
A Comprehensive Characterization of Mitochondrial Genome in Papillary Thyroid Cancer

Abstract
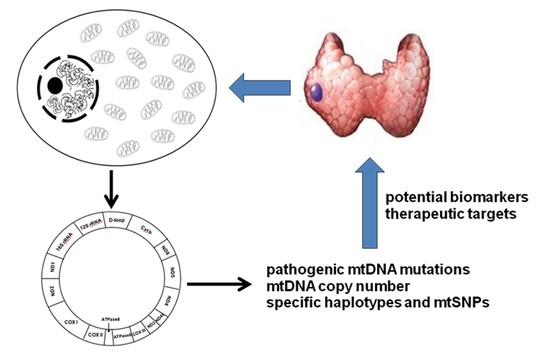
:
1. Introduction
2. Results
2.1. Distribution of mtDNA Variations
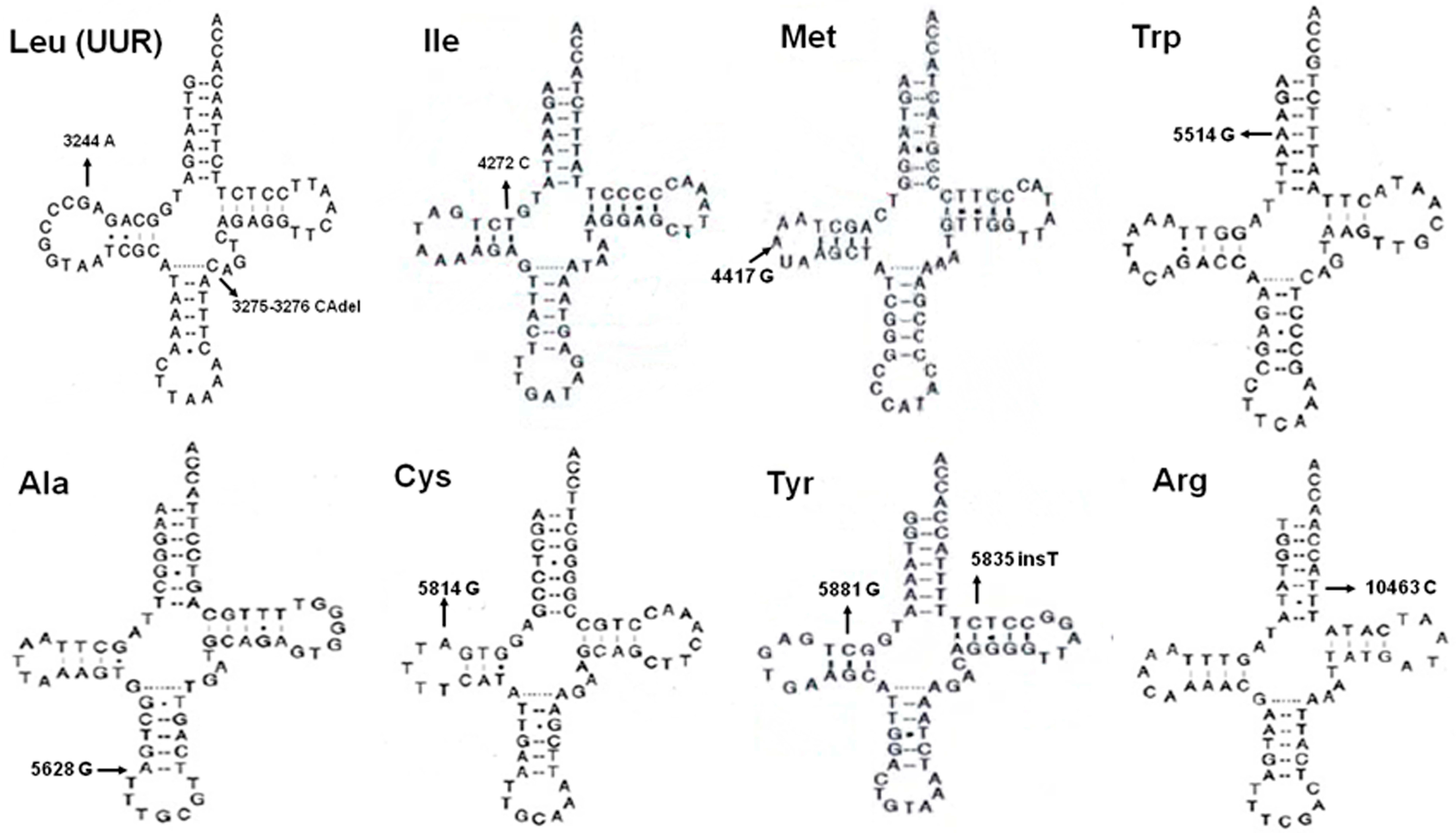
2.2. The mtDNA Variations in Non-Coding Region
2.3. The mtDNA Variations in Protein-Coding Region
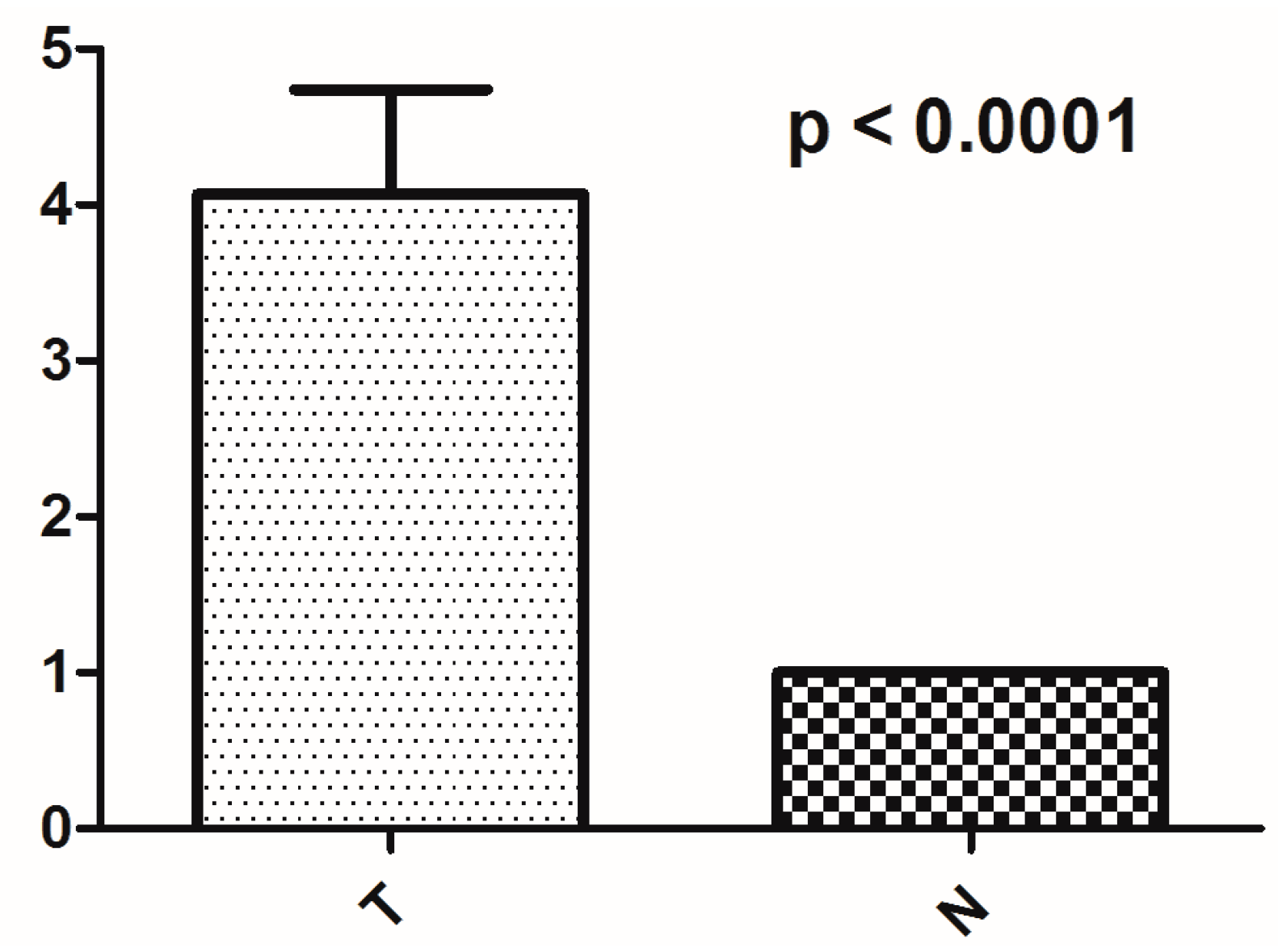
2.4. The Alteration of mtDNA Copy Number
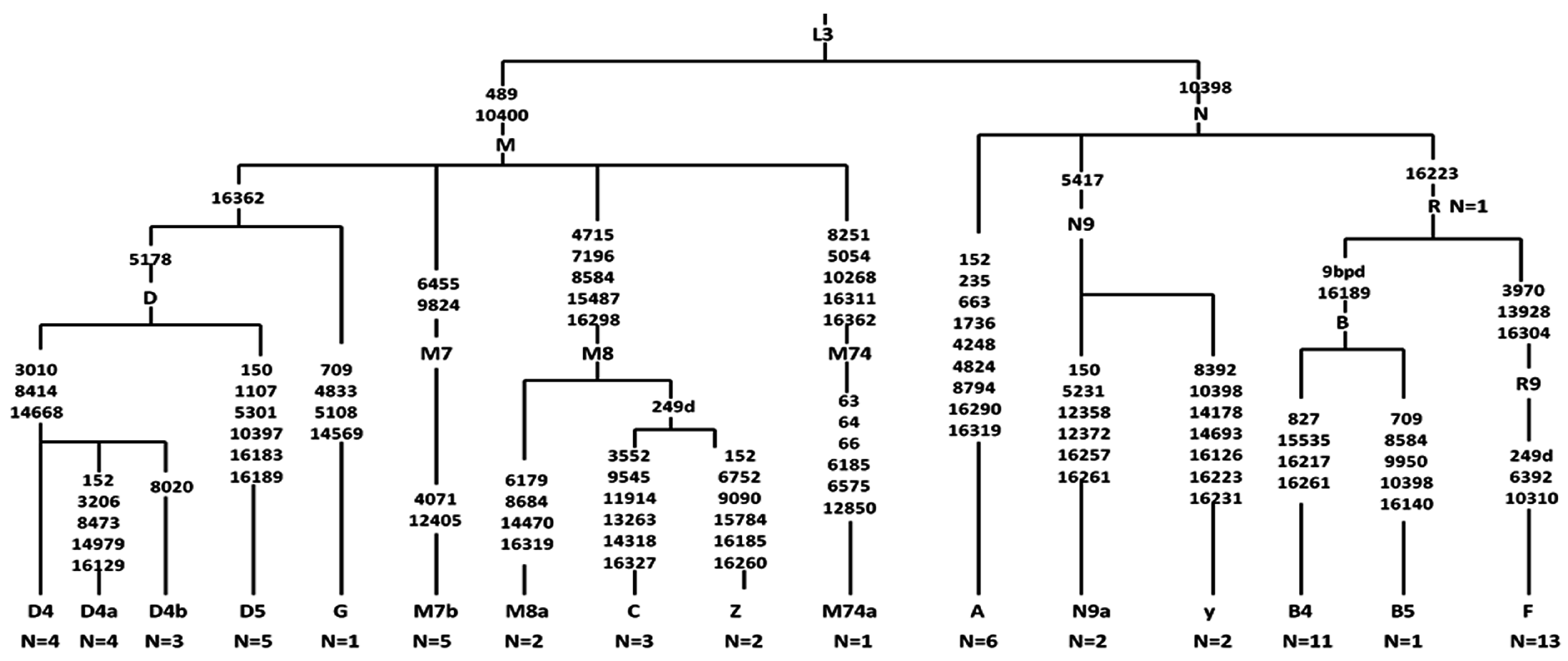
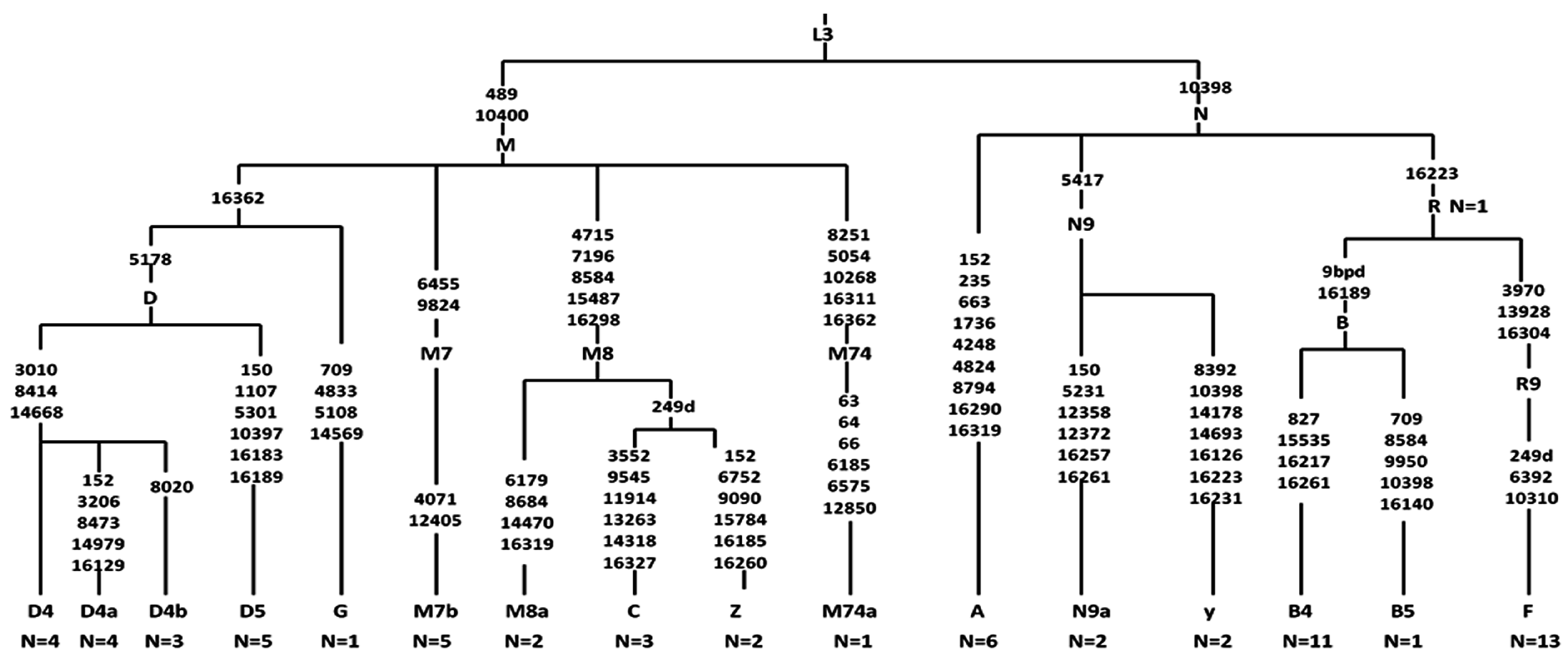
2.5. Analysis of Haplogroup and mtSNP
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. Sequencing of the Mitochondrial Genome
4.3. Sequence Analysis and Haplogroup Classification
4.4. Phylogenetic Conservation Analysis and Pathogenic Prediction
4.5. Determination of mtDNA Copy Number
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chan, D.C. Mitochondria: Dynamic organelles in disease, aging, and development. Cell 2006, 125, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G. Mitochondria in cancer. Oncogene 2006, 25, 4630–4632. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C.; Fan, W. Energetics, epigenetics, mitochondrial genetics. Mitochondrion 2010, 10, 12–31. [Google Scholar] [CrossRef] [PubMed]
- Larman, T.C.; DePalma, S.R.; Hadjipanayis, A.G.; Protopopov, A.; Zhang, J.; Gabriel, S.B.; Chin, L.; Seidman, C.E.; Kucherlapati, R.; Seidman, J.G. Spectrum of somatic mitochondrial mutations in five cancers. Proc. Natl. Acad. Sci. USA 2012, 109, 14087–14091. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Imanishi, H.; Takenaga, K.; Hayashi, J. Regulation of metastasis; mitochondrial DNA mutations have appeared on stage. J. Bioenerg. Biomembr. 2012, 44, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Markovina, S.; Grigsby, P.W.; Schwarz, J.K.; DeWees, T.; Moley, J.F.; Siegel, B.A.; Perkins, S.M. Treatment approach, surveillance, and outcome of well-differentiated thyroid cancer in childhood and adolescence. Thyroid 2014, 24, 1121–1126. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Miyauchi, A.; Ito, M.; Yabuta, T.; Masuoka, H.; Higashiyama, T.; Fukushima, M.; Kobayashi, K.; Kihara, M.; Miya, A. Prognosis and prognostic factors of differentiated thyroid carcinoma after the appearance of metastasis refractory to radioactive iodine therapy. Endocr. J. 2014, 61, 821–824. [Google Scholar] [CrossRef] [PubMed]
- Xing, M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat. Rev. Cancer 2013, 13, 184–199. [Google Scholar] [CrossRef] [PubMed]
