
Methodologies and Perspectives of Proteomics Applied to Filamentous Fungi: From Sample Preparation to Secretome Analysis

Abstract
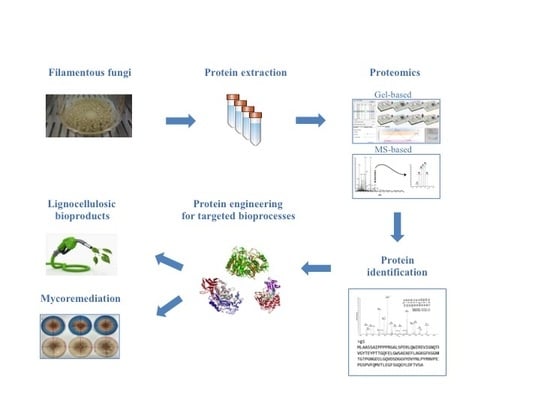
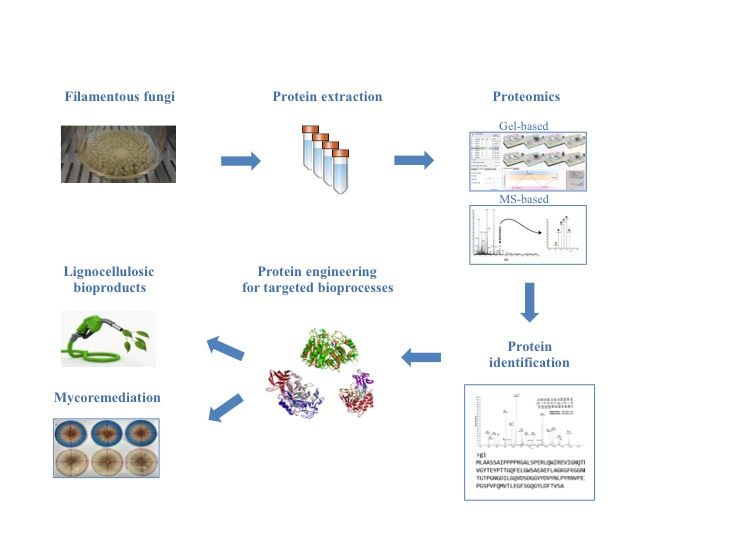
:
1. Introduction
2. Sample Preparation for Fungal Proteomics
3. Fungal Proteomics
3.1. Gel-Based Approaches
3.2. Gel-Free Approaches

{kind=link}
{kind=link}
| Labelling Strategy | Reaction Mechanism or Reagent Structure |
|---|---|
| (a) 18O chemical labelling The incorporation of two 18O occurs at C-terminus of lysine and arginine, during proteolytic digestion. |  |
| (b) ICAT The ICAT reagent consists of cysteine-directed reactive group, a polyether linker region containing deuteriums (heavy reagent) or hydrogens (light reagent), and a biotin group that allows purification of labeled peptides. |  |
| (c) iTRAQ |  |
| (d) TMT |  |
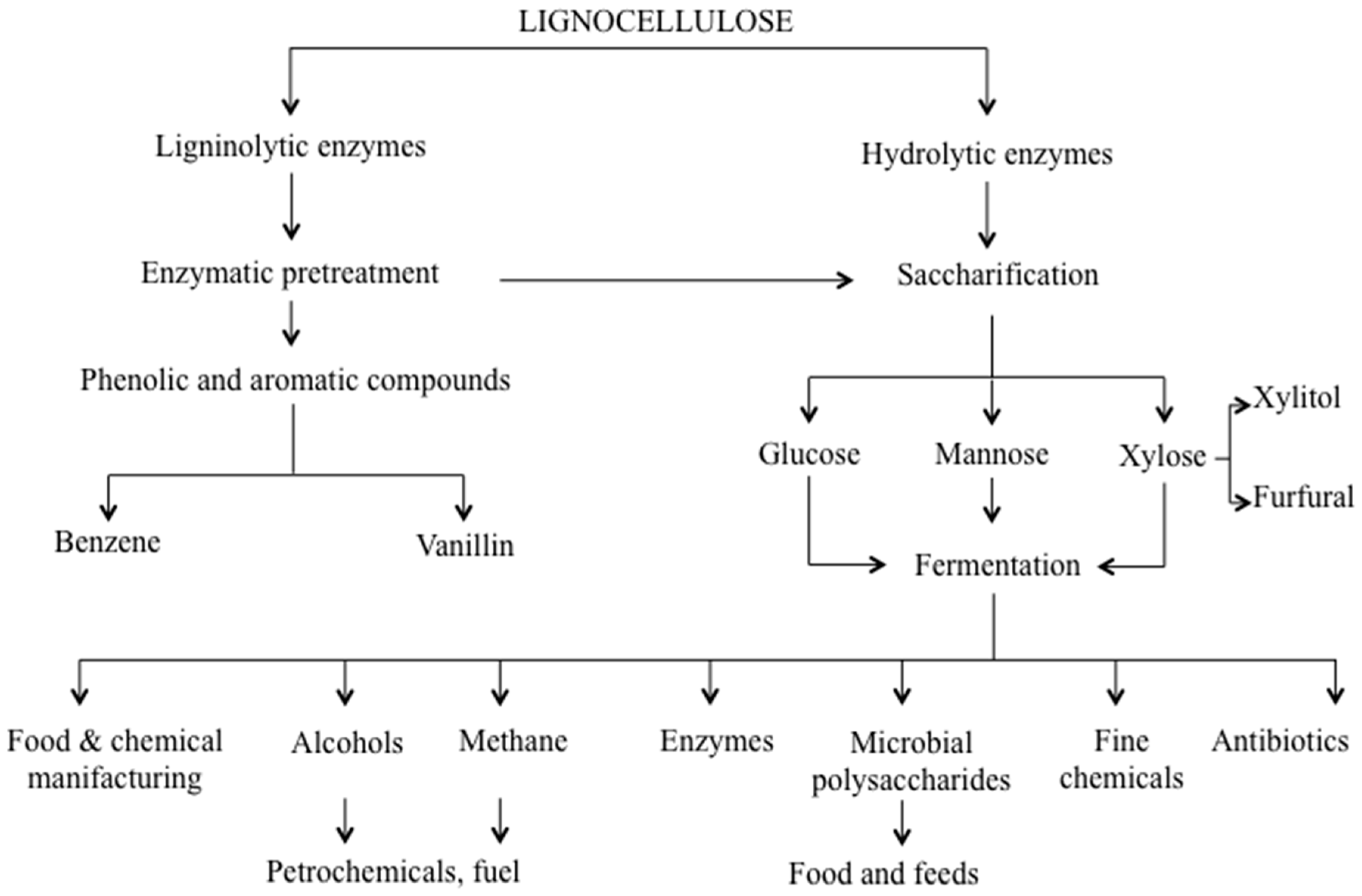
4. Secretome for Targeted Bioprocesses: From Efficient Biomass Deconstruction to Mycoremediation

5. Concluding Remarks
Author Contributions
Conflicts of Interest
References
- Camarero, S.; Jesús, M.M.; Martínez, A.T. Understanding lignin biodegradation for the improved utilization of plant biomass in modern biorefineries. Biofuels Bioprod. Biorefin. 2014, 8, 615–625. [Google Scholar] [CrossRef]
- Sanchez, C. Lignocellulosic residues: Biodegradation and bioconversion by fungi. Biotechnol. Adv. 2009, 27, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Kubicek, C.P. Fungi and Lignocellulosic Biomass; John Wiley & Sons: New York, NY, USA, 2012; pp. 29–44. [Google Scholar]
- Kubicek, C.P.; Mikus, M.; Schuster, A.; Schmoll, M.; Seiboth, B. Metabolic engineering strategies for the improvement of cellulase production by Hypocrea jecorina. Biotechnol. Biofuels 2009, 2, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Dashtban, M.; Schraf, T.H.; Syed, T.A.; Qin, W. Fungal biodegradation and enzymatic modification of lignin. Int. J. Biochem. Mol. Biol. 2010, 1, 36–50. [Google Scholar] [PubMed]
- Guerra, A.; Mendonca, R.; Ferraz, A.; Lu, F.; Ralph, J. Structural characterization of lignin during Pinus taeda wood treatment with Ceriporiopsis subvermispora. Appl. Environ. Microbiol. 2004, 70, 4073–4078. [Google Scholar] [CrossRef] [PubMed]
- Arora, D.S.; Sharma, R.K. Enhancement in in vitro digestibility of wheat straw obtained from different geographical regions during solid state fermentation by white rot fungi. BioResource 2009, 4, 909–920. [Google Scholar]
- Fackler, K.; Gradinger, C.; Hinterstoisser, B.; Messner, K.; Schwanninger, M. Lignin degradation by white rot fungi on spruce wood shavings during short-time solid-state fermentations monitored by near infrared spectroscopy. Enzym. Microb. Technol. 2006, 39, 1476–1483. [Google Scholar] [CrossRef]
- Hilden, K.; Hakala, T.K.; Maijala, P.; Lundell, T.K.; Hatakka, A. Novel thermotolerant laccases produced by the white-rot fungus Physisporinus rivulosus. Appl. Microbiol. Biotechnol. 2007, 77, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Itakura, S.; Enoki, A. Hydroxyl radical generation by an extracellular low-molecular-weight substance and phenol oxidase activity during wood degradation by the white-rot basidiomycete Trametes versicolor. J. Biotechnol. 1999, 75, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Ma, F.; Zhang, X. Lignocellulose degradation and enzyme production by Irpex lacteus CD2 during solid-state fermentation of corn stover. J. Biosci. Bioeng. 2009, 108, 372–375. [Google Scholar] [CrossRef] [PubMed]
- Anderson, W.F.; Akin, D.E. Structural and chemical properties of grass lignocelluloses related to conversion for biofuels. J. Ind. Microbiol. Biotechnol. 2008, 35, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Mai, C.; Kues, U.; Militz, H. Biotechnology in the wood industry. Appl. Microbiol. Biotechnol. 2004, 63, 477–494. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Hamid, A.M.; Solbiati, J.O.; Cann, I.K. Insights into lignin degradation and its potential industrial applications. Adv. Appl. Microbiol. 2013, 82, 1–28. [Google Scholar] [PubMed]
- Scherba, V.V.; Babitskaya, V.G. Polysaccharides of xylotrophic basidiomycetes. Appl. Biochem. Microbiol. 2008, 44, 78–83. [Google Scholar] [CrossRef]
