
PlGF and VEGF-A Regulate Growth of High-Risk MYCN-Single Copy Neuroblastoma Xenografts via Different Mechanisms


Abstract
:
1. Introduction
2. Results
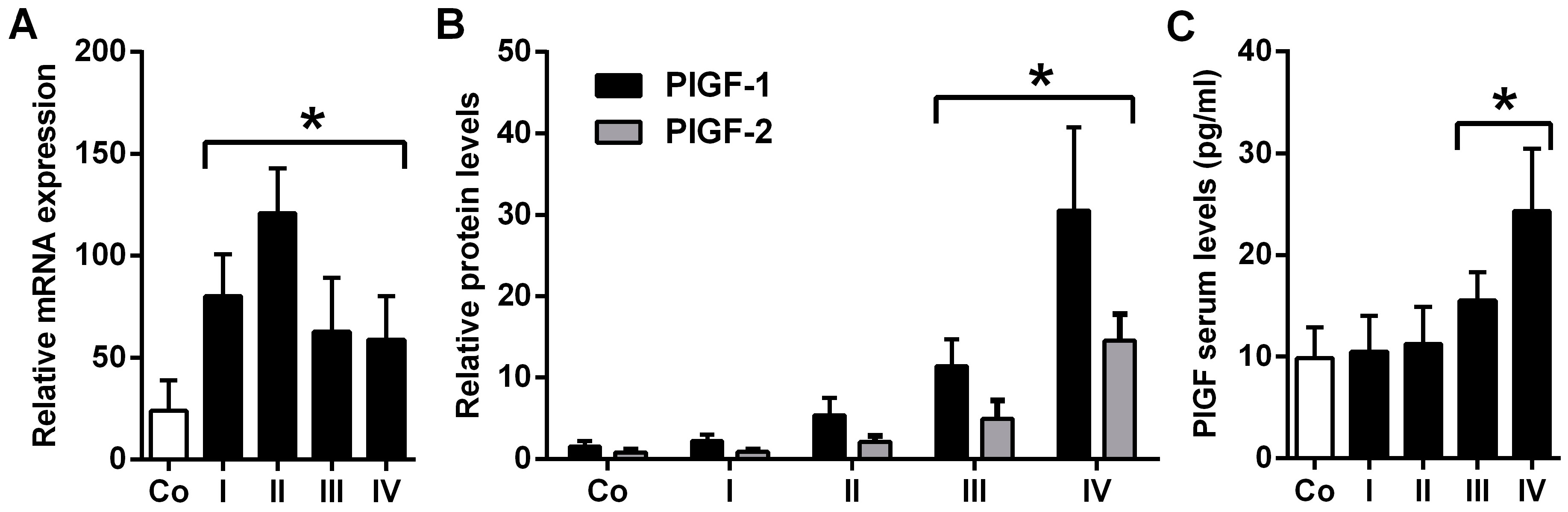
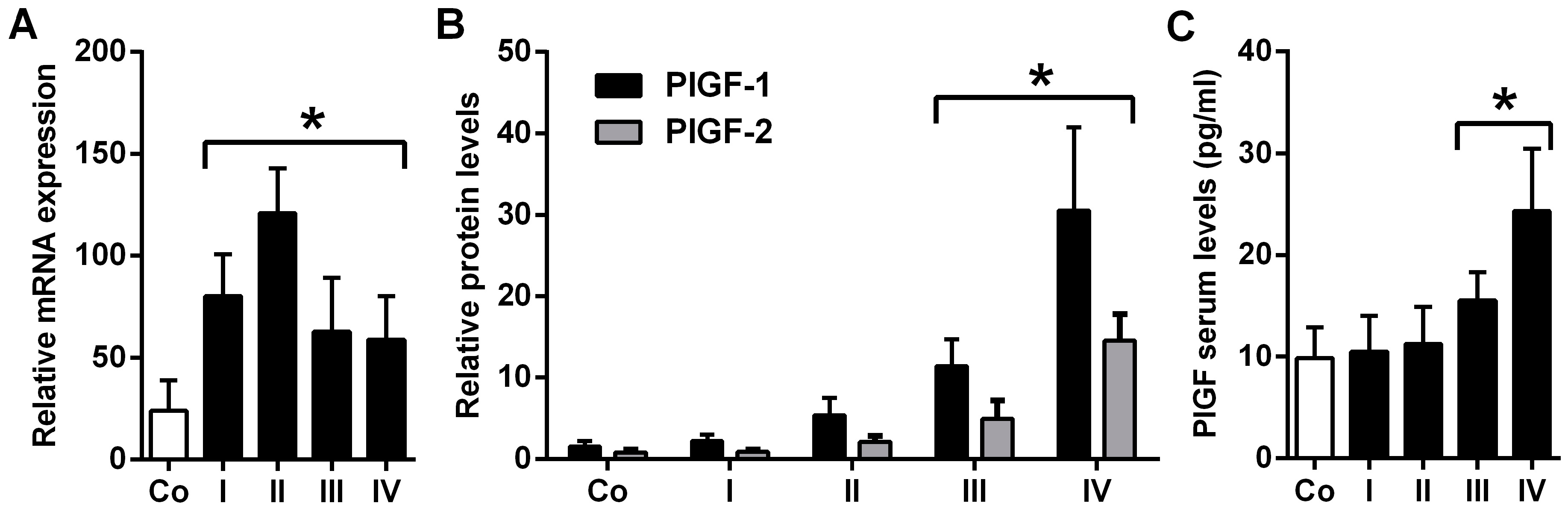
2.1. Placental Growth Factor (PlGF) Expression Is up-Rregulated in Neuroblastoma
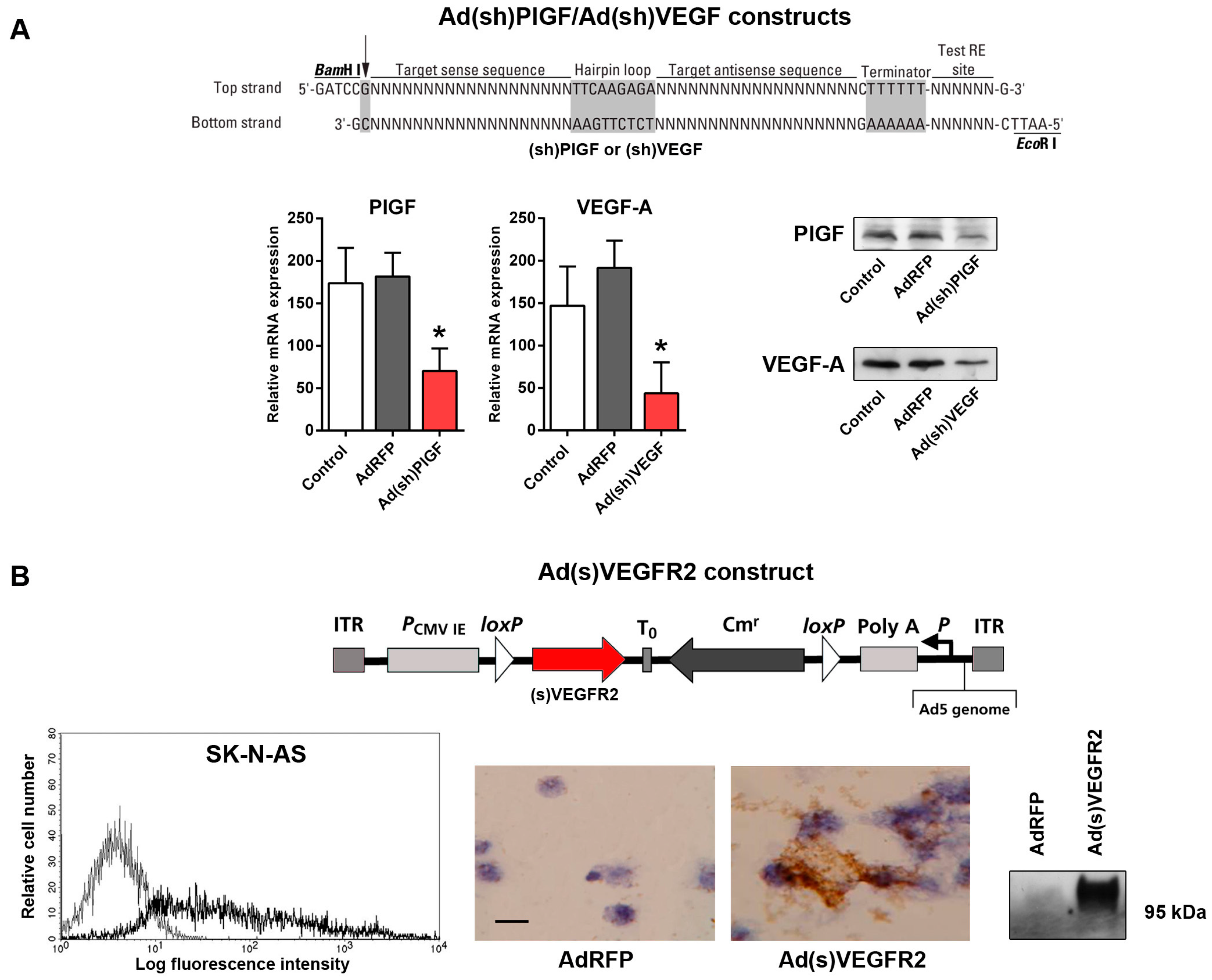
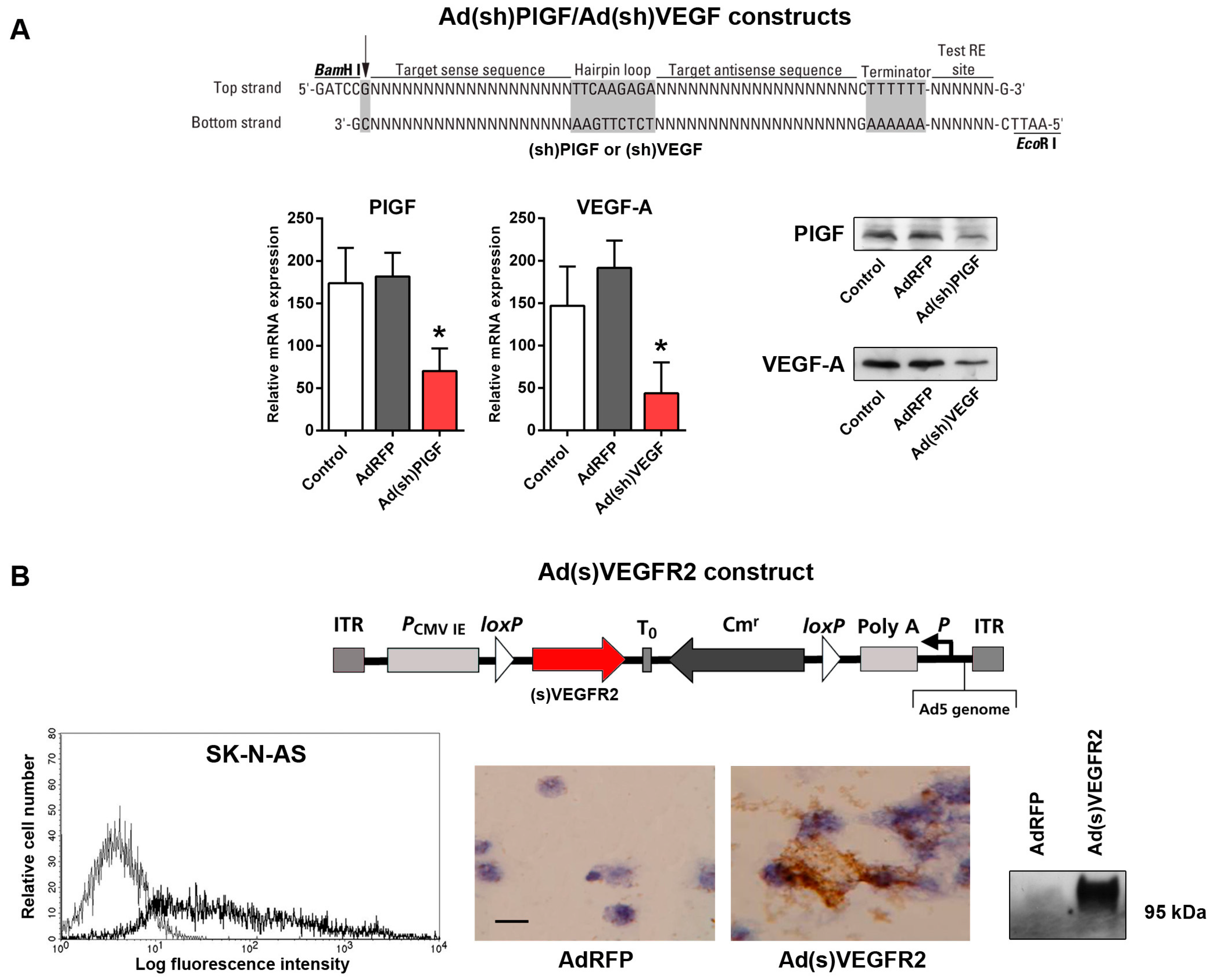
2.2. Generation of Replication-Incompetent Adenoviruses (Ads) Expressing Short Hairpin (shRNA) Specific to PlGF and VEGF and of Soluble (s)VEGFR2-Expressing Replication-Incompetent Ads
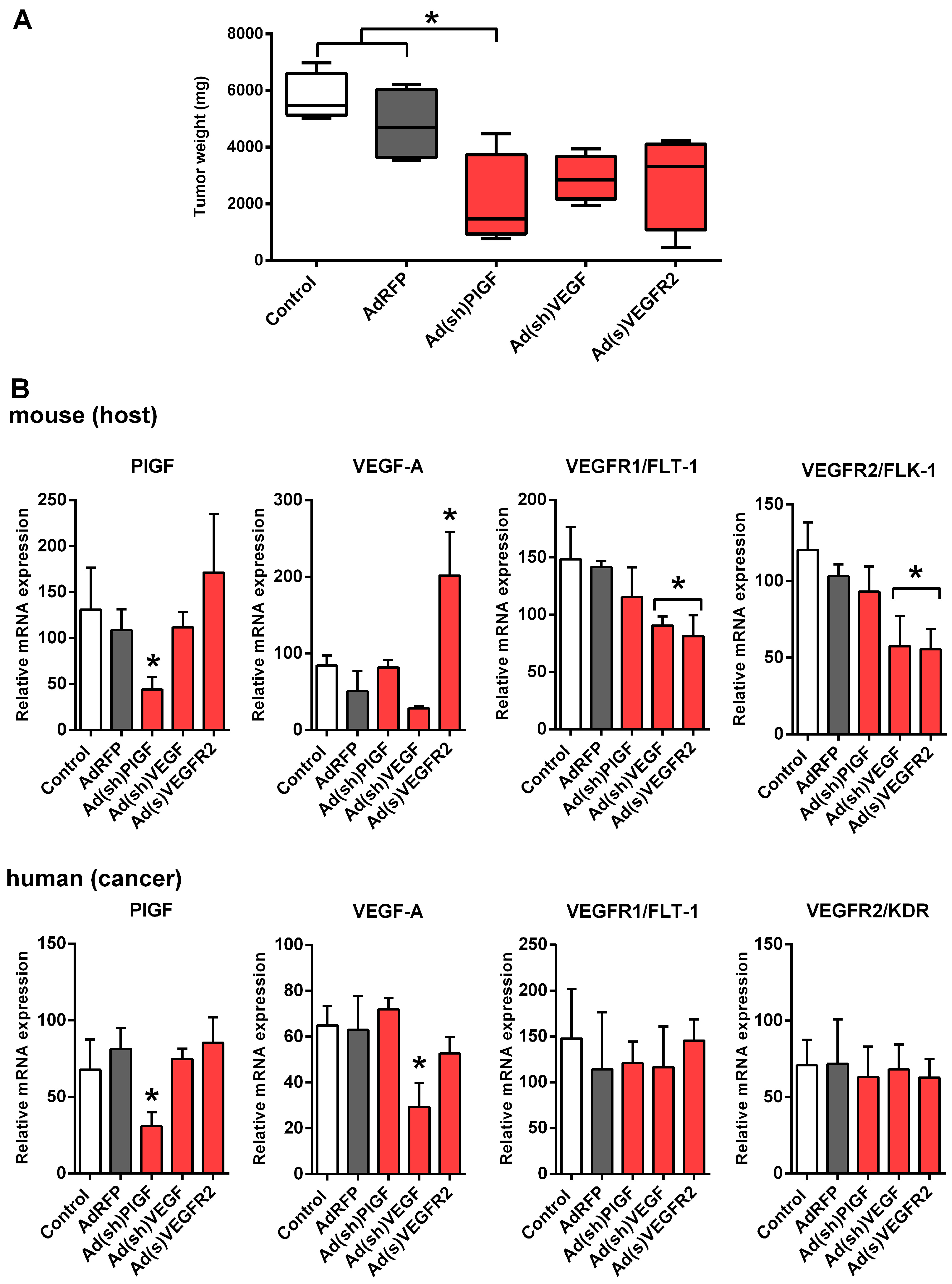
2.3. PlGF Blockade Reduces Growth of MYCN-Non-Amplified NB Xenografts
2.4. PlGF Gene Silencing Does Not Affect mRNA Levels of Its Receptor VEGFR1/FLT-1 in NB Xenografts
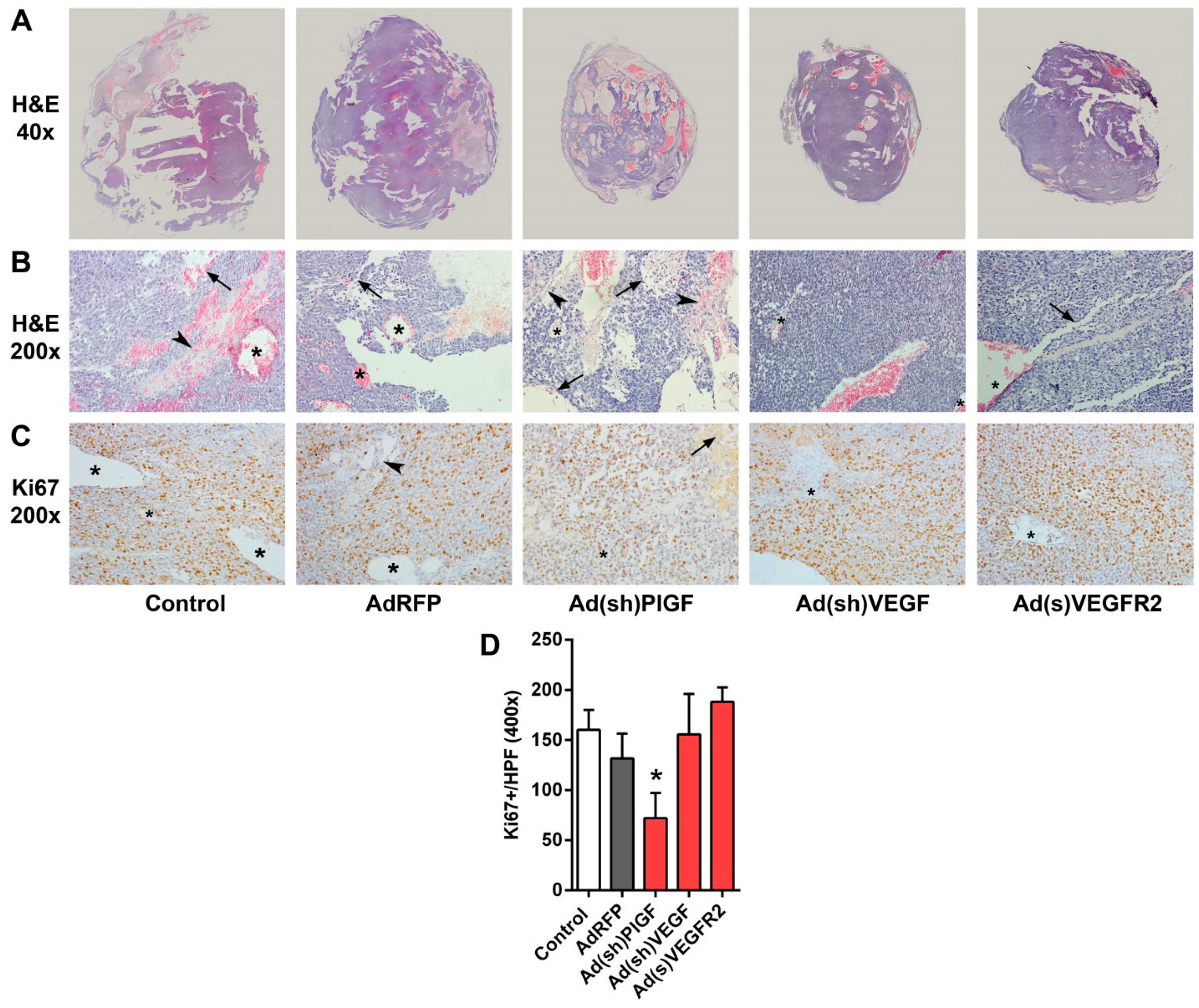
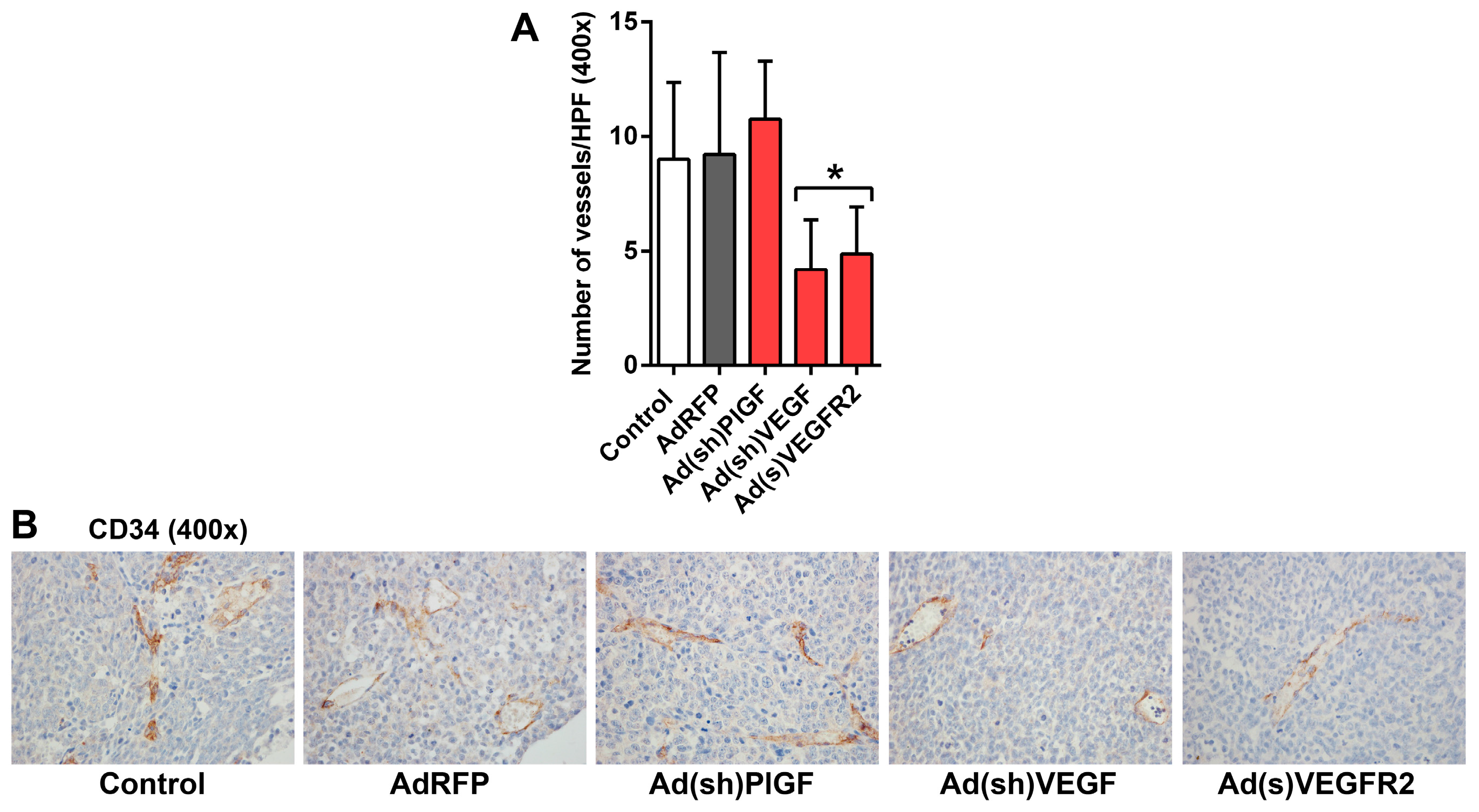
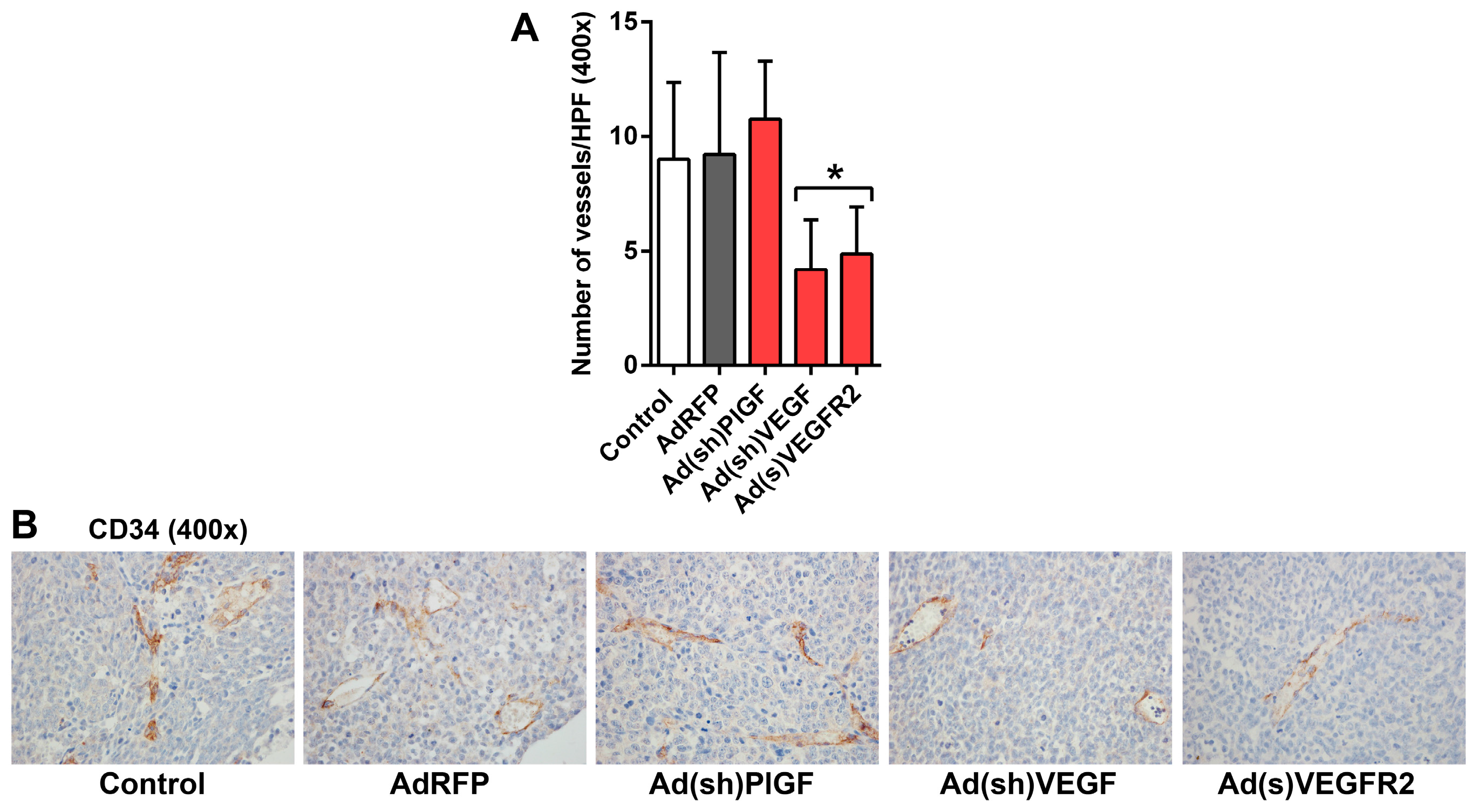
2.5. PlGF and VEGF Blockade Differentially Change the Histologic Appearance of NB Xenografts
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Cell Lines
4.3. Generation of PlGF- and VEGF-Specific shRNA-Expressing Ads
4.4. Construction of the Ad(s)VEGFR2 Adenoviral Vector
4.5. Quantitative Real-Time RT-PCR (qRT-PCR)
4.6. Protein Isolation and Western Blotting
4.7. Enzyme-Linked Immunosorbent Assay (ELISA)
4.8. Fluorescence-Activated Cell Sorting (FACS) Analysis
4.9. SK-N-AS Tumor Cell Implantation and Adenovirus Injection/Analysis of Adenoviruses in Vivo
4.10. Histology and Immunohistochemistry
4.11. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Schwab, M.; Westermann, F.; Hero, B.; Berthold, F. Neuroblastoma: Biology and molecular and chromosomal pathology. Lancet Oncol. 2003, 4, 472–480. [Google Scholar] [CrossRef]
