
In Vitro and in Vivo Antitumoral Effects of Combinations of Polyphenols, or Polyphenols and Anticancer Drugs: Perspectives on Cancer Treatment




Abstract
:1. Introduction
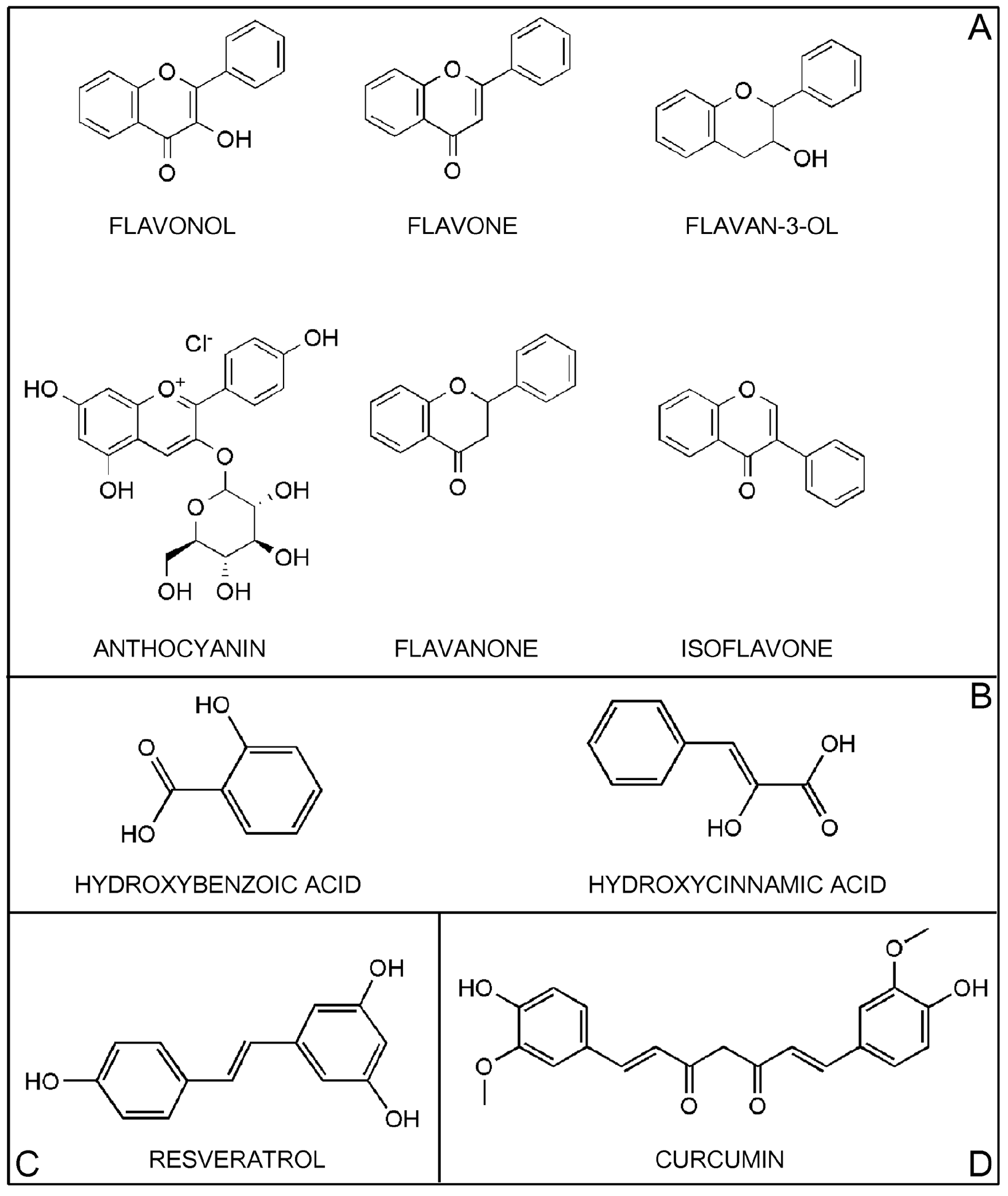
2. Classification of Polyphenols

2.1. Flavonoids
2.1.1. Flavonols
2.1.2. Flavones
2.1.3. Flavan-3-Ols
2.1.4. Anthocyanins
2.1.5. Flavanones
2.1.6. Isoflavones
2.1.7. Minor Subclass of Flavonoids
2.2. Phenolic Acids
2.3. Stilbenes
2.4. Lignans
2.5. Other Polyphenols
3. Polyphenols Target Signal Transduction Pathways Involved in Carcinogenesis
3.1. Modulation of ErbB Receptors Signaling Pathway by Polyphenols in Cancer Cells

{kind=link}
| Signaling Pathway | Treatment | In Vitro Model | In Vivo Model | Antitumoral Effects | Reference |
|---|---|---|---|---|---|
| ErbB receptors | CUR | MDA-MB-468 breast cancer cells (40 µM) | ↓ EGFR phosphorylation
↓ c-fos expression ↓ ERK, MKK4, JNK activity | [32] | |
| MDA-MB-231 breast cancer cells (30–50 µM) | ↓ Cell proliferation
↓ EGFR, ERK1/2, Akt, MAPK phosphorylation | [33,34] | |||
| Breast cancer cells (6–50 µM) | BALB-neuT transgenic mice (2 mg in 50 µL corn oil p.o. thrice weekly) | ↓ Tumor growth
↓ ERK1/2 activity ↑ Bax/Bcl-2 ratio ↑ PARP cleavage ↓ Tumor multiplicity | [29] | ||
| Gastric cancer cells (1–100 µM) | ↓ Cell proliferation
↓ ErbB2, cyclin D1 expression ↓ PAK1 activity | [35] | |||
| LNCaP, C4-2B prostate cancer cells (0–100 µM) | ↓ Cell proliferation
↓ EGFR, ErbB2 expression | [36] | |||
| Pancreatic and lung cancer cells (0–50 µM) | ↓ Cell proliferation
↓ COX-2, EGFR, phospho- ERK1/2 expression | [37] | |||
| HEY ovarian cancer cells (2.5–160 µM) | ↓ Bcl-2, Akt expression
↑ p38 activity | [38] | |||
| ErbB receptors | EGCG | MCF-7 breast cancer cells (5–20 µM) | ↓ ErbB2, ErbB3 phosphorylation
↓ MAPK pathway | [39] | |
| mammary tumor NF639 and SMF cells (0–80 µg/mL) | ↓ Cell proliferation
↓ ErbB2/neu phosphorylation ↓ NF-κB, MAPK pathways | [40] | |||
| HNSCC (10 µg/mL), breast cancer cells (30 µg/mL) | ↓ Cell proliferation
↓ EGFR, STAT3, Akt, c-fos activity | [41,42] | |||
| SW837 colon carcinoma cells (30 µg/mL) | ↓ EGFR, ErbB2 and ErbB3 cellular levels | [43] | |||
| RES | HepG2 liver cancer cells (50–300 µM) | ↓ Cell proliferation
↓ Cyclin D1, Akt, p38 kinase expression ↑ Phospho-ERK1/2 protein levels | [47] | ||
| A431 epidermoid carcinoma cells (0–100 µM) | ↓ Cyclin D1, MEK1, ERK1/2 expression | [48] | |||
| HT-29 colon cancer cells (25 µM) | ↓ JACK-STAT pathway
↓ iNOS, COX-2 expression | [49] | |||
| Quercetin | SKBR3 breast cancer cells (100–200 µM) | ↓ ErbB2 tyrosin kinase activity
↓ PI3K, Akt phosphorylation | [50] | ||
| HepG2 liver cancer cells (50 µM) | ↓ ERK1/2, Akt phosphorylation
↓ NF-κB pathway | [51] | |||
| A549 lung cancer cells (0–58 µM) | ↓ Cell proliferation
↓ Akt-1 activation ↑ ERK-MEK1/2 phosphorylation | [52] | |||
| Apigenin | PC-3, LNCaP prostate cancer cells (5–40 µM) | ↓ Cell proliferation
↑ Proportion of cells in G0/G1–phase ↓ Rb, p38 kinase and c-fos phosphorylation | [53] | ||
| HNSCC cells (6–100 µM) | ↓ Cell proliferation
↓ EGFR, ErbB2 phosphorylation | [55] | |||
| NF-κB | EGCG | A431 epidermoid carcinoma cells (10–40 µg/mL) | ↓ Cell proliferation
↓ NF-κB/p65 nuclear translocation | [57] | |
| Delphinidin | PC-3 prostate cancer cells (30–180 µM) | Athymic (nu/nu) nude mice bearing prostate cancer tumors (2 mg i.p. thrice weekly) | ↓ Tumor growth
↓ IκB kinase γ , IκB-α phosphorylation ↓ NF-κB DNA binding activity | [58,59] | |
| HCT-116 colon cancer cells (30–240 µM) | ↓ Cell proliferation
↓ IκB-α phosphorylation ↓ NF-κB activation | [60] | |||
| Anthocyanin | rats with esophagus tumor (3.8 μmol/g/day p.o.) | ↓ Tumor development
↓ NF-κB, COX-2 expression | [61] | ||
| CAL-27 oral cancer cells (0–500 µg/mL) | ↓ Cell proliferation, metastasis
↓ NF-κB, MMPs expression ↓ MAPK pathway | [62] | |||
| CA, CAPE | HepG2 liver cancer cells (CA 100 µg/mL; CAPE 5 µg/mL) | nude mice injected with HepG2 cells (CA + CAPE 5 mg/kg s.c thrice weekly; CA + CAPE 20 mg/kg/day p.o. for 5 weeks) | ↓ Tumor growth
↓ NF-κB, MMP-9 activity ↓ Liver metastasis | [63] | |
| CUR | Cervical cancer cells (5–60 µM) | ↓ IκB-α phosphorylation
↓ NF-κB activation | [64] | ||
| ICR mice (1–25 µM) | ↓ COX-2 expression
↓ NF-κB activation ↓ NF-κB nuclear translocation ↓ ERK1/2 activity | [65] | |||
| NF-κB | RES | MCF-7 breast cancer cells (50–150 µM) | ↓ Cell proliferation
↓ NF-κB activation ↓ Bcl-2 expression | [66] | |
| OCIM2, OCI/AML3 myeloid leukemia cells (5–75 µM) | ↓ Cell proliferation
↓ NF-κB activation ↑ PARP cleavage ↑ Proportion of cells in S-phase | [67] | |||
| HH/GLI | CUR | medulloblastoma cancer cells (40 µM) | ↓ SHH, GLI1, PTCH1 expression
↑ Proportion of cells in G2/M-phase | [68] | |
| EGCG | SW1353, CRL-7891 chondrosarcoma cells (0–4 µM) | ↓ Cell proliferation
↓ GLI1, PTCH1 expression | [69] | ||
| pancreatic cancer stem cells (20–60 µM) | ↓ Cell proliferation, invasion
↓ SMO, PTCH1, PTCH2, GLI1, GLI2 expression | [70] | |||
| Apigein, baicalein, CUR, RES EGCG, genistein, quercetin | Pancreatic cancer stem cells, prostate cancer cells (20–30 µM) | ↓ GLI1 expression | [71,72] |
3.2. Modulation of the NF-κB Pathway by Polyphenols in Cancer Cells
3.3. Modulation of HH/GLI Pathway by Polyphenols in Cancer Cells
3.4. Cross-Talk between ErbB Receptors and the HH/GLI and NF-κB Signaling Pathways in Cancer Cells
3.5. Other Signal Transduction Pathways Involved in Carcinogenesis
4. Bioavailability of Polyphenols
5. Combinations of Polyphenols: In Vitro and in Vivo Antitumoral Effects
| Treatment | In Vitro Model | In Vivo Model | Antitumoral Effects | Reference |
|---|---|---|---|---|
| Pterostilbene + quercetin (s) | B16M-F10 melanoma cells (40 µM pterostilbene + 20 µM quercetin) | C57BL/6J mice bearing B16M-F10 cells (20 mg/kg/day of each polyphenol i.v.) | ↓ Tumor growth
↓ Metastatic activity ↓ Bcl-2 expression ↑ Mice survival | [105] |
| Thearubigin + genistein (s) | PC-3 prostate cancer cells (0.125–0.5 µg/mL thearubricin + 5–20 µg/mL genistein) | ↓ Cell proliferation
↑ Proportion of cells in G2/M-phase | [106] | |
| Genistein + RES | SV40 rats bearing prostate cancer (83–250 mg/kg/day of each polyphenols p.o) | ↓ Tumor growth
↓ IGF-1 expression | [107] | |
| Quercetin + EGCG (a) | PC-3, LNCaP prostate cancer cells (10–20 µM of each polyphenol) | SCID mice bearing LAPC-4 prostate cancer cells (0.2%–0.4% of each polyphenol/day p.o) | ↓ Tumor growth
↓ AR expression ↓ PI3K/Akt pathway ↑ Bax/Bcl-2 ratio | [108,109] |
| CUR + EGCG (s) | A549, NCI-460NSCLC cells (10–20 µM of each polyphenol) | Lung cancer xenograft node mouse model (20 mg/kg/day of each polyphenol i.p.) | ↓ Tumor growth
↓ Cyclin D1 and B1 levels | [110] |
| MDA-MB-231 breast cancer cells (2–3 µM CUR + 20–25 µM EGCG) | Athymic nude mice implanted with MDA-MB-231 cells (200 mg/kg/day CUR p.o. + 25 mg/kg/day EGCG i.p.) | ↓ Tumor volume
↑ Proportion of cells in G2/M-phase | [111] | |
| Arc + CUR + EGCG (s) | LNCaP prostate cancer cells, MCF-7 breast cancer cells (1 μM Arc + 5–10 μM CUR + 40 μM EGCG) | ↓ Cell proliferation
↑ Proportion of cells inG0/G1-phase ↑ Bax/Bcl-2 ratio ↓ NF-κB, PI3K/Akt, STAT3 expression | [112] | |
| Luteolin + EGCG (s) | HNSCC and lung cancer cells (10 μM luteolin + 30 μM EGCG) | Athymic nude mice implanted with HNSCC and lung cancer cells (125 mg/kg luteolin + 10 mg/kg EGCG p.o. 5 days a week) | ↓ Tumor growth
↑ PARP, caspase-3 cleavage ↑ p53 phosphorylation ↓ Ki-67 expression | [113] |
