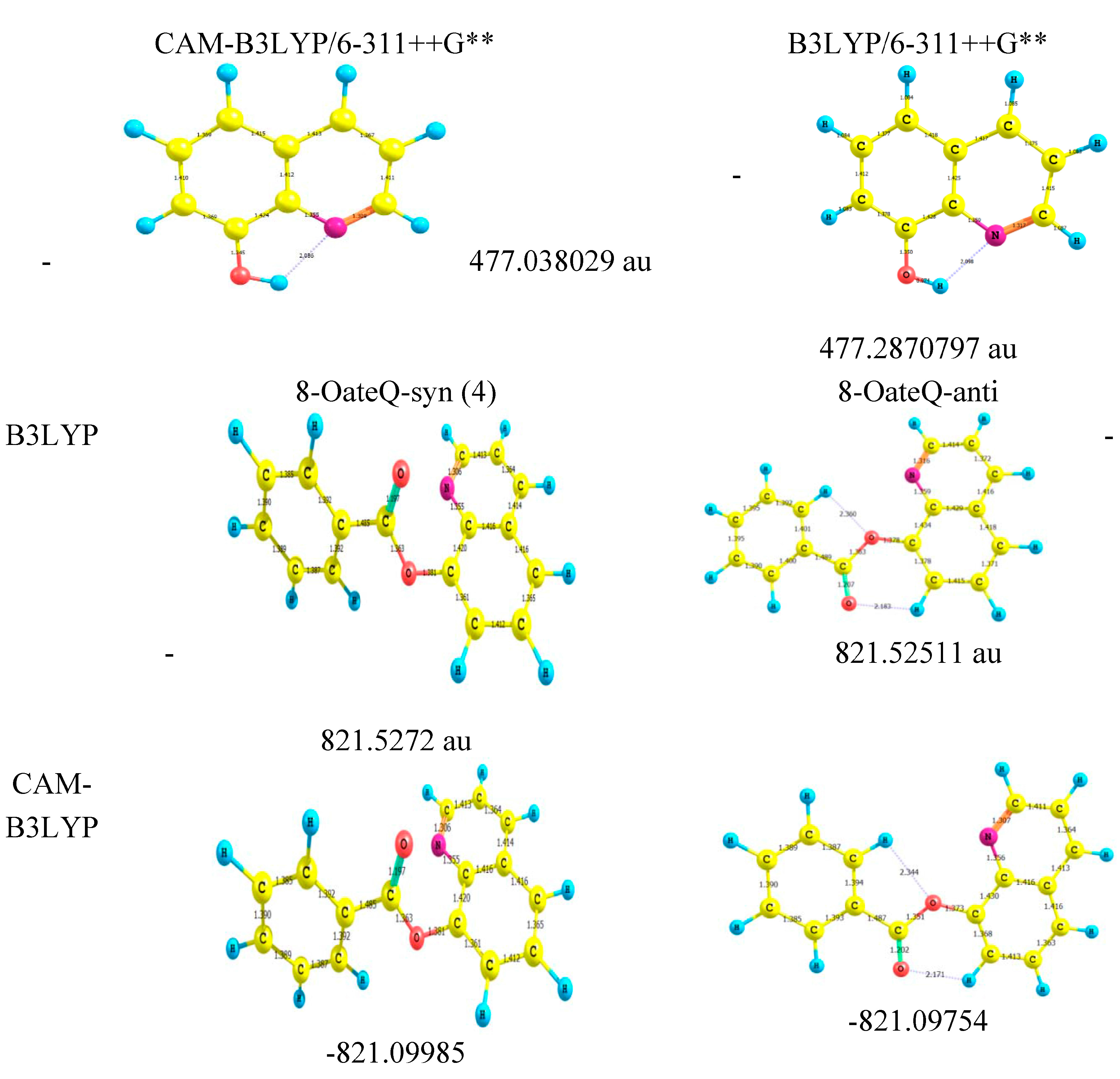
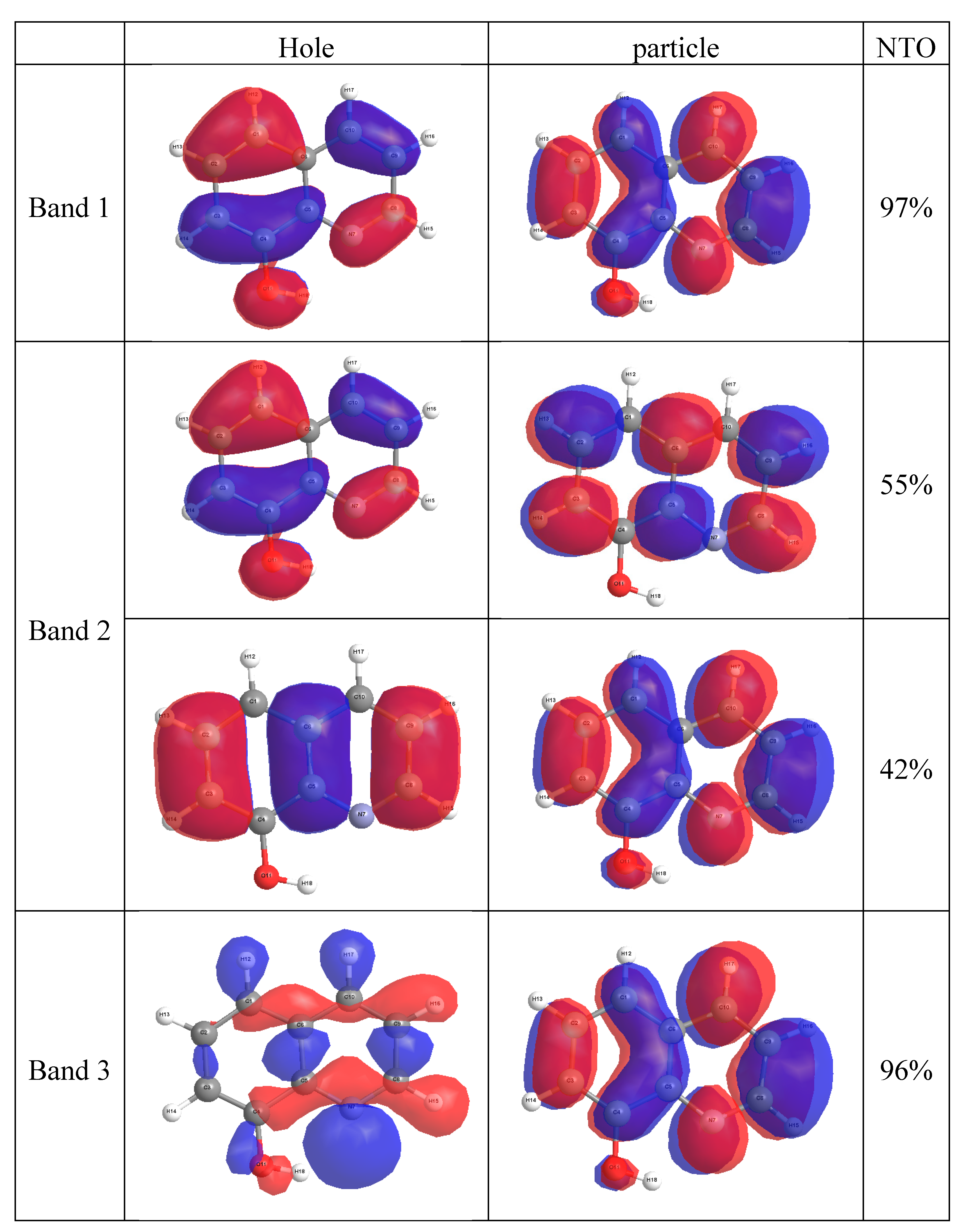
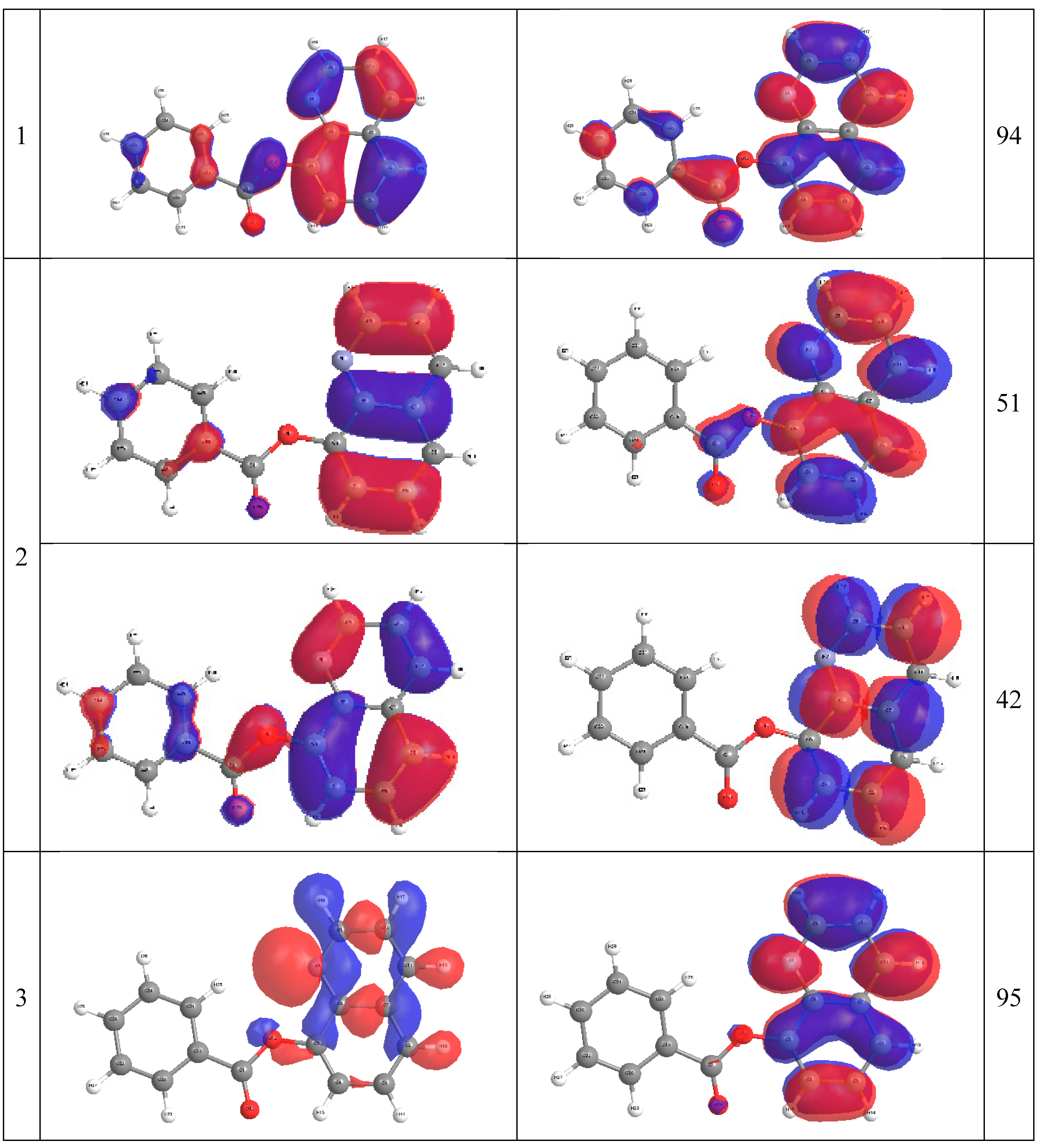
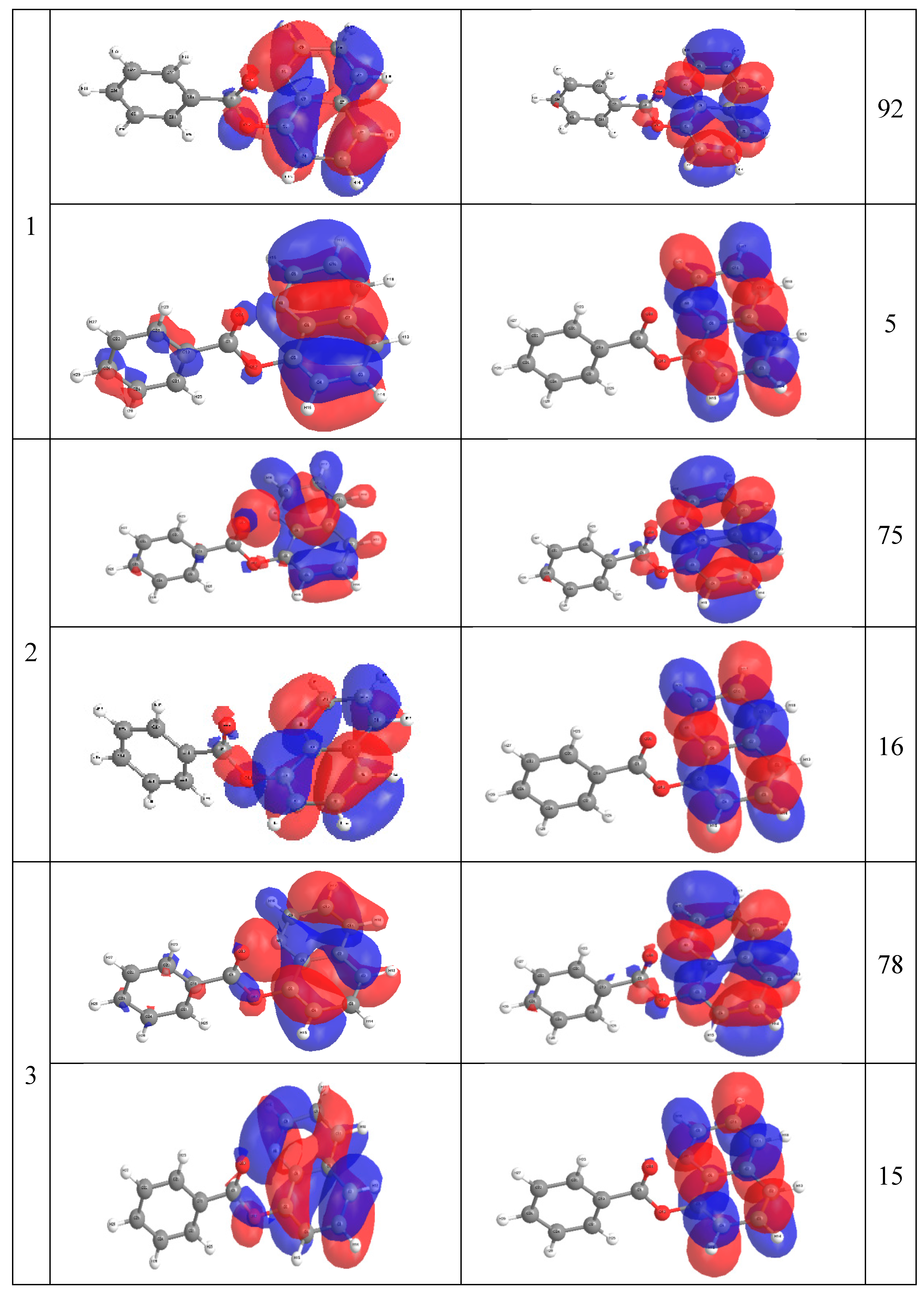
2.4. Molecular Orbital Calculations

| CAM-B3LYP/6-311++G** | |||
|---|---|---|---|
| λmax | f | MO Contribution | MO Coefficient |
| 302.49 | 0.06 | 38–39 | 0.70 |
| - | - | 37–40 | 0.11 |
| 275.58 | 0.002 | 38–40 | 0.53 |
| 248.83 | 0.003 | 36–39 | 0.69 |
| B3LYP/6-311++G** | |||
| 339.88 | 0.037 | 38–39 | 0.69 |
| 288.49 | 0.004 | 38–40 | 0.55 |
| 265.57 | 0.002 | 36–39 | 0.7 |
| CAM-B3LYP/6-311++G** | |||
|---|---|---|---|
| λmax | f | MO Contribution | MO Coefficient |
| 271.11 | 0.113 | 65–66 | 0.63 |
| - | - | 61–66 | 0.19 |
| 268.28 | 0.153 | 61–66 | 0.50 |
| - | - | 64–66 | 0.36 |
| - | - | 65–68 | 0.29 |
| 266.68 | 0.003 | 65–66 | 0.41 |
| B3LYP/6-311++G** | |||
| 292.04 | 0.084 | 65–66 | 0.64 |
| - | - | 61–66 | 0.2 |
| - | - | 62–66 | 0.14 |
| 288.4 | 0.14 | 61–66 | 0.57 |
| - | - | 62–66 | 0.37 |
| - | - | 64–66 | 0.17 |
| 277.02 | 0.002 | 65–67 | 0.69 |
| - | - | 60–67 | 0.11 |
| CAM-B3LYP/6-311++G** | |||
|---|---|---|---|
| λmax | f | MO Contribution | MO Coefficient |
| 281.21 | 0.225 | 65–66 | 0.66 |
| - | - | 64–68 | 0.13 |
| 268.94 | 0.01 | 64–66 | 0.47 |
| 268.80 | 0.002 | 61–66 | 060 |
| B3LYP/6-311++G** | |||
| 307.96 | 0.0221 | 65–66 | 0.7 |
| 290.96 | 0.002 | 62–66 | 0.66 |
