
Ceramide-Induced Apoptosis in Renal Tubular Cells: A Role of Mitochondria and Sphingosine-1-Phoshate

Abstract
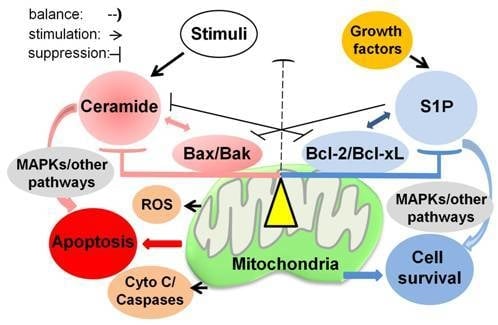
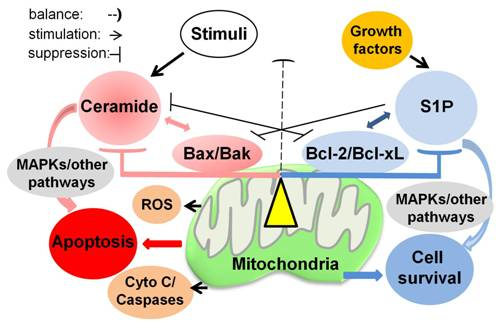
:
1. Introduction
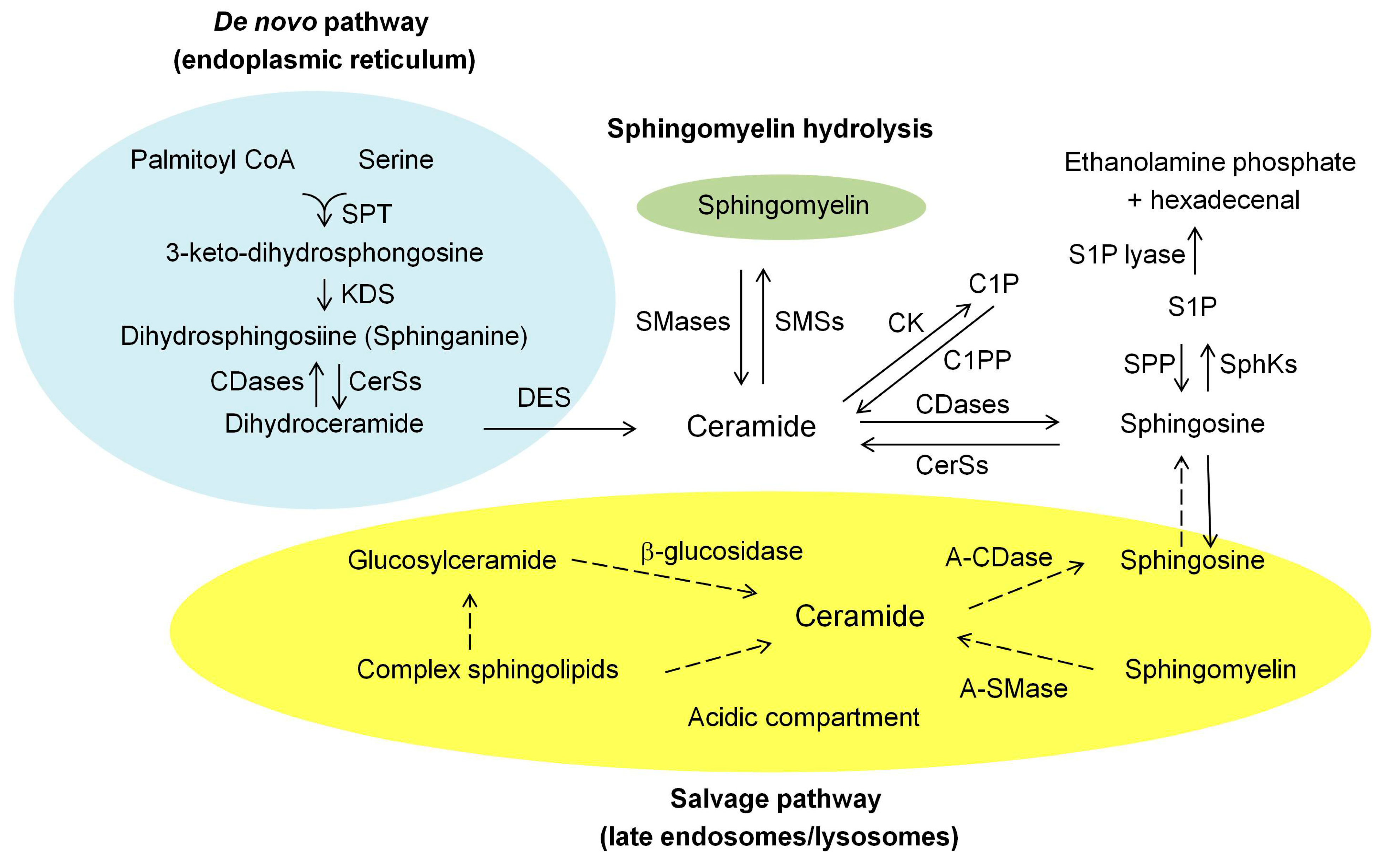
2. Ceramide Biosynthesis and Degrading Pathways in Mammalian Cells
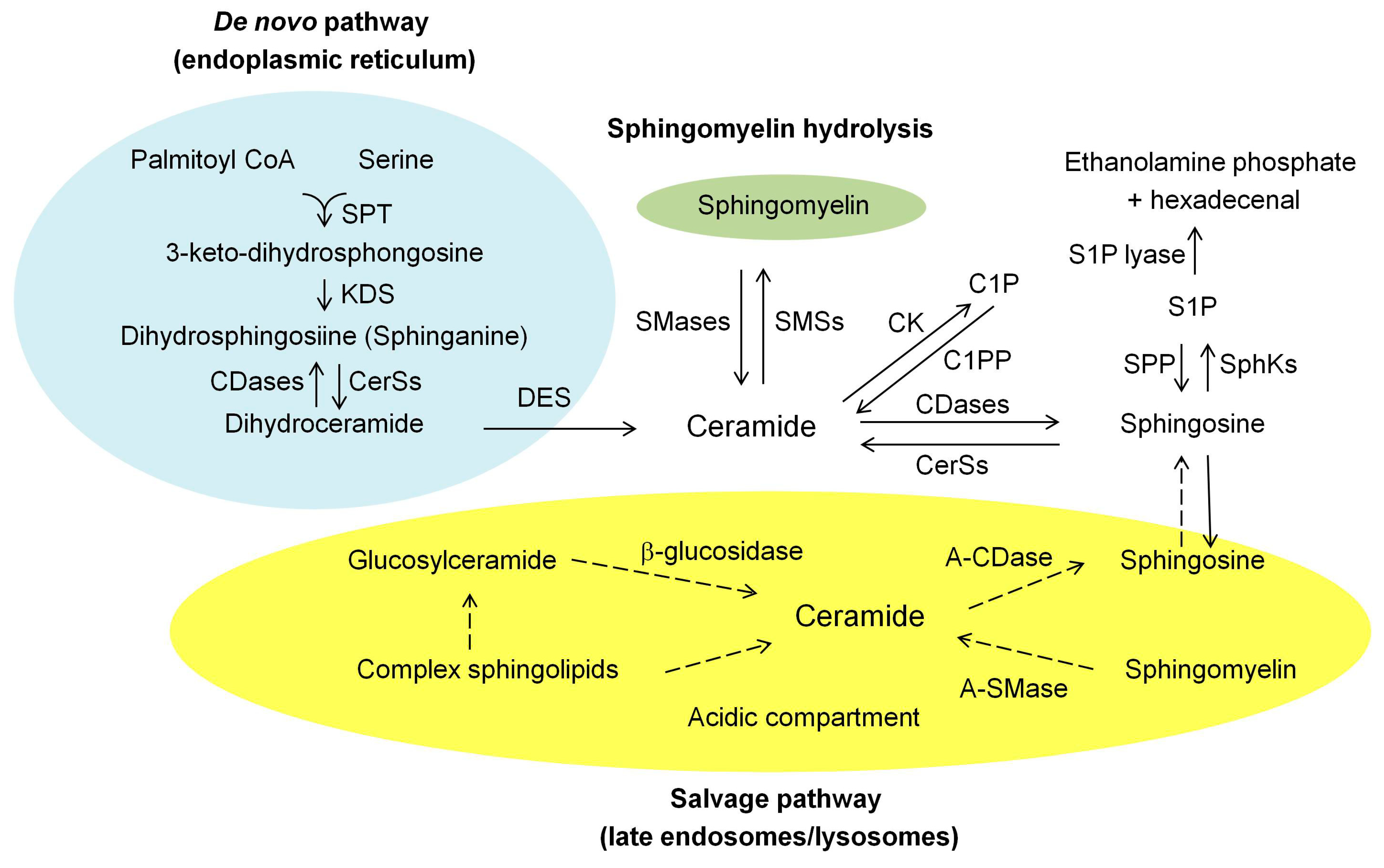
2.1. De Novo Synthesis Pathway
2.2. Hydrolysis of the Sphingomyelin (SM) Pathway
2.3. Salvage Pathway
2.4. Degrading Pathway
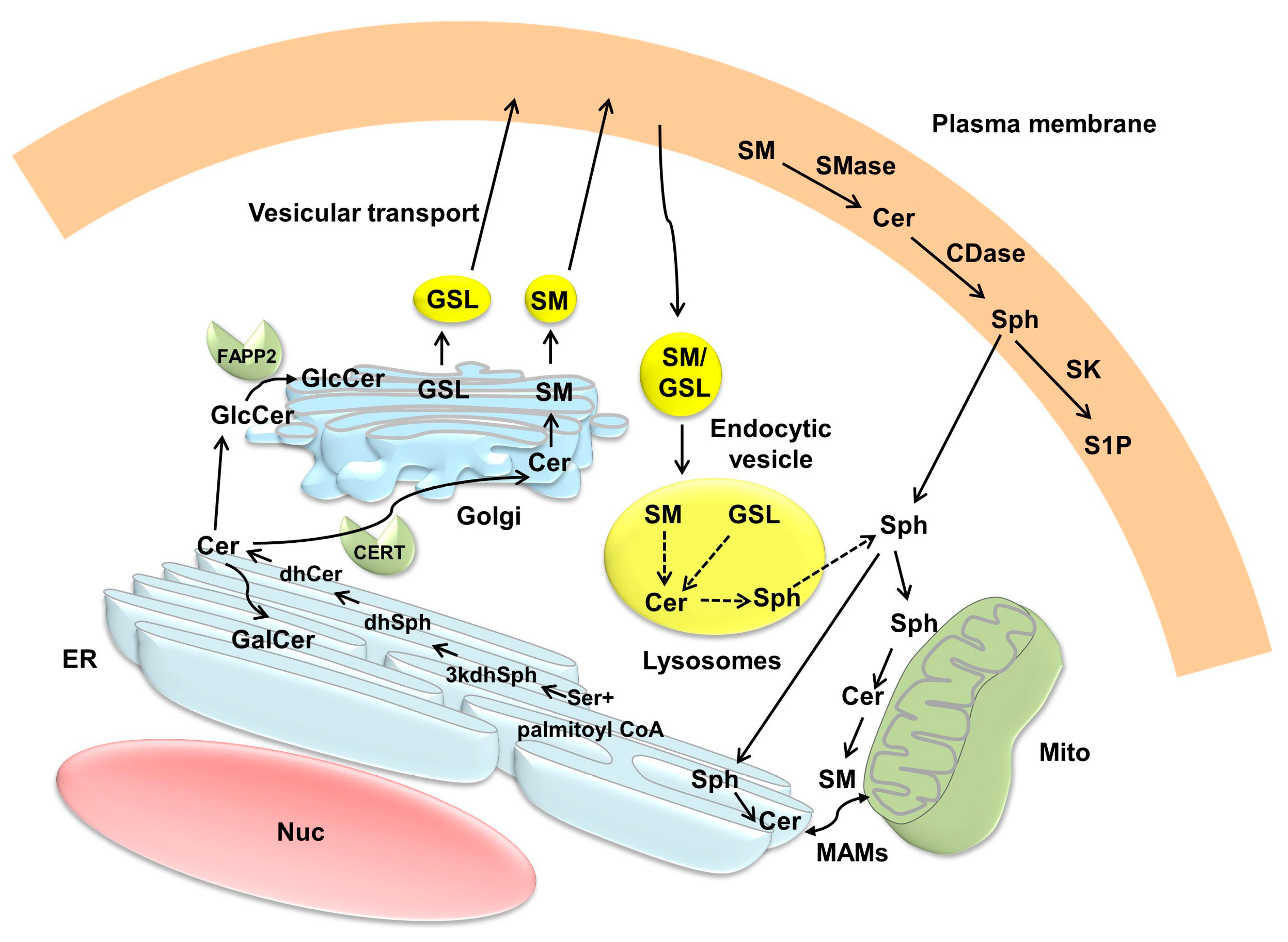
3. Compartmentalization of Ceramide Metabolism and Trafficking of Ceramide
3.1. Intracellular Localization of the Enzymes Involved in Ceramide Metabolism
3.2. Ceramide Compartmentalization and Trafficking
3.2.1. Endoplasmic Reticulum (ER) and Golgi
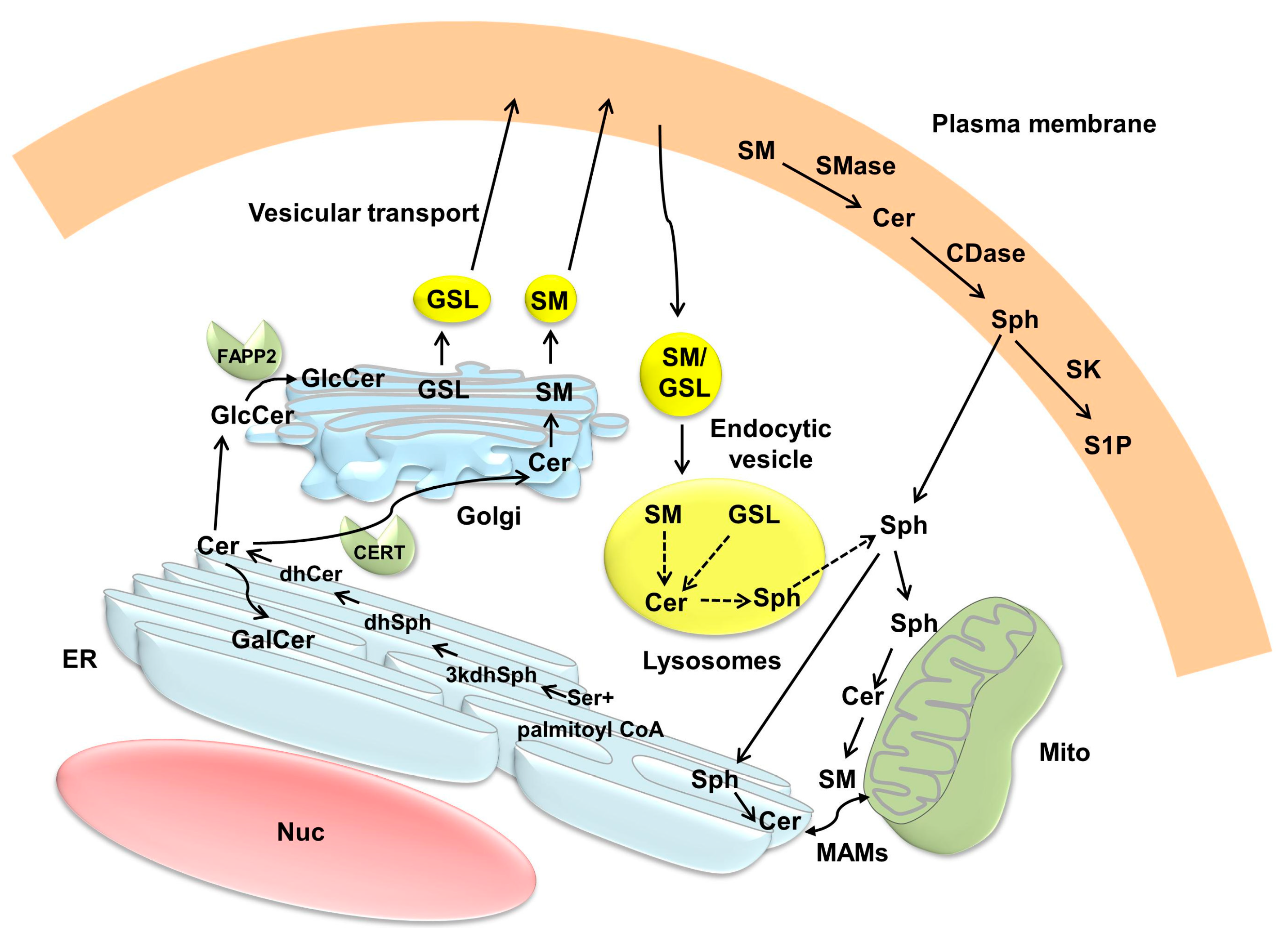
3.2.2. Mitochondria
3.2.3. Plasma Membrane and Lysosomes/Endosomes
3.2.4. Nuclei
3.3. Ceramide Compartmentalization and Trafficking in Renal Tubular Cells (RTCs)
4. Ceramide-Induced Apoptosis
4.1. Overview of Apoptosis
4.2. Ceramide-Induced Apoptosis in RTCs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
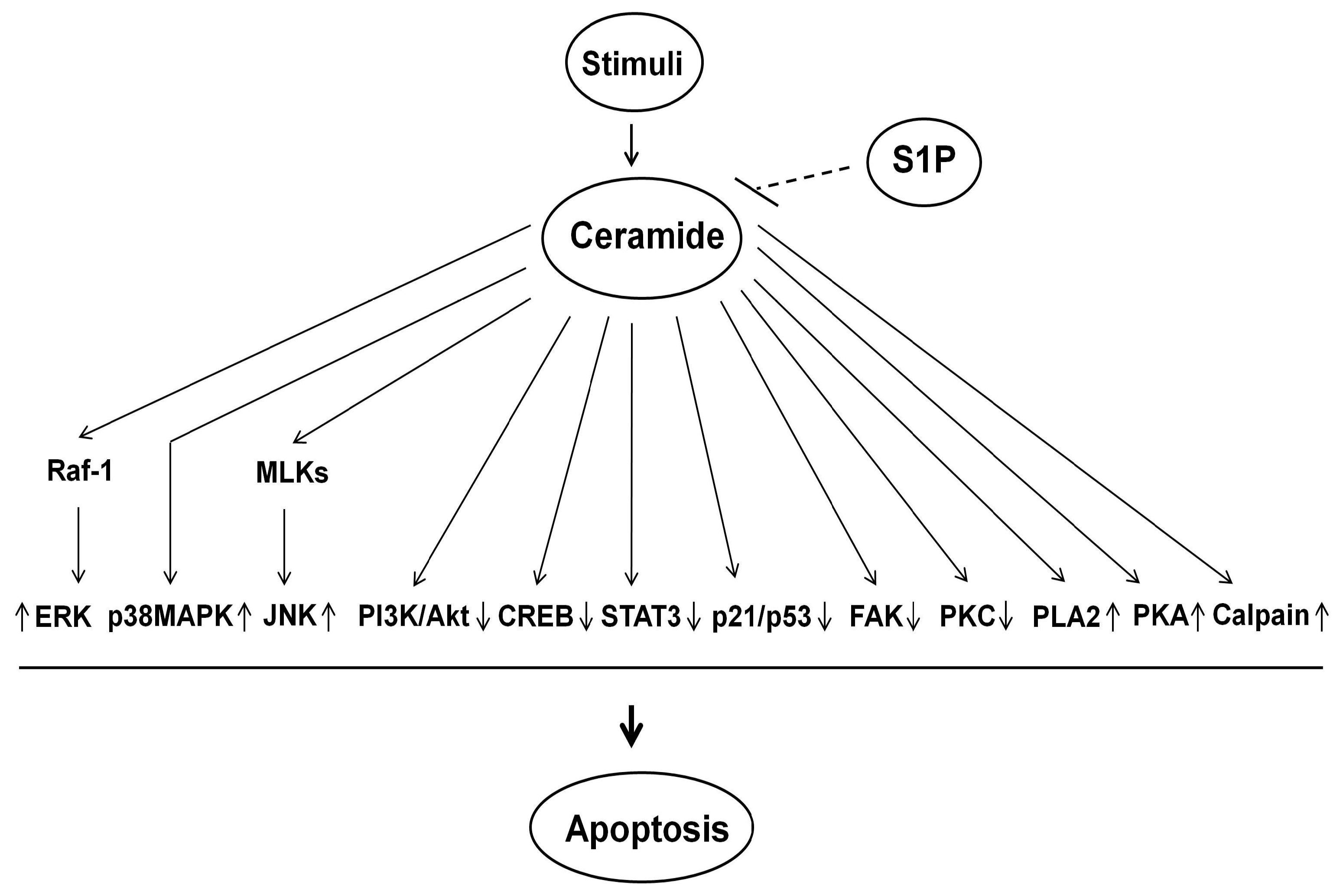{kind=link}
| Stimuli | Species | Cell Line for in Vitro */ Tissue for in Vivo | Increased Sphingolipid | Enzymes Involving Ceramide Metabolism | Ref. |
|---|---|---|---|---|---|
| In vitro study | |||||
| Cadmium | Rat | PTCs | Ceramide | CerSs↑ | [60,61] |
| Heat stress | Dog | MDCK cells | Ceramide | CerSs↑ | [56] |
| Hypoxia/reoxygenation | Pig/Rat | LLC-PK1 cells/NRK-52E cells | Ceramide | CerSs↑ | [31,32,50] |
| Interleukin-1β | Human | A498 cells ** | Ceramide | A-SMase↑, N-SMase↑ | [59] |
| Isoflurane | Mouse Human | PTCs/HK-2 cells | Ceramide/Sphingosine | A-SMase→, N-SMase→, CerS→ | [62] |
| Microcystin | Human | HEK293 cells | Ceramide | unknown | [63] |
| Nickel | Rat | PTCs | Ceramide | CerSs↑ | [64] |
| Oxalate | Pig/Dog | LLC-PK1 cells/MDCK cells | Ceramide | CerSs↑, SMases↑ | [54,57] |
| Oxidants | Human | HK-2 cells | Ceramide/Sphingosine↓ | N-CDases→, N-SMase↓, A-SMase→, CerSs→ | [53] |
| P-finbriae of E. coli | Human | A498cells ** | Ceramide | A-SMases→, N-SMase→, A-SMase↓, N-SMase→ | [58,59] |
| Radiocontrast | Pig | LLC-PK1 cells | Ceramide | CerSs↑ | [65] |
| Shiga-toxin B | Human | A498cells ** | Ceramide | A-SMase→, N-SMase→ | [58] |
| St. enterotoxin B | Human | PTCs | Ceramide | N-SMase↑ | [66] |
| TNF-α | Dog/ Human | MDCK cells/A498 cells ** | Ceramide | A-SMase↑, N-SMase↑ | [54,59] |