| Ellagic acid + quercetin; Ellagic acid + RES; quercetin + RES (s) | MOLT-4 leukemia cells (ellagic acid + quercetin 0–40 μM; Ellagic acid + RES, quercetin + RES 0–140 mM) | ↓ Cell proliferation
↑ Caspase-3 activity | [114,115] | |
| RES + CUR | NUB-7, LAN-5, IMR-32, SK-N-BE neuroblastoma cells (0–100 μM CUR + 0–200 μM RES) | ↓ Cell proliferation
↑ p53, Bax, p21 expression | [116] | |
| SJ-RH4, RD/18 rhabdomyosarcoma cells, Saos-2 osteosarcoma cells (6–50 μM of each polyphenol) | ↓ Cell proliferation
↑ Bax/Bcl-2 ratio ↓ ERK phosphorylation | [117] | ||
| CAL-27, SCC-15, FaDu, SALTO HNSCCcells (6–50 μM of each polyphenol) (a) | BALB/c mice implanted with SALTO cells (2 mg of each polyphenol in 50 μL of corn oil p.o. thrice weekly) | ↓ Tumor growth
↑ PARP cleavage ↑ Bax/Bcl-2 ratio ↓ ERK1/2 phosphorylation ↑ LC3 II expression | [118] | |
| HCT-116 colon cancer cells (0–50 μM of each polyphenol) (s) | SCID mice implanted with HCT-116 cells (150 mg/kg/day RES + 500 mg/kg/day CUR p.o. for 3 weeks) | ↓ Tumor growth
↓ NF-κB, EGFR, IGF-1R activity | [120] |
6. Combinations of Polyphenols and Anticancer Drugs: In Vitro and in Vivo Antitumoral Effects
| Treatment | In Vitro Model | In Vivo Model | Antitumoral Effects | Reference |
|---|---|---|---|---|
| CUR + 5-FU | HCT-116 colon cancer cells (5 µM CUR + 0–5 µM 5-FU) | ↓ IC50 of 5-FU
↑ Cytocrome c release ↑ PARP, caspase-3,-8,-9 cleavage ↓ Cyclin D1 expression ↓ NF-κB, PI-3K/Src activity | [122] | |
| CUR + cisplatin | UM-SCC-74B, UM-SCC-29 HNSCC cells (0.3–5 µM CUR + 3–50 µM cisplatin) | ↓ Cell proliferation
↓ STAT3 phosphorylation | [123] | |
| CUR + gemcitabine (a) | P34, Panc-1 pancreatic cancer cells. (10–15 µM CUR + 0.1–0.5 µM gemcitabine) | ↓ Cell proliferation
↓ COX-2 and p-ERK1/2expression | [124] | |
| BxPC-3, MIA PaCa-2, Panc-1 pancreatic cancer cells (10 µM CUR + 50 nM gemcitabine) | Mice bearing pancreatic tumors (1 g/kg/day CUR p.o. + 25 mg/kg gemcitabine i.p. twice weekly) | ↓ Tumor growth
↓ NF-κB activity ↓ Cyclin D1, c-myc, Bcl-2,Bcl-xL, COX-2, MMP, VEGF expression | [125] | |
| CUR + celecoxib (s) | P-34, MIA PaCa, Panc-1 pancreatic cancer cells (15 µM CUR + 25 µM celecoxib) | ↓ Cell proliferation
↓ COX-2 expression | [126] | |
| CUR + RSE + NLE + radiotherapy | BxPC-3, MIA PaCa-2, Panc-1 pancreatic cancer cells (100 nM CUR + 1 µg RSE+ 0.01% NLE + 10 Gy radiotherapy) | ↓ Cell proliferation
↑ Caspase-3,-7 activity ↓ NF-κB activity | [127] | |
| CUR + BCG | MBT-2, 253J-BV, KU-7, RT4V6 bladder cancer cells (0–25 µM CUR + 106 CFU BCG) | Syngeneic mice implanted with MBT-2 cells (1 g/kg/day CUR p.o. + 106 CFU BCG i.t. once weekly) | ↓ Tumor growth
↓ NF-κB activity ↑ TRIAL receptors ↓ Ki-67, CD31, cyclin D1, COX-2, c-myc, Bcl-2, VEGF expression | [128] |
| CUR + paclitaxel | MDA-MB-231breast cancer cells (0.01–10 µM CUR + 0.2–100 µM paclitaxel) | Athymic nude mice implanted with MDA-MB-231 cells (100 mg/kg/day CUR p.o. + 7 mg/kg paclitaxel i.p. weekly) | ↓ Tumor growth
↓ NF-κB activity ↓ MMP-9 expression | [129] |
| RES + CUR + CC | MCF-7, MDA-MB-231 breast cancer cells (10–100 µM RES + 10–30 µM CUR + 10 µM CC) | ↑ Proportion of cells in G0/G1-phase
↑ ROS generation ↑ p53 phosphorylation ↑ Bax/Bcl-2 ratio ↑ Caspase-9 expression | [130] | |
| RES + gemcitabine (s) | ASPC-1, MIA PaCa-2, Panc-1 pancreatic cancer cells (10 µM RES + 100 nM gemcitabine) | Athymic nude mice implanted with MIA PaCa-2 cells (40 mg/kg /day RES p.o. + 25 mg/kg gemcitabine i.p. twice weekly) | ↓ Tumor growth
↓ NF-κB activity ↓ Cyclin D1, Bcl-2, Bcl-xL, COX-2, MMP-9, VEGF, Ki-67, CD31 expression | [131] |
| RES + rapamycin (a) | MCF-7, MDA-MB-231, BT-549 breast cancer cells (10–50 µM RES + 0–10,000 nM rapamycin) | ↓ Cell proliferation
↓ PI3K/Akt pathway | [132] | |
| RES metabolites + SN38 or oxaliplatin (s) | SW480, SW620 colon cancercells (0–60 µM RES + 50 nM SN38 or 500 nM oxaliplatin) | ↓ Cell proliferation
↑ Proportion of cells in S-phase ↑ p53 phosphorylation | [133] | |
| EVOO + trastuzumab (s) | MCF-7, SKBR3 breast cancer cells (50 µM EVOO + 100 µg/mL trastuzumab) | ↓ Cell proliferation
↓ HER-2 expression | [134] | |
| EGCG + tamoxifen or sulindac (s) | PC-9 lung cancer cells (75 µM EGCG + 0–20 µM tamoxifen or 0–200 µM sulindac) | ↓ Cell proliferation
↓ TNF-α release | [135] | |
| EGCG+ celecoxib (s) | PC-9, A549, ChaGo K-1 lung cancer cells (100 µM EGCG + 1–50 µM celecoxib) | ↓ Cell proliferation
↑ GADD153 expression ↑ ERK1/2, p38 phosphorylation | [136] | |
| EGCG + NS38 or celecoxib (s) | LNCaP, PC-3, CWR22Rv1 prostate cancer cells (10–40 µM EGCG + 10 µMNS38) | Athymic nude mice implanted with CWR22Rv1 cells (0.1% EGCG in drinking water/day + 5 mg/kg/day celecoxib i.p. 5 days per week) | ↓ Tumor growth
↑ Bax/Bcl-2 ratio ↑ PARP cleavage ↑ Caspase-3, -9 expression ↓ NF-κB activity ↓ PPAR-γ expression ↓ PSA, IGF-1 serum levels | [137] |
| EGCG + paclitaxel or docetaxel (a) | PC-3ML prostate cancer cells (30 µM EGCG+ 6.25 nM paclitaxel or 3.12 nM docetaxel) | CB17 SCID mice implanted with PC-3ML cells (228 mg/kg/day EGCG + 20 mg/kg paclitaxel i.p.weekly) | ↓ Tumor growth
↑ p53, p73, p21, caspase-3 expression ↑ Mice survival rate ↓ Bone metastasis | [138] |
| EGCG + DOX | IBC-10a, PCa-20a, PC-3ML prostate cancer cells (0–60 µM EGCG + 2 nM or 1–6 µM DOX) | NOD-SCID mice implanted with PC-3ML cells (200 µM EGCG + 2 µM DOX) | ↓ Tumor growth
↑ PARP cleavage ↑ Mice survival rate | [139] |
| ECG + EGCG + DOX | BEL-7404/DOX liver cancer cells (60 mg/mL ECG or 14 mg/mL EGCG + 0.8–2.0 mg/mL DOX) | BALB/c nu/nu mice implanted with BEL-7404/DOX cells (40–160 mg/kg EGCG + 2 mg/kg DOX i.p.) | ↓ Tumor growth
↓ IC50 of DOX ↓ P-glycoprotein expression | [141] |
| EGCG + paclitaxel (s) | 4T1, MCF-7, MDA-MB-231 breast cancer cells (20 µM EGCG + 2 µM paclitaxel) | BALB/c mice implanted with 4T1 cells (30 mg/kg/day EGCG i.p. + 10 mg/kg paclitaxel i.p. thrice weekly) | ↓ Tumor growth
↑ JNK phosphorylation | [142] |
| EGCG + cisplatin | SKOV3, CAOV3, C200 ovarian cancer cells (0–20 µM EGCG + 1–350 µg/mL cisplatin) | ↓ Cell proliferation
↑ H202 levels | [143] | |
| EGCG+ gemcitabine or tasocitinib (s) | AsPC-1, PANC-1 pancreatic cancer cells (0–60 µM EGCG + 0.5 µM gemcitabine or tasocitinib) | ↓ Cell proliferation
↓ STAT3 pathway ↓ Cell migration ↑ PARP and caspase-3 cleavage | [144] | |
| Quercetin + DOX | MCF-7, MDA-231 breast cancer cells (5–10 µM quercetin + 10–100 nM DOX) | ↓ Cell proliferation
↓ DNA and protein synthesis ↓ Cell invasivity | [145] | |
| Quercetin + cisplatin | H520 NSCLC cells (40 µM quercetin + 5 µg/mL cisplatin) | ↑ Apoptotic rate
↑ Bax/Bcl-2 ratio ↑ Caspase-3 activity ↑ Cytochrome c release ↓ Bcl-xL expression | [146] | |
| HeP2 laryngeal cancer cells (40 µM quercetin + 2.5 µg/mL cisplatin) (s) | ↓ Akt phosphorylation
↑ JNK phosphorylation ↑ c-fos expression ↑ Bax/Bcl-2 ratio ↓ Bcl-xL, Ki-67 expression ↑ Cytochrome c release ↑ Caspase-8 ,-9 activity ↑ ROS production ↓ HSP70 activity | [147] | ||
| Genistein + cisplatin | BxPC-3 pancreatic cancer cells (25 µM genistein + 0.5 µM cisplatin) | SCID mice implanted with BxPC-3 cells (800 µg/kg/day genistein p.o. + 9 mg/kg cisplatin/day i.p.) | ↓ Tumor growth
↓ NF-κB activity | [148] |
| Panc-28, COLO-357, L3.6pl pancreatic cancer cells (30 µM genistein + 1–2 µM cisplatin) | SCID mice implanted with COLO-357 cells (1 mg/day genistein p.o. + 9 mg/kg cisplatin i.p.) | ↓ Tumor growth
↓ NF-κB activity ↓ Bcl-2 , Bcl-xL, MMP-9 expression ↑ PARP and caspase-3 cleavage ↓ Akt phosphorylation ↑ Cytochrome c release | [149] | |