| 283.17 | 0.052 | 65–67 | 0.7 |
| 8-HQ | ||
|---|---|---|
| Methanol | Acetonitrile | Chloroform |
| 296.78 | 296.84 | 299.31 |
| 273.47 | 273.47 | 274.17 |
| 246.66 | 246.67 | 247.39 |
| Experimental | ||
| 315 | 316 | 311 |
| 8-OateQ-syn | ||
| Methanol | Acetonitrile | Chloroform |
| 270.96 | 271.03 | 271.83 |
| 267.11 | 267.13 | 267.55 |
| 259.75 | 259.73 | 261.79 |
| Experimental | ||
| 314 | 276 | 279 |
| 8-OateQ-anti | ||
| Methanol | Acetonitrile | Chloroform |
| 282.03 | 282.12 | 283.31 |
| 268.83 | 268.39 | 268.78 |
| 262.87 | 262.86 | 264.43 |
| Experimental | ||
| 314 | 276 | 279 |



3. Experimental Section
3.1. General
3.2. Synthesis
3.2.1. Etherification, General Procedure
8-Methoxyquinoline (1)
8-Ethoxyquinoline (2)
8-(Octyloxy)quinoline (3)
8-Phenethoxyquinoline (4)
3.2.2. Esterification, General Procedure
Quinolin-8-yl benzoate (5)
Quinolin-8-yl benzenesulfonate (6)
3.3. Computational Methods
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Polanski, J.; Zouhiri, F.; Jeanson, L.; Desmaele, D.; D’Angelo, J.; Mouscadet, J.F.; Gasteiger, J.; le Bret, M. Use of the Kohonen neural network for rapid screening of ex vivo anti-HIV activity of styrylquinolines. J. Med. Chem. 2002, 45, 4647–4654. [Google Scholar]