- Rashid, M.H.; Siddiqui, K.S. The stability of extracellular beta-glucosidase from Aspergillus niger is significantly enhanced by non-covalently attached polysaccharides. Folia Microbiol. 1996, 41, 341–346. [Google Scholar] [CrossRef]
- Asgher, M.; Bhatti, H.N.; Ashraf, M.; Legge, R.L. Recent developments in biodegradation of industrial pollutants by white rot fungi and their enzyme system. Biodegradation 2008, 19, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Pointing, S.B. Feasibility of bioremediation by white rot fungi. Appl. Microbiol. Biotechnol. 2001, 57, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Maciel, M.J.; Silva, A.C.; Ribeiro, H.C.T. Industrial and biotechnological applications of ligninolytic enzymes of the basidiomycota: A review. Electron. J. Biotechnol. 2010, 13, 14–15. [Google Scholar]
- Gygi, S.P.; Rist, B.; Gerber, S.A.; Turecek, F.; Gelb, M.H.; Aebersold, R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 1999, 17, 994–999. [Google Scholar] [CrossRef] [PubMed]
- González-Fernandez, R.; Jorrin-Novo, J.V. Proteomic protocols for the study of filamentous fungi. In Laboratory Protocols in Fungal Biology: Current Methods in Fungal Biology; Gupta, V.K., Ed.; Springer: New York, NY, USA, 2013; pp. 299–308. [Google Scholar]
- Bowman, S.M.; Free, S.J. The structure and synthesis of the fungal cell wall. Bioessays 2006, 28, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, C.; García-Estrada, C.; Martín, J.F. Proteomics methodology applied to the analysis of filamentous fungi-new trends for an impressive diverse group of organisms. In Tandem Mass Spectrometry—Applications and Principles; Prasain, J., Ed.; InTech: Rijeka, Croatia, 2012; pp. 127–160. [Google Scholar]
- Grinyer, J.; Kautto, L.; Traini, M.; Willows, R.D.; Te’o, J.; Bergquist, P. Proteome mapping of the Trichoderma reesei 20S proteasome. Curr. Genet. 2007, 51, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Grinyer, J.; McKay, M.; Nevalainen, H.; Herbert, B.R. Fungal proteomics: Initial mapping of biological control strain Trichoderma harzianum. Curr. Genet. 2004, 45, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.; Hains, P.; Walsh, B.; Bergquist, P.; Nevalainen, H. Proteins associated with the cell envelope of Trichoderma reesei: A proteomic approach. Proteomics 2001, 1, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, M.P.; Marten, M.R. Comparison of lysis methods and preparation protocols for one- and two-dimensional electrophoresis of Aspergillus oryzae intracellular proteins. Electrophoresis 2002, 23, 2216–2222. [Google Scholar] [CrossRef] [PubMed]
- Grinyer, J.; Hunt, S.; McKay, M.; Herbert, B.R.; Nevalainen, H. Proteomic response of the biological control fungus Trichoderma atroviride to growth on the cell walls of Rhizoctonia solani. Curr. Genet. 2005, 47, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.D.; Saparno, A.; Blackwell, B.; Anoop, V.; Gleddie, S.; Tinker, N.A. Proteomic analyses of Fusarium graminearum grown under mycotoxininducing conditions. Proteomics 2008, 8, 2256–2265. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Acero, F.J.; Colby, T.; Harzen, A.; Cantoral, J.M.; Schmidt, J. Proteomic analysis of the phytopathogenic fungus Botrytis cinerea during cellulose degradation. Proteomics 2009, 9, 2892–2902. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Yu, S.; Kim, S.G.; Kim, H.J.; Kang, S.Y.; Hwang, D.H.; Jang, Y.S.; Kang, K.Y. Proteome analysis of rice blast fungus (Magnaporthe grisea) proteome during appressorium formation. Proteomics 2004, 4, 3579–3587. [Google Scholar] [CrossRef] [PubMed]
- Bhadauria, V.; Zhao, W.S.; Wang, L.X.; Zhang, Y.; Liu, J.H.; Yang, J.; Kong, L.A.; Peng, Y.L. Advances in fungal proteomics. Microbiol. Res. 2007, 162, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Bhadauria, V.; Peng, Y.L. Optimization of a protein extraction technique for fungal proteomics. Indian J. Microbiol. 2010, 50, 127–131. [Google Scholar]
- Shimizu, M.; Wariishi, H. Development of a sample preparation method for fungal proteomics. FEMS Microbiol. Lett. 2005, 247, 17–22. [Google Scholar]