| Genistein + gemcitabine | COLO-357, L3.6pl pancreatic cancer cells (25 µM genistein + 25 nM gemcitabine) | SCID mice implanted with COLO-357 and L3.6pl cells (1 mg/day genistein p.o. + 80 mg/kg/day gemcitabine i.v.) | ↓ Tumor growth
↓ NF-κB activity ↑ PARP and caspase-3 cleavage ↑ Cytochrome c release ↓ Bcl-2 , Bcl-xL expression ↓ Akt phosphorylation | [150] |
| Isoflavones + radiotherapy | PC-3 prostate cancer cells (0–15 µM isoflavones + 3 Gy radiotherapy) | Nude mice implanted with PC-3 cells (1 mg/day isoflavones p.o. + 5 Gy radiotherapy) | ↓ Tumor growth
↑ Bax expression ↑ PARP cleavage ↓ Bcl-xL, survivin expression ↓ Metastasis to para-aorticlymph nodes | [151] |
| Cur-NPs | CAL-27-cisplatin-resistent HNSCC cells (0–80 µM) | ↓ Cell proliferation
↑ Bax expression ↑ Caspase-3 ,-9 synthesis ↓ Bcl-2 , MDR1 expression ↑ ROS production | [152] | |
| GLUT1-PEG-PE micelles co-loaded with CUR and DOX | HCT-116 colon cancer cells (7.5–20 µM CUR + 0.1–0.4 µM DOX) | NU/NU nude mice implanted with HCT-116 cells (4 mg/kg/day CUR + 0.4 mg/kg/day DOX i.v.) | ↓ Cell viability
↓ Tumor growth ↑ Mice survival | [153] |
| MPEG-PCL micelles loaded with CUR and DOX (s) | LL/2, MS1 lung cancer cells (0–3 µg/mL CUR and DOX) | C57 mice implanted with LL/2 cells (5mg/kg CUR + 5 mg/kg DOX i.v. every five days) | ↓ Tumor growth
↑ Apoptosis ↓ Angiogenesis | [154] |
| Liposomal CUR + cisplatin | CAL-27, UM-SCC1 HNSCC cells (100 µM CUR + 10–20 µM cisplatin) | Athymic nude mice implanted with HNSCC cells (50 mg/kg CUR i.v. thrice weekly for three weeks + 0.75 µg/mL cisplatin i.p. after 4 weeks) | ↓ Tumor growth
↓ Cyclin D1expression ↓ NF-κB pathway ↑ p53 activity | [155] |
| PLGA-Nano-CUR particles + cisplatin or radiotherapy | cisplatin-resistant A2780CP ovarian cancer cells (2–20 µM CUR + 2.5–40 µM cisplatin; 2–8 µM CUR + 0–4 Gy radiotherapy) | ↓ Cell proliferation
↓ Bcl-xL, Mcl-1 expression ↑ PARP, caspase-3, -7, -9 cleavage ↓ β-Catenin activity | [156] |
7. Combinations of Polyphenols in Clinical Trials
8. Nanotechnology and Polyphenols
9. Perspective and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Manach, C.; Scalbert, A.; Morand, C.; Rémésy, C.; Jiménez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [PubMed]
- Scalbert, A.; Manach, C.; Morand, C.; Rémésy, C.; Jiménez, L. Dietary polyphenols and the prevention of diseases. Crit. Rev. Food Sci. Nutr. 2005, 45, 287–306. [Google Scholar] [CrossRef] [PubMed]
- Benvenuto, M.; Fantini, M.; Masuelli, L.; de Smaele, E.; Zazzeroni, F.; Tresoldi, I.; Calabrese, G.; Galvano, F.; Modesti, A.; Bei, R. Inhibition of ErbB receptors, Hedgehog and NF-kappaB signaling by polyphenols in cancer. Front. Biosci. (Landmark Ed.) 2013, 18, 1290–1310. [Google Scholar] [CrossRef]
- Marzocchella, L.; Fantini, M.; Benvenuto, M.; Masuelli, L.; Tresoldi, I.; Modesti, A.; Bei, R. Dietary flavonoids: Molecular mechanisms of action as anti- inflammatory agents. Recent Pat. Inflamm. Allergy Drug Discov. 2011, 5, 200–220. [Google Scholar] [CrossRef] [PubMed]
- Izzi, V.; Masuelli, L.; Tresoldi, I.; Sacchetti, P.; Modesti, A.; Galvano, F.; Bei, R. The effects of dietary flavonoids on the regulation of redox inflammatory networks. Front. Biosci. 2012, 17, 2396–2418. [Google Scholar] [CrossRef]
- Vallianou, N.G.; Evangelopoulos, A.; Schizas, N.; Kazazis, C. Potential anticancer properties and mechanisms of action of curcumin. Anticancer Res. 2015, 35, 645–651. [Google Scholar] [PubMed]
- Lall, R.K.; Syed, D.N.; Adhami, V.M.; Khan, M.I.; Mukhtar, H. Dietary polyphenols in prevention and treatment of prostate cancer. Int. J. Mol. Sci. 2015, 16, 3350–3376. [Google Scholar] [CrossRef] [PubMed]
- Chiurchiù, V.; Maccarrone, M. Chronic inflammatory disorders and their redox control: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2011, 15, 2605–2641. [Google Scholar] [CrossRef] [PubMed]
- Bertram, J.S. The molecular biology of cancer. Mol. Aspects Med. 2000, 21, 167–223. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Bei, R.; Masuelli, L.; Turriziani, M.; Li Volti, G.; Malaguarnera, M.; Galvano, F. Impaired expression and function of signaling pathway enzymes by anthocyanins: Role on cancer prevention and progression. Curr. Enzym. Inhib. 2009, 5, 184–197. [Google Scholar] [CrossRef]
- Bei, R.; Palumbo, C.; Masuelli, L.; Turriziani, M.; Frajese, G.V.; Li Volti, G.; Malaguarnera, M.; Galvano, F. Impaired expression and function of cancer-related enzymes by anthocyans: An update. Curr. Enzym. Inhib. 2012, 8, 2–21. [Google Scholar] [CrossRef]
- Kunnumakkara, A.B.; Anand, P.; Aggarwal, B.B. Curcumin inhibits proliferation, invasion, angiogenesis and metastasis of different cancers through interaction with multiple cell signaling proteins. Cancer Lett. 2008, 269, 199–225. [Google Scholar] [CrossRef] [PubMed]
- Mohan, A.; Narayanan, S.; Sethuraman, S.; Krishnan, U.M. Combinations of plant polyphenols & anti-cancer molecules: A novel treatment strategy for cancer chemotherapy. Anticancer Agents Med. Chem. 2013, 13, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Crozier, A.; Jaganath, I.B.; Clifford, M.N. Dietary phenolics: Chemistry, bioavailability and effects on health. Nat. Prod. Rep. 2009, 26, 1001–1043. [Google Scholar] [CrossRef] [PubMed]
- Williamson, G.; Manach, C. Bioavailability and bioefficacy of polyphenols in humans. II. Review of 93 intervention studies. Am. J. Clin. Nutr. 2005, 81, 243S–255S. [Google Scholar] [PubMed]
- Di Carlo, G.; Mascolo, N.; Izzo, A.A.; Capasso, F. Flavonoids: Old and new aspects of a class of natural therapeutic drugs. Life Sci. 1999, 65, 337–353. [Google Scholar] [CrossRef] [PubMed]
- Beecher, G.R. Overview of dietary flavonoids: Nomenclature, occurrence and intake. J. Nutr. 2003, 133, 3248S–3254S. [Google Scholar] [PubMed]
- Wang, L.S.; Stoner, G.D. Anthocyanins and their role in cancer prevention. Cancer Lett. 2008, 269, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Tomas-Barberan, F.A.; Clifford, M.N. Flavanones, chalcones and dihydrochalcones-nature, occurrence and dietary burden. J. Food Sci. Agric. 2000, 80, 1073–1080. [Google Scholar] [CrossRef]
- Sakai, T.; Kogiso, M. Soy isoflavones and immunity. J. Med. Investig. 2008, 55, 167–173. [Google Scholar] [CrossRef]
- Cassidy, A.; Hanley, B.; Lamuela-Raventos, R.M. Isoflavones, lignans and stilbenes-origins, metabolism and potential importance to human health. J. Sci. Food Agric. 2000, 80, 1044–1062. [Google Scholar] [CrossRef]
- Bishayee, A. Cancer prevention and treatment with resveratrol: From rodent studies to clinical trials. Cancer Prev. Res. (Phila) 2009, 2, 409–418. [Google Scholar] [CrossRef]
- Goswami, S.K.; Das, D.K. Resveratrol and chemoprevention. Cancer Lett. 2009, 284, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kundu, J.K.; Surh, Y.J. Cancer chemopreventive and therapeutic potential of resveratrol: Mechanistic perspectives. Cancer Lett. 2008, 269, 243–261. [Google Scholar] [CrossRef] [PubMed]
- Rowland, I.; Faughnan, M.; Hoey, L.; Wähälä, K.; Williamson, G.; Cassidy, A. Bioavailability of phyto-oestrogens. Br. J. Nutr. 2003, 89, S45–S58. [Google Scholar] [PubMed]
- Heinonen, S.; Nurmi, T.; Liukkonen, K.; Poutanen, K.; Wähälä, K.; Deyama, T.; Nishibe, S.; Adlercreutz, H. In vitro metabolism of plant lignans: New precursors of mammalian lignans enterolactone and enterodiol. J. Agric. Food Chem. 2001, 49, 3178–3186. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Tyagi, A.K.; Aggarwal, B.B. Recent developments in delivery, bioavailability, absorption and metabolism of curcumin: The golden pigment from golden spice. Cancer Res. Treat. 2014, 46, 2–18. [Google Scholar] [CrossRef] [PubMed]