- Brodeur, G.M.; Pritchard, J.; Berthold, F.; Carlsen, N.L.; Castel, V.; Castelberry, R.P.; de Bernardi, B.; Evans, A.E.; Favrot, M.; Hedborg, F.; et al. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J. Clin. Oncol. 1993, 11, 1466–1477. [Google Scholar] [PubMed]
- Maris, J.M. Recent advances in neuroblastoma. N. Engl. J. Med. 2010, 362, 2202–2211. [Google Scholar] [CrossRef] [PubMed]
- Rossler, J.; Taylor, M.; Geoerger, B.; Farace, F.; Lagodny, J.; Peschka-Suss, R.; Niemeyer, C.M.; Vassal, G. Angiogenesis as a target in neuroblastoma. Eur. J. Cancer 2008, 44, 1645–1656. [Google Scholar] [CrossRef] [PubMed]
- Meitar, D.; Crawford, S.E.; Rademaker, A.W.; Cohn, S.L. Tumor angiogenesis correlates with metastatic disease, n-myc amplification, and poor outcome in human neuroblastoma. J. Clin. Oncol. 1996, 14, 405–414. [Google Scholar] [PubMed]
- Ribatti, D. Anti-angiogenesis in neuroblastoma. Crit. Rev. Oncol. Hematol. 2013, 86, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Eggert, A.; Ikegaki, N.; Kwiatkowski, J.; Zhao, H.; Brodeur, G.M.; Himelstein, B.P. High-level expression of angiogenic factors is associated with advanced tumor stage in human neuroblastomas. Clin. Cancer Res. 2000, 6, 1900–1908. [Google Scholar] [PubMed]
- Kang, J.; Rychahou, P.G.; Ishola, T.A.; Mourot, J.M.; Evers, B.M.; Chung, D.H. N-myc is a novel regulator of PI3K-mediated VEGF expression in neuroblastoma. Oncogene 2008, 27, 3999–4007. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Raffaghello, L.; Pastorino, F.; Nico, B.; Brignole, C.; Vacca, A.; Ponzoni, M. In vivo angiogenic activity of neuroblastoma correlates with mycn oncogene overexpression. Int. J. Cancer 2002, 102, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Zaghloul, N.; Hernandez, S.L.; Bae, J.O.; Huang, J.; Fisher, J.C.; Lee, A.; Kadenhe-Chiweshe, A.; Kandel, J.J.; Yamashiro, D.J. Vascular endothelial growth factor blockade rapidly elicits alternative proangiogenic pathways in neuroblastoma. Int. J. Oncol. 2009, 34, 401–407. [Google Scholar] [PubMed]