- Vödisch, M.; Scherlach, K.; Winkler, R.; Hertweck, C.; Braun, H.P.; Roth, M.; Haas, H.; Werner, E.R.; Brakhage, A.A.; Kniemeyer, O. Analysis of the Aspergillus fumigatus proteome reveals metabolic changes and the activation of the pseurotin a biosynthesis gene cluster in response to hypoxia. J. Proteome Res. 2011, 10, 2508–2524. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, M.P.; Shen, J.; Raman, B.; Marten, M.R. Solubilization of trichloroacetic acid (TCA) precipitated microbial proteins via NaOH for one and two dimensional electrophoresis. J. Proteome Res. 2003, 2, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Rabilloud, T. Use of thiourea to increase the solubility of membrane proteins in two-dimensional electrophoresis. Electrophoresis 1998, 19, 758–760. [Google Scholar] [CrossRef] [PubMed]
- Everberg, H.; Gustavasson, N.; Tjerned, F. Enrichment of membrane proteins by partitioning in detergent/polymer aqueous two-phase systems. Methods Mol. Biol. 2008, 424, 403–412. [Google Scholar] [PubMed]
- Rabilloud, T.; Adessi, C.; Giraudel, A.; Lunardi, J. Improvement of the solubilization of proteins in twodimensional electrophoresis with immobilized pH gradients. Electrophoresis 1997, 18, 307–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbert, B.R.; Grinyer, J.; McCarthy, J.T.; Isaacs, M.; Harry, E.J.; Nevalainen, H.; Traini, M.D.; Hunt, S.; Schulz, B.; Laver, M.; et al. Improved 2-DE of microorganisms after acidic extraction. Electrophoresis 2006, 27, 1630–1640. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.W. The molds of Katrina. Update. N. Y. Acad. Sci. 2006, 126, 6–9. [Google Scholar]
- Kim, Y.; Nandakumar, M.P.; Marten, M.R. Proteomics of filamentous fungi. Trends Biotechnol. 2007, 25, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Fragner, D.; Zomorrodi, M.; Kues, U.; Majcherczyk, A. Optimized protocol for the 2-DE of extracellular proteins from higher basidiomycetes inhabiting lignocellulose. Electrophoresis 2009, 30, 2431–2441. [Google Scholar] [CrossRef] [PubMed]
- Kao, S.H.; Wong, H.K.; Chiang, C.Y.; Chen, H.M. Evaluating the compatibility of three colorimetric protein assays for two-dimensional electrophoresis experiments. Proteomics 2008, 8, 2178–2184. [Google Scholar] [CrossRef] [PubMed]
- Medina, M.L.; Francisco, WA. Isolation and enrichment of secreted proteins from filamentous fungi. In 2D PAGE: Sample Preparation and Fractionation; Humana Press: New York, NY, USA, 2008; pp. 275–285. [Google Scholar]
- Adav, S.S.; Li, A.A.; Manavalan, A.; Punt, P.; Sze, S.K. Quantitative iTRAQ secretome analysis of aspergillus niger reveals novel hydrolytic enzymes. J. Proteome Res. 2010, 9, 3932–3940. [Google Scholar] [CrossRef] [PubMed]
- Adav, S.S.; Chao, L.T.; Sze, S.K. Quantitative secretomic analysis of Trichoderma reesei strains reveals enzymatic composition for lignocellulosic biomass degradation. Mol. Cell Proteomics 2012, 11. [Google Scholar] [CrossRef] [PubMed]
- Salvachúa, D.; Martínez, A.T.; Tien, M.; López-Lucendo, M.F.; García, F.; de los Ríos, V.; Martínez, M.J.; Prieto, A. Differential proteomic analysis of the secretome of Irpex lacteus and other white-rot fungi during wheat straw pretreatment. Biotechnol. Biofuels 2013, 6, 115–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Vogel, A.; Eyzaguirre, J.; Oleas, G.; Callegari, E.; Navarrete, M. Proteomic analysis in non-denaturing condition of the secretome reveals the presence of multienzyme complexes in Penicillium purpurogenum. Appl. Microbiol. Biotechnol. 2011, 89, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.J.D.; Gómez-Mendoza, D.P.; Junqueira, M.; Domont, G.B.; Ferreira Filho, X.; de Sousa, M.V.; Ricart, C.A.O. Blue native-PAGE analysis of Trichoderma harzianum secretome reveals cellulases and hemicellulases working as multienzymatic complexes. Proteomics 2012, 12, 2729–2738. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, J.M.P.F.; de Graaff, L.H. Proteomics of industrial fungi: Trends and insights for biotechnology. Appl. Microbiol. Biotechnol. 2011, 89, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Macedo, M.L.; Ferraz, A.; Rodriguez, J.; Ottoboni, L.M.; de Mello, M.P. Iron-regulated proteins in Phanerochaete chrysosporium and Lentinula edodes: Differential analysis by sodium dodecyl sulfate polyacrylamide gel electrophoresis and two-dimensional polyacrylamide gel electrophoresis profiles. Electrophoresis 2002, 23, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, S.; Prokisch, H.; Schlunck, T.; Camp, D.G.; Ahting, U.; Waizenegger, T.; Scharfe, C.; Meitinger, T.; Imhof, A.; Neupert, W.; et al. Proteome analysis of mitochondrial outer membrane from Neurospora crassa. Proteomics 2006, 6, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.B.; Stuart, L.; Feener, E.P. Label-free quantitative analysis of one-dimensional PAGE LC/MS/MS proteome: Application on angiotensin II-stimulated smooth muscle cells secretome. Mol. Cell Proteomics 2008, 7, 2399–2409. [Google Scholar] [CrossRef] [PubMed]