| Ultraviolet light | Mouse | BMK cells | Ceramide | CerSs↑ | [38] |
| In vivo study | |||||
| Anti-GBM Ab-induced ARF | Mouse | Kidney | Ceramide | A-SMase↑, N-SMase↑ | [52] |
| Carbon tetrachloride | Rat | Kidney | Ceramide | A-SMase→, N-SMase↑ | [67] |
| Developing kidney | Rat | Kidney | Ceramide/S1P↓ | CerSs↑, SphKs↓ | [68,69] |
| Ischemia/reperfusion | Mouse | Kidney | Ceramide | A-SMase↓, N-SMase↓ | [51,52] |
| Isoflurane | Mouse | Kidney | Ceramide/Sphingosine | A-SMase→, ↑ #, N-SMase→, CDase↑ | [62] |
| Myohemoglobinuria | Mouse | Kidney | Ceramide | A-SMase↓, N-SMase↓ | [52] |
| Ureteral obstruction | Rat | Kidney | Ceramide | unknown | [70] |
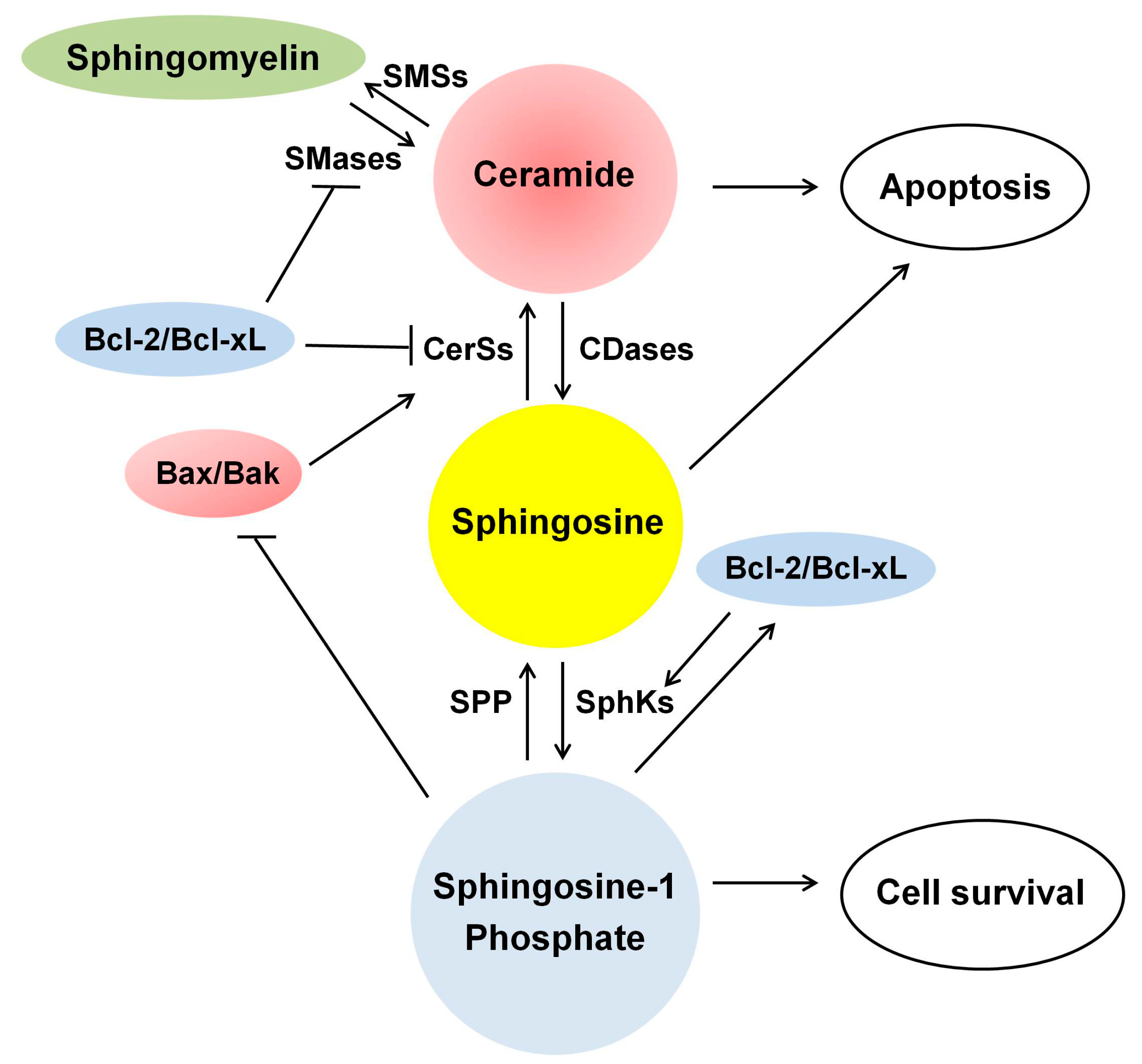
4.3. A Role of Balance between Ceramide and Sphingosine-1 Phosphate (S1P) in RTCs
4.3.1. Sphingosine/Sphinganine
4.3.2. Ceramide and S1P
Sphingosine Kinases (SphKs)
S1P/S1P Receptors as a Survival Factor
S1P Phosphatase and S1P Lyase as Pro-Apoptotic Factors
4.4. Ceramide Compartmentalization and Trafficking in Ceramide-Induced Apoptosis
5. Ceramide-Induced Signaling Pathway for Apoptosis
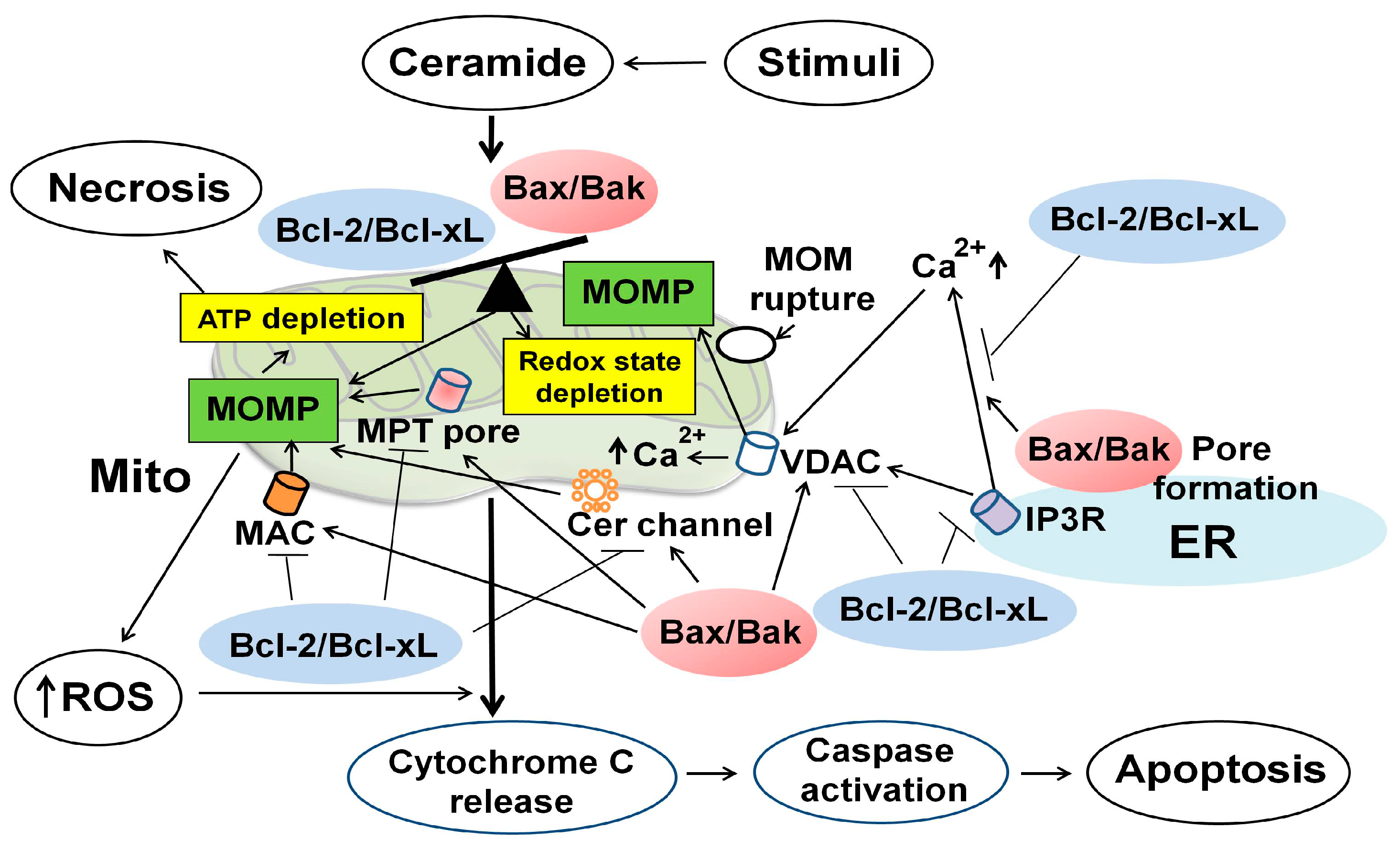
5.1. Overview of a Role of Mitochondria in Ceramide-Induced Apoptosis
5.2. Mitochondrial Outer Membrane Permeability (MOMP) and Bcl-2 Family Proteins
5.2.1. Mitochondrial Integrity Regulated by Bcl-2 Family Proteins


5.2.2. Ceramide-Induced Bax/Bak Pore Formation in Mitochondria
5.2.3. Ceramide Channel Regulated by Bcl-2 Family Proteins
5.2.4. Voltage-Dependent Anion Channel (VDAC) and Mitochondrial Permeability Transition (MPT) Pore
| Stimuli | Cell Line/Tissue | Enzymes Involving MOMP | Bcl-2 Proteins that Regulate Cer-Induced MOMP | Channel for Cer-Induced MOMP | Comments | Ref. |
|---|---|---|---|---|---|---|
| In other types of cells | ||||||
| t-Bid, N-SMase | Murine liver mo./HeLa cells | N-SMase↑ | tBid-induced Bax oligomerization/ conformational change | MAC | Bax cooperates with Hex and Bak cooperates with S1P to induce t-Bid-mediated MOMP. Bcl-xL and N-SMase inhibitor inhibit MOMP. | [108] |
| UV light | HeLa cells | A-SMase↑ | Bax conformational change | MAC | Bcl-2 inhibits MOMP. Bax change requires A-SMase activation. | [109] |
| C16-Cer | HeLa cells | NA | Bax conformational change | MAC | Cer but not UV induces Bax change in A-SMase-deficient cells. Bcl-2 prevents Cer-induced Bax change. | [109] |
| Irradiation | HeLa cells | CerS↑ in MAM | Bax↑ | MAC | Oligomeric Bax insertion into MOM causes MOMP. | [110] |