- Ross, R.A.; Walton, J.D.; Han, D.; Guo, H.F.; Cheung, N.K. A distinct gene expression signature characterizes human neuroblastoma cancer stem cells. Stem Cell Res. 2015, 15, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Moons, L.; Luttun, A.; Vincenti, V.; Compernolle, V.; De Mol, M.; Wu, Y.; Bon, F.; Devy, L.; Beck, H.; et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat. Med. 2001, 7, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Dewerchin, M.; Carmeliet, P. PLGF: A multitasking cytokine with disease-restricted activity. Cold Spring Harb. Perspect. Med. 2012, 2, a011056. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, S.K.; Giri, R.K.; Perelman, N.; Johnson, C.; Malik, P.; Kalra, V.K. Mechanism of monocyte activation and expression of proinflammatory cytochemokines by placenta growth factor. Blood 2003, 102, 1515–1524. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Allavena, P.; Mantovani, A. Tumor-associated macrophages: Functional diversity, clinical significance, and open questions. Semin. Immunopathol. 2013, 35, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Hilfenhaus, G.; Gohrig, A.; Pape, U.F.; Neumann, T.; Jann, H.; Zdunek, D.; Hess, G.; Stassen, J.M.; Wiedenmann, B.; Detjen, K.; et al. Placental growth factor supports neuroendocrine tumor growth and predicts disease prognosis in patients. Endocr. Relat. Cancer 2013, 20, 305–319. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Kerbel, R.S. Tumor angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Vacca, A.; Nico, B.; de Falco, G.; Giuseppe Montaldo, P.; Ponzoni, M. Angiogenesis and anti-angiogenesis in neuroblastoma. Eur. J. Cancer 2002, 38, 750–757. [Google Scholar] [CrossRef]
