
Retinal Cell Degeneration in Animal Models

 ,
,  ,
,
Abstract
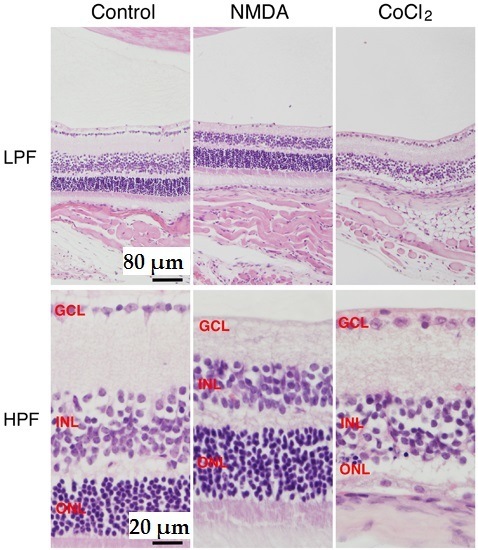
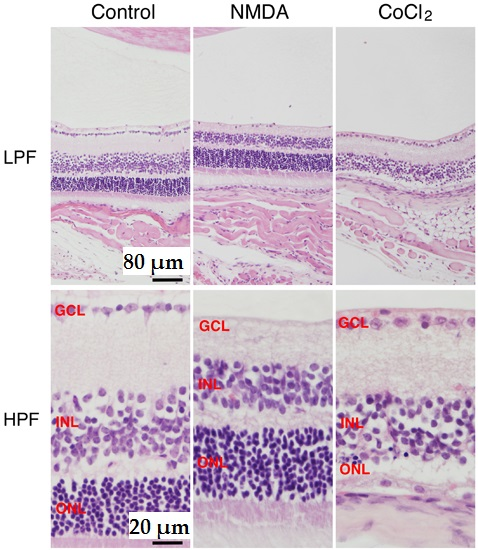
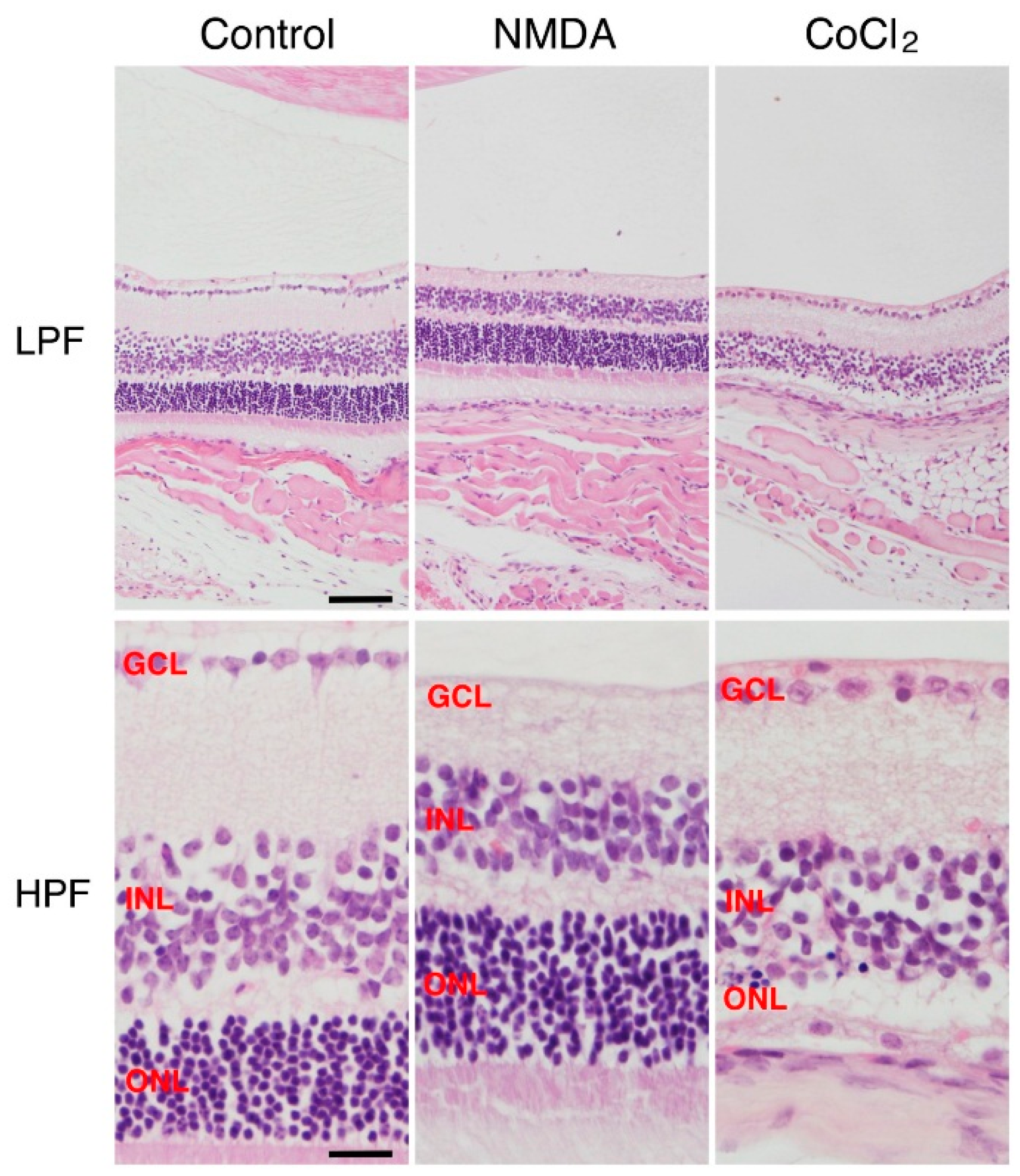
:
1. Introduction
2. NMDA-Induced Retinal Ganglion Cell Degeneration
3. CoCl2-Induced Retinal Photoreceptor Cell Degeneration

4. Demyelination and Retinal Cell Degeneration of Experimental Autoimmune Encephalomyelitis (EAE) as a Model for Multiple Sclerosis
5. Retinal Cell Degeneration Induced by Optic Nerve Crush Injury
6. Transient Ischemia-Induced Retinal Cell Degeneration
7. Light-Induced Retinal Cell Degeneration
8. Conclusions

{kind=link}
{kind=link}
| Target Region | Pathology | Proposed Mechanism | Reference | |
|---|---|---|---|---|
| NMDA-induced retinal cell degeneration | RGC | RGC loss | NMDAR, (GlunN2B and Glun2D) activation glutamate transporter deficit | [10,11,12,16,18] |
| CoCl2-induced retinal cell degeneration | Photoreceptor | photoreceptor cell loss | HIF-1alpha, hypoxia, septin8? | [59,61,66] |
| Experimental autoimmune encephalomyelitis (EAE) | Myeline | demyelination and axonal injurycaused RGC loss | dysfunctional NADH-dehydrogenase type-2, Dock3 | [87,88,93] |
| Optic nerve crush injury | Optic nerve | RGC loss | caspase activation | [95,96] |
| Transient ischemia-induced retinalcell degeneration | Retina | Degeneration of RGC, Photoreceptor cells andAmacrine cells | glutamate release and NMDA/non-NMDA receptor activation, Activation of TNF/TNF-R system, etc. | [139,140] |
| Light-induced retinal cell degeneration | RGC, photoreceptor | RGC loss photoreceptor cell loss | up-regulation of PKM2 for RGC loss increased expression of Gpr65 for photoreceptor loss | [144,145] |
Author Contributions
Conflicts of Interest
References
- Bresnick, G.H. Excitotoxins: A possible new mechanism for the pathogenesis of ischemic retinal damage. Arch. Ophthalmol. 1989, 107, 339–341. [Google Scholar] [CrossRef] [PubMed]
- Schumer, R.A.; Podos, S.M. The nerve of glaucoma! Arch. Ophthalmol. 1994, 112, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Osborne, N.N.; Ugarte, M.; Chao, M.; Chidlow, G.; Bae, J.H.; Wood, J.P.; Nash, M.S. Neuroprotection in relation to retinal ischemia and relevance to glaucoma. Surv. Ophthalmol. 1999, 43, S102–S128. [Google Scholar] [CrossRef]
- Carter-Dawson, L.; Crawford, M.L.; Harwerth, R.S.; Smith, E.L., 3rd; Feldman, R.; Shen, F.F.; Mitchell, C.K.; Whitetree, A. Vitreal glutamate concentration in monkeys with experimental glaucoma. Investig. Ophthalmol. Vis. Sci. 2002, 43, 2633–2637. [Google Scholar]
- Honkanen, R.A.; Baruah, S.; Zimmerman, M.B.; Khanna, C.L.; Weaver, Y.K.; Narkiewicz, J.; Waziri, R.; Gehrs, K.M.; Weingeist, T.A.; Boldt, H.C.; et al. Vitreous amino acid concentrations in patients with glaucoma undergoing vitrectomy. Arch. Ophthalmol. 2003, 121, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Gupta, S.K.; Agarwal, P.; Saxena, R.; Agrawal, S.S. Current concepts in the pathophysiology of glaucoma. Indian J. Ophthalmol. 2009, 57, 257–266. [Google Scholar] [PubMed]
- Klein, B.E.; Klein, R.; Sponsel, W.E.; Franke, T.; Cantor, L.B.; Martone, J.; Menage, M.J. Prevalence of glaucoma: The Beaver dam EYE Study. Ophthalmology 1992, 99, 1499–1504. [Google Scholar] [CrossRef]
- Bonomi, L.; Marchini, G.; Marraffa, M.; Bernardi, P.; de Franco, I.; Perfetti, S.; Varotto, A.; Tenna, V. Prevalence of glaucoma and intraocular pressure distribution in a defined population: The Egna-Neumarkt Study. Ophthalmology 1998, 105, 209–215. [Google Scholar] [CrossRef]
- Anderson, D.R.; Normal Tension Glaucoma Study. Collaborative normal tension glaucoma study. Curr. Opin. Ophthalmol. 2003, 14, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Lam, T.T.; Abler, A.S.; Kwong, J.M.; Tso, M.O. N-methyl-d-aspartate (NMDA)-induced apoptosis in rat retina. Investig. Ophthalmol. Vis. Sci. 1999, 40, 2391–2397. [Google Scholar]
- Izumi, Y.; Hammerman, S.B.; Kirby, C.O.; Benz, A.M.; Olney, J.W.; Zorumski, C.F. Involvement of glutamate in ischemic neurodegeneration in isolated retina. Vis. Neurosci. 2003, 20, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Bai, N.; Aida, T.; Yanagisawa, M.; Katou, S.; Sakimura, K.; Mishina, M.; Tanaka, K. NMDA receptor subunits have different roles in, NMDA-induced neurotoxicity in the retina. Mol. Brain 2013, 6, 34. [Google Scholar] [CrossRef] [PubMed]