- Polanski, J.; Niedbala, H.; Musiol, R.; Tabak, D.; Podeszwa, B.; Gieleciak, R.; Bak, A.; Palka, A.; Magdziarz, T. Analogues of the styrylquinoline and styrylquinazoline HIV-1 integrase inhibitors: Design and synthetic problems. Acta Pol. Pharm. Drug Res. 2004, 61, 3–4. [Google Scholar]
- Polanski, J.; Niedbala, H.; Musiol, R.; Podeszwa, B.; Tabak, D.; Palka, A.; Mencel, A.; Finster, J.; Mouscadet, J.F.; le Bret, M. 5-Hydroxy-6-quinaldic acid as a novel molecular scaffold for HIV-1 integrase inhibitors. Lett. Drugs Des. Discov. 2006, 3, 175–178. [Google Scholar]
- Polanski, J.; Niedbala, H.; Musiol, R.; Podeszwa, B.; Tabak, D.; Palka, A.; Mencel, A.; Mouscadet, J.F.; le Bret, M. Fragment based approach for the investigation of HIV-1 integrase inhibition. Lett. Drugs Des. Discov. 2007, 4, 99–105. [Google Scholar]
- Musiol, R.; Jampilek, J.; Kralova, K.; Richardson, D.R.; Kalinowski, D.; Podeszwa, B.; Finster, J.; Niedbala, H.; Palka, A.; Polanski, J. Investigating biological activity spectrum for novel quinoline analogues. Bioorg. Med. Chem. 2007, 15, 1280–1288. [Google Scholar]
- Podeszwa, B.; Niedbala, H.; Polanski, J.; Musiol, R.; Tabak, D.; Finster, J.; Serafin, K.; Wietrzyk, J.; Boryczka, S.; Mol, W.; et al. Investigating the antiproliferative activity of quinoline-5,8-diones and styrylquinolinecarboxylic acids on tumor cell lines. Bioorg. Med. Chem. Lett. 2007, 17, 6138–6141. [Google Scholar]