- Ribatti, D.; Ponzoni, M. Antiangiogenic strategies in neuroblastoma. Cancer Treat. Rev. 2005, 31, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Fakhari, M.; Pullirsch, D.; Paya, K.; Abraham, D.; Hofbauer, R.; Aharinejad, S. Upregulation of vascular endothelial growth factor receptors is associated with advanced neuroblastoma. J. Pediatr. Surg. 2002, 37, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Komuro, H.; Kaneko, S.; Kaneko, M.; Nakanishi, Y. Expression of angiogenic factors and tumor progression in human neuroblastoma. J. Cancer Res. Clin. Oncol. 2001, 127, 739–743. [Google Scholar] [PubMed]
- Becker, J.; Pavlakovic, H.; Ludewig, F.; Wilting, F.; Weich, H.A.; Albuquerque, R.; Ambati, J.; Wilting, J. Neuroblastoma progression correlates with downregulation of the lymphangiogenesis inhibitor sVEGFR-2. Clin. Cancer Res. 2010, 16, 1431–1441. [Google Scholar] [CrossRef] [PubMed]
- Canete, A.; Navarro, S.; Bermudez, J.; Pellin, A.; Castel, V.; Llombart-Bosch, A. Angiogenesis in neuroblastoma: Relationship to survival and other prognostic factors in a cohort of neuroblastoma patients. J. Clin. Oncol. 2000, 18, 27–34. [Google Scholar] [PubMed]
- Chlenski, A.; Liu, S.; Cohn, S.L. The regulation of angiogenesis in neuroblastoma. Cancer Lett. 2003, 197, 47–52. [Google Scholar] [CrossRef]
- Shusterman, S.; Maris, J.M. Prospects for therapeutic inhibition of neuroblastoma angiogenesis. Cancer Lett. 2005, 228, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Glade Bender, J.L.; Adamson, P.C.; Reid, J.M.; Xu, L.; Baruchel, S.; Shaked, Y.; Kerbel, R.S.; Cooney-Qualter, E.M.; Stempak, D.; Chen, H.X.; et al. Phase I trial and pharmacokinetic study of bevacizumab in pediatric patients with refractory solid tumors: A children’s oncology group study. J. Clin. Oncol. 