- Casson, R.J. Possible role of excitotoxicity in the pathogenesis of glaucoma. Clin. Exp. Ophthalmol. 2006, 34, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Kaur, C.; Foulds, W.S.; Ling, E.A. Hypoxia-ischemia and retinal ganglion cell damage. Clin. Ophthalmol. 2008, 2, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Hernández, C.; Simó, R. Neuroprotection in diabetic retinopathy. Curr. Diabetes Rep. 2012, 12, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, I.L.; Duarte, C.B.; Carvalho, A.P. Ca2+ influx through glutamate receptor-associated channels in retina cells correlates with neuronal cell death. Eur. J. Pharmacol. 1996, 302, 153–162. [Google Scholar] [CrossRef]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Harada, T.; Harada, C.; Nakamura, K.; Quah, H.M.; Okumura, A.; Namekata, K.; Saeki, T.; Aihara, M.; Yoshida, H.; Mitani, A.; et al. The potential role of glutamate transporters in the pathogenesis of normal tension glaucoma. J. Clin. Investig. 2007, 117, 1763–1770. [Google Scholar] [CrossRef] [PubMed]
- Kwong, J.M.; Lam, T.T.; Caprioli, J. Hyperthermic pre-conditioning protects retinal neurons from N-methyl-d-aspartate (NMDA)-induced apoptosis in rat. Brain Res. 2003, 970, 119–130. [Google Scholar] [CrossRef]
- Li, Y.; Schlamp, C.L.; Nickells, R.W. Experimental induction of retinal ganglion cell death in adult mice. Investig. Ophthalmol. Vis. Sci. 1999, 40, 1004–1008. [Google Scholar]
- Mattson, M.P.; Duan, W. ”Apoptotic” biochemical cascades in synaptic compartments: Roles in adaptive plasticity and neurodegenerative disorders. J. Neurosci. Res. 1999, 58, 152–166. [Google Scholar] [CrossRef]
- Macfarlane, B.V.; Wright, A.; Benson, H.A. Reversible blockade of retrograde axonal transport in the rat sciatic nerve by vincristine. J. Pharm. Pharmacol. 1997, 49, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Hara, A.; Niwa, M.; Kumada, M.; Kitaori, N.; Yamamoto, T.; Kozawa, O.; Mori, H. Fragmented, DNA transport in dendrites of retinal neurons during apoptotic cell death. Brain Res. 2004, 1007, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Hara, A.; Niwa, M.; Iwai, T.; Nakashima, M.; Bunai, Y.; Uematsu, T.; Yoshimi, N.; Mori, H. Neuronal apoptosis studied by a sequential, TUNEL technique: A method for tract-tracing. Brain Res. Brain Res. Protoc. 1999, 4, 140–146. [Google Scholar] [CrossRef]
- Kumada, M.; Niwa, M.; Wang, X.; Matsuno, H.; Hara, A.; Mori, H.; Matsuo, O.; Yamamoto, T.; Kozawa, O. Endogenous tissue type plasminogen activator facilitates, NMDA-induced retinal damage. Toxicol. Appl. Pharmacol. 2004, 200, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Kumada, M.; Niwa, M.; Hara, A.; Matsuno, H.; Mori, H.; Ueshima, S.; Matsuo, O.; Yamamoto, T.; Kozawa, O. Tissue type plasminogen activator facilitates, NMDA-receptor-mediated retinal apoptosis through an independent fibrinolytic cascade. Investig. Ophthalmol. Vis. Sci. 2005, 46, 1504–1507. [Google Scholar] [CrossRef] [PubMed]
- Mali, R.S.; Cheng, M.; Chintala, S.K. Plasminogen activators promote excitotoxicity-induced retinal damage. FASEB J. 2005, 19, 1280–1289. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Balsara, R.D.; Castellino, F.J. Synthetic conantokin peptides potently inhibit N-methyl-d-aspartate receptor-mediated currents of retinal ganglion cells. J. Neurosci. Res. 2014, 92, 1767–1774. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Suzuki, Y.; Kurauchi, Y.; Mori, A.; Nakahara, T.; Ishii, K. Hydrogen sulfide attenuates NMDA-induced neuronal injury via its anti-oxidative activity in the rat retina. Exp. Eye Res. 2014, 120, 90–96. [Google Scholar] [CrossRef] [PubMed]
- El-Azab, M.F.; Baldowski, B.R.; Mysona, B.A.; Shanab, A.Y.; Mohamed, I.N.; Abdelsaid, M.A.; Matragoon, S.; Bollinger, K.E.; Saul, A.; El-Remessy, A.B. Deletion of thioredoxin-interacting protein preserves retinal neuronal function by preventing inflammation and vascular injury. Br. J. Pharmacol. 2014, 171, 1299–1313. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Nakahara, T.; Mori, A.; Sakamoto, K.; Ishii, K. Protective effects of TGF-β inhibitors in a rat model of NMDA-induced retinal degeneration. Eur. J. Pharmacol. 2013, 699, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Jiang, L.; Shen, C.; Wan, H.; Xu, L.; Wang, N.; Jonas, J.B. Neuroprotective effect of epigallocatechin-3-gallate against N-methyl-d-aspartate-induced excitotoxicity in the adult rat retina. Acta Ophthalmol. 2012, 90, e609–e615. [Google Scholar] [CrossRef] [PubMed]
