
The Incremental Induction of Neuroprotective Properties by Multiple Therapeutic Strategies for Primary and Secondary Neural Injury


Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Risk Factors for Neural Injury
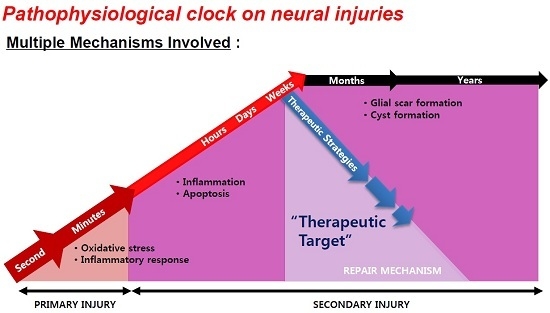
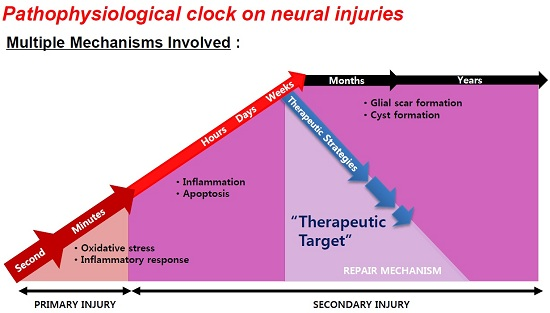
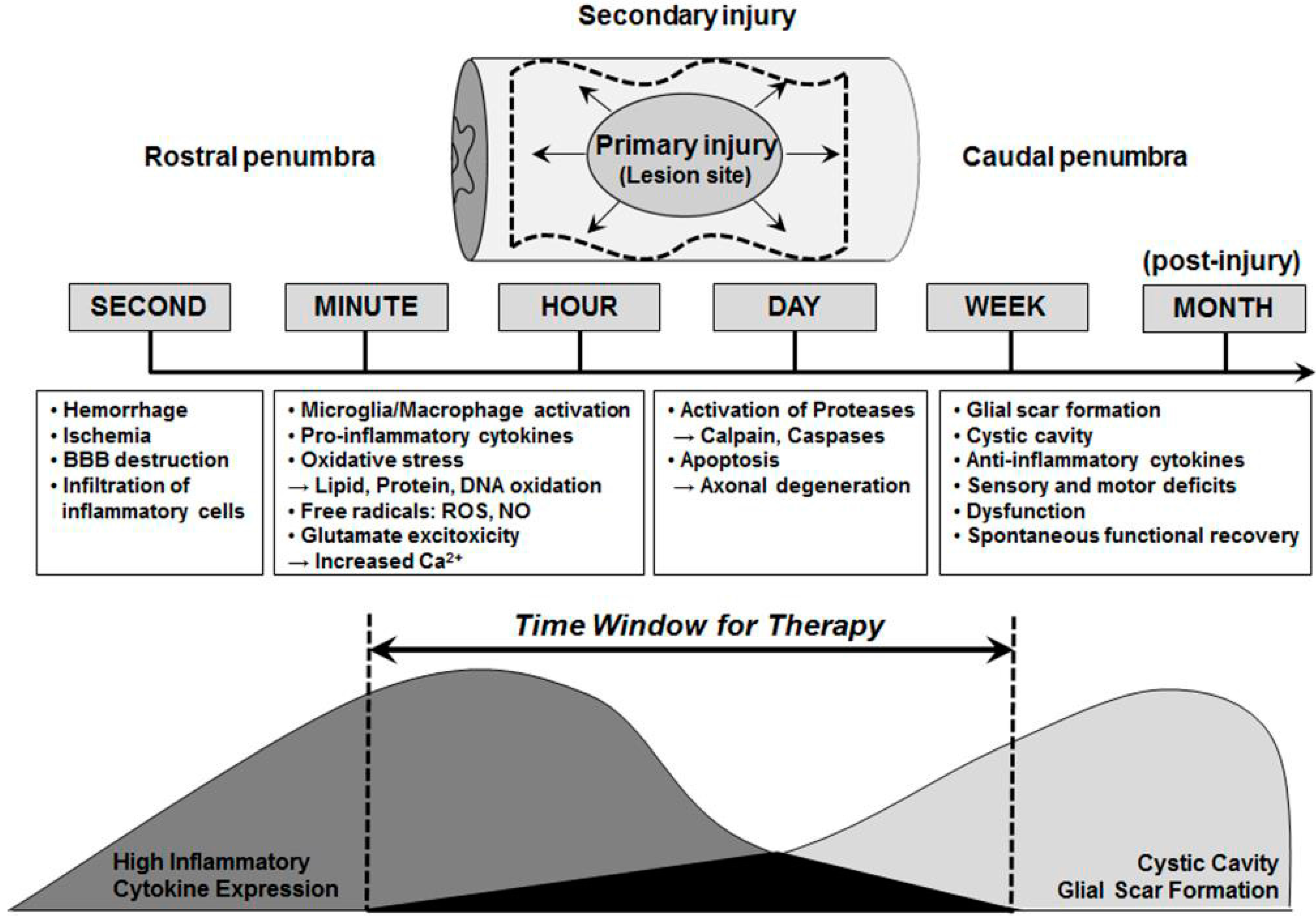
2. Window for Multi-Active Therapies: Primary and Secondary Injury

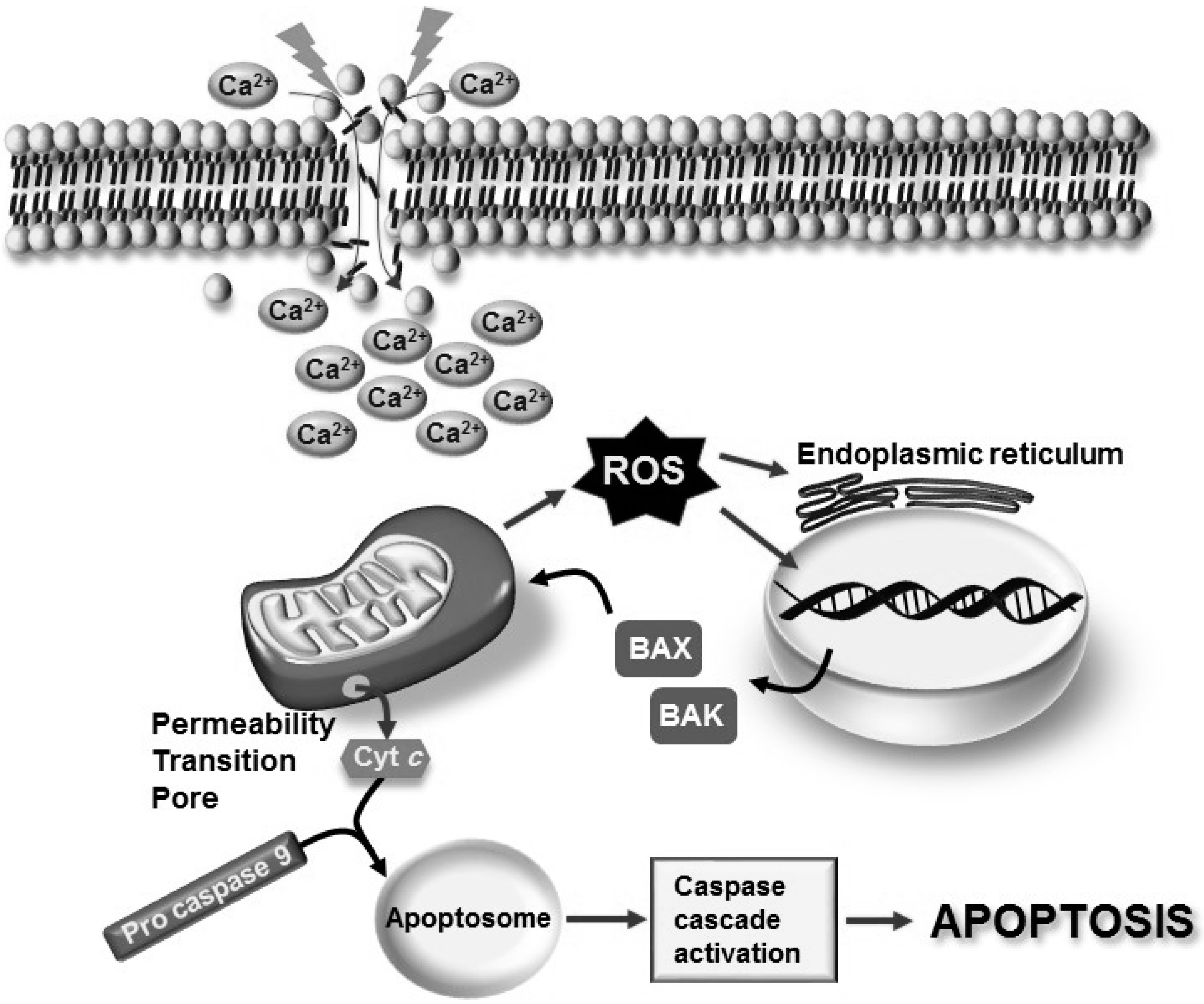
3. Brain Aging and Neurodegeneration

4. Role of Melatonin in Age-Related Neural Degeneration
5. Endogenous Factors Induced by Forced Exercise
Acknowledgments
Conflicts of Interest
References
- Rubinsztein, D.C. The roles of intracellular protein-degradation pathways in neuro-degeneration. Nature 2006, 443, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Bredesen, D.E.; Rao, R.V.; Mehlen, P. Cell death in the nervous system. Nature 2006, 443, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Bossy-Wetzel, E.; Schwarzenbacher, R.; Lipton, S.A. Molecular pathways to neuro-degeneration. Nat. Med. 2004, 10, S2–S9. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.J.; West, A.B.; Dawson, V.L.; Dawson, T.M. Molecular pathophysiology of Parkinson’s disease. Annu. Rev. Neurosci. 2005, 28, 57–87. [Google Scholar] [CrossRef] [PubMed]
- Niranjan, R. Molecular Basis of etiological implications in Alzheimer’s Disease: Focus on neuroinflammation. Mol. Neurobiol. 2013, 48, 412–428. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Park, S.; Krause, J.S.; Banik, N.L. Oxidative stress, DNA damage, and the telomeric complex as therapeutic targets in acute neurodegeneration. Neurochem. Int. 2013, 62, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Zhao, Z.; Goto, S.; Koltai, E. Age-associated neurodegeneration and oxidative damage to lipids, proteins and DNA. Mol. Asp. Med. 2011, 32, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Rajdev, S.; Hara, K.; Kokubo, Y.; Mestril, R.; Dillmann, W.; Weinstein, P.R.; Sharp, F.R. Mice overexpressing rat heat shock protein 70 are protected against cerebral infarction. Ann. Neurol. 2000, 47, 782–791. [Google Scholar] [CrossRef]
- Hu, B.R.; Martone, M.E.; Jones, Y.Z.; Liu, C.L. Protein aggregation after transient cerebral ischemia. J. Neurosci. 2000, 20, 3191–3199. [Google Scholar] [PubMed]
- Xu, J.; Kao, S.Y.; Lee, F.J.; Song, W.; Jin, L.W.; Yankner, B.A. Dopamine-dependent neurotoxicity of α-synuclein: A mechanism for selective neurodegeneration in Parkinson disease. Nat. Med. 2002, 8, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; et al. Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. 2001, 21, 3017–3023. [Google Scholar] [PubMed]
