
Stereoselective Blockage of Quinidine and Quinine in the hERG Channel and the Effect of Their Rescue Potency on Drug-Induced hERG Trafficking Defect

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
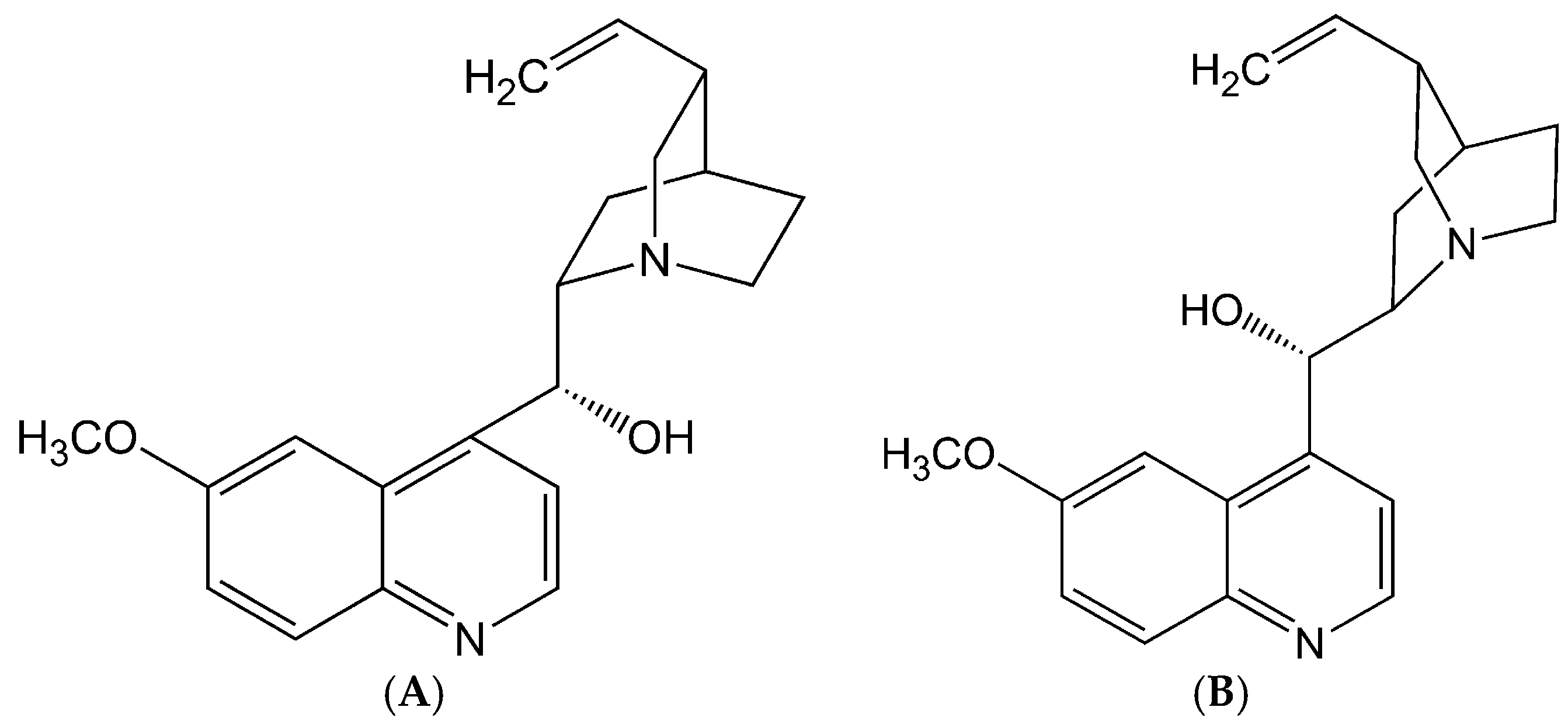
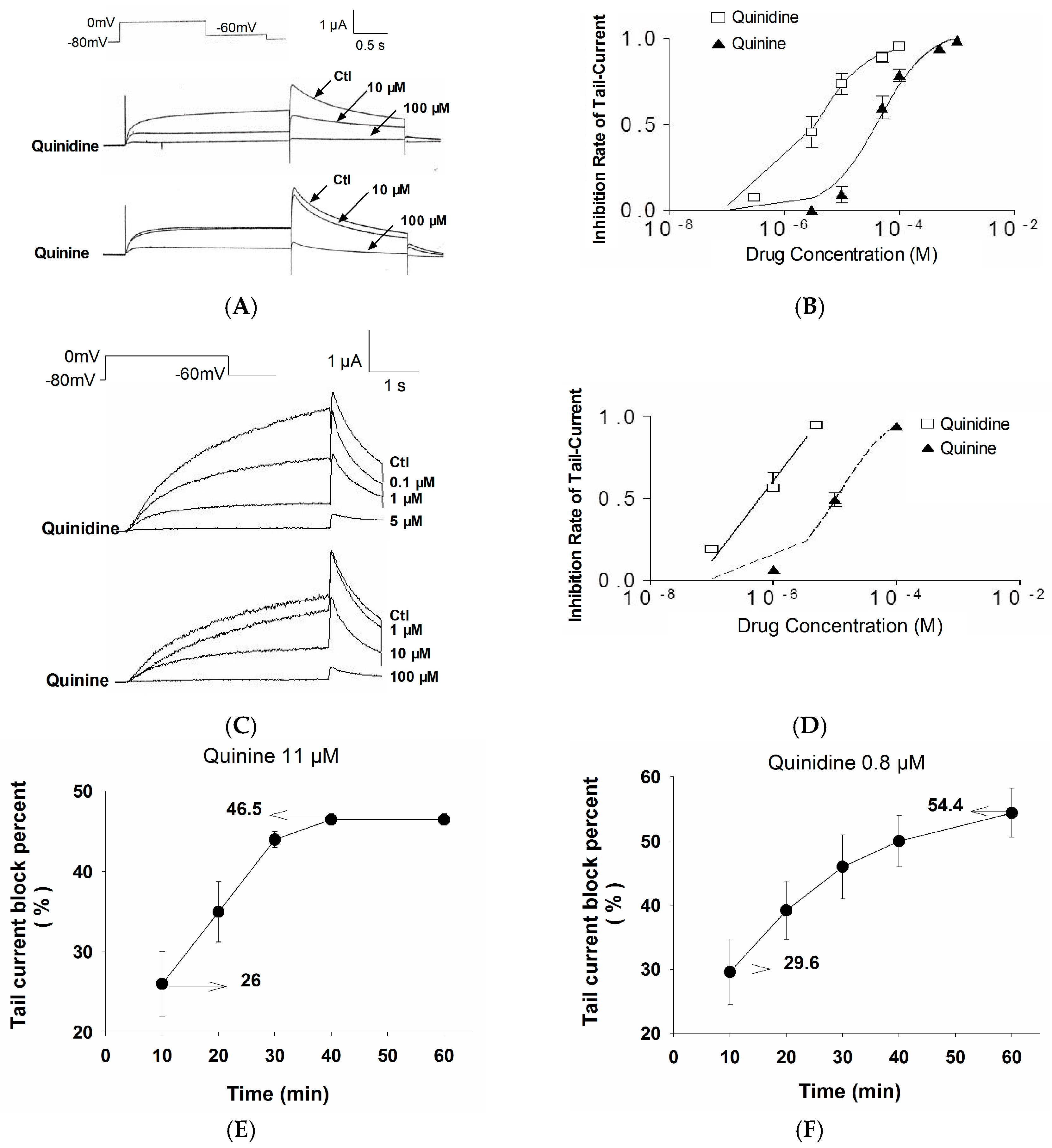
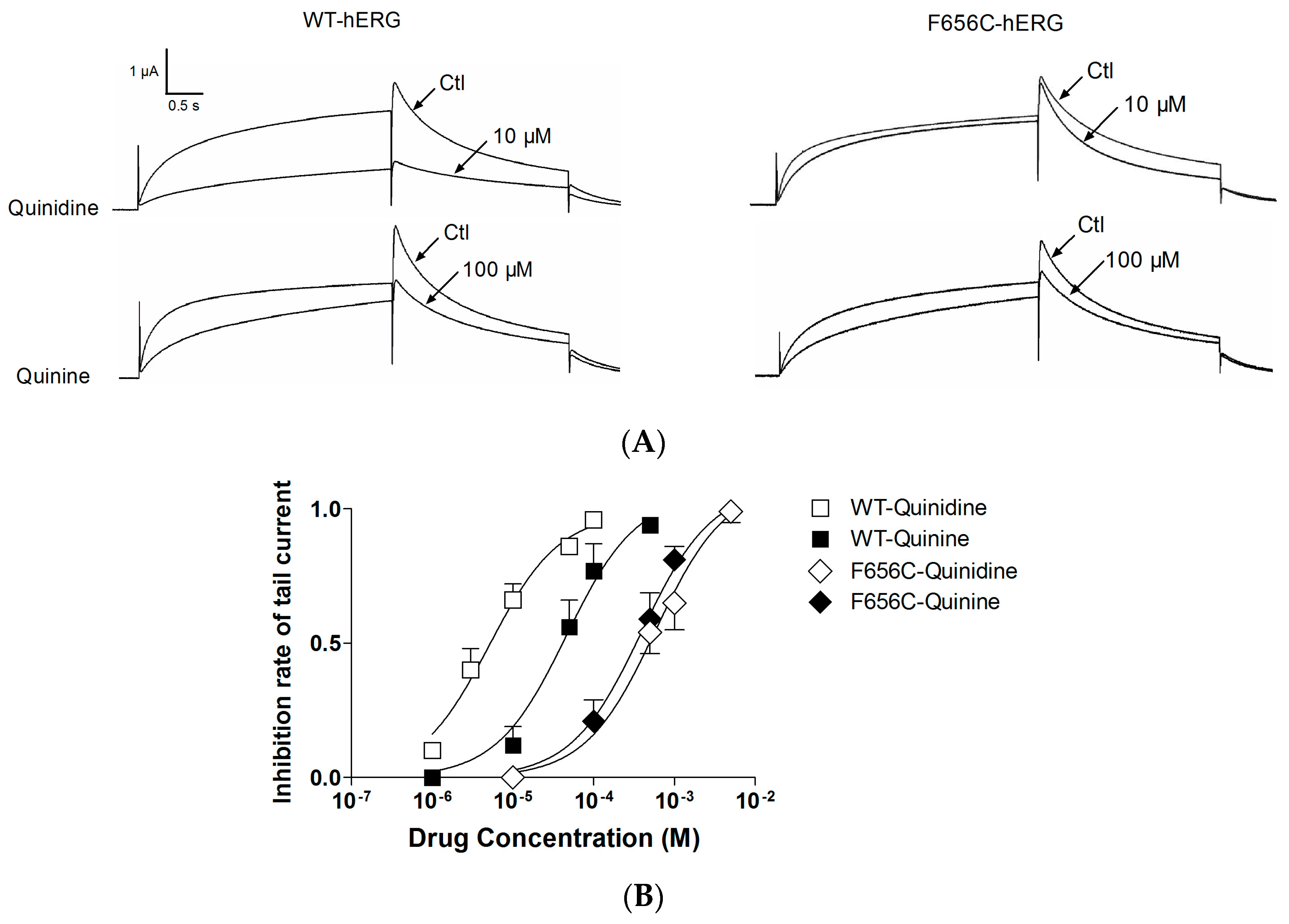