- Piersma, S.R.; Warmoes, M.O.; de Witm, M.; de Reus, I.; Knol, J.C.; Jiménez, C.R. Whole gel processing procedure for GeLC-MS/MS based proteomics. Proteome Sci. 2013, 11, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Guais, O.; Borderies, G.; Pichereaux, C.; Maestracci, M.; Neugnot, V.; Rossignol, M.; François, J.M. Proteomics analysis of “Rovabiot Excel”, a secreted protein cocktail from the filamentous fungus Penicillium funiculosum grown under industrial process fermentation. J. Ind. Microbiol. Biotechnol. 2008, 35, 1659–1668. [Google Scholar] [CrossRef] [PubMed]
- Braaksma, M.; Martens-Uzunova, E.S.; Punt, P.J.; Schaap, P.J. An inventory of the Aspergillus niger secretome by combining in silico predictions with shotgun proteomics data. BMC Genomics 2010, 11, 584–595. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, J.M.P.F.; van Passel, M.W.; Schaap, P.J.; de Graaff, L.H. Shotgun proteomics of Aspergillus niger microsomes upon d-xylose induction. Appl. Environ. Microbiol. 2010, 76, 4421–4429. [Google Scholar] [CrossRef] [PubMed]
- Couturier, M.; Navarro, D.; Olivé, C.; Chevret, D.; Haon, M.; Favel, A.; Lesage-Meessen, L.; Henrissat, B.; Coutinho, P.M.; Berrin, J.G. Post-genomic analyses of fungal lignocellulosic biomass degradation reveal the unexpected potential of the plant pathogen Ustilago maydis. BMC Genomics 2012, 13, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Wittig, I.; Braun, H.P.; Schägger, H. Blue native PAGE. Nat. Protoc. 2006, 1, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Jami, M.S.; Barreiro, C.; García-Estrada, C.; Martín, J.F. Proteome analysis of the penicillin producer Penicillium chrysogenum: Characterization of protein changes during the industrial strain improvement. Mol. Cell Proteomics 2010, 9, 1182–1198. [Google Scholar] [CrossRef] [PubMed]
- Jami, M.S.; García-Estrada, C.; Barreiro, C.; Cuadrado, A.A.; Salehi-Najafabadi, Z.; Martín, J.F. The Penicillium chrysogenum extracellular proteome. Conversionfrom a food-rotting strain to a versatile cell factory for white biotechnology. Mol. Cell Proteomics 2010, 9, 2729–2744. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, S.; Yildirim, V.; Kaya, L.; Albrecht, D.; Becher, D.; Hecker, M.; Ozcengiz, G. Phanerochaete chrysosporium soluble proteome as a prelude for the analysis of heavy metal stress response. Proteomics 2007, 7, 1249–1260. [Google Scholar] [CrossRef] [PubMed]
- Ravalason, H.; Jan, G.; Mollé, D.; Pasco, M.; Coutinho, P.M.; Lapierre, C.; Pollet, B.; Bertaud, F.; Petit-Conil, M.; Grisel, S.; et al. Secretome analysis of Phanerochaete chrysosporium strain CIRM-BRFM41 grown on softwood. Appl. Microbiol. Biotechnol. 2008, 80, 719–33. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Sun, J.; Nimtz, M.; Wissing, J.; Zeng, A.P.; Rinas, U. The intra- and extracellular proteome of Aspergillus niger growing on defined medium with xylose or maltose as carbon substrate. Microb. Cell Fact. 2010, 20, 23–36. [Google Scholar] [CrossRef]
- Shimizu, M.; Yuda, N.; Nakamura, T.; Tanaka, H.; Wariishi, H. Metabolic regulation at the tricarboxylic acid and glyoxylate cycles of the lignin-degrading basidiomycete Phanerochaete chrysosporium against exogenous addition of vanillin. Proteomics 2005, 5, 3919–3931. [Google Scholar] [CrossRef] [PubMed]
- Marouga, R.; David, S.; Hawkins, E. The development of the DIGE system: 2D fluorescence difference gel analysis technology. Anal. Bioanal. Chem. 2005, 382, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos Castro, L.; Pedersoli, W.R.; Antoniêto, A.C.C.; Steindorff, A.S.; Silva-Rocha, R.; Martinez-Rossi, N.M.; Brown, N.A.; Goldman, G.H.; Faça, V.M.; Persinoti, G.F.; et al. Comparative metabolism of cellulose, sophorose and glucose in Trichoderma reesei using high-throughput genomic and proteomic analyses. Biotechnol. Biofuels 2014, 7, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahmad, N.; Al-Obaidi, J.R.; Rashid, N.M.N.; Zean, N B.; Yusoff, M.H.Y.M.; Shaharuddin, N.S.; Mohd, J.N.A.; Saleh, N.M. Comparative proteomic analysis of different developmental stages of the edible mushroom Termitomyces heimii. Biol. Res. 2014, 47, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.R., III; Ruse, C.I.; Nakorchevsky, A. Proteomics by mass spectrometry: Approaches, advances, and applications. Annu. Rev. Biomed. Eng. 2009, 11, 49–79. [Google Scholar] [CrossRef] [PubMed]