- Masuelli, L.; Benvenuto, M.; Fantini, M.; Marzocchella, L.; Sacchetti, P.; di Stefano, E.; Tresoldi, I.; Izzi, V.; Bernardini, R.; Palumbo, C.; et al. Curcumin induces apoptosis in breast cancer cell lines and delays the growth of mammary tumors in neu transgenic mice. J. Biol. Regul. Homeost. Agents 2013, 27, 105–119. [Google Scholar]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell. Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- McKay, M.M.; Morrison, D.K. Integrating signals from RTKs to ERK/MAPK. Oncogene 2007, 26, 3113–3321. [Google Scholar] [CrossRef] [PubMed]
- Squires, M.S.; Hudson, E.A.; Howells, L.; Sale, S.; Houghton, C.E.; Jones, J.L.; Fox, L.H.; Dickens, M.; Prigent, S.A.; Manson, M.M. Relevance of mitogen activated protein kinase (MAPK) and phosphotidylinositol-3-kinase/protein kinase B (PI3K/PKB) pathways to induction of apoptosis by curcumin in breast cells. Biochem. Pharmacol. 2003, 65, 361–376. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.D.; Liu, X.E.; Huang, D.S. Curcumin induces apoptosis of triple-negative breast cancer cells by inhibition of EGFR expression. Mol. Med. Rep. 2012, 6, 1267–1270. [Google Scholar] [PubMed]
- Sun, S.H.; Huang, H.C.; Huang, C.; Lin, J.K. Cycle arrest and apoptosis in MDA-MB-231/Her2 cells induced by curcumin. Eur. J. Pharmacol. 2012, 690, 22–30. [Google Scholar] [CrossRef]
- Cai, X.Z.; Wang, J.; Li, X.D.; Wang, G.L.; Liu, F.N.; Cheng, M.S.; Li, F. Curcumin suppresses proliferation and invasion in human gastric cancer cells by downregulation of PAK1 activity and cyclin D1 expression. Cancer Biol. Ther. 2009, 8, 1360–1368. [Google Scholar] [CrossRef] [PubMed]
- Thangapazham, R.L.; Shaheduzzaman, S.; Kim, K.H.; Passi, N.; Tadese, A.; Vahey, M.; Dobi, A.; Srivastava, S.; Maheshwari, R.K. Androgen responsive and refractory prostate cancer cells exhibit distinct curcumin regulated transcriptome. Cancer Biol. Ther. 2008, 7, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Lev-Ari, S.; Starr, A.; Vexler, A.; Karaush, V.; Loew, V.; Greif, J.; Fenig, E.; Aderka, D.; Ben-Yosef, R. Inhibition of pancreatic and lung adenocarcinoma cell survival by curcumin is associated with increased apoptosis, down-regulation of COX-2 and EGFR and inhibition of Erk1/2 activity. Anticancer Res. 2006, 26, 4423–4430. [Google Scholar] [PubMed]
- Watson, J.L.; Greenshields, A.; Hill, R.; Hilchie, A.; Lee, P.W.; Giacomantonio, C.A.; Hoskin, D.W. Curcumin-induced apoptosis in ovarian carcinoma cells is p53-independent and involves p38 mitogen-activated protein kinase activation and downregulation of Bcl-2 and survivin expression and Akt signaling. Mol. Carcinog. 2010, 49, 13–24. [Google Scholar] [PubMed]
- Pan, M.H.; Lin, C.C.; Lin, J.K.; Chen, W.J. Tea polyphenol (−)-epigallocatechin 3-gallate suppresses heregulin-beta1-induced fatty acid synthase expression in human breast cancer cells by inhibiting phosphatidylinositol 3-kinase/Akt and mitogen-activated protein kinase cascade signaling. J. Agric. Food Chem. 2007, 55, 5030–5037. [Google Scholar] [CrossRef] [PubMed]
- Pianetti, S.; Guo, S.; Kavanagh, K.T.; Sonenshein, G.E. Green tea polyphenol epigallocatechin-3 gallate inhibits Her-2/neu signaling, proliferation, and transformed phenotype of breast cancer cells. Cancer Res. 2002, 62, 652–655. [Google Scholar] [PubMed]
- Masuda, M.; Suzui, M.; Lim, J.T.; Deguchi, A.; Soh, J.W.; Weinstein, I.B. Epigallocatechin-3-gallate decreases VEGF production in head and neck and breast carcinoma cells by inhibiting EGFR-related pathways of signal transduction. J. Exp. Ther. Oncol. 2002, 2, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Masuda, M.; Suzui, M.; Lim, J.T.; Weinstein, I.B. Epigallocatechin-3-gallate inhibits activation of HER-2/neu and downstream signaling pathways in human head and neck and breast carcinoma cells. Clin. Cancer Res. 2003, 9, 3486–3491. [Google Scholar] [PubMed]
- Shimizu, M.; Deguchi, A.; Joe, A.K.; Mckoy, J.F.; Moriwaki, H.; Weinstein, I.B. EGCG inhibits activation of HER3 and expression of cyclooxygenase-2 in human colon cancer cells. J. Exp. Ther. Oncol. 2005, 5, 69–78. [Google Scholar] [PubMed]
- Adachi, S.; Nagao, T.; Ingolfsson, H.; Maxfield, F.R.; Andersen, O.S.; Kopelovich, L.; Weinstein, I.B. The inhibitory effect of (−)-epigallocatechin gallate on activation of the epidermal growth factor receptor is associated with altered lipid order in HT29 colon cancer cells. Cancer Res. 2007, 67, 6493–6501. [Google Scholar] [CrossRef] [PubMed]
- Adachi, S.; Nagao, T.; To, S.; Joe, A.K.; Shimizu, M.; Matsushima-Nishiwaki, R.; Kozawa, O.; Moriwaki, H.; Maxfield, F.R.; Weinstein, I.B. (−)-Epigallocatechin gallate causes internalization of the epidermal growth factor receptor in human colon cancer cells. Carcinogenesis 2008, 29, 1986–1993. [Google Scholar] [CrossRef] [PubMed]
- Adachi, S.; Shimizu, M.; Shirakami, Y.; Yamauchi, J.; Natsume, H.; Matsushima-Nishiwaki, R.; To, S.; Weinstein, I.B.; Moriwaki, H.; Kozawa, O. (−)-Epigallocatechin gallate downregulates EGF receptor via phosphorylation at Ser1046/1047 by p38 MAPK in colon cancer cells. Carcinogenesis 2009, 30, 1544–1552. [Google Scholar] [CrossRef] [PubMed]
- Parekh, P.; Motiwale, L.; Naik, N.; Rao, K.V. Downregulation of cyclin D1 is associated with decreased levels of p38 MAP kinases, Akt/PKB and Pak1 during chemopreventive effects of resveratrol in liver cancer cells. Exp. Toxicol. Pathol. 2011, 63, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.L.; Zhu, Y.; Zhu, H.; Han, L.; Kopelovich, L.; Bickers, D.R.; Athar, M. Resveratrol inhibits proliferation of human epidermoid carcinoma A431 cells by modulating MEK1 and AP-1 signalling pathways. Exp. Dermatol. 2006, 15, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Serra, D.; Rufino, A.T.; Mendes, A.F.; Almeida, L.M.; Dinis, T.C. Resveratrol modulates cytokine-induced Jak/STAT activation more efficiently than 5-aminosalicylic acid: An in vitro approach. PLoS ONE 2014, 9, e109048. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.H.; An, J.Y.; Kwon, Y.T.; Li, L.Y.; Lee, Y.J. Quercetin-induced ubiquitination and down-regulation of Her-2/neu. J. Cell. Biochem. 2008, 105, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Granado-Serrano, A.B.; Martín, M.A.; Bravo, L.; Goya, L.; Ramos, S. Quercetin modulates NF-kappa B and AP-1/JNK pathways to induce cell death in human hepatoma cells. Nutr. Cancer 2010, 62, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Tran, E.; Nguyen, T.H.; Do, P.T.; Huynh, T.H.; Huynh, H. The role of activated MEK-ERK pathway in quercetin-induced growth inhibition and apoptosis in A549 lung cancer cells. Carcinogenesis 2004, 25, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Gupta, S. Apigenin-induced cell cycle arrest is mediated by modulation of MAPK, PI3K-Akt, and loss of cyclin D1 associated retinoblastoma dephosphorylation in human prostate cancer cells. Cell Cycle 2007, 6, 1102–1114. [Google Scholar] [CrossRef] [PubMed]
- Long, X.; Fan, M.; Bigsby, R.M.; Nephew, K.P. Apigenin inhibits antiestrogen-resistant breast cancer cell growth through estrogen receptor-alpha-dependent and estrogen receptor-alpha-independent mechanisms. Mol. Cancer Ther. 2008, 7, 2096–2108. [Google Scholar] [CrossRef] [PubMed]
- Masuelli, L.; Marzocchella, L.; Quaranta, A.; Palumbo, C.; Pompa, G.; Izzi, V.; Canini, A.; Modesti, A.; Galvano, F.; Bei, R. Apigenin induces apoptosis and impairs head and neck carcinomas EGFR/ErbB2 signaling. Front. Biosci. (Landmark Ed.) 2011, 16, 1060–1068. [Google Scholar] [CrossRef]