2008, 26, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Benesch, M.; Windelberg, M.; Sauseng, W.; Witt, V.; Fleischhack, G.; Lackner, H.; Gadner, H.; Bode, U.; Urban, C. Compassionate use of bevacizumab (Avastin®) in children and young adults with refractory or recurrent solid tumors. Ann. Oncol. 2008, 19, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D. Tumor refractoriness to anti-VEGF therapy. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Verheul, H.M.; Pinedo, H.M. Possible molecular mechanisms involved in the toxicity of angiogenesis inhibition. Nat. Rev. Cancer 2007, 7, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Cho, C.S.; Kim, W.U. Role of placenta growth factor in cancer and inflammation. Exp. Mol. Med. 2012, 44, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.H.; Lloyd, P.G. Post-transcriptional regulation of placenta growth factor mRNA by hydrogen peroxide. Microvasc. Res. 2012, 84, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Ahn, H.; Hinrichs, M.; Torry, R.J.; Torry, D.S. Evidence of a novel isoform of placenta growth factor (PLGF-4) expressed in human trophoblast and endothelial cells. J. Reprod. Immunol. 2003, 60, 53–60. [Google Scholar] [CrossRef]
- Yao, Y.G.; Yang, H.S.; Cao, Z.; Danielsson, J.; Duh, E.J. Upregulation of placental growth factor by vascular endothelial growth factor via a post-transcriptional mechanism. FEBS Lett. 2005, 579, 1227–1234. [Google Scholar] [CrossRef] [PubMed]