- Sipione, S.; Cattaneo, E. Modeling Huntington’s disease in cells, flies, and mice. Mol. Neurobiol. 2001, 23, 21–51. [Google Scholar] [PubMed]
- D’Amico, E.; Factor-Litvak, P.; Santella, R.M.; Mitsumoto, H. Clinical perspective on oxidative stress in sporadic amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2013, 65, 509–527. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.C.; Lucke-Wold, B.; Lucke-Wold, N.; Elliott, A.S.; Logsdon, A.F.; Rosen, C.L.; Huber, J.D. Neuroprotection for ischemic stroke: moving past shortcomings and identifying promising directions. Int. J. Mol. Sci. 2013, 14, 1890–1917. [Google Scholar] [CrossRef] [PubMed]
- Köhrmann, M.; Schellinger, P.D.; Schwab, S. The only evidence based neuroprotective therapy for acute ischemic stroke: Thrombolysis. Best. Pract. Res. Clin. Anaesthesiol. 2010, 24, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Amedei, A.; Prisco, D.; D’Elios, M.M. Multiple sclerosis: The role of cytokines in pathogenesis and in therapies. Int. J. Mol. Sci. 2012, 13, 13438–13460. [Google Scholar] [CrossRef] [PubMed]
- Khademi, M.; Dring, A.M.; Gilthorpe, J.D.; Wuolikainen, A.; Al Nimer, F.; Harris, R.A.; Andersson, M.; Brundin, L.; Piehl, F.; Olsson, T.; et al. Intense inflammation and nerve damage in early multiple sclerosis subsides at older age: A reflection by cerebrospinal fluid biomarkers. PLoS ONE 2013, 8, e63172. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Nozaki, K.; Guyton, M.K.; Smith, J.A.; Ray, S.K.; Banik, N.L. Calpain inhibition attenuated morphological and molecular changes in skeletal muscle of experimental allergic encephalomyelitis rats. J. Neurosci. Res. 2012, 90, 2134–2145. [Google Scholar] [CrossRef] [PubMed]
- Shields, D.C.; Tyor, W.R.; Deibler, G.E.; Hogan, E.L.; Banik, N.L. Increased calpain expression in activated glial and inflammatory cells in experimental allergic encephalomyelitis. Proc. Natl. Acad. Sci. USA. 1998, 95, 5768–5772. [Google Scholar] [CrossRef] [PubMed]
- Honorat, J.A.; Kinoshita, M.; Okuno, T.; Takata, K.; Koda, T.; Tada, S.; Shirakura, T.; Fujimura, H.; Mochizuki, H.; Sakoda, S.; et al. Xanthine oxidase mediates axonal and myelin loss in a murine model of multiple sclerosis. PLoS ONE 2013, 8, e71329. [Google Scholar] [CrossRef] [PubMed]
- Meraz-Ríos, M.A.; Toral-Rios, D.; Franco-Bocanegra, D.; Villeda-Hernández, J.; Campos-Peña, V. Inflammatory process in Alzheimer’s Disease. Front. Integr. Neurosci. 2013, 7. [Google Scholar] [CrossRef] [PubMed]