| C16-Cer | Mo. of HeLa cells/ mouse liver | NA | tBid-induced Bax↑ | MAC | Cer induces MCRM *, favoring Bax insertion to MOM and oligomerization. | [110] |
| C16-Cer | Rat liver mo. | NA | t-Bid-induced Bak↑ | Cer channel | Cer and Bax synergistically induce MOMP. Oligomeric Bax enhances Cer channel formation. | [112] |
| C16-Cer | Rat liver mo. | NA | Bcl-xL/CED-9 prevents MOMP | Cer channel | Bcl-xL/CED-9 prevents and disassembles Cer channel. | [114] |
| In renal tubular cells | ||||||
| Radiocontrast | LLC-PK1 cells | CerS↑ | Bax↑, Bcl-2↓ | probably MAC | CerS inhibition reverses the change in Bax/Bcl-2. | [65] |
| C2-Cer | HK-2 cells | NA | Bax↑ | probably MAC | [71] | |
| C16-Cer | BMK cells | NA | MOMP occurs in Bax−/−Bak−/− cells | Cer channel | Bax/Bak is dispensable for Cer channel formation. | [114] |
| C2-Cer | HEK293 cells | NA | Bad dephosphorylation | MPT pore | Bad/Bcl-xL/VDAC but not Bax/Bak regulate MPT pore opening. | [123] |
5.2.5. Ceramide-Induced Mitochondrial Calcium Uptake and Fission Regulate MOMP
5.2.6. Ceramide-Induced MOMP in the Regulation of Apoptosis of RTCs
5.2.7. Ceramide-Induced and Ceramide-Independent MOMP in Apoptosis
5.3. Regulation of the Enzyme Involved in Ceramide Metabolism by Bcl-2 Family Proteins
5.4. Ceramide-Induced Generation of Reactive Oxygen Species (ROS) and Its Regulation by Bcl-2 Family Proteins
5.4.1. Mitochondria and ROS Generation in Ceramide-Induced Apoptosis
5.4.2. Can ROS and Redox State Regulate the Enzymes Involved in Ceramide Metabolism?
5.4.3. A Role of Bcl-2 Family Proteins and ROS Production in Ceramide-Induced Apoptosis
| Stimuli | Cell Line/Tissue | Cer-Induced Alteration of ROS/AOS | Enzymes for Cer Production Regulated by ROS | Bcl-2 Proteins Regulated by ROS/AOS | Cell Death | Ref. |
|---|---|---|---|---|---|---|
| Hypoxia | LLC-PK1 cells, NRK-52E cells | ROS↑ | CerS↑, A-SMase→, N-SMase→ | unknown | Necrosis/Apoptosis | [31,32] |
| Oxidant | LLC-PK1 cells | Oxidant induces Cer | CerS↑, A-SMase→, N-SMase→ | unknown | Apoptosis/Necrosis | [33] |
| Oxidant | HK-2 cells | Oxidant induces Cer | N-SMase↓, A-SMase→, CerS→, GSH activates N-SMase. | unknown | Necrosis | [53] |
| Oxalate | MDCK cells, LLC-PK1 cells | ROS↑ | CerS↑, SMase↑ | unknown | Apoptosis | [54,57] |
| Cadmium | Rat RTCs | ROS↑ | unknown | unknown | Apoptosis | [61] |
| Carbon tetrachloride | Rat kidney | ROS↑, AOS↓ | N-SMase↑, A-SMase→ | unknown | Apoptosis | [67] |
| C2-Cer | HK-2 cells | ROS↑ | NA | Bax↑ | Apoptosis | [71] |
| Radiocontrast | LLC-PK1 cells | ROS↑ | CerS↑ | Bax↑, Bcl-2↓ | Apoptosis | [65,166] |