- Corver, W.E.; van Wezel, T.; Molenaar, K.; Schrumpf, M.; van den Akker, B.; van Eijk, R.; Ruano Neto, D.; Oosting, J.; Morreau, H. Near-haploidization significantly associates with oncocytic adrenocortical, thyroid, and parathyroid tumors but not with mitochondrial DNA mutations. Genes Chromosomes Cancer 2014, 53, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Ji, J.; Chen, G.; Fang, H.; Yan, S.; Shen, L.; Wei, J.; Yang, K.; Lu, J.; Bai, Y. Analysis of mitochondrial DNA mutations in D-loop region in thyroid lesions. Biochim. Biophys. Acta 2010, 1800, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Gasparre, G.; Porcelli, A.M.; Bonora, E.; Pennisi, L.F.; Toller, M.; Iommarini, L.; Ghelli, A.; Moretti, M.; Betts, C.M.; Martinelli, G.N.; et al. Disruptive mitochondrial DNA mutations in complex I subunits are markers of oncocytic phenotype in thyroid tumors. Proc. Natl. Acad. Sci. USA 2007, 104, 9001–9006. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Pesini, E.; Wallace, D.C. Evidence for adaptive selection acting on the tRNA and rRNA genes of human mitochondrial DNA. Hum. Mutat. 2006, 27, 1072–1081. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Kong, Y.; Zhang, H. Oxidative stress, mitochondrial dysfunction, and aging. J. Signal Transduct. 2012, 2011. [Google Scholar] [CrossRef] [PubMed]
- Kwok, C.S.N.; Quah, T.C.; Ariffin, H.; Tay, S.K.H.; Yeoh, A.E.J. Mitochondrial D-loop polymorphisms and mitochondrial DNA content in childhood acute lymphoblastic leukemia. J. Pediatr. Hematol. Oncol. 2011, 33, e239–e244. [Google Scholar] [CrossRef] [PubMed]
- Yu, M. Generation, function and diagnostic value of mitochondrial DNA copy number alterations in human cancers. Life Sci. 2011, 89, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, P.F.; Hudson, G. Mitochondrial genetics. Br. Med. Bull. 2013, 106, 135–159. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; Zhang, J.; Hancock, S.; Derbeneva, O.; Golhar, R.; Golik, P.; O’Hearn, S.; Levy, S.; Potluri, P.; Lvova, M.; et al. Progressive increase in mtDNA 3243A>G heteroplasmy causes abrupt transcriptional reprogramming. Proc. Natl. Acad. Sci. USA 2014, 111, E4033–E4042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Wu, J.; Dressman, D.C.; Iacobuzio-Donahue, C.; Markowitz, S.D.; Velculescu, V.E.; Diaz, L.A., Jr.; Kinzler, K.W.; Vogelstein, B.; Papadopoulos, N. Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nature 2010, 464, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Schon, E.A.; DiMauro, S.; Hirano, M. Human mitochondrial DNA: Roles of inherited and somatic mutations. Nat. Rev. Genet. 2012, 13, 878–890. [Google Scholar] [CrossRef] [PubMed]