- More, S.V.; Kumar, H.; Kim, I.S.; Song, S.Y.; Choi, D.K. Cellular and molecular mediators of neuroinflammation in the pathogenesis of Parkinson’s disease. Mediat. Inflamm. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Kumar, A.; Kulkarni, S.K. Targeting oxidative stress, mitochondrial dysfunction and neuroinflammatory signaling by selective cyclooxygenase (COX)-2 inhibitors mitigates MPTP-induced neurotoxicity in mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Beattie, M.S.; Farooqui, A.A.; Bresnahan, J.C. Review of current evidence for apoptosis after spinal cord injury. J. Neurotrauma 2000, 17, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Beattie, M.S.; Hermann, G.E.; Rogers, R.C.; Bresnahan, J.C. Cell death in models of spinal cord injury. Prog. Brain Res. 2002, 137, 37–47. [Google Scholar] [PubMed]
- Pan, J.Z.; Ni, L.; Sodhi, A.; Aguanno, A.; Young, W.; Hart, R.P. Cytokine activity contributes to induction of inflammatory cytokine mRNAs in spinal cord following contusion. J. Neurosci. Res. 2002, 68, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Sribnick, E.A.; Wingrave, J.M.; Matzelle, D.D.; Ray, S.K.; Banik, N.L. Estrogen as a neuroprotective agent in the treatment of spinal cord injury. Ann. N. Y. Acad. Sci. 2003, 993, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Paterniti, I.; Esposito, E.; Mazzon, E.; Bramanti, P.; Cuzzocrea, S. Evidence for the role of PI3-kinase-AKT-eNOS signalling pathway in secondary inflammatory process after spinal cord compression injury in mice. Eur. J. Neurosci. 2011, 33, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Okano, H. Stem cell biology of the central nervous system. J. Neurosci. Res. 2002, 69, 698–707. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, T.; Kaneko, N.; Zukin, R.S.; Castillo, P.E.; Etgen, A.M. Estradiol attenuates ischemia-induced death of hippocampal neurons and enhances synaptic transmission in aged, long-term hormone-deprived female rats. PLoS ONE 2012, 7, e38018. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Lee, S.K.; Park, K.; Lee, Y.; Hong, Y.; Lee, S.; Jeon, J.C.; Kim, J.H.; Lee, S.R.; Chang, K.T.; et al. Beneficial effects of endogenous and exogenous melatonin on neural reconstruction and functional recovery in an animal model of spinal cord injury. J. Pineal Res. 2012, 52, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Lee, Y.; Park, S.; Lee, S.; Hong, Y.; Lee, S.K.; Hong, Y. Synergistic effect of melatonin on exercise-induced neuronal reconstruction and functional recovery in a spinal cord injury animal model. J. Pineal Res. 2010, 48, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Prokai-Tatrai, K.; Perjesi, P.; Rivera-Portalatin, N.M.; Simpkins, J.W.; Prokai, L. Mechanistic investigations on the antioxidant action of a neuroprotective estrogen derivative. Steroids 2008, 73, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Losordo, D.W.; Isner, J.M. Estrogen and angiogenesis: A review. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.J.; Yuan, L.; Yang, D.; Han, W.N.; Li, Q.S.; Yang, W.; Liu, Q.S.; Qi, J.S. Melatonin protects against amyloid-β-induced impairments of hippocampal LTP and spatial learning in rats. Synapse 2013, 67, 626–636. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Dong, W.; Huang, F. Anti-amyloidogenic and anti-apoptotic role of melatonin in Alzheimer disease. Curr. Neuropharmacol. 2010, 8, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, S.; Benedusi, V.; Maggi, A.; Vegeto, E. Estrogen action in neuroprotection and brain inflammation. Ann. N. Y. Acad. Sci. 2006, 1089, 302–323. [Google Scholar] [CrossRef] [PubMed]
- Sheth, D.S.; Tajuddin, N.F.; Druse, M.J. Antioxidant neuroprotection against ethanol-induced apoptosis in HN2-5 cells. Brain Res. 2009, 1285, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Sotthibundhu, A.; Phansuwan-Pujito, P.; Govitrapong, P. Melatonin increases proliferation of cultured neural stem cells obtained from adult mouse subventricular zone. J. Pineal Res. 2010, 49, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, H.; Ii, M.; Jujo, K.; Thorne, T.; Ito, A.; Klyachko, E.; Hamada, H.; Kessler, J.A.; Tabata, Y.; Kawana, M.; et al. Estradiol promotes neural stem cell differentiation into endothelial lineage and angiogenesis in injured peripheral nerve. Angiogenesis 2013, 16, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Duan, W. Sirtuins: From metabolic regulation to brain aging. Front. Aging Neurosci. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Scahill, R.I.; Frost, C.; Jenkins, R.; Whitwell, J.L.; Rossor, M.N.; Fox, N.C. A longitudinal study of brain volume changes in normal aging using serial registered magnetic resonance imaging. Arch. Neurol. 2003, 60, 989–994. [Google Scholar] [CrossRef] [PubMed]
- Peters, R. Ageing and the brain. Postgrad. Med. J. 2006, 82, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Camins, A.; Sureda, F.X.; Junyent, F.; Verdaguer, E.; Folch, J.; Beas-Zarate, C.; Pallas, M. An overview of investigational antiapoptotic drugs with potential application for the treatment of neurodegenerative disorders. Expert Opin. Investig. Drugs 2010, 19, 587–604. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Blomgren, K.; Kroemer, G. Mitochondrial membrane permeabilization in neuronal injury. Nat. Rev. Neurosci. 2009, 10, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Ribe, E.M.; Serrano-Saiz, E.; Akpan, N.; Troy, C.M. Mechanisms of neuronal death in disease: Defining the models and the players. Biochem. J. 2008, 415, 165–182. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.S.; Lee, H.G.; Xiongwei, Z.; Perry, G.; Smith, M.A.; Castellani, R.J. Current approaches in the treatment of Alzheimer’s disease. Biomed. Pharmacother. 