5.4.4. Do Ceramide-Induced ROS Regulate the Expression of Bcl-2 Family Proteins in Ceramide-Induced Apoptosis?
5.4.5. A Role of Ceramide-Induced ROS in Apoptosis of RTCs
6. Interconnection between Mitochondria and ER in the Regulation of Calcium Homeostasis in Ceramide-Induced Apoptosis
6.1. Calcium Homeostasis in the ER and Ceramide-Induced Apoptosis
6.2. Regulation of Calcium Homeostasis between the ER and Mitochondria by Bcl-2 Family Proteins in Ceramide-Induced Apoptosis
7. Ceramide- and Sphingosine-1-Phosphate-Induced Cell Signaling Pathways in the Regulation of Apoptosis
7.1. Mitogen-Activated Protein Kinases (MAPKs) and Ceramide-Induced Apoptosis
7.1.1. Ceramide-Induced Regulation of MAPKs in Apoptosis

7.1.2. Mechanism of Ceramide-Induced Activation of MAPKs
7.1.3. MAPKs Regulate the Enzymes Involved in Ceramide Metabolism
7.1.4. A Crosstalk between MAPKs and Bcl-2 Family Proteins in the Regulation of Ceramide-Induced Apoptosis
7.2. Signaling Molecules Other than MAPKs Regulated by Ceramide
7.3. S1P-Induced Signaling Pathway in Apoptosis of RTCs
8. Strategy for Preventing Ceramide-Induced Apoptosis of RTCs by Growth Factors
9. Conclusions and Future Perspective
Acknowledgments
Abbreviations
| ACEC | apical ceramide-enriched compartment |
| AIF | apoptosis inducing factor |
| ANT | adenine nucleotide translocase |
| AP-1 | activator protein 1 |
| APF-1 | apoptosis protease-activating factor-1 |
| AR | adenosine receptor |
| ATP | adenosine 5'-triphosphate |
| BI-1 | Bax inhibitor-1 |
| BMK | baby mouse kidney |
| CDases | ceramidases |
| CDK5 | cyclin-dependent kinase 5 |
| CerSs | ceramide synthases |
| CERT | ceramide transfer protein |
| C1P | ceramide-1-phosphate |
| CREB | cAMP response element binding protein |
| Δψm | mitochondrial membrane potential |
| Drp1 | dynamin-related protein 1 |
| EGF | epidermal growth factor |
| EGFR | EGF receptor |
| ER | endoplasmic reticulum |
| ERK | extracellular signal-regulated kinase |
| FAK | focal adhesion kinase |
| GSH | glutathione |
| GSLs | glycosphingolipids |
| GSSG | oxidized GSH |
| HEK | human embryonic kidney |
| HIF | hypoxia-inducible factor |
| HSP | heat shock protein |
| IAP | inhibitor of apoptosis |
| IGF-1 | insulin-like growth factor-1 |
| IGF-II | insulin-like growth factor-II |
| IP3R | 1,4,5-trisphosphate receptor |
| IL | interleukin |
| I/R | ischemia/reperfusion |
| JNK | c-Jun N-terminal kinases |
| 3-keto-dihydro-Sph | 3-keto-dihydrosphingosine |
| LPS | lipopolysaccharide |
| MAMs | mitochondrial-associated membranes |
| MAPKKK | mitogen-activated protein kinase kinase kinases |
| MAPKs | mitogen-activated protein kinases |
| MDCK | Madin-Dabry canine kidney |
| MEKK | MAP/ERK kinase kinase |
| MIM | mitochondrial inner membrane |
| MLKs | mixed linage kinases |
| MOM | mitochondrial outer membrane |
| MOMP | mitochondrial outer membrane permeability |
| MPT | mitochondrial permeability transition |
| PDGF | platelet-derived growth factor |
| PDGFR | PDGF receptor |
| PERK | protein kinase R-like endoplasmic reticulum kinase |
| PI3K | phosphatidylinositol 3-kinase |
| PKA | protein kinase A |
| PKC | protein kinase C |
| PLA2 | phospholipase A2 |
| PLC | phospholipase C |
| PP2A | protein phosphatase 2A |
| ROS | reactive oxygen species |
| RTCs | renal tubular cells |
| SAPK | stress activated protein kinase |
| SERCA | sarcoplasmic-endoplasmic reticulum Ca2+-ATPase |
| SM | sphingomyelin |
| SMases | sphingomyelinases |
| SMSs | SM synthases |
| SphKs | sphingosine kinases |
| S1P | sphingosine-1 phosphate |
| S1PRs | S1P receptors |
| SPT | serine palmitoyl transferase |
| STAT | signal transducer and activator of transcription |
| tBid | truncated Bid |
| TNF | tumor necrosis factor |
| UV | ultraviolet |
| VDAC | voltage-dependent anion channel |
Conflicts of Interest
References
- Hait, N.C.; Oskeritzian, C.A.; Paugh, S.W.; Milstien, S.; Spiegel, S. Sphingosine kinases, sphingosine 1-phosphate, apoptosis and diseases. Biochim. Biophys. Acta 2006, 1758, 2016–2026. [Google Scholar] [CrossRef] [PubMed]
- Bartke, N.; Hannun, Y.A. Bioactive sphingolipids: Metabolism and function. J. Lipid Res. 2009, 50, S91–S96. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Many ceramides. J. Biol. Chem. 2011, 286, 27855–27862. [Google Scholar] [CrossRef] [PubMed]
- Siskind, L.J. Mitochondrial ceramide and the induction of apoptosis. J. Bioenerg. Biomembr. 2005, 37, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Colombini, M. Ceramide channels and their role in mitochondria-mediated apoptosis. Biochim. Biophys. Acta 2010, 1797, 1239–1244. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Novgorodov, S.A.; Chudakova, D.; Zhu, H.; Bielawska, A.; Bielawski, J.; Obeid, L.M.; Kindy, M.S.; Gudz, T.I. JNK3 signaling pathway activates ceramide synthase leading to mitochondrial dysfunction. J. Biol. Chem. 2007, 282, 25940–25949. [Google Scholar] [CrossRef]
- Gault, C.R.; Eblen, S.T.; Neumann, C.A.; Hannun, Y.A.; Obeid, L.M. Oncogenic K-Ras regulates bioactive sphingolipids in a sphingosine kinase 1-dependent manner. J. Biol. Chem. 2012, 287, 31794–31803. [Google Scholar] [CrossRef] [PubMed]
- Sheng, G.; Guo, J.; Warner, B.W. Epidermal growth factor receptor signaling modulates apoptosis via p38α MAPK-dependent activation of Bax in intestinal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G599–G606. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Zhou, C.; Sun, Y.; Di, W.; Scheffler, E.; Healey, S.; Wanebo, H.; Kouttab, N.; Chu, W.; Wan, Y. Paclitaxel and ceramide synergistically induce cell death with transient activation of EGFR and ERK pathway in pancreatic cancer cells. Oncol. Rep. 2006, 16, 907–913. [Google Scholar] [PubMed]
- Kitatani, K.; Idkowiak-Baldys, J.; Hannun, Y. The sphingolipid salvage pathway in ceramide metabolism and signalling. Cell. Signal. 2008, 20, 1010–1018. [Google Scholar] [CrossRef] [PubMed]
- Mullen, T.D.; Jenkins, R.W.; Clarke, C.J.; Bielawski, J.; Hannun, Y.A.; Obeid, L.M. Ceramide synthase-dependent ceramide generation and programmed cell death: Involvement of salvage pathway in regulating postmitochondrial events. J. Biol. Chem. 2011, 286, 15929–15942. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, Y.; Kohyama-Koganeya, A.; Hirabayashi, Y. New insights on glucosylated lipids: Metabolism and functions. Biochim. Biophys. Acta 2013, 1831, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Novgorodov, S.A.; Chudakova, D.A.; Wheeler, B.W.; Bielawski, J.; Kindy, M.S.; Obeid, L.M.; Gudz, T.I. Developmentally regulated ceramide synthase 6 increases mitochondrial Ca2+ loading capacity and promotes apoptosis. J. Biol. Chem. 2011, 286, 4644–4658. [Google Scholar] [CrossRef] [PubMed]
- Mesicek, J.; Lee, H.; Feldman, T.; Jiang, X.; Skobeleva, A.; Berdyshev, E.V.; Haimovitz-Friedman, A.; Fuks, Z.; Kolesnick, R. Ceramide synthases 2, 5, and 6 confer distinct roles in radiation-induced apoptosis in HeLa cells. Cell. Signal. 2010, 22, 1300–1307. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.X.; Rajagopalan, V.; Roddy, P.L.; Clarke, C.J.; Hannun, Y.A. Identification and characterization of murine mitochondria-associated neutral sphingomyelinase (MA-nSMase), the mammalian sphingomyelin phosphodiesterase 5. J. Biol. Chem. 2010, 285, 17993–18002. [Google Scholar] [CrossRef] [PubMed]
- Novgorodov, S.A.; Wu, B.X.; Gudz, T.I.; Bielawski, J.; Ovchinnikova, T.V.; Hannun, Y.A.; Obeid, L.M. Novel pathway of ceramide production in mitochondria: Thioesterase and neutral ceramidase produce ceramide from sphingosine and acyl-CoA. J. Biol. Chem. 2011, 286, 25352–25362. [Google Scholar] [CrossRef] [PubMed]
- Subathra, M.; Qureshi, A.; Luberto, C. Sphingomyelin synthases regulate protein trafficking and secretion. PLos One 2011, 6, e23644. [Google Scholar] [CrossRef] [PubMed]
- Ledeen, R.W.; Wu, G. Nuclear sphingolipids: Metabolism and signalling. J. Lipid Res. 2008, 49, 1176–1186. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, K.; Kawano-Kawada, M.; Hanada, K. Phosphoregulation of the ceramide transport protein CERT at serine 315 in the interaction with VAMP-associated protein (VAP) for inter-organelle trafficking of ceramide in mammalian cells. J. Biol. Chem. 2014, 289, 10748–10760. [Google Scholar] [CrossRef] [PubMed]
- Giussani, P.; Maceyka, M.; le Stunff, H.; Mikami, A.; Lépine, S.; Wang, E.; Kelly, S.; Merrill, A.H., Jr.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate phosphohydrolase regulates endoplasmic reticulum-to-Golgi trafficking of ceramide. Mol. Cell. Biol. 2006, 26, 5055–5069. [Google Scholar] [CrossRef] [PubMed]
- Stiban, J.; Caput, L.; Colombini, M. Ceramide synthesis in the endoplasmic reticulum can permeabilize mitochondria to proapoptotic proteins. J. Lipid Res. 2008, 49, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Babiychuk, E.B.; Atanassoff, A.P.; Monastyrskaya, K.; Brandenberger, C.; Studer, D.; Allemann, C.; Draeger, A. The targeting of plasmalemmal ceramide to mitochondria during apoptosis. PLoS One 2011, 6, e23706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomassini, B.; Testi, R. Mitochondria as sensors of sphingolipids. Biochimie 2002, 84, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gulbins, E.; Zhang, Y. Oxidative stress triggers Ca2+-dependent lysosome trafficking and activation of acid sphingomyelinase. Cell. Physiol. Biochem. 2012, 30, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Krishnamurthy, K.; Bieberich, E. Regulation of primary cilia formation by ceramide. J. Lipid Res. 2009, 50, 2103–2110. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Wang, G.; Dasgupta, S.; Dinkins, M.; Zhu, G.; Bieberich, E. Characterization of an apical ceramide-enriched compartment regulating ciliogenesis. Mol. Biol. Cell 2012, 23, 3156–3166. [Google Scholar] [CrossRef] [PubMed]
- Bakrac, B.; Kladnik, A.; Macek, P.; McHaffie, G.; Werner, A.; Lakey, J.H.; Anderluh, G. A toxin-based probe reveals cytoplasmic exposure of Golgi sphingomyelin. J. Biol. Chem. 2010, 285, 22186–22195. [Google Scholar] [CrossRef] [PubMed]
- Ueda, Y.; Makino, A.; Murase-Tamada, K.; Sakai, S.; Inaba, T.; Hullin-Matsuda, F.; Kobayashi, T. Sphingomyelin regulates the transbilayer movement of diacylglycerol in the plasma membrane of Madin–Darby canine kidney cells. FASEB J. 2013, 27, 3284–3297. [Google Scholar] [CrossRef] [PubMed]
- Kok, J.W.; Eskelinen, S.; Hoekstra, K.; Hoekstra, D. Salvage of glucosylceramide by recycling after internalization along the pathway of receptor-mediated endocytosis. Proc. Natl. Acad. Sci. USA 1989, 86, 9896–9900. [Google Scholar] [CrossRef] [PubMed]
- Ebel, P.; Vom Dorp, K.; Petrasch-Parwez, E.; Zlomuzica, A.; Kinugawa, K.; Mariani, J.; Minich, D.; Ginkel, C.; Welcker, J.; Degen, J.; et al. Inactivation of ceramide synthase 6 in mice results in an altered sphingolipid metabolism and behavioral abnormalities. J. Biol. Chem. 2013, 288, 21433–21447. [Google Scholar] [CrossRef] [PubMed]
- Basnakian, A.G.; Ueda, N.; Hong, X.; Galitovsky, V.E.; Yin, X.; Shah, S.V. Ceramide synthase is essential for endonuclease-mediated death of renal tubular epithelial cells induced by hypoxia-reoxygenation. Am. J. Physiol. Ren. Physiol. 2005, 288, F308–F314. [Google Scholar] [CrossRef]
- Ueda, N.; Kaushal, G.P.; Hong, X.; Shah, S.V. Role of enhanced ceramide generation in DNA damage and cell death in chemical hypoxic injury to LLC-PK1 cells. Kidney Int. 1998, 54, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Ueda, N.; Camargo, S.M.; Hong, X.; Basnakian, A.G.; Walker, P.D.; Shah, S.V. Role of ceramide synthase in oxidant injury to renal tubular epithelial cells. J. Am. Soc. Nephrol. 2001, 12, 2384–2391. [Google Scholar] [PubMed]
- Pewzner-Jung, Y.; Brenner, O.; Braun, S.; Laviad, E.L.; Ben-Dor, S.; Feldmesser, E.; Horn-Saban, S.; Amann-Zalcenstein, D.; Raanan, C.; Berkutzki, T.; et al. A critical role for ceramide synthase 2 in liver homeostasis: II. Insights into molecular changes leading to hepatopathy. J. Biol. Chem. 2010, 285, 10911–10923. [Google Scholar] [CrossRef] [PubMed]
- Mitsutake, S.; Tani, M.; Okino, N.; Mori, K.; Ichinose, S.; Omori, A.; Iida, H.; Nakamura, T.; Ito, M. Purification, characterization, molecular cloning, and subcellular distribution of neutral ceramidase of rat kidney. J. Biol. Chem. 2001, 276, 26249–26259. [Google Scholar] [CrossRef] [PubMed]