- Van de Veire, S.; Stalmans, I.; Heindryckx, F.; Oura, H.; Tijeras-Raballand, A.; Schmidt, T.; Loges, S.; Albrecht, I.; Jonckx, B.; Vinckier, S.; et al. Further pharmacological and genetic evidence for the efficacy of plgf inhibition in cancer and eye disease. Cell 2010, 141, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Bais, C.; Wu, X.M.; Yao, J.; Yang, S.Y.; Crawford, Y.; McCutcheon, K.; Tan, C.; Kolumam, G.; Vernes, J.M.; Eastham-Anderson, J.; et al. PLGF blockade does not inhibit angiogenesis during primary tumor growth. Cell 2010, 141, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.; Kharabi Masouleh, B.; Loges, S.; Cauwenberghs, S.; Fraisl, P.; Maes, C.; Jonckx, B.; de Keersmaecker, K.; Kleppe, M.; Tjwa, M.; et al. Loss or inhibition of stromal-derived PLGF prolongs survival of mice with imatinib-resistant Bcr-Abl1+ leukemia. Cancer Cell 2011, 19, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Bagley, R.G.; Ren, Y.; Weber, W.; Yao, M.; Kurtzberg, L.; Pinckney, J.; Bangari, D.; Nguyen, C.; Brondyk, W.; Kaplan, J.; et al. Placental growth factor upregulation is a host response to antiangiogenic therapy. Clin. Cancer Res. 2011, 17, 976–988. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Izycka-Swieszewska, E.; Drozynska, E.; Rzepko, R.; Dembowska, B.; Grajkowska, W.; Brozyna, A.; Perek, D.; Balcerska, A.; Jaskiewicz, K. Clinicopathological considerations on angiogenic potential in neuroblastoma schwannian stroma—Poor tumours. Folia Neuropathol. 2007, 45, 1–8. [Google Scholar] [PubMed]
- Rossler, J.; Breit, S.; Havers, W.; Schweigerer, L. Vascular endothelial growth factor expression in human neuroblastoma: Up-regulation by hypoxia. Int. J. Cancer 1999, 81, 113–117. [Google Scholar] [CrossRef]
- Seftor, R.E.; Hess, A.R.; Seftor, E.A.; Kirschmann, D.A.; Hardy, K.M.; Margaryan, N.V.; Hendrix, M.J. Tumor cell vasculogenic mimicry: From controversy to therapeutic promise. Am. J. Pathol. 2012, 181, 1115–1125. [Google Scholar] [CrossRef] [PubMed]