- Bonora, E.; Porcelli, A.M.; Gasparre, G.; Biondi, A.; Ghelli, A.; Carelli, V.; Baracca, A.; Tallini, G.; Martinuzzi, A.; Lenaz, G.; et al. Defective oxidative phosphorylation in thyroid oncocytic carcinoma is associated with pathogenic mitochondrial DNA mutations affecting complexes I and III. Cancer Res. 2006, 66, 6087–6096. [Google Scholar] [CrossRef] [PubMed]
- Lorenc, A.; Bryk, J.; Golik, P.; Kupryjanczyk, J.; Ostrowski, J.; Pronicki, M.; Semczuk, A.; Szolkowska, M.; Bartnik, E. Homoplasmic melas A3243G mtDNA mutation in a colon cancer sample. Mitochondrion 2003, 3, 119–124. [Google Scholar] [CrossRef]
- Mimaki, M.; Hatakeyama, H.; Ichiyama, T.; Isumi, H.; Furukawa, S.; Akasaka, M.; Kamei, A.; Komaki, H.; Nishino, I.; Nonaka, I.; et al. Different effects of novel mtDNA G3242A and G3244A base changes adjacent to a common A3243G mutation in patients with mitochondrial disorders. Mitochondrion 2009, 9, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Del Mar O’Callaghan, M.; Emperador, S.; López-Gallardo, E.; Jou, C.; Buján, N.; Montero, R.; Garcia-Cazorla, A.; Gonzaga, D.; Ferrer, I.; Briones, P.; et al. New mitochondrial DNA mutations in tRNA associated with three severe encephalopamyopathic phenotypes: Neonatal, infantile, and childhood onset. Neurogenetics 2012, 13, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, M.; Tomelleri, G.; Vattemi, G.; Filosto, M.; Rizzuto, N.; Tonin, P. A new mutation in the mitochondrial tRNAAla gene in a patient with ophthalmoplegia and dysphagia. Neuromuscul. Disord. 2001, 11, 481–484. [Google Scholar] [CrossRef]
- Mayr, J.A.; Meierhofer, D.; Zimmermann, F.; Feichtinger, R.; Kogler, C.; Ratschek, M.; Schmeller, N.; Sperl, W.; Kofler, B. Loss of complex I due to mitochondrial DNA mutations in renal oncocytoma. Clin. Cancer Res. 2008, 14, 2270–2275. [Google Scholar] [CrossRef] [PubMed]
- Jeronimo, C.; Nomoto, S.; Caballero, O.L.; Usadel, H.; Henrique, R.; Varzim, G.; Oliveira, J.; Lopes, C.; Fliss, M.S.; Sidransky, D. Mitochondrial mutations in early stage prostate cancer and bodily fluids. Oncogene 2001, 20, 5195–5198. [Google Scholar] [CrossRef] [PubMed]
- Alston, C.L.; Morak, M.; Reid, C.; Hargreaves, I.P.; Pope, S.A.; Land, J.M.; Heales, S.J.; Horvath, R.; Mundy, H.; Taylor, R.W. A novel mitochondrial MTND5 frameshift mutation causing isolated complex I deficiency, renal failure and myopathy. Neuromuscul. Disord. 2010, 20, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Tseng, L.M.; Yin, P.H.; Yang, C.W.; Tsai, Y.F.; Hsu, C.Y.; Chi, C.W.; Lee, H.C. Somatic mutations of the mitochondrial genome in human breast cancers. Genes Chromosomes Cancer 2011, 50, 800–811. [Google Scholar] [CrossRef] [PubMed]