- Inokuchi, Y.; Imai, S.; Nakajima, Y.; Shimazawa, M.; Aihara, M.; Araie, M.; Hara, H. Edaravone, a free radical scavenger, protects against retinal damage in vitro and in vivo. J. Pharmacol. Exp. Ther. 2009, 329, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Ju, W.K.; Kim, K.Y.; Angert, M.; Duong-Polk, K.X.; Lindsey, J.D.; Ellisman, M.H.; Weinreb, R.N. Memantine blocks mitochondrial OPA1 and cytochrome c release and subsequent apoptotic cell death in glaucomatous retina. Investig. Ophthalmol. Vis. Sci. 2009, 50, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Suemori, S.; Shimazawa, M.; Kawase, K.; Satoh, M.; Nagase, H.; Yamamoto, T.; Hara, H. Metallothionein, an endogenous antioxidant, protects against retinal neuron damage in mice. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3975–3982. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.J.; Reh, T.A. Exogenous growth factors stimulate the regeneration of ganglion cells in the chicken retina. Dev. Biol. 2002, 251, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Sattayasai, J.; Zappia, J.; Ehrlich, D. Differential effects of excitatory amino acids on photoreceptors of the chick retina: An electron-microscopical study using the zinc-iodide-osmium technique. Vis. Neurosci. 1989, 2, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Hara, A.; Niwa, M.; Kumada, M.; Aoki, H.; Kunisada, T.; Oyama, T.; Yamamoto, T.; Kozawa, O.; Mori, H. Intraocular injection of folate antagonist methotrexate induces neuronal differentiation of embryonic stem cells transplanted in the adult mouse retina. Brain Res. 2006, 1085, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Hara, A.; Taguchi, A.; Aoki, H.; Hatano, Y.; Niwa, M.; Yamada, Y.; Kunisada, T. Folate antagonist, methotrexate induces neuronal differentiation of human embryonic stem cells transplanted into nude mouse retina. Neurosci. Lett. 2010, 477, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Hara, A.; Niwa, M.; Kunisada, T.; Yoshimura, N.; Katayama, M.; Kozawa, O.; Mori, H. Embryonic stem cells are capable of generating a neuronal network in the adult mouse retina. Brain Res. 2004, 999, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Aoki, H.; Hara, A.; Niwa, M.; Motohashi, T.; Suzuki, T.; Kunisada, T. Transplantation of cells from eye-like structures differentiated from embryonic stem cells in vitro and in vivo regeneration of retinal ganglion-like cells. Graefes Arch. Clin. Exp. Ophthalmol. 2008, 246, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Aoki, H.; Hara, A.; Niwa, M.; Yamada, Y.; Kunisada, T. In vitro and in vivo differentiation of human embryonic stem cells into retina-like organs and comparison with that from mouse pluripotent epiblast stem cells. Dev. Dyn. 2009, 238, 2266–2279. [Google Scholar] [CrossRef] [PubMed]
- Berson, E.L. Retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 1993, 34, 1659–1676. [Google Scholar]
- Kennan, A.; Aherne, A.; Humphries, P. Light in retinitis pigmentosa. Trends Genet. 2005, 21, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Green, W.R. Histopathology of age-related macular degeneration. Mol. Vis. 1999, 5, 27. [Google Scholar] [PubMed]
- Zack, D.J.; Dean, M.; Molday, R.S.; Nathans, J.; Redmond, T.M.; Stone, E.M.; Swaroop, A.; Valle, D.; Weber, B.H. What can we learn about age-related macular degeneration from other retinal diseases? Mol. Vis. 1999, 5, 30. [Google Scholar] [PubMed]
- Delyfer, M.N.; Léveillard, T.; Mohand-Saïd, S.; Hicks, D.; Picaud, S.; Sahel, J.A. Inherited retinal degenerations: Therapeutic prospects. Biol. Cell 2004, 96, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.Y.; Cringle, S.J. Retinal degeneration and local oxygen metabolism. Exp. Eye Res. 2005, 80, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Bowes, C.; Li, T.; Danciger, M.; Baxter, L.C.; Applebury, M.L.; Farber, D.B. Retinal degeneration in the rd mouse is caused by a defect in the β subunit of rod cGMP-phosphodiesterase. Nature 1990, 347, 677–680. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, A.J.; García-Fernández, J.M.; González, B.; Foster, R.G. The spatio-temporal pattern of photoreceptor degeneration in the aged rd/rd mouse retina. Cell Tissue Res. 1996, 284, 193–202. [Google Scholar] [PubMed]
- Chang, B.; Heckenlively, J.R.; Hawes, N.L.; Roderick, T.H. New mouse primary retinal degeneration (rd-3). Genomics 1993, 16, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Hawes, N.L.; Chang, B.; Hageman, G.S.; Nusinowitz, S.; Nishina, P.M.; Schneider, B.S.; Smith, R.S.; Roderick, T.H.; Davisson, M.T.; Heckenlively, J.R. Retinal degeneration 6 (rd6): A new mouse model for human retinitis punctata albescens. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3149–3157. [Google Scholar]
- Maslim, J.; Valter, K.; Egensperger, R.; Holländer, H.; Stone, J. Tissue oxygen during a critical developmental period controls the death and survival of photoreceptors. Investig. Ophthalmol. Vis. Sci. 1997, 38, 1667–1677. [Google Scholar]
- Valter, K.; Maslim, J.; Bowers, F.; Stone, J. Photoreceptor dystrophy in the RCS rat: Roles of oxygen, debris, and bFGF. Investig. Ophthalmol. Vis. Sci. 1998, 39, 2427–2442. [Google Scholar]
- Badr, G.A.; Zhang, J.Z.; Tang, J.; Kern, T.S.; Ismail-Beigi, F. Glut1 and glut3 expression, but not capillary density, is increased by cobalt chloride in rat cerebrum and retina. Brain Res. Mol. Brain Res. 1999, 64, 24–33. [Google Scholar] [CrossRef]