- Sacket, S.J.; Chung, H.Y.; Okajima, F.; Im, D.S. Increase in sphingolipid catabolic enzyme activity during aging. Acta Pharmacol. Sin. 2009, 30, 1454–1461. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Kim, M.; Kim, J.Y.; Brown, K.M.; Haase, V.H.; D’Agati, V.D.; Lee, H.T. Proximal tubule sphingosine kinase-1 has a critical role in A1 adenosine receptor-mediated renal protection from ischemia. Kidney Int. 2012, 82, 878–891. [Google Scholar] [CrossRef] [PubMed]
- Siskind, L.J.; Mullen, T.D.; Romero Rosales, K.; Clarke, C.J.; Hernandez-Corbacho, M.J.; Edinger, A.L.; Obeid, L.M. The BCL-2 protein BAK is required for long-chain ceramide generation during apoptosis. J. Biol. Chem. 2010, 285, 11818–11826. [Google Scholar] [CrossRef] [PubMed]
- Sridevi, P.; Alexander, H.; Laviad, E.L.; Min, J.; Mesika, A.; Hannink, M.; Futerman, A.H.; Alexander, S. Stress-induced ER to Golgi translocation of ceramide synthase 1 is dependent on proteasomal processing. Exp. Cell. Res. 2010, 316, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Tomiuk, S.; Zumbansen, M.; Stoffel, W. Characterization and subcellular localization of murine and human magnesium-dependent neutral sphingomyelinase. J. Biol. Chem. 2000, 275, 5710–5717. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Chatterjee, S. Effects of gentamicin on sphingomyelinase activity in cultured human renal proximal tubular cells. J. Biol. Chem. 1987, 262, 12550–12556. [Google Scholar] [PubMed]
- Koch, A.; Pfeilschifter, J.; Huwiler, A. Sphingosine 1-phosphate in renal diseases. Cell. Physiol. Biochem. 2013, 31, 745–760. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, L.P.; Ren, S.; Schwalm, S.; Pfeilschifter, J.; Huwiler, A. Sphingosine kinase 1 and 2 regulate the capacity of mesangial cells to resist apoptotic stimuli in an opposing manner. Biol. Chem. 2008, 389, 1399–1407. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Kim, M.; Kim, N.; D’Agati, V.D.; Emala, C.W., Sr.; Lee, H.T. Isoflurane mediates protection from renal ischemia-reperfusion injury via sphingosine kinase and sphingosine-1-phosphate-dependent pathways. Am. J. Physiol. Ren. Physiol. 2007, 293, F1827–F1835. [Google Scholar] [CrossRef]
- Park, S.W.; Kim, J.Y.; Ham, A.; Brown, K.M.; Kim, M.; D’Agati, V.D.; Lee, H.T. A1 adenosine receptor allosteric enhancer PD-81723 protects against renal ischemia-reperfusion injury. Am. J. Physiol. Ren. Physiol. 2012, 303, F721–F732. [Google Scholar] [CrossRef]
- Ter Braak, M.; Danneberg, K.; Lichte, K.; Liphardt, K.; Ktistakis, N.T.; Pitson, S.M.; Hla, T.; Jakobs, K.H.; Meyer zu Heringdorf, D. Galpha(q)-mediated plasma membrane translocation of sphingosine kinase-1 and cross-activation of S1P receptors. Biochim. Biophys. Acta 2009, 1791, 357–370. [Google Scholar] [CrossRef] [PubMed]
- Maceyka, M.; Sankala, H.; Hait, N.C.; le Stunff, H.; Liu, H.; Toman, R.; Collier, C.; Zhang, M.; Satin, L.S.; Merrill, A.H., Jr.; et al. SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J. Biol. Chem. 2005, 280, 37118–37129. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, M.; Kornbluth, S. Caspases and kinases in a death grip. Cell 2009, 138, 838–854. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Moldoveanu, T.; Llambi, F.; Parsons, M.J.; Green, D.R. The BCL-2 family reunion. Mol. Cell 2010, 37, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Tsao, C.C.; Nica, A.F.; Kurinna, S.M.; Jiffar, T.; Mumby, M.; Ruvolo, P.P. Mitochondrial protein phosphatase 2A regulates cell death induced by simulated ischemia in kidney NRK-52E cells. Cell Cycle 2007, 6, 2377–2385. [Google Scholar] [CrossRef] [PubMed]
- Zager, R.A.; Iwata, M.; Conrad, D.S.; Burkhart, K.M.; Igarashi, Y. Altered ceramide and sphingosine expression during the induction phase of ischemic acute renal failure. Kidney Int. 1997, 52, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Zager, R.A.; Conrad, S.; Lochhead, K.; Sweeney, E.A.; Igarashi, Y.; Burkhart, K.M. Altered sphingomyelinase and ceramide expression in the setting of ischemic and nephrotoxic acute renal failure. Kidney Int. 1998, 53, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Zager, R.A.; Conrad, D.S.; Burkhart, K. Ceramide accumulation during oxidant renal tubular injury: Mechanisms and potential consequences. J. Am. Soc. Nephrol. 1998, 9, 1670–1680. [Google Scholar] [PubMed]
- Cao, L.C.; Honeyman, T.; Jonassen, J.; Scheid, C. Oxalate-induced ceramide accumulation in Madin–Darby canine kidney and LLC-PK1 cells. Kidney Int. 2000, 57, 2403–2411. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; Wu, S. Cell line dependent involvement of ceramide in ultraviolet light-induced apoptosis. Mol. Cell. Biochem. 2001, 219, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Niimura, Y.; Moue, T.; Takahashi, N.; Nagai, K. Modification of sphingoglycolipids and sulfolipids in kidney cell lines under heat stress: Activation of monohexosylceramide synthesis as a ceramide scavenger. Glycobiology 2010, 20, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.C.; Honeyman, T.W.; Cooney, R.; Kennington, L.; Scheid, C.R.; Jonassen, J.A. Mitochondrial dysfunction is a primary event in renal cell oxalate toxicity. Kidney Int. 2004, 66, 1890–1900. [Google Scholar] [CrossRef] [PubMed]
- Fischer, H.; Ellström, P.; Ekström, K.; Gustafsson, L.; Gustafsson, M.; Svanborg, C. Ceramide as a TLR4 agonist; A putative signalling intermediate between sphingolipid receptors for microbial ligands and TLR4. Cell. Microbiol. 2007, 9, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Hedlund, M.; Duan, R.D.; Nilsson, A.; Svanborg, C. Sphingomyelin, glycosphingolipids and ceramide signalling in cells exposed to P-fimbriated Escherichia coli. Mol. Microbiol. 1998, 29, 1297–1306. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.K.; Torchalski, B.; Thévenod, F. Cadmium-induced ceramide formation triggers calpain-dependent apoptosis in cultured kidney proximal tubule cells. Am. J. Physiol. Cell Physiol. 2007, 293, C839–C847. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.K.; Thévenod, F. Novel roles for ceramides, calpains and caspases in kidney proximal tubule cell apoptosis: Lessons from in vitro cadmium toxicity studies. Biochem. Pharmacol. 2008, 76, 1323–1332. [Google Scholar] [CrossRef] [PubMed]
- Lochhead, K.M.; Zager, R.A. Fluorinated anesthetic exposure “activates” the renal cortical sphingomyelinase cascade. Kidney Int. 1998, 54, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Ying, L.; Wang, H.; Li, N.; Fu, W.; Guo, Z.; Xu, L. Microcystin-LR induces ceramide to regulate PP2A and destabilize cytoskeleton in HEK293 cells. Toxicol. Sci. 2012, 128, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Dahdouh, F.; Raane, M.; Thévenod, F.; Lee, W.K. Nickel-induced cell death and survival pathways in cultured renal proximal tubule cells: Roles of reactive oxygen species, ceramide and ABCB1. Arch. Toxicol. 2014, 88, 881–892. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Yano, T.; Sendo, T.; Sueyasu, M.; Hirano, K.; Kanaide, H.; Oishi, R. Involvement of de novo ceramide synthesis in radiocontrast-induced renal tubular cell injury. Kidney Int. 2006, 69, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Neill, R.; Shupp, J.W.; Hammamieh, R.; Ionin, B.; Jett, M. Identification of staphylococcal enterotoxin B domains involved in binding to cultured human kidney proximal tubular cells: Imparting proliferation and death. Exp. Biol. Med. (Maywood) 2007, 232, 1142–1151. [Google Scholar] [CrossRef]
- Ichi, I.; Kamikawa, C.; Nakagawa, T.; Kobayashi, K.; Kataoka, R.; Nagata, E.; Kitamura, Y.; Nakazaki, C.; Matsura, T.; Kojo, S. Neutral sphingomyelinase-induced ceramide accumulation by oxidative stress during carbon tetrachloride intoxication. Toxicology 2009, 261, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Malik, R.K.; Thornhill, B.A.; Chang, A.Y.; Kiley, S.C.; Chevalier, R.L. Apoptosis parallels ceramide content in the developing rat kidney. Pediatr. Nephrol. 2000, 15, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Facchinetti, M.M.; Beuret, C.; Marquez, M.G.; Sterin-Speziale, N. Differential branching of the sphingolipid metabolic pathways with the stage of development. Involvement of sphingosine kinase. Biol. Neonate 2003, 84, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Malik, R.K.; Thornhill, B.A.; Chang, A.Y.; Kiley, S.C.; Chevalier, R.L. Renal apoptosis parallels ceramide content after prolonged ureteral obstruction in the neonatal rat. Am. J. Physiol. Ren. Physiol. 2001, 281, F56–F61. [Google Scholar]
- Iwayama, H.; Ueda, N. Role of mitochondrial Bax, caspases, and MAPKs for ceramide-induced apoptosis in renal proximal tubular cells. Mol. Cell. Biochem. 2013, 379, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Burlaka, I.; Liu, X.L.; Rebetz, J.; Arvidsson, I.; Yang, L.; Brismar, H.; Karpman, D.; Aperia, A. Ouabain protects against Shiga toxin-triggered apoptosis by reversing the imbalance between Bax and Bcl-xL. J. Am. Soc. Nephrol. 2013, 24, 1413–1423. [Google Scholar] [CrossRef] [PubMed]
- Idkowiak-Baldys, J.; Apraiz, A.; Li, L.; Rahmaniyan, M.; Clarke, C.J.; Kraveka, J.M.; Asumendi, A.; Hannun, Y.A. Dihydroceramide desaturase activity is modulated by oxidative stress. Biochem. J. 2010, 427, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Ghosh Choudhury, G.; Zhang, J.H.; Ghosh-Choudhury, N.; Abboud, H.E. Ceramide blocks PDGF-induced DNA synthesis in mesangial cells via inhibition of Akt kinase in the absence of apoptosis. Biochem. Biophys. Res. Commun. 2001, 286, 1183–1190. [Google Scholar] [CrossRef] [PubMed]
- Iwata, M.; Herrington, J.; Zager, R.A. Sphingosine: A mediator of acute renal tubular injury and subsequent cytoresistance. Proc. Natl. Acad. Sci. USA 1995, 92, 8970–8974. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; He, Q.; Sharma, R.P. Sphingosine kinase activity confers resistance to apoptosis by fumonisin B1 in human embryonic kidney (HEK-293) cells. Chem. Biol. Interact. 2004, 151, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Seefelder, W.; Humpf, H.U.; Schwerdt, G.; Freudinger, R.; Gekle, M. Induction of apoptosis in cultured human proximal tubule cells by fumonisins and fumonisin metabolites. Toxicol. Appl. Pharmacol. 2003, 192, 146–153. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Riley, R.T.; Sharma, R.P. Pharmacological antagonism of fumonisin B1 cytotoxicity in porcine renal epithelial cells (LLC-PK1): A model for reducing fumonisin-induced nephrotoxicity in vivo. Pharmacol. Toxicol. 2002, 90, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.K.; Bajwa, A.; Awad, A.S.; Lynch, K.R.; Okusa, M.D. Sphingosine-1-phosphate receptors: Biology and therapeutic potential in kidney disease. Kidney Int. 2008, 73, 1220–1230. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Kim, M.; Park, S.W.; Pitson, S.M.; Lee, H.T. Isoflurane protects human kidney proximal tubule cells against necrosis via sphingosine kinase and sphingosine-1-phosphate generation. Am. J. Nephrol. 2010, 31, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Bajwa, A.; Jo, S.K.; Ye, H.; Huang, L.; Dondeti, K.R.; Rosin, D.L.; Haase, V.H.; Macdonald, T.L.; Lynch, K.R.; Okusa, M.D. Activation of sphingosine-1-phosphate 1 receptor in the proximal tubule protects against ischemia-reperfusion injury. J. Am. Soc. Nephrol. 2010, 21, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Kim, M.; Brown, K.M.; D’Agati, V.D.; Lee, H.T. Inhibition of sphingosine 1-phosphate receptor 2 protects against renal ischemia-reperfusion injury. J. Am. Soc. Nephrol. 2012, 23, 266–280. [Google Scholar] [CrossRef] [PubMed]
- Ishizawa, S.; Takahashi-Fujigasaki, J.; Kanazawa, Y.; Matoba, K.; Kawanami, D.; Yokota, T.; Iwamoto, T.; Tajima, N.; Manome, Y.; Utsunomiya, K. Sphingosine-1-phosphate induces differentiation of cultured renal tubular epithelial cells under Rho kinase activation via the S1P2 receptor. Clin. Exp. Nephrol. 2014, 18, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Kim, M.; Kim, M.; D'Agati, V.D.; Lee, H.T. Sphingosine kinase 1 protects against renal ischemia-reperfusion injury in mice by sphingosine-1-phosphate1 receptor activation. Kidney Int. 2011, 80, 1315–1327. [Google Scholar] [CrossRef] [PubMed]