- Yin, P.H.; Wu, C.C.; Lin, J.C.; Chi, C.W.; Wei, Y.H.; Lee, H.C. Somatic mutations of mitochondrial genome in hepatocellular carcinoma. Mitochondrion 2010, 10, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Van Oven, M.; Kayser, M. Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum. Mutat. 2009, 30, E386–E394. [Google Scholar] [CrossRef] [PubMed]
- Booker, L.M.; Habermacher, G.M.; Jessie, B.C.; Sun, Q.C.; Baumann, A.K.; Amin, M.; Lim, S.D.; Fernandez-Golarz, C.; Lyles, R.H.; Brown, M.D.; et al. North american white mitochondrial haplogroups in prostate and renal cancer. J. Urol. 2006, 175, 468–472. [Google Scholar] [CrossRef]
- Bai, R.K.; Leal, S.M.; Covarrubias, D.; Liu, A.; Wong, L.J. Mitochondrial genetic background modifies breast cancer risk. Cancer Res. 2007, 67, 4687–4694. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Zheng, K.; Clark, J.; Swerdlow, R.H.; Pulst, S.M.; Sutton, J.P.; Shinobu, L.A.; Simon, D.K. Rapamycin drives selection against a pathogenic heteroplasmic mitochondrial DNA mutation. Hum. Mol. Genet. 2014, 23, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Livingstone, E.; Swann, S.; Lilla, C.; Schadendorf, D.; Roesch, A. Combining BRAF V 600E inhibition with modulators of the mitochondrial bioenergy metabolism to overcome drug resistance in metastatic melanoma. Exp. Dermatol. 2015, 24, 709–710. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, F.; Ghiorzo, P.; Queirolo, P. Overcoming resistance to BRAF inhibition in BRAF-mutated metastatic melanoma. Oncotarget 2014, 5, 10206–10221. [Google Scholar] [CrossRef] [PubMed]
- Hedinger, C.; Williams, E.D.; Sobin, L.H. The who histological classification of thyroid tumors: A commentary on the second edition. Cancer 1989, 63, 908–911. [Google Scholar] [CrossRef]
- Rieder, M.J.; Taylor, S.L.; Tobe, V.O.; Nickerson, D.A. Automating the identification of DNA variations using quality-based fluorescence re-sequencing: Analysis of the human mitochondrial genome. Nucleic Acids Res. 1998, 26, 967–973. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.M.; Kubacka, I.; Chinnery, P.F.; Lightowlers, R.N.; Turnbull, D.M.; Howell, N. Reanalysis and revision of the Cambridge Reference Sequence for human mitochondrial DNA. Nat. Genet. 1999, 23. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | Gene | Replacement | Amino-Acid Change or Watson-Crick Base-Pairing a | Conservation Index (%) b | Number of 66 PTC Patients (%) | Number of 376 Healthy Controls (%) | Heter/Homo c |