- Walther, T.C.; Mann, M. Mass spectrometry-based proteomics in cell biology. J. Cell Biol. 2010, 190, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Bantscheff, M.; Lemeer, S.; Savitski, M.M.; Kuster, B. Quantitative mass spectrometry in proteomics: Critical review update from 2007 to the present. Anal. Bioanal. Chem. 2012, 404, 939–965. [Google Scholar] [CrossRef] [PubMed]
- Washburn, M.P.; Wolters, D.; Yates, J.R., III. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 2001, 19, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Cooper, B.; Neelam, A.; Campbell, K.B.; Lee, J.; Liu, G.; Garrett, W.M.; Scheffler, B.; Tucker, M.L. Protein accumulation in the germinating Uromyces appendiculatus uredospore. Mol. Plant Microbe Interact. 2007, 20, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Beeson, W.T.; Iavarone, A.T.; Sun, J.; Marletta, M.A.; Cate, J.H.; Glass, N.L. Systems analysis of plant cell wall degradation by the model filamentous fungus Neurospora crassa. Proc. Natl. Acad. Sci. USA 2009, 106, 22157–22162. [Google Scholar] [CrossRef] [PubMed]
- Scully, E.D.; Hoover, K.; Carlson, J.; Tien, M.; Geib, S.M. Proteomic analysis of Fusarium solani isolated from the asian longhorned beetle, Anoplophora glabripennis. PLoS One 2012, 7, e32990. [Google Scholar] [CrossRef] [PubMed]
- De Godoy, L.M.; Olsen, J.V.; Cox, J.; Nielsen, M.L.; Hubner, N.C.; Fröhlich, F.; Walther, T.C.; Mann, M. Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature 2008, 455, 1251–1254. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, N.; Kulak, N.A.; Cox, J.; Neuhauser, N.; Mayr, K.; Hoerning, O.; Vorm, O.; Mann, M. System-wide perturbation analysis with nearly complete coverage of the yeast proteome by single-shot ultra HPLC runs on a bench top Orbitrap. Mol. Cell Proteomics 2012, 11. [Google Scholar] [CrossRef] [PubMed]
- Picotti, P.; Clément-Ziza, M.; Lam, H.; Campbell, D.S.; Schmidt, A.; Deutsch, E.W.; Röst, H.; Sun, Z.; Rinner, O.; Reiter, L.; et al. A complete mass-spectrometric map of the yeast proteome applied to quantitative trait analysis. Nature 2013, 494, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Webb, K.J.; Xu, T.; Park, S.K.; Yates, J.R., III. Modified MuDPIT separation identified 4488 proteins in a system-wide analysis of quiescence in yeast. J. Proteome Res. 2013, 12, 2177–2184. [Google Scholar] [CrossRef] [PubMed]
- Picotti, P.; Bodenmiller, B.; Mueller, L.N.; Domon, B.; Aebersold, R. Full dynamic range proteome analysis of S. cerevisiae by targeted proteomics. Cell 2009, 138, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S.S.; Geiger, T.; Chatterjee, B.; Bandilla, P.; Fröhlich, F.; Cox, J.; Mann, M. Deep and highly sensitive proteome coverage by LC-MS/MS without prefractionation. Mol. Cell Proteomics 2011. [Google Scholar] [CrossRef]
- Kelstrup, C.D.; Jersie-Christensen, R.R.; Batth, T.S.; Arrey, T.N.; Kuehn, A.; Kellmann, M.; Olsen, J.V. Rapid and deep proteomes by faster sequencing on a benchtop quadrupole ultra-high-field orbitrap mass spectrometer. J. Proteome Res. 2014, 13, 6187–6195. [Google Scholar] [CrossRef] [PubMed]
- Scheltema, R.A.; Hauschild, J.P.; Lange, O.; Hornburg, D.; Denisov, E.; Damoc, E.; Kuehn, A.; Makarov, A.; Mann, M. The Q exactive HF, a benchtop mass spectrometer with a pre-filter, high-performance quadrupole and an ultra-high-field orbitrap analyzer. Mol. Cell Proteomics 2014, 13, 3698–3708. [Google Scholar] [CrossRef] [PubMed]
- Stewart, I.I.; Thomson, T.; Figeys, D. 18O labeling: A tool for proteomics. Rapid Commun. Mass Spectrom. 2001, 15, 2456–2465. [Google Scholar] [CrossRef] [PubMed]
- Ross, P.L.; Huang, Y.N.; Marchese, J.N.; Williamson, B.; Parker, K.; Hattan, S.; Khainovski, N.S.; Dey, S.; Daniels, S.; Purkayastha, S.; et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell Proteomics 2004, 3, 1154–1169. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.; Schäfer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, T.; Johnstone, R.; Mohammed, A.K.; Hamon, C. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904. [Google Scholar] [CrossRef] [PubMed]