2008, 62, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.; Wei, Q.; Feng, L.; Dong, G.; Tao, Y.; Mei, L.; Xie, Z.J.; Dong, Z. Bak regulates mitochondrial morphology and pathology during apoptosis by interacting with mitofusins. Proc. Natl. Acad. Sci. USA 2007, 104, 11649–11654. [Google Scholar] [CrossRef] [PubMed]
- Caroppi, P.; Sinibaldi, F.; Fiorucci, L.; Santucci, R. Apoptosis and human disease: Mitochondrion damage and lethal role of released cytochrome C as proapoptotic protein. Curr. Med. Chem. 2009, 16, 4058–4065. [Google Scholar] [CrossRef] [PubMed]
- Skinner, D.C.; Malpaux, B. High melatonin concentrations in third ventricular cerebrospinal fluid are not due to Galen vein blood recirculating through the choroid plexus. Endocrinology 1999, 140, 4399–4405. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Maestroni, G.J.M. Melatonin in relation to the antioxidative defense and immune systems: Possible implications for cell and organ transplantation. J. Mol. Med. 1999, 77, 36–39. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.X.; Qi, W.B.; Manchester, L.C.; Karbownik, M.; Calvo, J.R. Pharmacology and physiology of melatonin in the reduction of oxidative stress in vivo. Biol. Signals Recept. 2000, 9, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Palaksha, K.J.; Park, K.; Park, S.; Kim, H.D.; Reiter, R.J.; Chang, K.T. Melatonin plus exercise-based neurorehabilitative therapy for spinal cord injury. J. Pineal Res. 2010, 49, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Reiter, R.J.; Manchester, L.C.; Yan, M.T.; El-Sawi, M.; Sainz, R.M.; Mayo, J.C.; Kohen, R.; Allegra, M.; Hardeland, R. Chemical and physical properties and potential mechanisms: Melatonin as a broad spectrum antioxidant and free radical scavenger. Curr. Top. Med. Chem. 2002, 2, 181–197. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.X.; Osuna, C.; Gitto, E. Actions of melatonin in the reduction of oxidative stress: A review. J. Biomed. Sci. 2000, 7, 444–458. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.X.; Gitto, E.; Sainz, R.M.; Mayo, J.C.; Leon, J.; Manchester, L.C.; Vijayalaxmi; Kilic, E.; Kilic, U. Pharmacological utility of melatonin in reducing oxidative cellular and molecular damage. Pol. J. Pharmacol. 2004, 56, 159–170. [Google Scholar] [PubMed]
- Rodriguez, C.; Mayo, J.C.; Sainz, R.M.; Antolín, I.; Herrera, F.; Martín, V.; Reiter, R.J. Regulation of antioxidant enzymes: a significant role for melatonin. J. Pineal Res. 2004, 36, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Chen, L.D.; Poeggeler, B.; Manchester, L.C.; Reiter, R.J. Melatonin: A potent, endogenous hydroxyl radical scavenger. Endocr. J. 1993, 1, 57–60. [Google Scholar]