- Wang, G.L.; Semenza, G.L. Desferrioxamine induces erythropoietin gene expression and hypoxia-inducible factor 1DNA-binding activity: Implications for models of hypoxia signal transduction. Blood 1993, 82, 3610–3615. [Google Scholar] [PubMed]
- Ji, Z.; Yang, G.; Shahzidi, S.; Tkacz-Stachowska, K.; Suo, Z.; Nesland, J.M.; Peng, Q. Induction of hypoxia-inducible factor-1α overexpression by cobalt chloride enhances cellular resistance to photodynamic therapy. Cancer Lett. 2006, 244, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.G.; Lee, H.; Rho, H.M. Transcriptional repression of the human p53 gene by cobalt chloride mimicking hypoxia. FEBS Lett. 2001, 507, 259–263. [Google Scholar] [CrossRef]
- Vengellur, A.; Woods, B.G.; Ryan, H.E.; Johnson, R.S.; LaPres, J.J. Gene expression profiling of the hypoxia signaling pathway in hypoxia-inducible factor 1α null mouse embryonic fibroblasts. Gene Expr. 2003, 11, 181–197. [Google Scholar] [CrossRef] [PubMed]
- Roessner, C.A.; Santander, P.J.; Scott, A.I. Multiple biosynthetic pathways for vitamin B12: Variations on a central theme. Vitam. Horm. 2001, 61, 267–297. [Google Scholar] [PubMed]
- Yuan, Y.; Hilliard, G.; Ferguson, T.; Millhorn, D.E. Cobalt inhibits the interaction between hypoxia-inducible factor-alpha and von Hippel-Lindau protein by direct binding to hypoxia-inducible factor-α. J. Biol. Chem. 2003, 278, 15911–15916. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.H.; Kirschenbaum, A.; Yao, S.; Stearns, M.E.; Holland, J.F.; Claffey, K.; Levine, A.C. Upregulation of vascular endothelial growth factor by cobalt chloride-simulated hypoxia is mediated by persistent induction of cyclooxygenase-2 in a metastatic human prostate cancer cell line. Clin. Exp. Metastasis 1999, 17, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Minchenko, A.; Bauer, T.; Salceda, S.; Caro, J. Hypoxic stimulation of vascular endothelial growth factor expression in vitro and in vivo. Lab. Investig. 1994, 71, 374–379. [Google Scholar] [PubMed]
- Matsumoto, M.; Makino, Y.; Tanaka, T.; Tanaka, H.; Ishizaka, N.; Noiri, E.; Fujita, T.; Nangaku, M. Induction of renoprotective gene expression by cobalt ameliorates ischemic injury of the kidney in rats. J. Am. Soc. Nephrol. 2003, 14, 1825–1832. [Google Scholar] [CrossRef] [PubMed]
- Hara, A.; Niwa, M.; Aoki, H.; Kumada, M.; Kunisada, T.; Oyama, T.; Yamamoto, T.; Kozawa, O.; Mori, H. A new model of retinal photoreceptor cell degeneration induced by a chemical hypoxia-mimicking agent, cobalt chloride. Brain Res. 2006, 1109, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Hara, A.; Taguchi, A.; Niwa, M.; Aoki, H.; Yamada, Y.; Ito, H.; Nagata, K.; Kunisada, T.; Mori, H. Localization of septin 8 in murine retina, and spatiotemporal expression of septin 8 in a murine model of photoreceptor cell degeneration. Neurosci. Lett. 2007, 423, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Kwan, T.; Kurimoto, Y.; Shatos, M.; Lund, R.D.; Young, M.J. Transplantation of EGF-responsive neurospheres from GFP transgenic mice into the eyes of rd mice. Brain Res. 2002, 943, 292–300. [Google Scholar] [CrossRef]
- Meyer, J.S.; Katz, M.L.; Maruniak, J.A.; Kirk, M.D. Neural differentiation of mouse embryonic stem cells in vitro and after transplantation into eyes of mutant mice with rapid retinal degeneration. Brain Res. 2004, 1014, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.C.; Chang, C.Y.; Kao, A.; Hsi, B.; Lee, S.H.; Chen, Y.H.; Wang, I.J. Hypoxia-induced retinal neovascularization in zebrafish embryos: A potential model of retinopathy of prematurity. PLoS ONE 2015, 10, e0126750. [Google Scholar] [CrossRef] [PubMed]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef]
- Arnold, A.C. Evolving management of optic neuritis and multiple sclerosis. Am. J. Ophthalmol. 2005, 139, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Beck, R.W.; Gal, R.L.; Bhatti, M.T.; Brodsky, M.C.; Buckley, E.G.; Chrousos, G.A.; Corbett, J.; Eggenberger, E.; Goodwin, J.A.; Katz, B.; et al. Visual function more than 10 years after optic neuritis: Experience of the optic neuritis treatment trial. Am. J. Ophthalmol. 2004, 137, 77–83. [Google Scholar] [PubMed]
- Talman, L.S.; Bisker, E.R.; Sackel, D.J.; Long, D.A.; Galetta, K.M., Jr.; Ratchford, J.N.; Lile, D.J.; Farrell, S.K.; Loguidice, M.J.; Remington, G.; et al. Longitudinal study of vision and retinal nerve fiber layer thickness in multiple sclerosis. Ann. Neurol. 2010, 67, 749–760. [Google Scholar] [PubMed]
- Lidster, K.; Jackson, S.J.; Ahmed, Z.; Munro, P.; Coffey, P.; Giovannoni, G.; Baker, M.D.; Baker, D. Neuroprotection in a novel mouse model of multiple sclerosis. PLoS ONE 2013, 8, e79188. [Google Scholar] [CrossRef] [PubMed]