- Cagas, S.E.; Jain, M.R.; Li, H.; Perlin, D.S. The proteomic signature of Aspergillus fumigatus during early development. Mol. Cell Proteomics 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Cagas, S.E.; Jain, M.R.; Li, H.; Perlin, D.S. Profiling the Aspergillus fumigatus proteome in response to caspofungin. Antimicrob. Agents Chemother. 2011, 55, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Li, J.; Zhao, S.; Zhang, R.; Wang, M.; Miao, Y.; Shen, Y.; Shen, Q. Secretome diversity and quantitative analysis of cellulolytic Aspergillus fumigatus Z5 in the presence of different carbon sources. Biotechnol. Biofuels 2013, 6, 149–165. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, A.; Adav, S.S.; Sze, S.K. TRAQ-based quantitative secretome analysis of Phanerochaete chrysosporium. J. Proteomics 2011, 75, 642–654. [Google Scholar] [CrossRef] [PubMed]
- Adav, S.S.; Ravindran, A.; Sze, S.K. Quantitative proteomic analysis of lignocellulolytic enzymes by Phanerochaete chrysosporium on different lignocellulosic biomass. J. Proteomics 2012, 75, 1493–1504. [Google Scholar] [CrossRef] [PubMed]
- Adav, S.S.; Chao, L.T.; Sze, S.K. Protein abundance in multiplexed samples (PAMUS) for quantitation of Trichoderma reesei secretome. J. Proteomics 2013, 83, 180–196. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, Y.; Li, S. Deuterium isobaric amine-reactive tags for quantitative proteomics. Anal. Chem. 2010, 82, 7588–7595. [Google Scholar] [CrossRef] [PubMed]
- Ramsubramaniam, N.; Tao, F.; Li, S.; Marten, M.R. Novel and cost-effective 6-plex isobaric tagging reagent, DiART, is effective for identification and relative quantification of complex protein mixtures using PQD fragmentation. J. Mass Spectrom. 2013, 48, 1032–1041. [Google Scholar] [CrossRef] [PubMed]
- Mann, M. Functional and quantitative proteomics using SILAC. Nat. Rev. Mol. Cell Biol. 2006, 7, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Oda, Y.; Huang, K.; Cross, F.R.; Cowburn, D.; Chait, B.T. Accurate quantitation of protein expression and site-specific phosphorylation. Proc. Natl. Acad. Sci. USA 1999, 96, 6591–6596. [Google Scholar] [CrossRef] [PubMed]
- De Godoy, L.M. SILAC yeast: From labeling to comprehensive proteome quantification. In Shotgun Proteomics; Martins-de-Souza, D., Ed.; Springer: New York, NY, USA, 2014; pp. 81–109. [Google Scholar]
- Phillips, C.M.; Iavarone, A.T.; Marletta, M.A. Quantitative proteomic approach for cellulose degradation by Neurospora crassa. J. Proteome Res. 2011, 10, 4177–4185. [Google Scholar] [CrossRef] [PubMed]
- Wasinger, V.C.; Zeng, M.; Yau, Y. Current status and advances in quantitative proteomic mass spectrometry. Int. J. Proteomics 2013. [Google Scholar] [CrossRef]
- Liu, H.; Sadygov, R.G.; Yates, J.R., III. A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 2004, 76, 4193–4201. [Google Scholar] [CrossRef] [PubMed]
- Zybailov, B.; Coleman, M.K.; Florens, L.; Washburn, M.P. Correlation of relative abundance ratios derived from peptide ion chromatograms and spectrum counting for quantitative proteomic analysis using stable isotope labeling. Anal. Chem. 2005, 77, 6218–6224. [Google Scholar] [CrossRef] [PubMed]
- Zybailov, B.; Mosley, A.L.; Sardiu, M.E.; Coleman, M.K.; Florens, L.; Washburn, M.P. Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J. Proteome Res. 2006, 5, 2339–2347. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Vogel, C.; Wang, R.; Yao, X.; Marcotte, E.M. Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat. Biotechnol. 2006, 25, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Ishihama, Y.; Oda, Y.; Tabata, T.; Sato, T.; Nagasu, T.; Rappsilber, J.; Mann, M. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol. Cell Proteomics 2005, 4, 1265–1272. [Google Scholar] [CrossRef] [PubMed]
- Adav, S.S.; Cheow, E.S.H.; Ravindran, A.; Dutta, B.; Sze, S.K. Label free quantitative proteomic analysis of secretome by Thermobifida fusca on different lignocellulosic biomass. J. Proteomics 2012, 75, 3694–3706. [Google Scholar] [CrossRef] [PubMed]