- Musiol, R.; Tabak, D.; Niedbala, H.; Podeszwa, B.; Jampilek, J.; Kralova, K.; Dohnal, J.; Finster, J.; Mencel, A.; Polanski, J. Investigating biological activity spectrum for novel quinoline analogues 2: Hydroxyquinolinecarboxamides with photosynthesis-inhibiting activity. Bioorg. Med. Chem. 2008, 16, 4490–4499. [Google Scholar]
- Jampilek, J.; Musiol, R.; Pesko, M.; Kralova, K.; Vejsova, M.; Carroll, J.; Coffey, A.; Finster, J.; Tabak, D.; Niedbala, H.; et al. Ring-substituted 4-hydroxy-1h-quinolin-2-ones: Preparation and biological activity. Molecules 2009, 14, 1145–1159. [Google Scholar]
- Jampilek, J.; Musiol, R.; Finster, J.; Pesko, M.; Carroll, J.; Kralova, K.; Vejsova, M.; O’Mahony, J.; Coffey, A.; Dohnal, J.; et al. Investigating biological activity spectrum for novel styrylquinazoline analogue. Molecules 2009, 14, 4246–4265. [Google Scholar]
- Jampilek, J.; Dolezal, M.; Kunes, J.; Buchta, V.; Kralova, K. Quinaldine derivatives: Preparation and biological activity. Med. Chem. 2005, 1, 591–599. [Google Scholar]
- Shchavlev, A.E.; Pankratov, A.N.; Shalabay, A.V. DFT computational studies on rotation barriers, tautomerism, intramolecular hydrogen bond, and solvent effects in 8-hydroxyquinoline. Int. J. Quantum Chem. 2006, 106, 876–886. [Google Scholar]