- Lublin, F.D. Role of myelin antigens in murine relapsing experimental allergic encephalomyelitis. J. Clin. Lab. Immunol. 1984, 13, 179–182. [Google Scholar] [PubMed]
- Quinn, T.A.; Dutt, M.; Shindler, K.S. Optic neuritis and retinal ganglion cell loss in a chronic murine model of multiple sclerosis. Front. Neurol. 2011, 2, 50. [Google Scholar] [CrossRef] [PubMed]
- Horstmann, L.; Schmid, H.; Heinen, A.P.; Kurschus, F.C.; Dick, H.B.; Joachim, S.C. Inflammatory demyelination induces glia alterations and ganglion cell loss in the retina of an experimental autoimmune encephalomyelitis model. J. Neuroinflamm. 2013, 10, 120. [Google Scholar] [CrossRef] [PubMed]
- Shindler, K.S.; Ventura, E.; Dutt, M.; Rostami, A. Inflammatory demyelination induces axonal injury and retinal ganglion cell apoptosis in experimental optic neuritis. Exp. Eye Res. 2008, 87, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Dutt, M.; Tabuena, P.; Ventura, E.; Rostami, A.; Shindler, K.S. Timing of corticosteroid therapy is critical to prevent retinal ganglion cell loss in experimental optic neuritis. Investig. Ophthalmol. Vis. Sci. 2010, 51, 1439–1445. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.S.; Dine, K.; Luna, E.; Ahlem, C.; Shindler, K.S. HE3286 reduces axonal loss and preserves retinal ganglion cell function in experimental optic neuritis. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5744–5751. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.W.; Das, A.; Guyton, M.K.; Ray, S.K.; Rohrer, B.; Banik, N.L. Calpain inhibition attenuates apoptosis of retinal ganglion cells in acute optic neuritis. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4935–4941. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Guyton, M.K.; Smith, A.; Wallace, G., IV; McDowell, M.L.; Matzelle, D.D.; Ray, S.K.; Banik, N.L. Calpain inhibitor attenuated optic nerve damage in acute optic neuritis in rats. J. Neurochem. 2013, 124, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, R.; Dvoriantchikova, G.; Barakat, D.; Ivanov, D.; Bethea, J.R.; Shestopalov, V.I. Transgenic inhibition of astroglial NF-κB protects from optic nerve damage and retinal ganglion cell loss in experimental optic neuritis. J. Neuroinflamm. 2012, 9, 213. [Google Scholar] [CrossRef] [PubMed]
- Sühs, K.W.; Fairless, R.; Williams, S.K.; Heine, K.; Cavalié, A.; Diem, R. N-Methyl-d-aspartate receptor blockade is neuroprotective in experimental autoimmune optic neuritis. J. Neuropathol. Exp. Neurol. 2014, 73, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Gadjanski, I.; Boretius, S.; Williams, S.K.; Lingor, P.; Knöferle, J.; Sättler, M.B.; Fairless, R.; Hochmeister, S.; Sühs, K.W.; Michaelis, T.; et al. Role of n-type voltage-dependent calcium channels in autoimmune optic neuritis. Ann. Neurol. 2009, 66, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Shirani, A.; Zhao, Y.; Karim, M.E.; Evans, C.; Kingwell, E.; van der Kop, M.L.; Oger, J.; Gustafson, P.; Petkau, J.; Tremlett, H. Association between use of interferon β and progression of disability in patients with relapsing-remitting multiple sclerosis. JAMA 2012, 308, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Talla, V.; Yu, H.; Chou, T.H.; Porciatti, V.; Chiodo, V.; Boye, S.L.; Hauswirth, W.W.; Lewin, A.S.; Guy, J. NADH-dehydrogenase type-2 suppresses irreversible visual loss and neurodegeneration in the EAE animal model of MS. Mol. Ther. 2013, 21, 1876–1888. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.; McDonough, J.; Yin, X.; Peterson, J.; Chang, A.; Torres, T.; Gudz, T.; Macklin, W.B.; Lewis, D.A.; Fox, R.J.; et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann. Neurol. 2006, 59, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, G. A new colour reaction on copper and ceratin carbonyl compounds. Acta Chem. Scand. 1950, 4, 205–208. [Google Scholar] [CrossRef]
- Praet, J.; Guglielmetti, C.; Berneman, Z.; van der Linden, A.; Ponsaerts, P. Cellular and molecular neuropathology of the cuprizone mouse model: Clinical relevance for multiple sclerosis. Neurosci. Biobehav. Rev. 2014, 485–505. [Google Scholar] [CrossRef] [PubMed]
- Payne, T.; Newmark, J.; Reid, K.H. The focally demyelinated rat fimbria: A new in vitro model for the study of acute demyelination in the central nervous system. Exp. Neurol. 1991, 114, 66–72. [Google Scholar] [CrossRef]
- Beckmann, D.V.; Carvalho, F.B.; Mazzanti, C.M.; dos Santos, R.P.; Andrades, A.O.; Aiello, G.; Rippilinger, A.; Graça, D.L.; Abdalla, F.H.; Oliveira, L.S.; et al. Neuroprotective role of quercetin in locomotor activities and cholinergic neurotransmission in rats experimentally demyelinated with ethidium bromide. Life Sci. 2014, 103, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Namekata, K.; Kimura, A.; Harada, C.; Yoshida, H.; Matsumoto, Y.; Harada, T. Dock3 protects myelin in the cuprizone model for demyelination. Cell Death Dis. 2014, 5, e1395. [Google Scholar] [CrossRef] [PubMed]