- Arike, L.; Peil, L. Spectral counting label-free proteomics. In Shotgun Proteomics; Martins-de-Souza, D., Ed.; Springer: New York, NY, USA, 2014; pp. 213–222. [Google Scholar]
- Tjalsma, H.; Bolhuis, A.; Jongbloed, J.D.H.; Bron, S.; van Dijl, J.M. Signal peptidedependent protein transport in Bacillus subtilis: A genome-based survey of the secretome. Microb. Mol. Biol. Rev. 2000, 64, 515–547. [Google Scholar] [CrossRef]
- Alfaro, M.; Oguiza, J.A.; Ramírez, L.; Pisabarro, A.G. Comparative analysis of secretomes in basidiomycete fungi. J. Proteomics 2014, 102, 28–43. [Google Scholar] [CrossRef] [PubMed]
- Nickel, W.; Seedorf, M. Unconventional mechanisms of protein transport to the cell surface of eukaryotic cells. Annu. Rev. Cell Dev. Biol. 2008, 24, 287–308. [Google Scholar] [CrossRef] [PubMed]
- Bouws, H.; Wattenberg, A.; Zorn, H. Fungal secretomes—Nature’s toolbox for white biotechnology. Appl. Microbiol. Biotechnol. 2008, 80, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Adav, S.S.; Sze, S.K. Fungal secretome for biorefinery: Recent advances in proteomic technology. Mass Spectrom. Lett. 2013, 4, 1–9. [Google Scholar] [CrossRef]
- Kubicek, C.P. Systems biological approaches towards understanding cellulase production by Trichoderma reesei. J. Biotechnol. 2013, 163, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Martinez, D.; Berka, R.; Henrissat, B.; Saloheimo, M.; Arvas, M.; Baker, S.E.; Chapman, J.; Chertkov, O.; Coutinho, P.M.; Cullen, D.; et al. Genome sequencing and analysis of the biomass-degrading fungus Trichoderma reesei (syn. Hypocrea jecorina). Nat. Biotechnol. 2008, 26, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.R.; Fidantsef, A.L. Directed evolution of industrial enzymes: An update. Curr. Opin. Biotechnol. 2003, 14, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Adav, S.S.; Ravindran, A.; Sze, S.K. Proteomic analysis of temperature dependent extracellular proteins from Aspergillus fumigatus grown under solid-state culture condition. J. Proteome Res. 2013, 12, 2715–2731. [Google Scholar] [CrossRef] [PubMed]
- Chundawat, S.P.; Lipton, M.S.; Purvine, S.O.; Uppugundla, N.; Gao, D.; Balan, V.; Dale, B.E. Proteomics-based compositional analysis of complex cellulase-hemicellulase mixture. J. Proteome Res. 2011, 10, 4365–4372. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, T.; Yazaki, S.; Ui, S.; Mimura, A. Production of large multienzyme complex by aerobic thermophilic fungus Chaetomium sp. nov. MS-017 grown on palm oil mill fibre. Lett. Appl. Microbiol. 2005, 40, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Callegari, E.; Navarrete, N. The use of mass spectrometry for characterization of fungal secretomes. In Tandem Mass Spectrometry—Applications and Principles; Prasain, K.J., Ed.; InTech: Rijeka, Croatia, 2012. [Google Scholar] [CrossRef]
- Langston, J.A.; Shaghasi, T.; Abbate, E.; Xu, F.; Vlasenko, E.; Sweeney, M.D. Oxidoreductive cellulose depolymerization by the enzymes cellobiose dehydrogenase and glycoside hydrolase 61. Appl. Environ. Microbiol. 2011, 77, 7007–7015. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.L.; Thitikorn-amorn, J.; Hsieh, J.F.; Ou, B.M.; Chen, S.H.; Ratanakhanokchai, K.; Huang, P.J.; Chen, S.T. Enhanced enzymatic conversion with freeze pretreatment of rice straw. Biomass Bioenergy 2011, 35, 90–95. [Google Scholar] [CrossRef]
- Sun, Y.; Cheng, J.Y. Hydrolysis of lignocellulosic materials for ethanol production: A review. Bioresour. Technol. 2002, 83, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Palmqvist, E.; Hahn-Hägerdal, B. Fermentation of lignocellulosic hydrolysates. II: Inhibitors and mechanisms of inhibition. Bioresour. Technol. 2000, 74, 25–33. [Google Scholar] [CrossRef]
- Martinez, D.; Larrondo, L.F.; Putnam, N.; Gelpke, M.D.S.; Huang, K.; Chapman, J.; Helfenbein, K.G.; Ramaiya, P.; Detter, J.C.; Larimer, F.; et al. Genome sequence of the lignocellulose degrading fungus Phanerochaete chrysosporium strain RP78. Nat. Biotechnol. 2004, 22, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Wymelenberg, A.V.; Sabat, G.; Martinez, D.; Rajangam, A.S.; Teeri, T.T.; Gaskell, J.; Kersten, P.J.; Cullen, D. The Phanerochaete chrysosporium secretome: Database predictions and initial mass spectrometry peptide identifications in cellulose-grown medium. J. Biotechnol. 2005, 118, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Wymelenberg, A.V.; Minges, P.; Sabat, G.; Martinez, D.; Aerts, A.; Salamov, A.; Grigoriev, I.; Shapiro, H.; Putnam, N.; Belinky, P.; et al. Computational analysis of the Phanerochaete chrysosporium v2.0 genome database and mass spectrometry identification of peptides in ligninolytic cultures reveal complex mixtures of secreted proteins. Fungal Genet. Biol. 2006, 43, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Liu, F.; Koc, H.; Tien, M. Expression analysis of extracellular proteins from Phanerochaete chrysosporium grown on different liquid and solid substrates. Microbiology 2007, 153, 3023–3033. [Google Scholar] [CrossRef] [PubMed]