- Lee, W.S.; Yi, S.M.; Yun, J.W.; Jung, J.H.; Kim, D.H.; Kim, H.J.; Chang, S.H.; Kim, G.; Ryu, C.H.; Shin, S.C.; et al. Polyphenols isolated from Allium cepa L. induces apoptosis by induction of p53 and suppression of Bcl-2 through inhibiting PI3K/Akt signaling pathway in AGS human cancer cells. J. Cancer Prev. 2014, 19, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Hastak, K.; Afaq, F.; Ahmad, N.; Mukhtar, H. Essential role of caspases in epigallocatechin-3-gallate-mediated inhibition of nuclear factor kappa B and induction of apoptosis. Oncogene 2004, 23, 2507–2522. [Google Scholar] [CrossRef] [PubMed]
- Hafeez, B.B.; Siddiqui, I.A.; Asim, M.; Malik, A.; Afaq, F.; Adhami, V.M.; Saleem, M.; Din, M.; Mukhtar, H. A dietary anthocyanidin delphinidin induces apoptosis of human prostate cancer PC3 cells in vitro and in vivo: Involvement of nuclear factor-kappaB signaling. Cancer Res. 2008, 68, 8564–8572. [Google Scholar] [CrossRef] [PubMed]
- Hafeez, B.B.; Asim, M.; Siddiqui, I.A.; Adhami, V.M.; Murtaza, I.; Mukhtar, H. Delphinidin, a dietary anthocyanidin in pigmented fruits and vegetables: A new weapon to blunt prostate cancer growth. Cell Cycle 2008, 7, 3320–3326. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.M.; Afaq, F.; Khan, N.; Mukhtar, H. Delphinidin, an anthocyanidin in pigmented fruits and vegetables, induces apoptosis and cell cycle arrest in human colon cancer HCT116 cells. Mol. Carcinog. 2009, 48, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.S.; Hecht, S.S.; Carmella, S.G.; Yu, N.; Larue, B.; Henry, C.; McIntyre, C.; Rocha, C.; Lechner, J.F.; Stoner, G.D. Anthocyanins in black raspberries prevent esophageal tumors in rats. Cancer Prev. Res. (Phila) 2009, 2, 84–93. [Google Scholar] [CrossRef]
- Fan, M.J.; Wang, I.C.; Hsiao, Y.T.; Lin, H.Y.; Tang, N.Y.; Hung, T.C.; Quan, C.; Lien, J.C.; Chung, J.G. Anthocyanins from black rice (Oryza sativa L.) demonstrate antimetastatic properties by reducing MMPs and NF-κB expressions in human oral cancer CAL 27 cells. Nutr. Cancer 2015, 67, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Chung, T.W.; Moon, S.K.; Chang, Y.C.; Ko, J.H.; Lee, Y.C.; Cho, G.; Kim, S.H.; Kim, J.G.; Kim, C.H. Novel and therapeutic effect of caffeic acid and caffeic acid phenyl ester on hepatocarcinoma cells: Complete regression of hepatoma growth and metastasis by dual mechanism. FASEB J. 2004, 18, 1670–1681. [Google Scholar] [CrossRef] [PubMed]
- Divya, C.S.; Pillai, M.R. Antitumor action of curcumin in human papillomavirus associated cells involves downregulation of viral oncogenes, prevention of NFkB and AP-1 translocation, and modulation of apoptosis. Mol. Carcinog. 2006, 45, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Chun, K.S.; Keum, Y.S.; Han, S.S.; Song, Y.S.; Kim, S.H.; Surh, Y.J. Curcumin inhibits phorbol ester-induced expression of cyclooxygenase-2 in mouse skin through suppression of extracellular signal-regulated kinase activity and NF-kappaB activation. Carcinogenesis 2003, 24, 1515–1524. [Google Scholar] [CrossRef] [PubMed]
- Pozo-Guisado, E.; Merino, J.M.; Mulero-Navarro, S.; Lorenzo-Benayas, M.J.; Centeno, F.; Alvarez-Barrientos, A.; Fernandez-Salguero, P.M. Resveratrol-induced apoptosis in MCF-7 human breast cancer cells involves a caspase-independent mechanism with downregulation of Bcl-2 and NF-kappaB. Int. J. Cancer 2005, 115, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Estrov, Z.; Shishodia, S.; Faderl, S.; Harris, D.; Van, Q.; Kantarjian, H.M.; Talpaz, M.; Aggarwal, B.B. Resveratrol blocks interleukin-1beta-induced activation of the nuclear transcription factor NF-kappaB, inhibits proliferation, causes S-phase arrest, and induces apoptosis of acute myeloid leukemia cells. Blood 2003, 102, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Elamin, M.H.; Shinwari, Z.; Hendrayani, S.F.; Al-Hindi, H.; al-Shail, E.; Khafaga, Y.; al-Kofide, A.; Aboussekhra, A. Curcumin inhibits the Sonic Hedgehog signaling pathway and triggers apoptosis in medulloblastoma cells. Mol. Carcinog. 2010, 49, 302–314. [Google Scholar] [PubMed]
- Tang, G.Q.; Yan, T.Q.; Guo, W.; Ren, T.T.; Peng, C.L.; Zhao, H.; Lu, X.C.; Zhao, F.L.; Han, X. (−)-Epigallocatechin-3-gallate induces apoptosis and suppresses proliferation by inhibiting the human Indian Hedgehog pathway in human chondrosarcoma cells. J. Cancer Res. Clin. Oncol. 2010, 136, 1179–1185. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.N.; Fu, J.; Nall, D.; Rodova, M.; Shankar, S.; Srivastava, R.K. Inhibition of sonic hedgehog pathway and pluripotency maintaining factors regulate human pancreatic cancer stem cell characteristics. Int. J. Cancer 2012, 131, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, L.; Jiao, M.; Wu, D.; Wu, K.; Li, X.; Zhu, G.; Yang, L.; Wang, X.; Hsieh, J.T.; et al. Genistein inhibits the stemness properties of prostate cancer cells through targeting Hedgehog-Gli1 pathway. Cancer Lett. 2012, 323, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Slusarz, A.; Shenouda, N.S.; Sakla, M.S.; Drenkhahn, S.K.; Narula, A.S.; MacDonald, R.S.; Besch-Williford, C.L.; Lubahn, D.B. Common botanical compounds inhibit the hedgehog signaling pathway in prostate cancer. Cancer Res. 2010, 70, 3382–3390. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, M.D.; Harrison, S.C. Structure of an IkappaBalpha/NF-kappaB complex. Cell 1998, 95, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Naugler, W.E.; Karin, M. NF-kappaB and cancer-identifying targets and mechanisms. Curr. Opin. Genet. Dev. 2008, 18, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, F.H.; Li, Y.; Wang, Z.; Kong, D. NF-kappaB signaling pathway and its therapeutic implications in human diseases. Int. Rev. Immunol. 2008, 27, 293–319. [Google Scholar] [CrossRef] [PubMed]
- Bei, R.; Masuelli, L.; Palumbo, C.; Modesti, M.; Modesti, A. A common repertoire of autoantibodies is shared by cancer and autoimmune disease patients: Inflammation in their induction and impact on tumor growth. Cancer Lett. 2009, 281, 8–23. [Google Scholar] [CrossRef] [PubMed]
- Sethi, G.; Sung, B.; Aggarwal, B.B. Nuclear factor-kappaB activation: From bench to bedside. Exp. Biol. Med. (Maywood) 2008, 233, 21–31. [Google Scholar] [CrossRef]
- Mimeault, M.; Johansson, S.L.; Henichart, J.P.; Depreux, P.; Batra, S.K. Cytotoxic effects induced by docetaxel, gefitinib, and cyclopamine on side population and nonside population cell fractions from human invasive prostate cancer cells. Mol. Cancer Ther. 2010, 9, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Mangelberger, D.; Kern, D.; Loipetzberger, A.; Eberl, M.; Aberger, F. Cooperative Hedgehog-EGFR signaling. Front. Biosci. (Landmark Ed.) 2012, 17, 90–99. [Google Scholar] [CrossRef]
- Kubo, M.; Nakamura, M.; Tasaki, A.; Yamanaka, N.; Nakashima, H.; Nomura, M.; Kuroki, S.; Katano, M. Hedgehog signaling pathway is a new therapeutic target for patients with breast cancer. Cancer Res. 2004, 64, 6071–6074. [Google Scholar] [CrossRef] [PubMed]
- Stecca, B.; Mas, C.; Clement, V.; Zbinden, M.; Correa, R.; Piguet, V.; Beermann, F.; Ruiz, I.; Altaba, A. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 5895–5900. [Google Scholar] [CrossRef] [PubMed]