- Pezzolo, A.; Parodi, F.; Corrias, M.V.; Cinti, R.; Gambini, C.; Pistoia, V. Tumor origin of endothelial cells in human neuroblastoma. J. Clin. Oncol. 2007, 25, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.M.; Maris, J.M.; Hogarty, M.D.; Seeger, R.C.; Reynolds, C.P.; Brodeur, G.M.; White, P.S. Homozygous deletion of cdkn2a (p16INK4a/p14ARF) but not within 1p36 or at other tumor suppressor loci in neuroblastoma. Cancer Res. 2001, 61, 679–686. [Google Scholar] [PubMed]
- Lucas, T.; Abraham, D.; Untergasser, G.; Zins, K.; Hofer, E.; Gunsilius, E.; Aharinejad, S. Adenoviral-mediated endothelial precursor cell delivery of soluble CD115 suppresses human prostate cancer xenograft growth in mice. Stem Cells 2009, 27, 2342–2352. [Google Scholar] [CrossRef] [PubMed]
- Zins, K.; Thomas, A.; Lucas, T.; Sioud, M.; Aharinejad, S.; Abraham, D. Inhibition of stromal PLGF suppresses the growth of prostate cancer xenografts. Int. J. Mol. Sci. 2013, 14, 17958–17971. [Google Scholar] [CrossRef] [PubMed]
- Paulus, P.; Stanley, E.R.; Schafer, R.; Abraham, D.; Aharinejad, S. Colony-stimulating factor-1 antibody reverses chemoresistance in human MCF-7 breast cancer xenografts. Cancer Res. 2006, 66, 4349–4356. [Google Scholar] [CrossRef] [PubMed]
- Zins, K.; Schafer, R.; Paulus, P.; Dobler, S.; Fakhari, N.; Sioud, M.; Aharinejad, S.; Abraham, D. Frizzled2 signaling regulates growth of high-risk neuroblastomas by interfering with β-catenin-dependent and β-catenin-independent signaling pathways. Oncotarget 2016. [Google Scholar] [CrossRef]
- Zins, K.; Gunawardhana, S.; Lucas, T.; Abraham, D.; Aharinejad, S. Targeting Cdc42 with the small molecule drug AZA197 suppresses primary colon cancer growth and prolongs survival in a preclinical mouse xenograft model by downregulation of PAK1 activity. J. Transl. Med. 2013, 11, 295. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Age, Months | Sex, M/F | Tumor Location |