|---|---|---|---|---|---|---|---|
| RNA Region | |||||||
| 1629 | tRNAVal | A–T | A–U↓ | 24.4% | 1 (1.52%) | 0 (0.00%) | Homo |
| 2274 | 16S rRNA | A–G | 100% | 1 (1.52%) | 0 (0.00%) | Heter | |
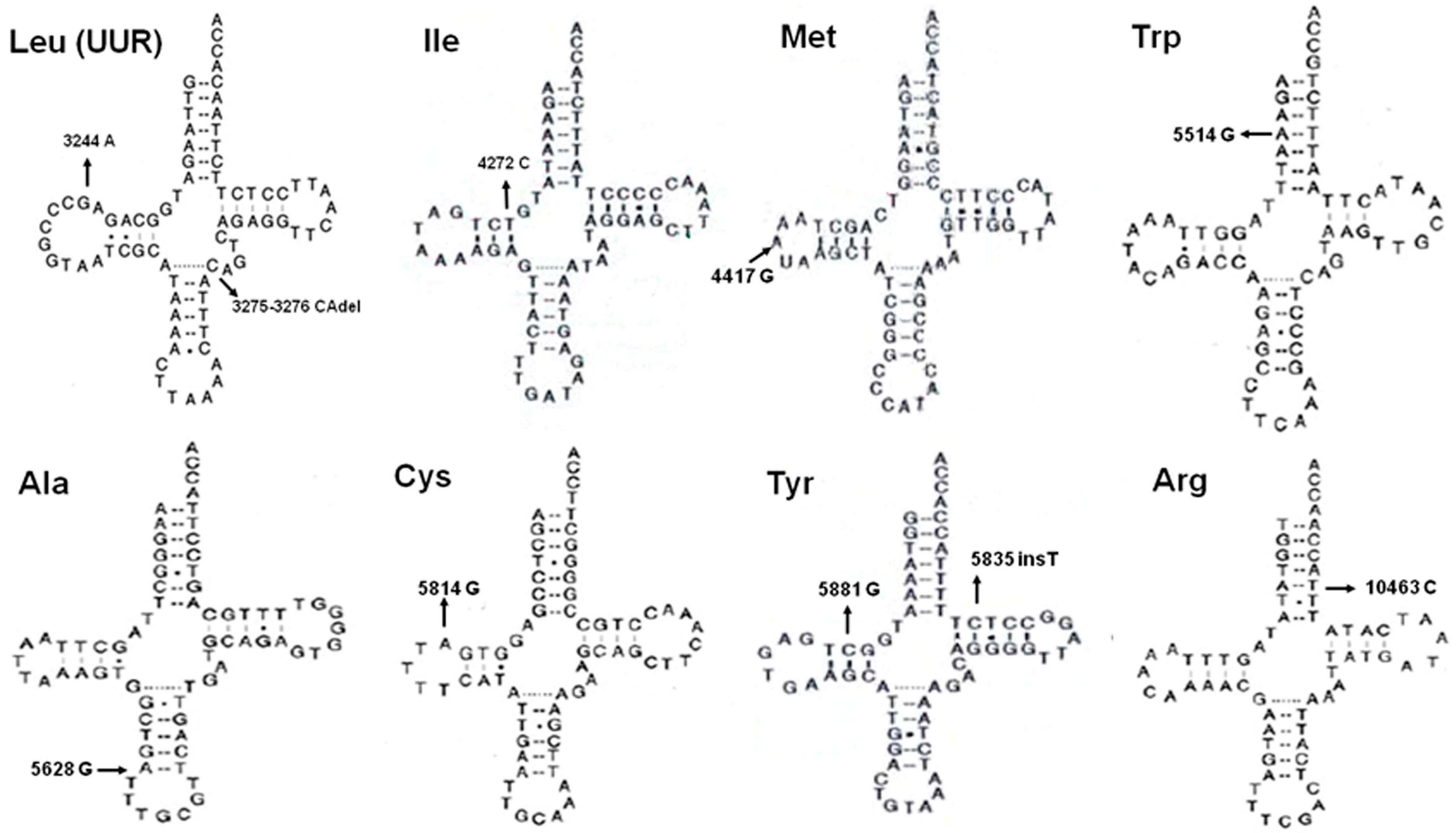
| 3275–3276 | tRNALeu(UUR) | Del CA | - | 1 (1.52%) | 0 (0.00%) | Heter | |
| 4272 | tRNAIle | T–C | A–U↓ | 100% | 1 (1.52%) | 0 (0.00%) | Homo |
| 5835 | tRNATyr | Ins T | - | 1 (1.52%) | 0 (0.00%) | Homo | |
| 5881 | tRNATyr | G–C | C–G↓ | 100% | 1 (1.52%) | 0 (0.00%) | Homo |
| 10040 | tRNAGly | C–A | 43.9% | 1 (1.52%) | 0 (0.00%) | Homo | |
| Protein-Coding Region | |||||||
| 4520–4521 | ND2 | Del AC | - | - | 1 (1.52%) | 0 (0.00%) | Homo |
| 4875 | ND2 | C–T | Leu -> Leu | 100% | 1 (1.52%) | 0 (0.00%) | Homo |
| 4969 | ND2 | G–A | No: Trp -> Ter d | 100% | 1 (1.52%) | 0 (0.00%) | Homo |
| 4971 | ND2 | G–A | No: Gly -> Ser | 100% | 1 (1.52%) | 0 (0.00%) | Homo |
| 5977 | COI | G–A | No: Trp -> Ter | 100% | 1 (1.52%) | 0 (0.00%) | Heter |
| 6238 | COI | T–C | No: Leu -> Pro | 100% | 1 (1.52%) | 0 (0.00%) | Heter |
| 7104 | COI | T–C | No: Ser -> Pro | 100% | 1 (1.52%) | 0 (0.00%) | Heter |
| 7750 | COII | C–A | No: Ile -> Met | 58.5% | 1 (1.52%) | 0 (0.00%) | Homo |
| 7928 | COII | G–A | No: Gly -> Ter | 56.1% | 1 (1.52%) | 0 (0.00%) | Homo |
| 9253 | COIII | G–A | No: Trp -> Ter | 100% | 1 (1.52%) | 0 (0.00%) | Heter |
| 10521 | ND4L | G–A | No: Gly -> Ter | 100% | 1 (1.52%) | 0 (0.00%) | Homo |
| 10622 | ND4L | C–T | Thr -> Thr | 36.6% | 1 (1.52%) | 0 (0.00%) | Homo |
| 11646 | ND4 | Ins T | - | - | 1 (1.52%) | 0 (0.00%) | Homo |
| 11673–11677 | ND4 | C5–C4 | - | - | 1 (1.52%) | 0 (0.00%) | Heter |
| 11673–11677 | ND4 | C5–C6 | - | - | 1 (1.52%) | 0 (0.00%) | Homo |
| 12794 | ND5 | T–A | No: Leu -> Ter | 100% | 1 (1.52%) | 0 (0.00%) | Heter |
| 12858 | ND5 | Ins T | - | - | 1 (1.52%) | 0 (0.00%) | Heter |
| 12943 | ND5 | C–T | No: Leu -> Phe | 24.4% | 1 (1.52%) | 0 (0.00%) | Heter |
| 13128–13132 | ND5 | C5–4 | - | - | 1 (1.52%) | 0 (0.00%) | Homo |
| 13170 | ND5 | Del A | - | - | 1 (1.52%) | 0 (0.00%) | Homo |
| 13621 | ND5 | C–T | No: Leu -> Phe | 51.2% | 1 (1.52%) | 0 (0.00%) | Homo |
| 13825 | ND5 | G–A | No: Gly -> Ter | 100% | 1 (1.52%) | 0 (0.00%) | Homo |
| 14310 | ND6 | C–A | No: Gly -> Trp | 70.7% | 1 (1.52%) | 0 (0.00%) | Heter |
| 14495–14502 | ND6 | (AAAT)2–1 | - | - | 1 (1.52%) | 0 (0.00%) | Homo |