- Allegra, M.; Reiter, R.J.; Tan, D.X.; Gentile, C.; Tesoriere, L.; Livrea, M.A. The chemistry of melatonins interaction with reactive species. J. Pineal Res. 2003, 34, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Cuzzocrea, S.; Reiter, R.J. Pharmacological actions of melatonin in acute and chronic inflammation. Curr. Top. Med. Chem. 2002, 2, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, J.M.; Reiter, R.J. Melatonin-immune system relationships. Curr. Top. Med. Chem. 2002, 2, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Shinozuka, K.; Staples, M.; Borlongan, C.V. Melatonin-based therapeutics for neuroprotection in stroke. Int. J. Mol. Sci. 2013, 14, 8924–8947. [Google Scholar] [CrossRef] [PubMed]
- Menendez-Pelaez, A.; Reiter, R.J. Distribution of melatonin in mammalian tissues: The relative importance of nuclear versus cytosolic localization. J. Pineal Res. 1993, 15, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Kuwana, T.; Newmeyer, D.D. Bcl-2 family proteins and the role of mitochondria in apoptosis. Curr. Opin. Cell Biol. 2003, 15, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Borner, C. The Bcl-2 protein family: sensors and checkpoints for life-or-death decisions. Mol. Immunol. 2003, 39, 615–647. [Google Scholar] [CrossRef]
- Juknat, A.A.; Méndez Mdel, V.; Quaglino, A.; Fameli, C.I.; Mena, M.; Kotler, M.L. Melatonin prevents hydrogen peroxide-induced Bax expression in cultured rat astrocytes. J. Pineal Res. 2005, 38, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Andrabi, S.A.; Sayeed, I.; Siemen, D.; Wolf, G.; Horn, T.F. Direct inhibition of the mitochondrial permeability transition pore: A possible mechanism responsible for anti-apoptotic effects of melatonin. FASEB J. 2004, 18, 869–871. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Huang, Q.X.; Yang, S.S.; Chu, J.; Wang, J.Z.; Tian, Q. Melatonin in Alzheimer’s disease. Int. J. Mol. Sci. 2013, 14, 14575–14593. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.Q.; Xu, G.G.; Duan, P.; Zhang, Q.; Wang, J.Z. Effects of melatonin on wortmannin-induced tau hyperphosphorylation. Acta Pharmacol. Sin. 2005, 26, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Li, X.C.; Wang, Z.F.; Zhang, J.X.; Wang, Q.; Wang, J.Z. Effect of melatonin on calyculin A-induced tau hyperphosphorylation. Eur. J. Pharmacol. 2005, 510, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Li, S.P.; Deng, Y.Q.; Wang, X.C.; Wang, Y.P.; Wang, J.Z. Melatonin protects SH-SY5Y neuroblastoma cells from calyculin A-induced neurofilament impairment and neurotoxicity. J. Pineal Res. 2004, 36, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, Y.; Fu, Z.; Li, Y.; Feng, J.; Luo, J.; Zhang, Q.; Wang, Q.; Tian, Q. Melatonin ameliorates Alzheimer-like pathological changes and spatial memory retention impairment induced by calyculin A. J. Psychopharmacol. 