- Henriksson, G.; Zhang, L.; Li, J.; Ljungquist, P.; Reitberger, T.; Pettersson, G.; Johansson, G. Is cellobiose dehydrogenase from Phanerochaete chrysosporium a lignin degrading enzyme? Biochim. Biophys. Acta 2000, 1480, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Ander, P.; Marzullo, L. Sugar oxidoreductases and veratryl alcohol oxidase as related to lignin degradation. J. Biotechnol. 1997, 53, 115–131. [Google Scholar] [CrossRef] [PubMed]
- Hori, C.; Gaskell, J.; Igarashi, K.; Kersten, P.; Mozuch, M.; Samejima, M.; Cullen, D. Temporal alterations in the secretome of the selective ligninolytic fungus Ceriporiopsis subvermispora during growth on aspen wood reveal this organism’s strategy for degrading lignocellulose. Appl. Environ. Microbiol. 2014, 80, 2062–2070. [Google Scholar] [CrossRef] [PubMed]
- Perrotta, G.; Bianco, L.; Carbone, F.; Daddiego, L.; Facella, P.; Lopez, L. Functional metagenomic and proteomic characterization of soil microbial community associated with decomposing reeds. New Biotechnol. 2014, 31, S170–S171. [Google Scholar] [CrossRef]
- Baldrian, P.; López-Mondéjar, R. Microbial genomics, transcriptomics and proteomics: New discoveries in decomposition research using complementary methods. Appl. Microbiol. Biotechnol. 2014, 98, 1531–1537. [Google Scholar] [CrossRef] [PubMed]
- Adav, S.S.; Ravindran, A.; Cheow, E.S.H.; Sze, S.K. Quantitative proteomic analysis of secretome of microbial consortium during saw dust utilization. J. Proteomics 2012, 75, 5590–5603. [Google Scholar] [CrossRef] [PubMed]
- Novotný, Č.; Svobodová, K.; Erbanová, P.; Cajthaml, T.; Kasinath, A.; Lang, E.; Šašek, V. Ligninolytic fungi in bioremediation: extracellular enzyme production and degradation rate. Soil Biol. Biochem. 2004, 36, 1545–1551. [Google Scholar] [CrossRef]
- More, T.T.; Yan, S.; Tyagi, R.D.; Surampalli, R.Y. Potential use of filamentous fungi for wastewater sludge treatment. Bioresour. Technol. 2010, 101, 7691–7700. [Google Scholar] [CrossRef] [PubMed]
- Gadd, G.M. Fungi in Bioremediation; Cambridge University Press: Cambridge, UK, 2001; p. 496. [Google Scholar]
- Reina, R.; Kellner, H.; Jehmlich, N.; Ullrich, R.; García-Romera, I.; Aranda, E.; Liers, C. Differences in the secretion pattern of oxidoreductases from Bjerkandera adusta induced by a phenolic olive mill extract. Fungal Genet. Biol. 2014, 72, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Korniłłowicz-Kowalska, T.; Rybczyńska, K. Anthraquinone dyes decolorization capacity of anamorphic Bjerkandera adusta CCBAS 930 strain and its HRP-like negative mutants. World J. Microbiol. Biotechnol. 2014, 30, 1725–1736. [Google Scholar] [CrossRef] [PubMed]
- Anastasi, A.; Spina, F.; Prigione, V.; Tigini, V.; Giansanti, P.; Varese, G.C. Scale-up of a bioprocess for textile wastewater treatment using Bjerkandera adusta. Bioresour. Technol. 2010, 101, 3067–3075. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bianco, L.; Perrotta, G. Methodologies and Perspectives of Proteomics Applied to Filamentous Fungi: From Sample Preparation to Secretome Analysis. Int. J. Mol. Sci. 2015, 16, 5803-5829. https://doi.org/10.3390/ijms16035803
Bianco L, Perrotta G. Methodologies and Perspectives of Proteomics Applied to Filamentous Fungi: From Sample Preparation to Secretome Analysis. International Journal of Molecular Sciences. 2015; 16(3):5803-5829. https://doi.org/10.3390/ijms16035803
Chicago/Turabian StyleBianco, Linda, and Gaetano Perrotta. 2015. "Methodologies and Perspectives of Proteomics Applied to Filamentous Fungi: From Sample Preparation to Secretome Analysis" International Journal of Molecular Sciences 16, no. 3: 5803-5829. https://doi.org/10.3390/ijms16035803