- Bakar, A.M.; Park, S.W.; Kim, M.; Lee, H.T. Isoflurane protects against human endothelial cell apoptosis by inducing sphingosine kinase-1 via ERK MAPK. Int. J. Mol. Sci. 2012, 13, 977–993. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Park, S.W.; Kim, M.; D’Agati, V.D.; Lee, H.T. Isoflurane activates intestinal sphingosine kinase to protect against renal ischemia-reperfusion-induced liver and intestine injury. Anesthesiology 2011, 114, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.T.; Park, S.W.; Kim, M.; Ham, A.; Anderson, L.J.; Brown, K.M.; D’Agati, V.D.; Cox, G.N. Interleukin-11 protects against renal ischemia and reperfusion injury. Am. J. Physiol. Ren. Physiol. 2012, 303, F1216–F1224. [Google Scholar] [CrossRef]
- Nieto, F.L.; Pescio, L.G.; Favale, N.O.; Adamo, A.M.; Sterin-Speziale, N.B. Sphingolipid metabolism is a crucial determinant of cellular fate in nonstimulated proliferating Madin–Darby canine kidney (MDCK) cells. J. Biol. Chem. 2008, 283, 25682–25691. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.R.; Johnson, K.Y.; Becker, K.P.; Bielawski, J.; Mao, C.; Obeid, L.M. Role of human sphingosine-1-phosphate phosphatase 1 in the regulation of intra- and extracellular sphingosine-1-phosphate levels and cell viability. J. Biol. Chem. 2003, 278, 34541–34547. [Google Scholar] [CrossRef] [PubMed]
- Mandala, S.M.; Thornton, R.; Galve-Roperh, I.; Poulton, S.; Peterson, C.; Olivera, A.; Bergstrom, J.; Kurtz, M.B.; Spiegel, S. Molecular cloning and characterization of a lipid phosphohydrolase that degrades sphingosine-1-phosphate and induces cell death. Proc. Natl. Acad. Sci. USA 2000, 97, 7859–7864. [Google Scholar] [CrossRef] [PubMed]
- Oskouian, B.; Sooriyakumaran, P.; Borowsky, A.D.; Crans, A.; Dillard-Telm, L.; Tam, Y.Y.; Bandhuvula, P.; Saba, J.D. Sphingosine-1-phosphate lyase potentiates apoptosis via p53- and p38-dependent pathways and is down-regulated in colon cancer. Proc. Natl. Acad. Sci. USA 2006, 103, 17384–17389. [Google Scholar] [CrossRef] [PubMed]
- Kirby, R.J.; Jin, Y.; Fu, J.; Cubillos, J.; Swertfeger, D.; Arend, L.J. Dynamic regulation of sphingosine-1-phosphate homeostasis during development of mouse metanephric kidney. Am. J. Physiol. Ren. Physiol. 2009, 296, F634–F641. [Google Scholar] [CrossRef]
- Chandran, S.; Machamer, C.E. Inactivation of ceramide transfer protein during pro-apoptotic stress by Golgi disassembly and caspase cleavage. Biochem. J. 2012, 442, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Milhas, D.; Clarke, C.J.; Idkowiak-Baldys, J.; Canals, D.; Hannun, Y.A. Anterograde and retrograde transport of neutral sphingomyelinase-2 between the Golgi and the plasma membrane. Biochim. Biophys. Acta 2010, 1801, 1361–1374. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Yi, F.; Zhang, F.; Poklis, J.L.; Li, P.L. Lysosomal targeting and trafficking of acid sphingomyelinase to lipid raft platforms in coronary endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 2056–2062. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, C.; Elks, C.M.; Kruger, C.; Cleland, E.; Addison, K.; Noland, R.C.; Stadler, K. Albumin-bound fatty acids but not albumin itself alter redox balance in tubular epithelial cells and induce a peroxide-mediated redox-sensitive apoptosis. Am. J. Physiol. Ren. Physiol. 2014, 306, F896–F906. [Google Scholar] [CrossRef]
- Aschrafi, A.; Franzen, R.; Shabahang, S.; Fabbro, D.; Pfeilschifter, J.; Huwiler, A. Ceramide induces translocation of protein kinase C-α to the Golgi compartment of human embryonic kidney cells by interacting with the C2 domain. Biochim. Biophys. Acta 2003, 1634, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Serlachius, E.; Svennilson, J.; Schalling, M.; Aperia, A. Protein kinase C in the developing kidney: Isoform expression and effects of ceramide and PKC inhibitors. Kidney Int. 1997, 52, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Dong, Z. Primary cilia and kidney injury: Current research status and future perspectives. Am. J. Physiol. Ren. Physiol. 2013, 305, F1085–F1098. [Google Scholar] [CrossRef]
- Nicotera, P.; Melino, G. Regulation of the apoptosis-necrosis switch. Oncogene 2004, 23, 2757–2765. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Bratton, S.B. Regulation of the intrinsic apoptosis pathway by reactive oxygen species. Antioxid. Redox Signal. 2013, 19, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Quillet-Mary, A.; Jaffrézou, J.P.; Mansat, V.; Bordier, C.; Naval, J.; Laurent, G. Implication of mitochondrial hydrogen peroxide generation in ceramide-induced apoptosis. J. Biol. Chem. 1997, 272, 21388–21395. [Google Scholar] [CrossRef] [PubMed]
- Sawada, M.; Nakashima, S.; Banno, Y.; Yamakawa, H.; Takenaka, K.; Shinoda, J.; Nishimura, Y.; Sakai, N.; Nozawa, Y. Influence of Bax or Bcl-2 overexpression on the ceramide-dependent apoptotic pathway in glioma cells. Oncogene 2000, 19, 3508–3520. [Google Scholar] [CrossRef] [PubMed]
- Leber, B.; Lin, J.; Andrews, D.W. Embedded together: The life and death consequences of interaction of the Bcl-2 family with membranes. Apoptosis 2007, 12, 897–911. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, E.V.; Priault, M.; Pietkiewicz, D.; Cheng, E.H.; Antonsson, B.; Manon, S.; Korsmeyer, S.J.; Mannella, C.A.; Kinnally, K.W. A novel, high conductance channel of mitochondria linked to apoptosis in mammalian cells and Bax expression in yeast. J. Cell Biol. 2001, 155, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Dejean, L.M.; Martinez-Caballero, S.; Guo, L.; Hughes, C.; Teijido, O.; Ducret, T.; Ichas, F.; Korsmeyer, S.J.; Antonsson, B.; Jonas, E.A.; Kinnally, K.W. Oligomeric Bax is a component of the putative cytochrome c release channel MAC, mitochondrial apoptosis-induced channel. Mol. Biol. Cell. 2005, 16, 2424–2432. [Google Scholar] [CrossRef] [PubMed]
- Dejean, L.M.; Martinez-Caballero, S.; Kinnally, K.W. Is MAC the knife that cuts cytochrome C from mitochondria during apoptosis? Cell Death Differ. 2006, 13, 1387–1395. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; McStay, G.P.; Bharti, A.; Kuwana, T.; Clarke, C.J.; Siskind, L.J.; Obeid, L.M.; Green, D.R. Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell 2012, 148, 988–1000. [Google Scholar] [CrossRef] [PubMed]
- Kashkar, H.; Wiegmann, K.; Yazdanpanah, B.; Haubert, D.; Krönke, M. Acid sphingomyelinase is indispensable for UV light-induced Bax conformational change at the mitochondrial membrane. J. Biol. Chem. 2005, 280, 20804–20813. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Rotolo, J.A.; Mesicek, J.; Penate-Medina, T.; Rimner, A.; Liao, W.C.; Yin, X.; Ragupathi, G.; Ehleiter, D.; Gulbins, E.; et al. Mitochondrial ceramide-rich macrodomains functionalize Bax upon irradiation. PLoS One 2011, 6, e19783. [Google Scholar] [CrossRef] [PubMed]
- Sawai, H.; Kawai, S.; Domae, N. Reduced expression of Bax in ceramide-resistant HL-60 subline. Biochem. Biophys. Res. Commun. 2004, 319, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, V.; Perera, M.N.; Colombini, D.; Datskovskiy, D.; Chadha, K.; Colombini, M. Ceramide and activated Bax act synergistically to permeabilize the mitochondrial outer membrane. Apoptosis 2010, 15, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Siskind, L.J.; Colombini, M. The lipids C2- and C16-ceramide form large stable channels. Implications for apoptosis. J. Biol. Chem. 2000, 275, 38640–38644. [Google Scholar] [CrossRef] [PubMed]
- Siskind, L.J.; Feinstein, L.; Yu, T.; Davis, J.S.; Jones, D.; Choi, J.; Zuckerman, J.E.; Tan, W.; Hill, R.B.; Hardwick, J.M.; et al. Anti-apoptotic Bcl-2 family proteins disassemble ceramide channels. J. Biol. Chem. 2008, 283, 6622–6630. [Google Scholar]
- Perera, M.N.; Lin, S.H.; Peterson, Y.K.; Bielawska, A.; Szulc, Z.M.; Bittman, R.; Colombini, M. Bax and Bcl-xL exert their regulation on different sites of the ceramide channel. Biochem. J. 2012, 445, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Elrick, M.J.; Fluss, S.; Colombini, M. Sphingosine, a product of ceramide hydrolysis, influences the formation of ceramide channels. Biophys. J. 2006, 91, 1749–1756. [Google Scholar] [CrossRef] [PubMed]
- Stiban, J.; Fistere, D.; Colombini, M. Dihydroceramide hinders ceramide channel formation: Implications on apoptosis. Apoptosis 2006, 11, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Baines, C.P.; Kaiser, R.A.; Sheiko, T.; Craigen, W.J.; Molkentin, J.D. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat. Cell. Biol. 2007, 9, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Rapizzi, E.; Pinton, P.; Szabadkai, G.; Wieckowski, M.R.; Vandecasteele, G.; Baird, G.; Tuft, R.A.; Fogarty, K.E.; Rizzuto, R. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J. Cell Biol. 2002, 159, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Scharstuhl, A.; Mutsaers, H.A.; Pennings, S.W.; Russel, F.G.; Wagener, F.A. Involvement of VDAC, Bax and ceramides in the efflux of AIF from mitochondria during curcumin-induced apoptosis. PLoS One 2009, 4, e6688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, M.; Andrade-Navarro, M.A.; Gross, A. Mitochondrial carriers and pores: Key regulators of the mitochondrial apoptotic program? Apoptosis 2007, 12, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Abundis, E.; Correa, F.; Pavón, N.; Zazueta, C. Bax distribution into mitochondrial detergent-resistant microdomains is related to ceramide and cholesterol content in postischemic hearts. FEBS J. 2009, 276, 5579–5588. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.S.; Madesh, M.; Davies, E.; Antonsson, B.; Danial, N.; Hajnóczky, G. Bad targets the permeability transition pore independent of Bax or Bak to switch between Ca2+-dependent cell survival and death. Mol. Cell 2009, 33, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Novgorodov, S.A.; Szulc, Z.M.; Luberto, C.; Jones, J.A.; Bielawski, J.; Bielawska, A.; Hannun, Y.A.; Obeid, L.M. Positively charged ceramide is a potent inducer of mitochondrial permeabilization. J. Biol. Chem. 2005, 280, 16096–16105. [Google Scholar] [CrossRef] [PubMed]
- Darios, F.; Lambeng, N.; Troadec, J.D.; Michel, P.P.; Ruberg, M. Ceramide increases mitochondrial free calcium levels via caspase 8 and Bid: Role in initiation of cell death. J. Neurochem. 2003, 84, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Ferrari, D.; Rapizzi, E.; Di Virgilio, F.; Pozzan, T.; Rizzuto, R. The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: Significance for the molecular mechanism of Bcl-2 action. EMBO J. 2001, 20, 2690–2701. [Google Scholar] [CrossRef] [PubMed]
- Parra, V.; Eisner, V.; Chiong, M.; Criollo, A.; Moraga, F.; Garcia, A.; Härtel, S.; Jaimovich, E.; Zorzano, A.; Hidalgo, C.; et al. Changes in mitochondrial dynamics during ceramide-induced cardiomyocyte early apoptosis. Cardiovasc. Res. 2008, 77, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Arnoult, D.; Rismanchi, N.; Grodet, A.; Roberts, R.G.; Seeburg, D.P.; Estaquier, J.; Sheng, M.; Blackstone, C. Bax/Bak-dependent release of DDP/TIMM8a promotes Drp1-mediated mitochondrial fission and mitoptosis during programmed cell death. Curr. Biol. 2005, 15, 2112–2118. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.; Wei, Q.; Feng, L.; Dong, G.; Tao, Y.; Mei, L.; Xie, Z.J.; Dong, Z. Bak regulates mitochondrial morphology and pathology during apoptosis by interacting with mitofusins. Proc. Natl. Acad. Sci. USA 2007, 104, 11649–11654. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, C.; Delivani, P.; Cullen, S.P.; Martin, S.J. Bax- or Bak-induced mitochondrial fission can be uncoupled from cytochrome C release. Mol. Cell 2008, 31, 570–585. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.; Kennington, L.; Cooney, R.; Kohjimoto, Y.; Cao, L.C.; Honeyman, T.; Pullman, J.; Jonassen, J.; Scheid, C. Oxalate toxicity in renal epithelial cells: Characteristics of apoptosis and necrosis. Toxicol. Appl. Pharmacol. 2000, 162, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Dagher, P.C. Apoptosis in ischemic renal injury: Roles of GTP depletion and p53. Kidney Int. 2004, 66, 506–509. [Google Scholar] [CrossRef] [PubMed]
- Cabral, L.M.; Wengert, M.; da Ressurreição, A.A.; Feres-Elias, P.H.; Almeida, F.G.; Vieyra, A.; Caruso-Neves, C.; Einicker-Lamas, M. Ceramide is a potent activator of plasma membrane Ca2+-ATPase from kidney-promixal tubule cells with protein kinase A as an intermediate. J. Biol. Chem. 2007, 282, 24599–24606. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.G.; Du, Q.; Huang, S.; Dong, Z. Drp1 dephosphorylation in ATP depletion-induced mitochondrial injury and tubular cell apoptosis. Am. J. Physiol. Ren. Physiol. 2010, 299, F199–F206. [Google Scholar] [CrossRef]