- Berson, D.M.; Dunn, F.A.; Takao, M. Phototransduction by retinal ganglion cells that set the circadian clock. Science 2002, 295, 1070–1073. [Google Scholar] [CrossRef] [PubMed]
- Berkelaar, M.; Clarke, D.B.; Wang, Y.C.; Bray, G.M.; Aguayo, A.J. Axotomy results in delayed death and apoptosis of retinal ganglion cells in adult rats. J. Neurosci. 1994, 14, 4368–4374. [Google Scholar] [PubMed]
- Choudhury, S.; Liu, Y.; Clark, A.F.; Pang, I.H. Caspase-7: A critical mediator of optic nerve injury-induced retinal ganglion cell death. Mol. Neurodegener. 2015, 10, 40. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Z.; Kalinski, H.; Berry, M.; Almasieh, M.; Ashush, H.; Slager, N.; Brafman, A.; Spivak, I.; Prasad, N.; Mett, I.; et al. Ocular neuroprotection by siRNA targeting caspase-2. Cell Death Dis. 2011, 2, e173. [Google Scholar] [CrossRef] [PubMed]
- Kermer, P.; Klocker, N.; Labes, M.; Thomsen, S.; Srinivasan, A.; Bahr, M. Activation of caspase-3 in axotomized rat retinal ganglion cells in vivo. FEBS Lett. 1999, 453, 361–364. [Google Scholar] [CrossRef]
- Kermer, P.; Ankerhold, R.; Klocker, N.; Krajewski, S.; Reed, J.C.; Bahr, M. Caspase-9: Involvement in secondary death of axotomized rat retinal ganglion cells in vivo. Mol. Brain Res. 2000, 85, 144–150. [Google Scholar] [CrossRef]
- Weishaupt, J.H.; Diem, R.; Kermer, P.; Krajewski, S.; Reed, J.C.; Bahr, M. Contribution of caspase-8 to apoptosis of axotomized rat retinal ganglion cells in vivo. Neurobiol. Dis. 2003, 13, 124–135. [Google Scholar] [CrossRef]
- Cheung, Z.H.; Chan, Y.M.; Siu, F.K.; Yip, H.K.; Wu, W.; Leung, M.C.; So, K.F. Regulation of caspase activation in axotomized retinal ganglion cells. Mol. Cell. Neurosci. 2004, 25, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Monnier, P.P.; D’Onofrio, P.M.; Magharious, M.; Hollander, A.C.; Tassew, N.; Szydlowska, K.; Tymianski, M.; Koeberle, P.D. Involvement of caspase-6 and caspase-8 in neuronal apoptosis and the regenerative failure of injured retinal ganglion cells. J. Neurosci. 2011, 31, 10494–10505. [Google Scholar] [CrossRef] [PubMed]
- Hänninen, V.A.; Pantcheva, M.B.; Freeman, E.E.; Poulin, N.R.; Grosskreutz, C.L. Activation of caspase 9 in a rat model of experimental glaucoma. Curr. Eye Res. 2002, 25, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Tahzib, N.G.; Ransom, N.L.; Reitsamer, H.A.; McKinnon, S.J. Alpha-fodrin is cleaved by caspase-3 in a chronic ocular hypertension (COH) rat model of glaucoma. Brain Res. Bull. 2004, 62, 491–495. [Google Scholar] [CrossRef]
- Kim, H.S.; Park, C.K. Retinal ganglion cell death is delayed by activation of retinal intrinsic cell survival program. Brain Res. 2005, 1057, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Lam, T.T.; Abler, A.S.; Tso, M.O. Apoptosis and caspases after ischemia-reperfusion injury in rat retina. Investig. Ophthalmol. Vis. Sci. 1999, 40, 967–975. [Google Scholar]
- Produit-Zengaffinen, N.; Pournaras, C.J.; Schorderet, D.F. Retinal ischemia-induced apoptosis is associated with alteration in Bax and Bcl-x(L) expression rather than modifications in Bak and Bcl-2. Mol. Vis. 2009, 15, 2101–2110. [Google Scholar] [PubMed]
- Grosskreutz, C.L.; Hänninen, V.A.; Pantcheva, M.B.; Huang, W.; Poulin, N.R.; Dobberfuhl, A.P. FK506 blocks activation of the intrinsic caspase cascade after optic nerve crush. Exp. Eye Res. 2005, 80, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Tsai, R.K.; Chang, C.H.; Wang, H.Z. Neuroprotective effects of recombinant human granulocyte colony-stimulating factor (G-CSF) in neurodegeneration after optic nerve crush in rats. Exp. Eye Res. 2008, 87, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, A.; Naka, M.; Fukuda, M.; Nakamura, M.; Negi, A. Tafluprost protects rat retinal ganglion cells from apoptosis in vitro and in vivo. Graefes Arch. Clin. Exp. Ophthalmol. 2009, 247, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Biermann, J.; Grieshaber, P.; Goebel, U.; Martin, G.; Thanos, S.; Di Giovanni, S.; Lagrèze, W.A. Valproic acid-mediated neuroprotection and regeneration in injured retinal ganglion cells. Investig. Ophthalmol. Vis. Sci. 2010, 51, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Semba, K.; Namekata, K.; Kimura, A.; Harada, C.; Katome, T.; Yoshida, H.; Mitamura, Y.; Harada, T. Dock3 overexpression and p38 MAPK inhibition synergistically stimulate neuroprotection and axon regeneration after optic nerve injury. Neurosci. Lett. 2014, 581, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Mansour-Robaey, S.; Clarke, D.B.; Wang, Y.C.; Bray, G.M.; Aguayo, A.J. Effects of ocular injury and administration of brain-derived neurotrophic factor on survival and regrowth of axotomized retinal ganglion cells. Proc. Natl. Acad. Sci. USA 1994, 91, 1632–1636. [Google Scholar] [CrossRef] [PubMed]