| 14774 | Cytb | C–A | No: Leu -> Ile | 63.4% | 1 (1.52%) | 0 (0.00%) | Heter |
| 15018 | Cytb | T–A | No: Phe -> Tyr | 100% | 1 (1.52%) | 0 (0.00%) | Heter |
| Position | Gene | Change | Reported a | Number of 66 PTC Patients (%) | Number of 16 Normal Thyroid Tissues (%) | Number of 376 Healthy Controls (%) | Heter/Homo b |
|---|---|---|---|---|---|---|---|
| Nonsense Mutation | |||||||
| 4969 | ND2 | G–A | N | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Homo |
| 5977 | COI | G–A | N | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Heter |
| 7928 | COII | G–A | N | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Homo |
| 9253 | COIII | G–A | N | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Heter |
| 10521 | ND4L | G–A | N | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Homo |
| 12794 | ND5 | T–A | N | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Heter |
| 13825 | ND5 | G–A | N | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Homo |
| Frameshift Mutation | |||||||
| 4520–4521 | ND2 | Del AC | N | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Homo |
| 10952 | ND4 | Ins C | Y | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Homo |
| 11032–11038 | ND4 | A7–6 | Y | 4 (6.06%) | 0 (0.00%) | 0 (0.00%) | Homo + Heter |
| 11646 | ND4 | Ins T | N | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Homo |
| 11673–11677 | ND4 | C5–C4 | N | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Heter |
| 11673–11677 | ND4 | C5–C6 | N | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Homo |
| 12418–12425 | ND5 | Del A | Y | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Heter |
| 12858 | ND5 | Ins T | N | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Heter |
| 13128–13132 | ND5 | C5–4 | N | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Homo |
| 13170 | ND5 | Del A | N | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Homo |
| 14495–14502 | ND6 | (AAAT)2–1 | N | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Homo |
| Position | Gene | Change | Amino-Acid Change | Conservation Index (%) a | Reported b | Number of 66 PTC Patients (%) | Number of 16 Normal Thyroid Tissues (%) | Number of 376 Healthy Controls (%) | Polyphen-2 c | SIFT | Mutation Assesor | Provean | SNP&GO | Align GVGD d | PANTHER (Pdeleterious) e |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3392 f | ND1 | G–A | No: Gly -> Asp | 100.00% | Y | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Probably | Not Tolerated | High | Deleterious | Disease | C65 | NA g |