- National Toxicology Program (NTP). National Toxicology Program (NTP). Toxicology and Carcinogenesis Studies of 8-Hydroxyquinoline in F344/N Rats and B6C3F1 Mice, NTP TR 276; Technical Report; U.S. Department of Health and Human Services: Research Triangle Park, NC, USA, 1985. [Google Scholar]
- Kovtun, Y.P.; Prostota, Y.O.; Tolmachev, A.I. Metallochromic merocyanines of 8-hydroxyquinoline series. Dyes Pigment. 2003, 58, 83–91. [Google Scholar]
- Pearce, D.A.; Jotterand, N.; Carrico, I.S.; Imperiali, B. Derivatives of 8-hydroxy-2-methylquinoline are powerful prototypes for zinc sensors in biological systems. J. Am. Chem. Soc. 2001, 123, 5160–5161. [Google Scholar]
- Elroby, S.A.K.; El-Shishtawy, R.M.; Makki, M.S.I. Influence of the protonation, deprotonation and transition metal ions on the fluorescence of 8-hydroxyquinoline: A computational study. Mol. Simul. 2011, 37, 940–952. [Google Scholar]
- Leung, L.M.; Lo, W.Y.; So, S.K.; Lee, K.M.; Choi, W.K. A high-efficiency blue emitter for small molecule-based organic light- emitting diode. J. Am. Chem. Soc. 2000, 122, 5640–5641. [Google Scholar]
- Devol, I.; Bardez, E. J. Complexation of Al(III) by 8-hydroxyquinoline and drastic fluorescence enhancement in reverse micelles. Colloid Interface Sci. 1998, 200, 241–248. [Google Scholar]
- Pohl, R.; Montes, V.A.; Shinar, J.; Anzenbacher, P., Jr. Red-green-blue emission from tris(5-aryl-8-quinolinolate)Al(III) complexes. J. Org. Chem. 2004, 69, 1723–1725. [Google Scholar]