- Harada, C.; Azuchi, Y.; Noro, T.; Guo, X.; Kimura, A.; Namekata, K.; Harada, T. TrkB Signaling in Retinal Glia Stimulates Neuroprotection after Optic Nerve Injury. Am. J. Pathol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Harada, C.; Guo, X.; Namekata, K.; Kimura, A.; Nakamura, K.; Tanaka, K.; Parada, L.F.; Harada, T. Glia- and neuron-specific functions of TrkB signalling during retinal degeneration and regeneration. Nat. Commun. 2011, 2, 189. [Google Scholar] [CrossRef] [PubMed]
- Lenkowski, J.R.; Raymond, P.A. Müller glia: Stem cells for generation and regeneration of retinal neurons in teleost fish. Prog. Retin. Eye Res. 2014, 40, 94–123. [Google Scholar] [CrossRef] [PubMed]
- Koriyama, Y.; Homma, K.; Kato, S. Activation of cell survival signals in the goldfish retinal ganglion cells after optic nerve injury. Adv. Exp. Med. Biol. 2006, 572, 333–337. [Google Scholar] [PubMed]
- Tamura, A.; Graham, D.I.; McCulloch, J.; Teasdale, G.M. Focal cerebral ischaemia in the rat: 1. Description of technique and early neuropathological consequences following middle cerebral artery occlusion. J. Cereb. Blood Flow Metab. 1981, 1, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Longa, E.Z.; Weinstein, P.R.; Carlson, S.; Cummins, R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989, 20, 84–91. [Google Scholar] [CrossRef] [PubMed]
- D’Onofrio, P.M.; Koeberle, P.D. What can we learn about stroke from retinal ischemia models? Acta Pharmacol. Sin. 2013, 34, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, J.; Yoshida, Y.; Nakazawa, T.; Ooneda, G. Experimental studies of ischemic brain edema: A new experimental model of cerebral embolism in rats in which recirculation can be introduced in the ischemic area. Jpn. J. Stroke 1986, 8, 1–8. [Google Scholar] [CrossRef]
- Steele, E.C.; Guo, Q., Jr.; Namura, S. Filamentous middle cerebral artery occlusion causes ischemic damage to the retina in mice. Stroke 2008, 39, 2099–2104. [Google Scholar] [CrossRef] [PubMed]
- Muthaian, R.; Minhas, G.; Anand, A. Pathophysiology of stroke and stroke-induced retinal ischemia: Emerging role of stem cells. J. Cell. Physiol. 2012, 227, 1269–1679. [Google Scholar] [CrossRef] [PubMed]
- Joo, C.K.; Choi, J.S.; Ko, H.W.; Park, K.Y.; Sohn, S.; Chun, M.H.; Oh, Y.J.; Gwag, B.J. Necrosis and apoptosis after retinal ischemia: Involvement of NMDA-mediated excitotoxicity and p53. Investig. Ophthalmol. Vis. Sci. 1999, 40, 713–772. [Google Scholar]
- Kuroiwa, S.; Katai, N.; Shibuki, H.; Kurokawa, T.; Umihira, J.; Nikaido, T.; Kametani, K.; Yoshimura, N. Expression of cell cycle-related genes in dying cells in retinal ischemic injury. Investig. Ophthalmol. Vis. Sci. 1998, 39, 610–617. [Google Scholar]
- Perlman, J.I.; McCole, S.M.; Pulluru, P.; Chang, C.J.; Lam, T.T.; Tso, M.O. Disturbances in the distribution of neurotransmitters in the rat retina after ischemia. Curr. Eye Res. 1996, 15, 589–596. [Google Scholar] [CrossRef]
- Halder, S.K.; Matsunaga, H.; Yamaguchi, H.; Ueda, H. Novel neuroprotective action of prothymosin α-derived peptide against retinal and brain ischemic damages. J. Neurochem. 2013, 125, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Niwa, M.; Hara, A.; Matsuno, H.; Kawase, K.; Kozawa, O.; Mow, H.; Uematsu, T. Neuronal degradation in mouse retina after a transient ischemia and protective effect of hypothermia. Neurol. Res. 2002, 24, 730–735. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.K.; Ke, C.Y.; Khor, C.N.; Cai, Y.J.; Lee, Y.J. Involvement of SDF1a and STAT3 in granulocyte colony-stimulating factor rescues optic ischemia-induced retinal function loss by mobilizing hematopoietic stem cells. Investig. Ophthalmol. Vis. Sci. 2013, 54, 1920–1930. [Google Scholar] [CrossRef] [PubMed]
- Miyaki, K.; Matsubara, A.; Nishiwaki, A.; Tomida, K.; Morita, H.; Yoshida, M.; Ogura, Y. Pitavastatin attenuates leukocyte-endothelial interactions induced by ischemia-reperfusion injury in the rat retina. Curr. Eye Res. 2009, 34, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Daugeliene, L.; Niwa, M.; Hara, A.; Matsuno, H.; Yamamoto, T.; Kitazawa, Y.; Uematsu, T. Transient ischemic injury in the rat retina caused by thrombotic occlusion-thrombolytic reperfusion. Investig. Ophthalmol. Vis. Sci. 2000, 41, 2743–2747. [Google Scholar]
- Wong, T.Y.; Scott, I.U. Retinal-vein occlusion. N. Engl. J. Med. 2010, 363, 2135–2144. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, A.; Imai, K.; Kim, H.C.; de Juan, E., Jr. Activation of protein tyrosine phosphorylation after retinal branch vein occlusion in cats. Investig. Ophthalmol. Vis. Sci. 1997, 38, 372–380. [Google Scholar]