| 3644 | ND1 | T–C | No: Val -> Ala | 97.60% | Y | 1 (1.52%) | 0 (0.00%) | 2 (0.53%) | Benign | Not Tolerated | Medium | Deleterious | Neutral | C65 | 0.29125 |
| 3679 | ND1 | T–C | No: Ser -> Pro | 100.00% | Y | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Probably | Not Tolerated | High | Deleterious | Disease | C65 | 0.74261 |
| 3745 | ND1 | G–A | No: Ala -> Thr | 92.70% | Y | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Benign | Not Tolerated | Low | Neutral | Neutral | C55 | 0.21113 |
| 4971 | ND2 | G–A | No: Gly -> Ser | 100.00% | N | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Probably | Not Tolerated | Medium | Deleterious | Neutral | C55 | 0.36251 |
| 6238 | COI | T–C | No: Leu -> Pro | 100.00% | N | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Probably | Not Tolerated | High | Deleterious | Disease | C65 | 0.87509 |
| 6340 | COI | C–T | No: Thr -> Ile | 82.90% | Y | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Benign | Not Tolerated | Medium | Neutral | Neutral | C65 | 0.21096 |
| 6681 | COI | T–C | No: Tyr -> His | 85.40% | Y | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Benign | Tolerated | Neutral | Neutral | Neutral | C65 | 0.32881 |
| 7104 | COI | T–C | No: Ser -> Pro | 100.00% | N | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Possibly | Not Tolerated | Neutral | Neutral | Disease | C65 | 0.5134 |
| 7329 | COI | T–C | No: Phe ->Leu | 100.00% | N | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Benign | Tolerated | Low | Neutral | Neutral | C15 | 0.16379 |
| 8156 | COII | G–A | No: Val -> Met | 75.61% | N | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Probably | Not Tolerated | Medium | Neutral | Neutral | C15 | 0.53442 |
| 8989 | ATP6 | G–A | No: Ala -> Thr | 100.00% | Y | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Probably | Not Tolerated | Low | Deleterious | Neutral | C55 | 0.47286 |
| 9187 | ATP6 | T–C | No: Tyr -> His | 100.00% | Y | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Probably | Not Tolerated | High | Deleterious | Disease | C65 | NA |
| 9355 | COIII | A–G | No: Asn -> Ser | 82.90% | Y | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Benign | Tolerated | Neutral | Neutral | Neutral | C45 | 0.14014 |
| 10573 | ND4L | G–A | No: Gly -> Glu | 97.60% | Y | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Probably | Not Tolerated | High | Deleterious | Neutral | C65 | 0.40946 |
| 12850 | ND5 | A–G | No: Ile -> Val | 90.20% | Y | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Possibly | Tolerated | Neutral | Neutral | Neutral | C25 | 0.50297 |