- Valeur, B.; Leray, I. Design principles of fluorescent molecular sensors for cation recognition. Coord. Chem. Rev. 2000, 205, 3–40. [Google Scholar]
- Abdelaal, M.Y.; Sobahi, T.R.; El-Shishtawy, R.M. Chromophoric thin film based on cellulose triacetate blends for sensing metal ions. CR Chim. 2014, 17, 557–562. [Google Scholar]
- Valeur, B. Molecular Fluorescence, Principles and Applications. Wiley-VCH: Weinheim, Germany, 2002. [Google Scholar]
- Lakowics, J.R. Principles of Fluorescence Spectroscopy, 2nd ed.; Kluwer, Academic/Plenum Publishers: New York, NY, USA, 1999. [Google Scholar]
- Burke, K.; Werschnik, J.; Gross, E.K.U. Time-dependent density functional theory: Past, present, and future. J. Chem. Phys. 2005, 123, 062206. [Google Scholar] [CrossRef]
- Foresman, J.B.; Head-Gordon, M.; Pople, J.A. Toward a systematic molecular orbital theory for excited states. J. Phys. Chem. 1992, 96, 135–149. [Google Scholar]
- Putz, M.V.; Ionaşcu, C.; Putz, A.M.; Ostafe, V. Alert-QSAR. Implications for electrophilic theory of chemical carcinogenesis. Int. J. Mol. Sci. 2011, 12, 5098–5134. [Google Scholar]
- Putz, M.V. Residual-QSAR. Implications for genotoxic carcinogenesis. Chem. Cent. J. 2011, 5, 29. [Google Scholar] [CrossRef]
- Putz, M.V. Density functionals of chemical bonding. Int. J. Mol. Sci. 2008, 9, 1050–1095. [Google Scholar]
- Miertus, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar]