2011, 25, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.P.; Li, X.T.; Liu, S.J.; Zhou, X.W.; Wang, X.C.; Wang, J.Z. Melatonin ameliorated okadaic-acid induced Alzheimer-like lesions. Acta Pharmacol. Sin. 2004, 25, 276–280. [Google Scholar] [PubMed]
- Radák, Z.; Ihasz, F.; Koltai, E.; Goto, S.; Taylor, A.W.; Boldogh, I. The redox-associated adaptive response of brain to physical exercise. Free Radic. Res. 2014, 48, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Radák, Z.; Hart, N.; Sarga, L.; Koltai, E.; Atalay, M.; Ohno, H.; Boldogh, I. Exercise plays a preventive role against Alzheimer’s disease. J. Alzheimers Dis. 2010, 20, 777–783. [Google Scholar] [PubMed]
- Rothman, S.M.; Mattson, M.P. Activity-dependent, stress-responsive BDNF signaling and the quest for optimal brain health and resilience throughout the lifespan. Neuroscience 2013, 239, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Radák, Z.; Zhao, Z.; Koltai, E.; Ohno, H.; Atalay, M. Oxygen consumption and usage during physical exercise: the balance between oxidative stress and ROS-dependent adaptive signaling. Antioxid. Redox Signal. 2013, 18, 1208–1246. [Google Scholar] [CrossRef] [PubMed]
- Radák, Z.; Chung, H.Y.; Goto, S. Exercise and hormesis: oxidative stress-related adaptation for successful aging. Biogerontology 2005, 6, 278–294. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Magnus, T. Ageing and neuronal vulnerability. Nat. Rev. Neurosci. 2006, 7, 278–294. [Google Scholar] [CrossRef] [PubMed]
- Cotman, C.W.; Berchtold, N.C. Exercise: A behavioral intervention to enhance brain health and plasticity. Trends. Neurosci. 2002, 25, 295–301. [Google Scholar] [CrossRef]
- Cotman, C.W.; Engesser-Cesar, C. Exercise enhances and protects brain function. Exerc. Sport Sci. Rev. 2002, 30, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.; Iigo, M.; Ohtani-Kaneko, R.; Nakamura, N.; Suzuki, T.; Reiter, R.J.; Hirata, K. Administration of melatonin and related indoles prevents exercise-induced cellular oxidative changes in rats. Biol. Signals 1997, 6, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Kishi, T.; Hirooka, Y.; Katsuki, M.; Ogawa, K.; Shinohara, K.; Isegawa, K.; Sunagawa, K. Exercise training causes sympathoinhibition through antioxidant effect in the rostral ventrolateral medulla of hypertensive rats. Clin. Exp. Hypertens. 