- Saito, M.; Korsmeyer, S.J.; Schlesinger, P.H. BAX-dependent transport of cytochrome c reconstituted in pure liposomes. Nat. Cell. Biol. 2000, 2, 553–555. [Google Scholar] [CrossRef] [PubMed]
- Antonsson, B.; Montessuit, S.; Lauper, S.; Eskes, R.; Martinou, J.C. Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release from mitochondria. Biochem. J. 2000, 345, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Roucou, X.; Rostovtseva, T.; Montessuit, S.; Martinou, J.C.; Antonsson, B. Bid induces cytochrome c-impermeable Bax channels in liposomes. Biochem. J. 2002, 363, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Listenberger, L.L.; Ory, D.S.; Schaffer, J.E. Palmitate-induced apoptosis can occur through a ceramide-independent pathway. J. Biol. Chem. 2001, 276, 14890–14895. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, J.; Akahane, K.; Nakamura, J.; Naruse, K.; Kamiya, H.; Himeno, T.; Nakamura, N.; Shibata, T.; Kondo, M.; Nagasaki, H.; et al. Palmitate induces apoptosis in Schwann cells via both ceramide-dependent and independent pathways. Neuroscience 2011, 176, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Chan, C. Hydrogen peroxide and hydroxyl radicals mediate palmitate-induced cytotoxicity to hepatoma cells: Relation to mitochondrial permeability transition. Free Radic. Res. 2007, 41, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Beverly, L.J.; Howell, L.A.; Hernandez-Corbacho, M.; Casson, L.; Chipuk, J.E.; Siskind, L.J. BAK activation is necessary and sufficient to drive ceramide synthase-dependent ceramide accumulation following inhibition of BCL2-like proteins. Biochem. J. 2013, 452, 111–119. [Google Scholar] [PubMed]
- Ravid, T.; Tsaba, A.; Gee, P.; Rasooly, R.; Medina, E.A.; Goldkorn, T. Ceramide accumulation precedes caspase-3 activation during apoptosis of A549 human lung adenocarcinoma cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L1082–L1092. [Google Scholar] [CrossRef] [PubMed]
- Bektas, M.; Jolly, P.S.; Müller, C.; Eberle, J.; Spiegel, S.; Geilen, C.C. Sphingosine kinase activity counteracts ceramide-mediated cell death in human melanoma cells: Role of Bcl-2 expression. Oncogene 2005, 24, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Zigdon, H.; Kogot-Levin, A.; Park, J.W.; Goldschmidt, R.; Kelly, S.; Merrill, A,H., Jr.; Scherz, A.; Pewzner-Jung, Y.; Saada, A.; Futerman, A.H. Ablation of ceramide synthase 2 causes chronic oxidative stress due to disruption of the mitochondrial respiratory chain. J. Biol. Chem. 2013, 288, 4947–4956. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.C.; Allen, K.; Griffiths, H.R. Synthetic ceramides induce growth arrest or apoptosis by altering cellular redox status. Arch. Biochem. Biophys. 2002, 407, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Pautz, A.; Franzen, R.; Dorsch, S.; Böddinghaus, B.; Briner, V.A.; Pfeilschifter, J.; Huwiler, A. Cross-talk between nitric oxide and superoxide determines ceramide formation and apoptosis in glomerular cells. Kidney Int. 2002, 61, 790–796. [Google Scholar] [CrossRef] [PubMed]
- Huwiler, A.; Pfeilschifter, J.; van den Bosch, H. Nitric oxide donors induce stress signaling via ceramide formation in rat renal mesangial cells. J. Biol. Chem. 1999, 274, 7190–7195. [Google Scholar] [CrossRef] [PubMed]
- Grammatikos, G.; Teichgräber, V.; Carpinteiro, A.; Trarbach, T.; Weller, M.; Hengge, U.R.; Gulbins, E. Overexpression of acid sphingomyelinase sensitizes glioma cells to chemotherapy. Antioxid. Redox Signal. 2007, 9, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- Cinq-Frais, C.; Coatrieux, C.; Grazide, M.H.; Hannun, Y.A.; Nègre-Salvayre, A.; Salvayre, R.; Augé, N. A signaling cascade mediated by ceramide, Src and PDGFRβ coordinates the activation of the redox-sensitive neutral sphingomyelinase-2 and sphingosine kinase-1. Biochim. Biophys. Acta 2013, 1831, 1344–1356. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Castillo, S.S.; Goldkorn, T. nSMase2 activation and trafficking are modulated by oxidative stress to induce apoptosis. Biochem. Biophys. Res. Commun. 2006, 344, 900–905. [Google Scholar] [CrossRef] [PubMed]
- Maceyka, M.; Milstien, S.; Spiegel, S. Shooting the messenger: Oxidative stress regulates sphingosine-1-phosphate. Circ. Res. 2007, 100, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Pyszko, J.; Strosznajder, J.B. Sphingosine kinase 1 and sphingosine-1-phosphate in oxidative stress evoked by 1-methyl-4-phenylpyridinium (MPP+) in human dopaminergic neuronal cells. Mol. Neurobiol. 2014, 50, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Kizhakkayil, J.; Thayyullathil, F.; Chathoth, S.; Hago, A.; Patel, M.; Galadari, S. Glutathione regulates caspase-dependent ceramide production and curcumin-induced apoptosis in human leukemic cells. Free Radic. Biol. Med. 2012, 52, 1854–1864. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Khan, E.; Careaga, M.; Goldkorn, T. Neutral sphingomyelinase 2 is activated by cigarette smoke to augment ceramide-induced apoptosis in lung cell death. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L125–L133. [Google Scholar] [CrossRef] [PubMed]
- Charruyer, A.; Jean, C.; Colomba, A.; Jaffrézou, J.P.; Quillet-Mary, A.; Laurent, G.; Bezombes, C. PKCζ protects against UV-C-induced apoptosis by inhibiting acid sphingomyelinase-dependent ceramide production. Biochem. J. 2007, 405, 77–83. [Google Scholar] [PubMed]
- Bruce, C.R.; Risis, S.; Babb, J.R.; Yang, C.; Kowalski, G.M.; Selathurai, A.; Lee-Young, R.S.; Weir, J.M.; Yoshioka, K.; Takuwa, Y.; et al. Overexpression of sphingosine kinase 1 prevents ceramide accumulation and ameliorates muscle insulin resistance in high-fat diet-fed mice. Diabetes 2012, 61, 3148–3155. [Google Scholar] [CrossRef] [PubMed]
- Franzen, R.; Pfeilschifter, J.; Huwiler, A. Nitric oxide induces neutral ceramidase degradation by the ubiquitin/proteasome complex in renal mesangial cell cultures. FEBS Lett. 2002, 532, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Liu, W.; Xie, X.; Xu, S.; Huang, K.; Peng, J.; Shen, X.; Liu, P.; Wang, L.; Xia, P.; Huang, H. Sphingosine kinase-1 pathway mediates high glucose-induced fibronectin expression in glomerular mesangial cells. Mol. Endocrinol. 2011, 25, 2094–2105. [Google Scholar] [CrossRef] [PubMed]
- Esposti, M.D.; Hatzinisiriou, I.; McLennan, H.; Ralph, S. Bcl-2 and mitochondrial oxygen radicals. New approaches with reactive oxygen species-sensitive probes. J. Biol. Chem. 1999, 274, 29831–29837. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.; Low, I.C.; Pervaiz, S. Regulation of mitochondrial metabolism: Yet another facet in the biology of the oncoprotein Bcl-2. Biochem. J. 2011, 435, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Amarante-Mendes, G.P.; Naekyung Kim, C.; Liu, L.; Huang, Y.; Perkins, C.L.; Green, D.R.; Bhalla, K. Bcr-Abl exerts its antiapoptotic effect against diverse apoptotic stimuli through blockage of mitochondrial release of cytochrome c and activation of caspase-3. Blood 1998, 91, 1700–1705. [Google Scholar] [PubMed]
- Kirkland, R.A.; Saavedra, G.M.; Cummings, B.S.; Franklin, J.L. Bax regulates production of superoxide in both apoptotic and nonapoptotic neurons: Role of caspases. J. Neurosci. 2010, 30, 16114–16127. [Google Scholar] [CrossRef] [PubMed]
- D’Errico, I.; lo Sasso, G.; Salvatore, L.; Murzilli, S.; Martelli, N.; Cristofaro, M.; Latorre, D.; Villani, G.; Moschetta, A. Bax is necessary for PGC1α pro-apoptotic effect in colorectal cancer cells. Cell Cycle 2011, 10, 2937–2945. [Google Scholar] [CrossRef] [PubMed]
- Henke, N.; Lisak, D.A.; Schneider, L.; Habicht, J.; Pergande, M.; Methne, A. The ancient cell death suppressor BAX inhibitor-1. Cell Calcium 2011, 50, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Nie, C.; Tian, C.; Zhao, L.; Petit, P.X.; Mehrpour, M.; Chen, Q. Cysteine 62 of Bax is critical for its conformational activation and its proapoptotic activity in response to H2O2-induced apoptosis. J. Biol. Chem. 2008, 283, 15359–15369. [Google Scholar] [CrossRef]
- Yang, D.; Yang, D.; Jia, R.; Ding, G. Selective inhibition of the reverse mode of Na+/Ca2+ exchanger attenuates contrast-induced cell injury. Am. J. Nephrol. 2013, 37, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.L.; Seok, J.Y.; Kwon, C.H.; Kang, S.K.; Kim, Y.K. Role of MAPK in ceramide-induced cell death in primary cultured astrocytes from mouse embryonic brain. Neurotoxicology 2006, 27, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Flowers, M.; Fabriás, G.; Delgado, A.; Casas, J.; Abad, J.L.; Cabot, M.C. C6-ceramide and targeted inhibition of acid ceramidase induce synergistic decreases in breast cancer cell growth. Breast Cancer Res. Treat. 2012, 133, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Thévenod, F.; Lee, W.K. Cadmium and cellular signaling cascades: Interactions between cell death and survival pathways. Arch. Toxicol. 2013, 87, 1743–1786. [Google Scholar] [CrossRef] [PubMed]
- Morales, M.C.; Pérez-Yarza, G.; Rementería, N.N.; Boyano, M.D.; Apraiz, A.; Gómez-Muñoz, A.; Pérez-Andrés, E.; Asumendi, A. 4-HPR-mediated leukemia cell cytotoxicity is triggered by ceramide-induced mitochondrial oxidative stress and is regulated downstream by Bcl-2. Free Radic. Res. 2007, 41, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Kobrinsky, E.; Spielman, A.I.; Rosenzweig, S.; Marks, A.R. Ceramide triggers intracellular calcium release via the IP(3) receptor in Xenopus laevis oocytes. Am. J. Physiol. Cell Physiol. 1999, 277, C665–C672. [Google Scholar]
- Heath-Engel, H.M.; Chang, N.C.; Shore, G.C. The endoplasmic reticulum in apoptosis and autophagy: Role of the BCL-2 protein family. Oncogene 2008, 27, 6419–6433. [Google Scholar] [CrossRef] [PubMed]
- Senkal, C.E.; Ponnusamy, S.; Manevich, Y.; Meyers-Needham, M.; Saddoughi, S.A.; Mukhopadyay, A.; Dent, P.; Bielawski, J.; Ogretmen, B. Alteration of ceramide synthase 6/C16-ceramide induces activating transcription factor 6-mediated endoplasmic reticulum (ER) stress and apoptosis via perturbation of cellular Ca2+ and ER/Golgi membrane network. J. Biol. Chem. 2011, 286, 42446–42458. [Google Scholar] [CrossRef] [PubMed]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; de Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Darios, F.; Muriel, M.P.; Khondiker, M.E.; Brice, A.; Ruberg, M. Neurotoxic calcium transfer from endoplasmic reticulum to mitochondria is regulated by cyclin-dependent kinase 5-dependent phosphorylation of tau. J. Neurosci. 2005, 25, 4159–4168. [Google Scholar] [CrossRef] [PubMed]
- Yacoub, A.; Hamed, H.A.; Allegood, J.; Mitchell, C.; Spiegel, S.; Lesniak, M.S.; Ogretmen, B.; Dash, R.; Sarkar, D.; Broaddus, W.C.; et al. PERK-dependent regulation of ceramide synthase 6 and thioredoxin play a key role in mda-7/IL-24-induced killing of primary human glioblastoma multiforme cells. Cancer Res. 2010, 70, 1120–1129. [Google Scholar] [CrossRef] [PubMed]
- Scorrano, L.; Oakes, S.A.; Opferman, J.T.; Cheng, E.H.; Sorcinelli, M.D.; Pozzan, T.; Korsmeyer, S.J. BAX and BAK regulation of endoplasmic reticulum Ca2+: A control point for apoptosis. Science 2003, 300, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Logue, S.E.; Cleary, P.; Saveljeva, S.; Samali, A. New directions in ER stress-induced cell death. Apoptosis 2013, 18, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Fiebig, A.A.; Zhu, W.; Hollerbach, C.; Leber, B.; Andrews, D.W. Bcl-XL is qualitatively different from and ten times more effective than Bcl-2 when expressed in a breast cancer cell line. BMC Cancer 2006, 6, 213. [Google Scholar] [CrossRef] [PubMed]