|---|---|---|---|
| Control | 21 ± 17 | 3/4 | – |
| Stage I * | 7 ± 4 | 5/3 | Adrenal |
| Stage II | 9 ± 3 | 7/3 | 9 Adrenal/1 Vagus nerve |
| Stage III | 10 ± 4 | 4/5 | 8 Adrenal/1 Sympathetic trunk |
| Stage IV | 14 ± 11 | 6/4 | Adrenal |
| Gene | Primer Sense 5′–3′ | Primer Antisense 5′–3′ |
|---|---|---|
| human PlGF | GTTCAGCCCATCCTGTGTCT | CTTCATCTTCTCCCGCAGAG |
| human VEGF-A | AGCCTTGCCTTGCTGCTCTA | GTGCTGGCCTTGGTGAGG |
| human FLT-1 | GTCACAGAAGAGGATGAAGGTGTC | CACAGTCCGGCACGTAGGTGATT |
| human KDR | GCATCTCATCTGTTACAGC | CTTCATCAATCTTTACCCC |
| mouse PlGF | GAGCTTCGGCTTGGGAAGAAG | GTTCCAGAGAGGGGACAAAGG |
| mouse VEGF-A | GCAGAAGTCCCATGAAGTGATC | CTCCAGGGCTTCATCGTTACAG |
| mouse FLT-1 | AAACCTGTCCAACTACCTC | TAATCCTCGTCTCCTTCC |
| mouse KDR | GGAGATTGAAAGAAGGAAC | ACTTCCTCTTCCTCCATAC |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zins, K.; Kovatchki, D.; Lucas, T.; Abraham, D. PlGF and VEGF-A Regulate Growth of High-Risk MYCN-Single Copy Neuroblastoma Xenografts via Different Mechanisms. Int. J. Mol. Sci. 2016, 17, 1613. https://doi.org/10.3390/ijms17101613
Zins K, Kovatchki D, Lucas T, Abraham D. PlGF and VEGF-A Regulate Growth of High-Risk MYCN-Single Copy Neuroblastoma Xenografts via Different Mechanisms. International Journal of Molecular Sciences. 2016; 17(10):1613. https://doi.org/10.3390/ijms17101613
Chicago/Turabian StyleZins, Karin, Daniel Kovatchki, Trevor Lucas, and Dietmar Abraham. 2016. "PlGF and VEGF-A Regulate Growth of High-Risk MYCN-Single Copy Neuroblastoma Xenografts via Different Mechanisms" International Journal of Molecular Sciences 17, no. 10: 1613. https://doi.org/10.3390/ijms17101613