| 13535 | ND5 | A–G | No: Asn -> Ser | 87.80% | Y | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Benign | Not Tolerated | Low | Deleterious | Neutral | C45 | NA |
| 13748 | ND5 | A–G | No: Asn -> Ser | 85.40% | Y | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Benign | Tolerated | Neutral | Neutral | Neutral | C45 | 0.5082 |
| 14310 | ND6 | C–A | No: Gly -> Trp | 78.05% | N | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Probably | Not Tolerated | Medium | Deleterious | Disease | C65 | 0.71527 |
| 14463 | ND6 | T–C | No: Thr -> Ala | 90.20% | Y | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Benign | Tolerated | Neutral | Deleterious | Neutral | C55 | 0.15283 |
| 15018 | Cytb | T–A | No: Phe -> Tyr | 100.00% | N | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Possibly | Not Tolerated | High | Deleterious | Disease | C15 | 0.68543 |
| 15045 | Cytb | G–A | No: Arg -> Gln | 100.00% | Y | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Probably | Not Tolerated | High | Deleterious | Disease | C35 | 0.59378 |
| 15090 | Cytb | T–C | No: Ile -> Thr | 85.40% | Y | 1 (1.52%) | 0 (0.00%) | 1 (0.27%) | Possibly | Tolerated | Low | Deleterious | Neutral | C65 | 0.42865 |
| 15479 | Cytb | T–C | No: Phe -> Leu | 80.50% | Y | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Benign | Tolerated | Low | Deleterious | Neutral | C15 | 0.39962 |
| 15483 | Cytb | C–T | No: Ser -> Leu | 80.50% | Y | 1 (1.52%) | 0 (0.00%) | 0 (0.00%) | Possibly | Tolerated | Low | Deleterious | Neutral | C65 | 0.45816 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, X.; Wang, W.; Ruan, G.; Liang, M.; Zheng, J.; Chen, Y.; Wu, H.; Fahey, T.J., III; Guan, M.; Teng, L. A Comprehensive Characterization of Mitochondrial Genome in Papillary Thyroid Cancer. Int. J. Mol. Sci. 2016, 17, 1594. https://doi.org/10.3390/ijms17101594
Su X, Wang W, Ruan G, Liang M, Zheng J, Chen Y, Wu H, Fahey TJ III, Guan M, Teng L. A Comprehensive Characterization of Mitochondrial Genome in Papillary Thyroid Cancer. International Journal of Molecular Sciences. 2016; 17(10):1594. https://doi.org/10.3390/ijms17101594
Chicago/Turabian StyleSu, Xingyun, Weibin Wang, Guodong Ruan, Min Liang, Jing Zheng, Ye Chen, Huiling Wu, Thomas J. Fahey, III, Minxin Guan, and Lisong Teng. 2016. "A Comprehensive Characterization of Mitochondrial Genome in Papillary Thyroid Cancer" International Journal of Molecular Sciences 17, no. 10: 1594. https://doi.org/10.3390/ijms17101594