- Hsin, Y.H.; Cheng, C.H.; Tzen, J.T.; Wu, M.J.; Shu, K.H.; Chen, H,C. Effect of aristolochic acid on intracellular calcium concentration and its links with apoptosis in renal tubular cells. Apoptosis 2006, 11, 2167–2177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dremina, E.S.; Sharov, V.S.; Kumar, K.; Zaidi, A.; Michaelis, E.K.; Schöneich, C. Anti-apoptotic protein Bcl-2 interacts with and destabilizes the sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA). Biochem. J. 2004, 383, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Monaco, G.; Beckers, M.; Ivanova, H.; Missiaen, L.; Parys, J.B.; de Smedt, H.; Bultynck, G. Profiling of the Bcl-2/Bcl-XL-binding sites on type 1 IP3 receptor. Biochem. Biophys. Res. Commun. 2012, 428, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Camandola, S.; Cutler, R.G.; Gary, D.S.; Milhavet, O.; Mattson, M.P. Suppression of calcium release from inositol 1,4,5-trisphosphate-sensitive stores mediates the anti-apoptotic function of nuclear factor-κB. J. Biol. Chem. 2005, 280, 22287–22296. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Xu, W.; Palmer, A.E.; Reed, J.C. BI-1 regulates endoplasmic reticulum Ca2+ homeostasis downstream of Bcl-2 family proteins. J. Biol. Chem. 2008, 283, 11477–11484. [Google Scholar] [CrossRef] [PubMed]
- Willaime, S.; Vanhoutte, P.; Caboche, J.; Lemaigre-Dubreuil, Y.; Mariani, J.; Brugg, B. Ceramide-induced apoptosis in cortical neurons is mediated by an increase in p38 phosphorylation and not by the decrease in ERK phosphorylation. Eur. J. Neurosci. 2001, 13, 2037–2046. [Google Scholar] [CrossRef] [PubMed]
- Stoica, B.A.; Movsesyan, V.A.; Knoblach, S.M.; Faden, A.I. Ceramide induces neuronal apoptosis through mitogen-activated protein kinases and causes release of multiple mitochondrial proteins. Mol. Cell. Neurosci. 2005, 29, 355–371. [Google Scholar] [CrossRef] [PubMed]
- Falluel-Morel, A.; Aubert, N.; Vaudry, D.; Basille, M.; Fontaine, M.; Fournier, A.; Vaudry, H.; Gonzalez, B.J. Opposite regulation of the mitochondrial apoptotic pathway by C2-ceramide and PACAP through a MAP-kinase-dependent mechanism in cerebellar granule cells. J. Neurochem. 2004, 91, 1231–1243. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Fox, T.; Adhikary, G.; Kester, M.; Pearlman, E. Inhibition of corneal inflammation by liposomal delivery of short-chain, C-6 ceramide. J. Leukoc. Biol. 2008, 83, 1512–1521. [Google Scholar] [CrossRef] [PubMed]
- Willaime-Morawek, S.; Brami-Cherrier, K.; Mariani, J.; Caboche, J.; Brugg, B. c-Jun N-terminal kinases/c-Jun and p38 pathways cooperate in ceramide-induced neuronal apoptosis. Neuroscience 2003, 119, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Donato, N.J.; Klostergaard, J. Distinct stress and cell destruction pathways are engaged by TNF and ceramide during apoptosis of MCF-7 cells. Exp. Cell Res. 2004, 294, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, S.; Yan, Y.; Daubert, R.A.; Han, J.; Schnellmann, R.G. ERK promotes hydrogen peroxide-induced apoptosis through caspase-3 activation and inhibition of Akt in renal epithelial cells. Am. J. Physiol. Ren. Physiol. 2007, 292, F440–F447. [Google Scholar] [CrossRef]
- Bourbon, N.A.; Yun, J.; Berkey, D.; Wang, Y.; Kester, M. Inhibitory actions of ceramide upon PKC-ε/ERK interactions. Am. J. Physiol. Cell Physiol. 2001, 280, C1403–C1411. [Google Scholar] [PubMed]
- Sathyanarayana, P.; Barthwal, M.K.; Kundu, C.N.; Lane, M.E.; Bergmann, A.; Tzivion, G.; Rana, A. Activation of the Drosophila MLK by ceramide reveals TNF-α and ceramide as agonists of mammalian MLK3. Mol. Cell 2002, 10, 1527–1533. [Google Scholar] [CrossRef] [PubMed]
- Huwiler, A.; Xin, C.; Brust, A.K.; Briner, V.A.; Pfeilschifter, J. Differential binding of ceramide to MEKK1 in glomerular endothelial and mesangial cells. Biochim. Biophys. Acta 2004, 1636, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.L.; Lin, C.F.; Chang, W.T.; Huang, W.C.; Teng, C.F.; Lin, Y.S. Ceramide induces p38 MAPK and JNK activation through a mechanism involving a thioredoxin-interacting protein-mediated pathway. Blood 2008, 111, 4365–4374. [Google Scholar] [CrossRef] [PubMed]
- Park, M.A.; Walker, T.; Martin, A.P.; Allegood, J.; Vozhilla, N.; Emdad, L.; Sarkar, D.; Rahmani, M.; Graf, M.; Yacoub, A.; et al. MDA-7/IL-24-induced cell killing in malignant renal carcinoma cells occurs by a ceramide/CD95/PERK-dependent mechanism. Mol. Cancer Ther. 2009, 8, 1280–1291. [Google Scholar] [CrossRef] [PubMed]
- Pfeilschifter, J.; Huwiler, A. Identification of ceramide targets in interleukin-1 and tumor necrosis factor-α signaling in mesangial cells. Kidney Int. Suppl. 1998, 67, S34–S39. [Google Scholar] [CrossRef] [PubMed]
- Bourbon, N.A.; Yun, J.; Kester, M. Ceramide directly activates protein kinase Cζ to regulate a stress-activated protein kinase signaling complex. J. Biol. Chem. 2000, 275, 35617–35623. [Google Scholar] [CrossRef] [PubMed]
- Yabu, T.; Shiba, H.; Shibasaki, Y.; Nakanishi, T.; Imamura, S.; Touhata, K.; Yamashita, M. Stress-induced ceramide generation and apoptosis via the phosphorylation and activation of nSMase1 by JNK signalling. Cell Death Differ. 2015, 22, 258–273. [Google Scholar] [CrossRef] [PubMed]
- Bizzozero, L.; Cazzato, D.; Cervia, D.; Assi, E.; Simbari, F.; Pagni, F.; de Palma, C.; Monno, A.; Verdelli, C.; Querini, P.R.; et al. Acid sphingomyelinase determines melanoma progression and metastatic behaviour via the microphtalmia-associated transcription factor signalling pathway. Cell Death Differ. 2014, 21, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Sridevi, P.; Alexander, H.; Laviad, E.L.; Pewzner-Jung, Y.; Hannink, M.; Futerman, A.H.; Alexander, S. Ceramide synthase 1 is regulated by proteasomal mediated turnover. Biochim. Biophys. Acta 2009, 1793, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Franzen, R.; Pautz, A.; Bräutigam, L.; Geisslinger, G.; Pfeilschifter, J.; Huwiler, A. Interleukin-1β induces chronic activation and de novo synthesis of neutral ceramidase in renal mesangial cells. J. Biol. Chem. 2001, 276, 35382–35389. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Oh, J.E.; Kim, S.W.; Chun, Y.J.; Kim, M.Y. Ceramide induces p38 MAPK-dependent apoptosis and Bax translocation via inhibition of Akt in HL-60 cells. Cancer Lett. 2008, 260, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, J.L.; Menter, D.G.; Beham, A.; von Eschenbach, A.; McDonnell, T.J. Regulation of lipid signaling pathways for cell survival and apoptosis by bcl-2 in prostate carcinoma cells. Exp. Cell Res. 1997, 234, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Manna, S.K.; Haridas, V.; Aggarwal, B.B. Bcl-xL suppresses TNF-mediated apoptosis and activation of nuclear factor-kB, activation protein-1, and c-Jun N-terminal kinase. J. Interferon Cytokine Res. 2000, 20, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Crager, M.; Pugazhenthi, S. Modulation of apoptosis pathways by oxidative stress and autophagy in β cells. Exp. Diabetes Res. 2012, 2012, 647914. [Google Scholar] [PubMed]
- Kurinna, S.M.; Tsao, C.C.; Nica, A.F.; Jiffar, T.; Ruvolo, P.P. Ceramide promotes apoptosis in lung cancer-derived A549 cells by a mechanism involving c-Jun NH2-terminal kinase. Cancer Res. 2004, 64, 7852–7856. [Google Scholar] [CrossRef] [PubMed]
- Asakuma, J.; Sumitomo, M.; Asano, T.; Asano, T.; Hayakawa, M. Selective Akt inactivation and tumor necrosis factor-related apoptosis-inducing ligand sensitization of renal cancer cells by low concentrations of paclitaxel. Cancer Res. 2003, 63, 1365–1370. [Google Scholar] [PubMed]
- Gao, P.; Peterson, Y.K.; Smith, R.A.; Smith, C.D. Characterization of isoenzyme-selective inhibitors of human sphingosine kinases. PLoS One 2012, 7, e44543. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, C.; Childs, S.; Ohotski, J.; McGlynn, L.; Riddick, M.; MacFarlane, S.; Tasker, D.; Pyne, S.; Pyne, N.J.; Edwards, J.; et al. Regulation of cell survival by sphingosine-1-phosphate receptor S1P1 via reciprocal ERK-dependent suppression of Bim and PI-3-kinase/protein kinase C-mediated upregulation of Mcl-1. Cell Death Dis. 2013, 4, e927. [Google Scholar] [CrossRef] [PubMed]
- Betito, S.; Cuvillier, O. Regulation by sphingosine 1-phosphate of Bax and Bad activities during apoptosis in a MEK-dependent manner. Biochem. Biophys. Res. Commun. 2006, 340, 1273–1277. [Google Scholar] [CrossRef] [PubMed]
- El-Shewy, H.M.; Sohn, M.; Wilson, P.; Lee, M.H.; Hammad, S.M.; Luttrell, L.M.; Jaffa, A.A. Low-density lipoprotein induced expression of connective tissue growth factor via transactivation of sphingosine 1-phosphate receptors in mesangial cells. Mol. Endocrinol. 2012, 26, 833–845. [Google Scholar] [CrossRef] [PubMed]
- Pitson, S.M.; Moretti, P.A.; Zebol, J.R.; Lynn, H.E.; Xia, P.; Vadas, M.A.; Wattenberg, B.W. Activation of sphingosine kinase 1 by ERK1/2-mediated phosphorylation. EMBO J. 2003, 22, 5491–5500. [Google Scholar] [CrossRef] [PubMed]
- Gibson, S.B. Epidermal growth factor and trail interactions in epithelial-derived cells. Vitam. Horm. 2004, 67, 207–227. [Google Scholar] [PubMed]
- Mimeault, M.; Pommery, N.; Hénichart, J.P. Synergistic antiproliferative and apoptotic effects induced by epidermal growth factor receptor and protein kinase A inhibitors in human prostatic cancer cell lines. Int. J. Cancer 2003, 106, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Auge, N.; Garcia, V.; Maupas-Schwalm, F.; Levade, T.; Salvayre, R.; Negre-Salvayre, A. Oxidized LDL-induced smooth muscle cell proliferation involves the EGF receptor/PI-3 kinase/Akt and the sphingolipid signaling pathways. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1990–1995. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Du, X.Y.; Chen, J.S.; Zhou, Y.C.; Song, J.G. Secretory phospholipase A2 inhibits epidermal growth factor-induced receptor activation. Exp. Cell Res. 2002, 279, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Reinehr, R.; Becker, S.; Eberle, A.; Grether-Beck, S.; Häussinger, D. Involvement of NADPH oxidase isoforms and Src family kinases in CD95-dependent hepatocyte apoptosis. J. Biol. Chem. 2005, 280, 27179–27194. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, E.D.; Mackova, M.; Das, S.; Payne, S.G.; Lowen, B.; Sibley, C.P.; Chan, G.; Guilbert, L.J. Multiple anti-apoptotic pathways stimulated by EGF in cytotrophoblasts. Placenta 2005, 26, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Maceyka, M.; Hait, N.C.; Paugh, S.W.; Sankala, H.; Milstien, S.; Spiegel, S. Sphingosine kinase 1 is required for migration, proliferation and survival of MCF-7 human breast cancer cells. FEBS Lett. 2005, 579, 5313–5317. [Google Scholar] [CrossRef] [PubMed]
- Yogi, A.; Callera, G.E.; Aranha, A.B.; Antunes, T.T.; Graham, D.; McBride, M.; Dominiczak, A.; Touyz, R.M. Sphingosine-1-phosphate-induced inflammation involves receptor tyrosine kinase transactivation in vascular cells: Upregulation in hypertension. Hypertension 2011, 57, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Meyer zu Heringdorf, D.; Lass, H.; Kuchar, I.; Alemany, R.; Guo, Y.; Schmidt, M.; Jakobs, K.H. Role of sphingosine kinase in Ca2+ signalling by epidermal growth factor receptor. FEBS Lett. 1999, 461, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Iwayama, H.; Sakamoto, T.; Nawa, A.; Ueda, N. Crosstalk between Smad and mitogen-activated protein kinases for the regulation of apoptosis in cyclosporine A-induced renal tubular injury. Nephron Extra 2011, 1, 178–189. [Google Scholar] [CrossRef] [PubMed]
- El-Shewy, H.M.; Abdel-Samie, S.A.; al Qalam, A.M.; Lee, M.H.; Kitatani, K.; Anelli, V.; Jaffa, A.A.; Obeid, L.M.; Luttrell, L.M. Phospholipase C and protein kinase C-β 2 mediate insulin-like growth factor II-dependent sphingosine kinase 1 activation. Mol. Endocrinol. 2011, 25, 2144–2156. [Google Scholar] [CrossRef] [PubMed]
- Fatatis, A.; Miller, R.J. Cell cycle control of PDGF-induced Ca2+ signaling through modulation of sphingolipid metabolism. FASEB J. 1999, 13, 1291–1301. [Google Scholar] [PubMed]
- Olivera, A.; Edsall, L.; Poulton, S.; Kazlauskas, A.; Spiegel, S. Platelet-derived growth factor-induced activation of sphingosine kinase requires phosphorylation of the PDGF receptor tyrosine residue responsible for binding of PLCγ. FASEB J. 1999, 13, 1593–1600. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ueda, N. Ceramide-Induced Apoptosis in Renal Tubular Cells: A Role of Mitochondria and Sphingosine-1-Phoshate. Int. J. Mol. Sci. 2015, 16, 5076-5124. https://doi.org/10.3390/ijms16035076
Ueda N. Ceramide-Induced Apoptosis in Renal Tubular Cells: A Role of Mitochondria and Sphingosine-1-Phoshate. International Journal of Molecular Sciences. 2015; 16(3):5076-5124. https://doi.org/10.3390/ijms16035076
Chicago/Turabian StyleUeda, Norishi. 2015. "Ceramide-Induced Apoptosis in Renal Tubular Cells: A Role of Mitochondria and Sphingosine-1-Phoshate" International Journal of Molecular Sciences 16, no. 3: 5076-5124. https://doi.org/10.3390/ijms16035076