- Riobó, N.A.; Haines, G.M.; Emerson, C.P., Jr. Protein kinase C-delta and mitogen-activated protein/extracellular signal-regulated kinase-1 control GLI activation in hedgehog signaling. Cancer Res. 2006, 66, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Riobó, N.A.; Lu, K.; Ai, X.; Haines, G.M.; Emerson, C.P., Jr. Phosphoinositide 3-kinase and Akt are essential for Sonic Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 4505–4510. [Google Scholar] [CrossRef] [PubMed]
- Schnidar, H.; Eberl, M.; Klingler, S.; Mangelberger, D.; Kasper, M.; Hauser-Kronberger, C.; Regl, G.; Kroismayr, R.; Moriggl, R.; Sibilia, M.; et al. Epidermal growth factor receptor signaling synergizes with Hedgehog/GLI in oncogenic transformation via activation of the MEK/ERK/JUN pathway. Cancer Res. 2009, 69, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Seto, M.; Ohta, M.; Asaoka, Y.; Ikenoue, T.; Tada, M.; Miyabayashi, K.; Mohri, D.; Tanaka, Y.; Ijichi, H.; Tateishi, K.; et al. Regulation of the hedgehog signaling by the mitogen-activated protein kinase cascade in gastric cancer. Mol. Carcinog. 2009, 48, 703–712. [Google Scholar]
- Merkhofer, E.C.; Cogswell, P.; Baldwin, A.S. Her2 activates NF-kappaB and induces invasion through the canonical pathway involving IKKalpha. Oncogene 2010, 29, 1238–1248. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Ju, X.; Willmarth, N.E.; Casimiro, M.C.; Ojeifo, J.; Sakamaki, T.; Katiyar, S.; Jiao, X.; Popov, V.M.; Yu, Z.; et al. Nuclear factor-kappaB enhances ErbB2-induced mammary tumorigenesis and neoangiogenesis in vivo. Am. J. Pathol. 2009, 174, 1910–1920. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Luo, J.L.; Karin, M. IkappaB kinase alpha kinase activity is required for self-renewal of ErbB2/Her2-transformed mammary tumor-initiating cells. Proc. Natl. Acad. Sci. USA 2007, 104, 15852–15857. [Google Scholar] [CrossRef] [PubMed]
- Makino, K.; Day, C.P.; Wang, S.C.; Li, Y.M.; Hung, M.C. Upregulation of IKKalpha/IKKbeta by integrin-linked kinase is required for HER2/neu-induced NF-kappaB antiapoptotic pathway. Oncogene 2004, 23, 3883–3887. [Google Scholar] [CrossRef] [PubMed]
- Elledge, S.J. Cell cycle checkpoints: Preventing an identity crisis. Science 1996, 274, 1664–1672. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.W.; Ruley, H.E.; Jacks, T.; Housman, D.E. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell 1993, 74, 957–867. [Google Scholar] [CrossRef] [PubMed]
- Aas, T.; Børresen, A.L.; Geisler, S.; Smith-Sørensen, B.; Johnsen, H.; Varhaug, J.E.; Akslen, L.A.; Lønning, P.E. Specific P53 mutations are associated with de novo resistance to doxorubicin in breast cancer patients. Nat. Med. 1996, 2, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Børresen-Dale, A.L.; Johnsen, H.; Aas, T.; Geisler, J.; Akslen, L.A.; Anker, G.; Lønning, P.E. TP53 gene mutations predict the response to neoadjuvant treatment with 5-fluorouracil and mitomycin in locally advanced breast cancer. Clin. Cancer Res. 2003, 9, 5582–5588. [Google Scholar] [PubMed]
- Plati, J.; Bucur, O.; Khosravi-Far, R. Apoptotic cell signaling in cancer progression and therapy. Integr. Biol. (Camb.) 2011, 3, 279–296. [Google Scholar] [CrossRef]
- Cavallaro, U.; Christofori, G. Molecular mechanisms of tumor angiogenesis and tumor progression. J. Neurooncol. 2000, 50, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Hahnfeldt, P.; Panigrahy, D.; Folkman, J.; Hlatky, L. Tumor development under angiogenic signaling: A dynamical theory of tumor growth, treatment response, and postvascular dormancy. Cancer Res. 1999, 59, 4770–4775. [Google Scholar] [PubMed]
- Alesiani, D.; Cicconi, R.; Mattei, M.; Montesano, C.; Bei, R.; Canini, A. Cell cycle arrest and differentiation induction by 5,7-dimethoxycoumarin in melanoma cell lines. Int. J. Oncol. 2008, 32, 425–434. [Google Scholar] [PubMed]
- Alesiani, D.; Cicconi, R.; Mattei, M.; Bei, R.; Canini, A. Inhibition of Mek 1/2 kinase activity and stimulation of melanogenesis by 5,7-dimethoxycoumarin treatment of melanoma cells. Int. J. Oncol. 2009, 34, 1727–1735. [Google Scholar] [PubMed]
- Renis, M.; Calandra, L.; Scifo, C.; Tomasello, B.; Cardile, V.; Vanella, L.; Bei, R.; la Fauci, L.; Galvano, F. Response of cell cycle/stress-related protein expression and DNA damage upon treatment of CaCo2 cells with anthocyanins. Br. J. Nutr. 2008, 100, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Manach, C.; Williamson, G.; Morand, C.; Scalbert, A.; Rémésy, C. Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. Am. J. Clin. Nutr. 2005, 81, 230S–242S. [Google Scholar] [PubMed]
- Scalbert, A.; Williamson, G. Dietary intake and bioavailability of polyphenols. J. Nutr. 2000, 130, 2073S–2085S. [Google Scholar] [PubMed]
- Ginsburg, I.; Kohen, R.; Koren, E. Microbial and host cells acquire enhanced oxidant-scavenging abilities by binding polyphenols. Arch. Biochem. Biophys. 2011, 506, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Bohn, T. Dietary factors affecting polyphenol bioavailability. Nutr. Rev. 2014, 72, 429–452. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, P.; Asensi, M.; Segarra, R.; Ortega, A.; Benlloch, M.; Obrador, E.; Varea, M.T.; Asensio, G.; Jordá, L.; Estrela, J.M. Association between pterostilbene and quercetin inhibits metastatic activity of B16 melanoma. Neoplasia 2005, 7, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K. Synergistic effects of thearubigin and genistein on human prostate tumor cell (PC-3) growth via cell cycle arrest. Cancer Lett. 2000, 151, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Harper, C.E.; Cook, L.M.; Patel, B.B.; Wang, J.; Eltoum, I.A.; Arabshahi, A.; Shirai, T.; Lamartiniere, C.A. Genistein and resveratrol, alone and in combination, suppress prostate cancer in SV-40 tag rats. Prostate 2009, 69, 1668–1682. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Heber, D.; Henning, S.M. Quercetin increased the antiproliferative activity of green tea polyphenol (−)-epigallocatechin gallate in prostate cancer cells. Nutr. Cancer 2012, 64, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Vadgama, J.V.; Said, J.W.; Magyar, C.E.; Doan, N.; Heber, D.; Henning, S.M. Enhanced inhibition of prostate cancer xenograft tumor growth by combining quercetin and green tea. J. Nutr. Biochem. 2014, 25, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.H.; Wang, X.; Yang, M.; Shi, X.; Huang, W.; Feng, Q. Combination of low concentration of (−)-epigallocatechin gallate (EGCG) and curcumin strongly suppresses the growth of non-small cell lung cancer in vitro and in vivo through causing cell cycle arrest. Int. J. Mol. Sci. 2013, 14, 12023–12036. [Google Scholar] [CrossRef] [PubMed]
- Somers-Edgar, T.J.; Scandlyn, M.J.; Stuart, E.C.; le Nedelec, M.J.; Valentine, S.P.; Rosengren, R.J. The combination of epigallocatechin gallate and curcumin suppresses ER alpha-breast cancer cell growth in vitro and in vivo. Int. J. Cancer 2008, 122, 1966–1971. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wang, B.; Chung, S.; Wu, Y.; Henning, S.M.; Vadgama, J.V. Increased chemopreventive effect by combining arctigenin, green tea polyphenol and curcumin in prostate and breast cancer cells. RSC Adv. 2014, 4, 35242–35250. [Google Scholar] [CrossRef] [PubMed]
- Amin, A.R.; Wang, D.; Zhang, H.; Peng, S.; Shin, H.J.; Brandes, J.C.; Tighiouart, M.; Khuri, F.R.; Chen, Z.G.; Shin, D.M. Enhanced anti-tumor activity by the combination of the natural compounds (−)-epigallocatechin-3-gallate and luteolin: Potential role of p53. J. Biol. Chem. 2010, 285, 34557–34665. [Google Scholar] [CrossRef] [PubMed]