2012, 34, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Zaggar, M.; Dao, A.; Alhaider, I.; Alkadhi, K. Regular treadmill exercise prevents sleep deprivation-induced disruption of synaptic plasticity and associated signaling cascade in the dentate gyrus. Mol. Cell. Neurosci. 2013, 56, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Kaneko, T.; Tahara, S.; Nakamoto, H.; Pucsok, J.; Sasvari, M.; Nyakas, C.; Goto, S. Regular exercise improves cognitive function and decreases oxidative damage in rat brain. Neurochem. Int. 2001, 38, 17–23. [Google Scholar] [CrossRef]
- Ogonovszky, H.; Berkes, I.; Kumagai, S.; Kaneko, T.; Tahara, S.; Goto, S.; Radák, Z. The effects of moderate-, strenuous- and over-training on oxidative stress markers, DNA repair, and memory, in rat brain. Neurochem. Int. 2005, 46, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Van Praag, H.; Kempermann, G.; Gage, F.H. Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nat. Neurosci. 1999, 2, 266–270. [Google Scholar] [PubMed]
- Marosi, K.; Felszeghy, K.; Mehra, R.D.; Radak, Z.; Nyakas, C. Are the neuroprotective effects of estradiol and physical exercise comparable during ageing in female rats? Biogerontology 2012, 13, 413–427. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Maudsley, S.; Martin, B. A neural signaling triumvirate that influences ageing and age-related disease: Insulin/IGF-1, BDNF and serotonin. Ageing Res. Rev. 2004, 3, 445–464. [Google Scholar] [CrossRef] [PubMed]
- Anlar, B.; Sullivan, K.A.; Feldman, E.L. Insulin-like growth factor-1 and central nervous system development. Horm. Metab. Res. 1999, 32, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Elfving, B.; Christensen, T.; Ratner, C.; Wienecke, J.; Klein, A.B. Transient activation of mTOR following forced treadmill exercise in rats. Synapse 2013, 67, 620–625. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.; Park, S.; Won, J.; Lee, S.-R.; Chang, K.-T.; Hong, Y. The Incremental Induction of Neuroprotective Properties by Multiple Therapeutic Strategies for Primary and Secondary Neural Injury. Int. J. Mol. Sci. 2015, 16, 19657-19670. https://doi.org/10.3390/ijms160819657
Lee S, Park S, Won J, Lee S-R, Chang K-T, Hong Y. The Incremental Induction of Neuroprotective Properties by Multiple Therapeutic Strategies for Primary and Secondary Neural Injury. International Journal of Molecular Sciences. 2015; 16(8):19657-19670. https://doi.org/10.3390/ijms160819657
Chicago/Turabian StyleLee, Seunghoon, Sookyoung Park, Jinyoung Won, Sang-Rae Lee, Kyu-Tae Chang, and Yonggeun Hong. 2015. "The Incremental Induction of Neuroprotective Properties by Multiple Therapeutic Strategies for Primary and Secondary Neural Injury" International Journal of Molecular Sciences 16, no. 8: 19657-19670. https://doi.org/10.3390/ijms160819657