- Miertus, S.; Tomasi, J. Approximate evaluations of the electrostatic free energy and internal energy changes in solution processes. Chem. Phys. 1982, 65, 239–245. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A.J.; Vreven, T.; Kudin, K.N.; Burant, J.C. Gaussian 03; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Reichardt, C. Solvatochromic dyes as solvent polarity indicators. Chem. Rev. 1994, 94, 2319–2358. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zayed, M.E.M.; El-Shishtawy, R.M.; Elroby, S.A.; Obaid, A.Y.; Al-amshany, Z.M. Experimental and Theoretical Study of O-Substituent Effect on the Fluorescence of 8-Hydroxyquinoline. Int. J. Mol. Sci. 2015, 16, 3804-3819. https://doi.org/10.3390/ijms16023804
Zayed MEM, El-Shishtawy RM, Elroby SA, Obaid AY, Al-amshany ZM. Experimental and Theoretical Study of O-Substituent Effect on the Fluorescence of 8-Hydroxyquinoline. International Journal of Molecular Sciences. 2015; 16(2):3804-3819. https://doi.org/10.3390/ijms16023804
Chicago/Turabian StyleZayed, Mohie E. M., Reda M. El-Shishtawy, Shaaban A. Elroby, Abdullah Y. Obaid, and Zahra M. Al-amshany. 2015. "Experimental and Theoretical Study of O-Substituent Effect on the Fluorescence of 8-Hydroxyquinoline" International Journal of Molecular Sciences 16, no. 2: 3804-3819. https://doi.org/10.3390/ijms16023804