- Mertens-Talcott, S.U.; Talcott, S.T.; Percival, S.S. Low concentrations of quercetin and ellagic acid synergistically influence proliferation, cytotoxicity and apoptosis in MOLT-4 human leukemia cells. J. Nutr. 2003, 133, 2669–2774. [Google Scholar] [PubMed]
- Mertens-Talcott, S.U.; Percival, S.S. Ellagic acid and quercetin interact synergistically with resveratrol in the induction of apoptosis and cause transient cell cycle arrest in human leukemia cells. Cancer Lett. 2005, 218, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Liontas, A.; Yeger, H. Curcumin and resveratrol induce apoptosis and nuclear translocation and activation of p53 in human neuroblastoma. Anticancer Res. 2004, 24, 987–998. [Google Scholar] [PubMed]
- Masuelli, L.; Marzocchella, L.; Focaccetti, C.; Tresoldi, I.; Palumbo, C.; Izzi, V.; Benvenuto, M.; Fantini, M.; Lista, F.; Tarantino, U.; et al. Resveratrol and diallyl disulfide enhance curcumin-induced sarcoma cell apoptosis. Front. Biosci. (Landmark Ed.) 2012, 17, 498–508. [Google Scholar] [CrossRef]
- Masuelli, L.; di Stefano, E.; Fantini, M.; Mattera, R.; Benvenuto, M.; Marzocchella, L.; Sacchetti, P.; Focaccetti, C.; Bernardini, R.; Tresoldi, I.; et al. Resveratrol potentiates the in vitro and in vivo anti-tumoral effects of curcumin in head and neck carcinomas. Oncotarget 2014, 5, 10745–10762. [Google Scholar] [PubMed]
- Elattar, T.M.; Virji, A.S. The effect of red wine and its components on growth and proliferation of human oral squamous carcinoma cells. Anticancer Res. 1999, 19, 5407–5414. [Google Scholar] [PubMed]
- Majumdar, A.P.; Banerjee, S.; Nautiyal, J.; Patel, B.B.; Patel, V.; Du, J.; Yu, Y.; Elliott, A.A.; Levi, E.; Sarkar, F.H. Curcumin synergizes with resveratrol to inhibit colon cancer. Nutr. Cancer 2009, 61, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.M.; Fong, D. Overcoming drug resistance by phytochemicals. In Drug Resistance in Cancer Cells; Metha, K., Bates, S.E., Siddik, Z.H., Eds.; Springer Science+Business Media, LLC: New York, NY, USA, 2009; pp. 315–342. [Google Scholar]
- Shakibaei, M.; Mobasheri, A.; Lueders, C.; Busch, F.; Shayan, P.; Goel, A. Curcumin enhances the effect of chemotherapy against colorectal cancer cells by inhibition of NF-κB and Src protein kinase signaling pathways. PLoS ONE 2013, 8, e57218. [Google Scholar] [CrossRef] [PubMed]
- Abuzeid, W.M.; Davis, S.; Tang, A.L.; Saunders, L.; Brenner, J.C.; Lin, J.; Fuchs, J.R.; Light, E.; Bradford, C.R.; Prince, M.E.; et al. Sensitization of head and neck cancer to cisplatin through the use of a novel curcumin analog. Arch. Otolaryngol. Head Neck Surg. 2011, 137, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Lev-Ari, S.; Vexler, A.; Starr, A.; Ashkenazy-Voghera, M.; Greif, J.; Aderka, D.; Ben-Yosef, R. Curcumin augments gemcitabine cytotoxic effect on pancreatic adenocarcinoma cell lines. Cancer Investig. 2007, 25, 411–418. [Google Scholar] [CrossRef]
- Kunnumakkara, A.B.; Guha, S.; Krishnan, S.; Diagaradjane, P.; Gelovani, J.; Aggarwal, B.B. Curcumin potentiates antitumor activity of gemcitabine in an orthotopic model of pancreatic cancer through suppression of proliferation, angiogenesis, and inhibition of nuclear factor-kappaB-regulated gene products. Cancer Res. 2007, 67, 3853–3861. [Google Scholar] [CrossRef] [PubMed]
- Lev-Ari, S.; Zinger, H.; Kazanov, D.; Yona, D.; Ben-Yosef, R.; Starr, A.; Figer, A.; Arber, N. Curcumin synergistically potentiates the growth inhibitory and pro-apoptotic effects of celecoxib in pancreatic adenocarcinoma cells. Biomed. Pharmacother. 2005, 59, S276–S280. [Google Scholar] [CrossRef] [PubMed]
- Veeraraghavan, J.; Natarajan, M.; Lagisetty, P.; Awasthi, V.; Herman, T.S.; Aravindan, N. Impact of curcumin, raspberry extract, and neem leaf extract on rel protein-regulated cell death/radiosensitization in pancreatic cancer cells. Pancreas 2011, 40, 1107–1119. [Google Scholar] [CrossRef] [PubMed]
- Kamat, A.M.; Tharakan, S.T.; Sung, B.; Aggarwal, B.B. Curcumin potentiates the antitumor effects of Bacillus Calmette-Guerin against bladder cancer through the downregulation of NF-kappaB and upregulation of TRAIL receptors. Cancer Res. 2009, 69, 8958–8966. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.J.; Lee, S.H.; Price, J.E.; Kim, L.S. Curcumin suppresses the paclitaxel-induced nuclear factor-kappaB in breast cancer cells and potentiates the growth inhibitory effect of paclitaxel in a breast cancer nude mice model. Breast J. 2009, 15, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Zaidi, D.; Shyam, H.; Sharma, R.; Balapure, A.K. Polyphenols sensitization potentiates susceptibility of MCF-7 and MDA MB-231 cells to Centchroman. PLoS ONE 2012, 7, e37736. [Google Scholar] [CrossRef] [PubMed]
- Harikumar, K.B.; Kunnumakkara, A.B.; Sethi, G.; Diagaradjane, P.; Anand, P.; Pandey, M.K.; Gelovani, J.; Krishnan, S.; Guha, S.; Aggarwal, B.B. Resveratrol, a multitargeted agent, can enhance antitumor activity of gemcitabine in vitro and in orthotopic mouse model of human pancreatic cancer. Int. J. Cancer 2010, 127, 257–268. [Google Scholar] [PubMed]
- He, X.; Wang, Y.; Zhu, J.; Orloff, M.; Eng, C. Resveratrol enhances the anti-tumor activity of the mTOR inhibitor rapamycin in multiple breast cancer cell lines mainly by suppressing rapamycin-induced AKT signaling. Cancer Lett. 2011, 301, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Aires, V.; Limagne, E.; Cotte, A.K.; Latruffe, N.; Ghiringhelli, F.; Delmas, D. Resveratrol metabolites inhibit human metastatic colon cancer cells progression and synergize with chemotherapeutic drugs to induce cell death. Mol. Nutr. Food Res. 2013, 57, 1170–1181. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Vazquez-Martin, A.; Colomer, R.; Brunet, J.; Carrasco-Pancorbo, A.; Garcia-Villalba, R.; Fernandez-Gutierrez, A.; Segura-Carretero, A. Olive oil’s bitter principle reverses acquired autoresistance to trastuzumab (Herceptin) in HER2-overexpressing breast cancer cells. BMC Cancer 2007, 7, 80. [Google Scholar] [CrossRef] [PubMed]
- Suganuma, M.; Okabe, S.; Kai, Y.; Sueoka, N.; Sueoka, E.; Fujiki, H. Synergistic effects of (−)-epigallocatechin gallate with (−)-epicatechin, sulindac, or tamoxifen on cancer-preventive activity in the human lung cancer cell line PC-9. Cancer Res. 1999, 59, 44–47. [Google Scholar] [PubMed]
- Suganuma, M.; Kurusu, M.; Suzuki, K.; Tasaki, E.; Fujiki, H. Green tea polyphenol stimulates cancer preventive effects of celecoxib in human lung cancer cells by upregulation of GADD153 gene. Int. J. Cancer 2006, 119, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Adhami, V.M.; Malik, A.; Zaman, N.; Sarfaraz, S.; Siddiqui, I.A.; Syed, D.N.; Afaq, F.; Pasha, F.S.; Saleem, M.; Mukhtar, H. Combined inhibitory effects of green tea polyphenols and selective cyclooxygenase-2 inhibitors on the growth of human prostate cancer cells both in vitro and in vivo. Clin. Cancer Res. 2007, 13, 1611–1619. [Google Scholar] [CrossRef] [PubMed]
- Stearns, M.E.; Wang, M. Synergistic effects of the green tea extract epigallocatechin-3-gallate and taxane in eradication of malignant human prostate tumors. Transl. Oncol. 2011, 4, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Stearns, M.E.; Amatangelo, M.D.; Varma, D.; Sell, C.; Goodyear, S.M. Combination therapy with epigallocatechin-3-gallate and doxorubicin in human prostate tumor modeling studies: Inhibition of metastatic tumor growth in severe combined immunodeficiency mice. Am. J. Pathol. 2010, 177, 3169–3179. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ye, H.L.; Zhang, G.; Yao, W.M.; Chen, X.Z.; Zhang, F.C.; Liang, G. Autophagy inhibition contributes to the synergistic interaction between EGCG and doxorubicin to kill the hepatoma Hep3B cells. PLoS ONE 2014, 9, e85771. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Tang, A.; Lin, X.; Li, L.; Zhang, S.; Huang, Z.; Tang, H.; Li, Q.Q. Green tea catechins augment the antitumor activity of doxorubicin in an in vivo mouse model for chemoresistant liver cancer. Int. J. Oncol. 2010, 37, 111–123. [Google Scholar] [PubMed]