- Takei, K.; Sato, T.; Nonoyama, T.; Miyauchi, T.; Goto, K.; Hommura, S. A new model of transient complete obstruction of retinal vessels induced by endothelin-1 injection into the posterior vitreous body in rabbits. Graefes Arch. Clin. Exp. Ophthalmol. 1993, 231, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Linner, E. Occlusion of the retinal veins in rabbits induced by light coagulation. Acta Ophthalmol. 1961, 39, 739–740. [Google Scholar] [CrossRef]
- Zhang, Y.; Fortune, B.; Atchaneeyasakul, L.O.; McFarland, T.; Mose, K.; Wallace, P.; Main, J.; Wilson, D.; Appukuttan, B.; Stout, J.T. Natural history and histology in a rat model of laser-induced photothrombotic retinal vein occlusion. Curr. Eye Res. 2008, 33, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wu, Y.; Zheng, M.; Gu, Q.; Zheng, Z.; Xia, X. Establishing an experimental rat model of photodynamically-induced retinal vein occlusion using erythrosin B. Int. J. Ophthalmol. 2014, 7, 232–238. [Google Scholar] [PubMed]
- Pournaras, C.J.; Petropoulos, I.K.; Pournaras, J.A.; Stangos, A.N.; Gilodi, N.; Rungger-Brändle, E. The rationale of retinal endovascular fibrinolysis in the treatment of retinal vein occlusion: From experimental data to clinical application. Retina 2012, 32, 1566–1573. [Google Scholar] [CrossRef] [PubMed]
- Osborne, N.N.; Casson, R.J.; Wood, J.P.; Chidlow, G.; Graham, M.; Melena, J. Retinal ischemia: Mechanisms of damage and potential therapeutic strategies. Prog. Retin. Eye Res. 2004, 23, 91–147. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, V.; Mohand-Said, S.; Hanoteau, N.; Fuchs, C.; Pfizenmaier, K.; Eisel, U. Neurodegenerative and neuroprotective effects of tumor Necrosis factor (TNF) in retinal ischemia: Opposite roles of TNF receptor 1 and TNF receptor 2. J. Neurosci. 2002, 22, 216. [Google Scholar]
- Randolph, S.A. Age-related macular degeneration. Workplace Health Saf. 2014, 62, 352. [Google Scholar] [CrossRef] [PubMed]
- Marc, R.E.; Jones, B.W.; Watt, C.B.; Vazquez-Chona, F.; Vaughan, D.K.; Organisciak, D.T. Extreme retinal remodeling triggered by light damage: Implications for age related macular degeneration. Mol. Vis. 2008, 14, 782–806. [Google Scholar] [PubMed]
- Wenzel, A.; Grimm, C.; Samardzija, M.; Remé, C.E. Molecular mechanisms of light-induced photoreceptor apoptosis and neuroprotection for retinal degeneration. Prog. Retin. Eye Res. 2005, 24, 275–306. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Chen, H.; Zhu, M.; Zhu, R.; Qin, B.; Fang, H.; Dai, M.; Sang, A.; Liu, X. Up-Regulation of PKM2 Relates to Retinal Ganglion Cell Apoptosis After Light-Induced Retinal Damage in Adult Rats. Cell. Mol. Neurobiol. 2015, 35, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Ail, D.; Rüfenacht, V.; Caprara, C.; Samardzija, M.; Kast, B.; Grimm, C. Increased expression of the proton-sensing G protein-coupled receptor Gpr65 during retinal degeneration. Neuroscience 2015, 301, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Perusek, L.; Maeda, A. Autophagy in light-induced retinal damage. Exp. Eye Res. 2016, 144, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Faktorovich, E.G.; Steinberg, R.H.; Yasumura, D.; Matthes, M.T.; LaVail, M.M. Basic fibroblast growth factor and local injury protect photoreceptors from light damage in the rat. J. Neurosci. 1992, 12, 3554–3567. [Google Scholar] [PubMed]
- Liu, C.; Peng, M.; Laties, A.M.; Wen, R. Preconditioning with bright light evokes a protective response against light damage in the rat retina. J. Neurosci. 1998, 18, 1337–1344. [Google Scholar] [PubMed]
- Casson, R.J.; Wood, J.P.; Melena, J.; Chidlow, G.; Osborne, N.N. The effect of ischemic preconditioning on light-induced photoreceptor injury. Investig. Ophthalmol. Vis. Sci. 2003, 44, 1348–1354. [Google Scholar] [CrossRef]
- Casson, R.J.; Chidlow, G.; Wood, J.P.; Vidal-Sanz, M.; Osborne, N.N. The effect of retinal ganglion cell injury on light-induced photoreceptor degeneration. Investig. Ophthalmol. Vis. Sci. 2004, 45, 685–693. [Google Scholar] [CrossRef]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niwa, M.; Aoki, H.; Hirata, A.; Tomita, H.; Green, P.G.; Hara, A. Retinal Cell Degeneration in Animal Models. Int. J. Mol. Sci. 2016, 17, 110. https://doi.org/10.3390/ijms17010110
Niwa M, Aoki H, Hirata A, Tomita H, Green PG, Hara A. Retinal Cell Degeneration in Animal Models. International Journal of Molecular Sciences. 2016; 17(1):110. https://doi.org/10.3390/ijms17010110
Chicago/Turabian StyleNiwa, Masayuki, Hitomi Aoki, Akihiro Hirata, Hiroyuki Tomita, Paul G. Green, and Akira Hara. 2016. "Retinal Cell Degeneration in Animal Models" International Journal of Molecular Sciences 17, no. 1: 110. https://doi.org/10.3390/ijms17010110