- Luo, T.; Wang, J.; Yin, Y.; Hua, H.; Jing, J.; Sun, X.; Li, M.; Zhang, Y.; Jiang, Y. (−)-Epigallocatechin gallate sensitizes breast cancer cells to paclitaxel in a murine model of breast carcinoma. Breast Cancer Res. 2010, 12, R8. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.M.; Soprano, K.J.; Weinstein, K.; Fong, D. Epigallocatechin-3-gallate delivers hydrogen peroxide to induce death of ovarian cancer cells and enhances their cisplatin susceptibility. J. Cell Physiol. 2006, 207, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.N.; Fu, J.; Shankar, S.; Srivastava, R.K. EGCG enhances the therapeutic potential of gemcitabine and CP690550 by inhibiting STAT3 signaling pathway in human pancreatic cancer. PLoS ONE 2012, 7, e31067. [Google Scholar] [CrossRef] [PubMed]
- Staedler, D.; Idrizi, E.; Kenzaoui, B.H.; Juillerat-Jeanneret, L. Drug combinations with quercetin: Doxorubicin plus quercetin in human breast cancer cells. Cancer Chemother. Pharmacol. 2011, 68, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Kuhar, M.; Sen, S.; Singh, N. Role of mitochondria in quercetin-enhanced chemotherapeutic response in human non-small cell lung carcinoma H-520 cells. Anticancer Res. 2006, 26, 1297–1303. [Google Scholar] [PubMed]
- Sharma, H.; Sen, S.; Singh, N. Molecular pathways in the chemosensitization of cisplatin by quercetin in human head and neck cancer. Cancer Biol. Ther. 2005, 4, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, R.M.; Banerjee, S.; Li, Y.; Aboukameel, A.; Kucuk, O.; Sarkar, F.H. Cisplatin-induced antitumor activity is potentiated by the soy isoflavone genistein in BxPC-3 pancreatic tumor xenografts. Cancer 2006, 106, 1260–1268. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Zhang, Y.; Wang, Z.; Che, M.; Chiao, P.J.; Abbruzzese, J.L.; Sarkar, F.H. In vitro and in vivo molecular evidence of genistein action in augmenting the efficacy of cisplatin in pancreatic cancer. Int. J. Cancer 2007, 120, 906–917. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Zhang, Y.; Ali, S.; Bhuiyan, M.; Wang, Z.; Chiao, P.J.; Philip, P.A.; Abbruzzese, J.; Sarkar, F.H. Molecular evidence for increased antitumor activity of gemcitabine by genistein in vitro and in vivo using an orthotopic model of pancreatic cancer. Cancer Res. 2005, 65, 9064–9072. [Google Scholar] [CrossRef] [PubMed]
- Raffoul, J.J.; Banerjee, S.; Che, M.; Knoll, Z.E.; Doerge, D.R.; Abrams, J.; Kucuk, O.; Sarkar, F.H.; Hillman, G.G. Soy isoflavones enhance radiotherapy in a metastatic prostate cancer model. Int. J. Cancer 2007, 120, 2491–2498. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.Y.; Peng, S.F.; Lee, C.Y.; Lu, C.C.; Tsai, S.C.; Shieh, T.M.; Wu, T.S.; Tu, M.G.; Chen, M.Y.; Yang, J.S. Curcumin-loaded nanoparticles induce apoptotic cell death through regulation of the function of MDR1 and reactive oxygen species in cisplatin-resistant CAR human oral cancer cells. Int. J. Oncol. 2013, 43, 1141–1150. [Google Scholar] [PubMed]
- Abouzeid, A.H.; Patel, N.R.; Rachman, I.M.; Senn, S.; Torchilin, V.P. Anti-cancer activity of anti-GLUT1 antibody-targeted polymeric micelles co-loaded with curcumin and doxorubicin. J. Drug Target. 2013, 21, 994–1000. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.L.; Shen, Y.M.; Zhang, Q.W.; Li, Y.L.; Luo, M.; Liu, Z.; Li, Y.; Qian, Z.Y.; Gao, X.; Shi, H.S. Codelivery of curcumin and doxorubicin by MPEG-PCL results in improved efficacy of systemically administered chemotherapy in mice with lung cancer. Int. J. Nanomed. 2013, 8, 3521–3531. [Google Scholar]
- Duarte, V.M.; Han, E.; Veena, M.S.; Salvado, A.; Suh, J.D.; Liang, L.J.; Faull, K.F.; Srivatsan, E.S.; Wang, M.B. Curcumin enhances the effect of cisplatin in suppression of head and neck squamous cell carcinoma via inhibition of IKKbeta protein of the NFkappaB pathway. Mol. Cancer Ther. 2010, 9, 2665–2675. [Google Scholar] [CrossRef] [PubMed]
- Yallapu, M.M.; Maher, D.M.; Sundram, V.; Bell, M.C.; Jaggi, M.; Chauhan, S.C. Curcumin induces chemo/radio-sensitization in ovarian cancer cells and curcumin nanoparticles inhibit ovarian cancer cell growth. J. Ovarian Res. 2010, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Spagnuolo, C.; Tedesco, I.; Russo, G.L. Phytochemicals in cancer prevention and therapy: Truth or dare? Toxins (Basel) 2010, 2, 517–551. [Google Scholar] [CrossRef]
- Saldanha, S.N.; Tollefsbol, T.O. The role of nutraceuticals in chemoprevention and chemotherapy and their clinical outcomes. J. Oncol. 2012, 2012, 192464. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, J.; Taskeen, M.; Mohammad, I.; Huo, C.; Chan, T.H.; Dou, Q.P. Recent advances on tea polyphenols. Front. Biosci. (Elite Ed.) 2012, 4, 111–131. [Google Scholar] [CrossRef]
- Gupta, S.C.; Patchva, S.; Aggarwal, B.B. Therapeutic roles of curcumin: Lessons learned from clinical trials. AAPS J. 2013, 15, 195–218. [Google Scholar] [CrossRef] [PubMed]
- Bayet-Robert, M.; Kwiatkowski, F.; Leheurteur, M.; Gachon, F.; Planchat, E.; Abrial, C.; Mouret-Reynier, M.A.; Durando, X.; Barthomeuf, C.; Chollet, P. Phase I dose escalation trial of docetaxel plus curcumin in patients with advanced and metastatic breast cancer. Cancer Biol. Ther. 2010, 9, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Wang, Y.; Zhang, Y.; Wan, X.; Li, J.; Liu, K.; Wang, F.; Liu, K.; Liu, Q.; Yang, C.; et al. Anti-cancer activities of tea epigallocatechin-3-gallate in breast cancer patients under radiotherapy. Curr. Mol. Med. 2012, 12, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Ulbricht, C.E.; Chao, W. Phytochemicals in the oncology setting. Curr. Treat. Opt. Oncol. 2010, 11, 95–106. [Google Scholar] [CrossRef]
- Tabrez, S.; Priyadarshini, M.; Urooj, M.; Shakil, S.; Ashraf, G.M.; Khan, M.S.; Kamal, M.A.; Alam, Q.; Jabir, N.R.; Abuzenadah, A.M.; et al. Cancer chemoprevention by polyphenols and their potential application as nanomedicine. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2013, 31, 67–98. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fantini, M.; Benvenuto, M.; Masuelli, L.; Frajese, G.V.; Tresoldi, I.; Modesti, A.; Bei, R. In Vitro and in Vivo Antitumoral Effects of Combinations of Polyphenols, or Polyphenols and Anticancer Drugs: Perspectives on Cancer Treatment. Int. J. Mol. Sci. 2015, 16, 9236-9282. https://doi.org/10.3390/ijms16059236
Fantini M, Benvenuto M, Masuelli L, Frajese GV, Tresoldi I, Modesti A, Bei R. In Vitro and in Vivo Antitumoral Effects of Combinations of Polyphenols, or Polyphenols and Anticancer Drugs: Perspectives on Cancer Treatment. International Journal of Molecular Sciences. 2015; 16(5):9236-9282. https://doi.org/10.3390/ijms16059236
Chicago/Turabian StyleFantini, Massimo, Monica Benvenuto, Laura Masuelli, Giovanni Vanni Frajese, Ilaria Tresoldi, Andrea Modesti, and Roberto Bei. 2015. "In Vitro and in Vivo Antitumoral Effects of Combinations of Polyphenols, or Polyphenols and Anticancer Drugs: Perspectives on Cancer Treatment" International Journal of Molecular Sciences 16, no. 5: 9236-9282. https://doi.org/10.3390/ijms16059236