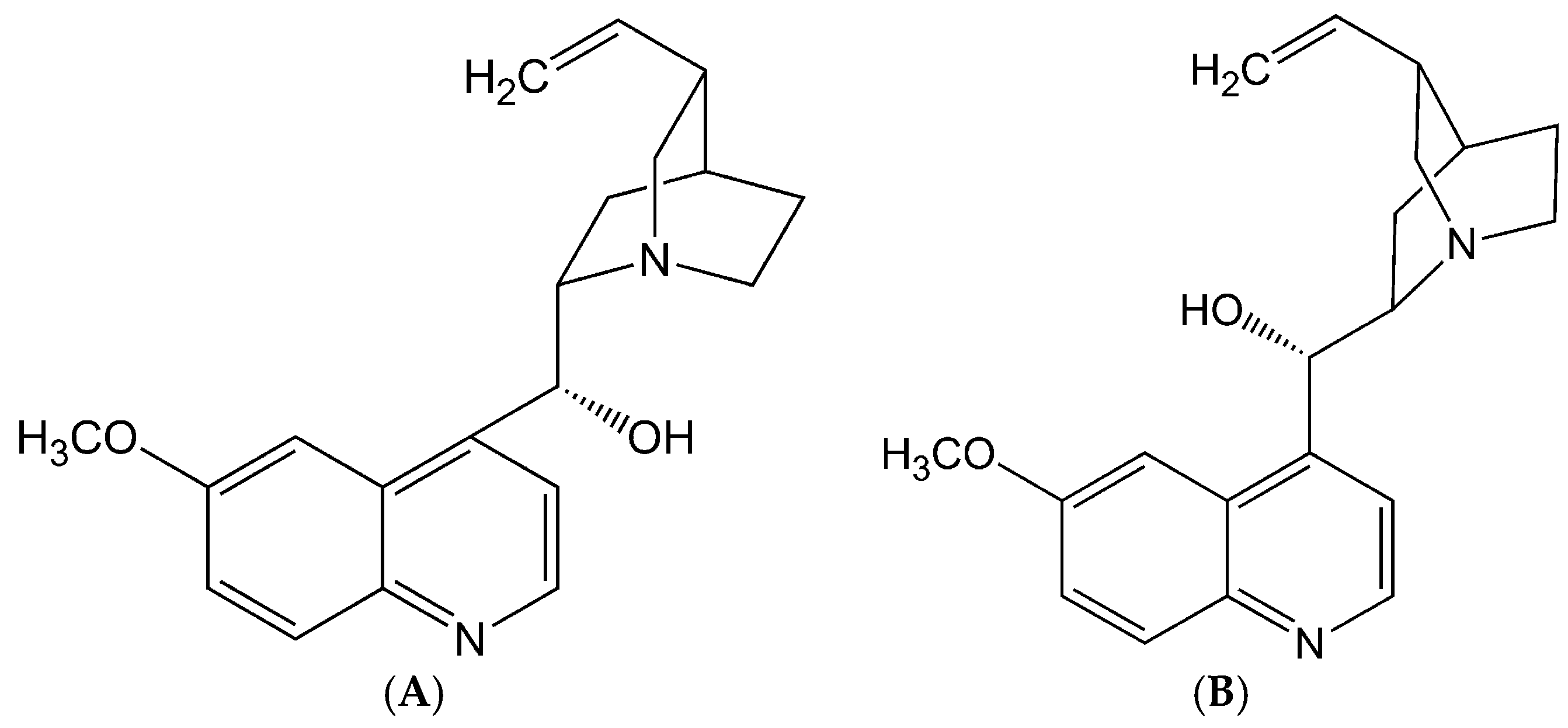
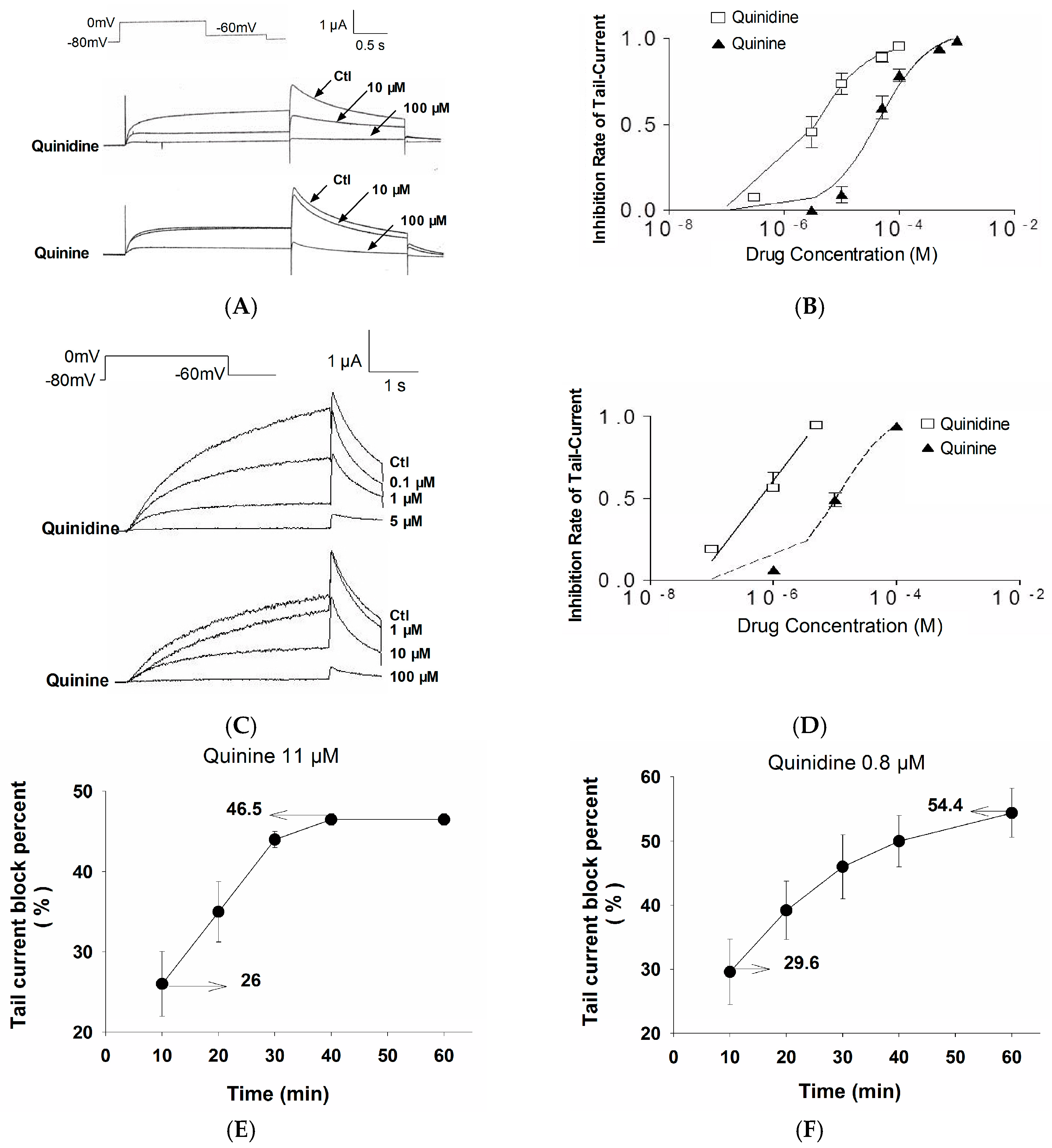
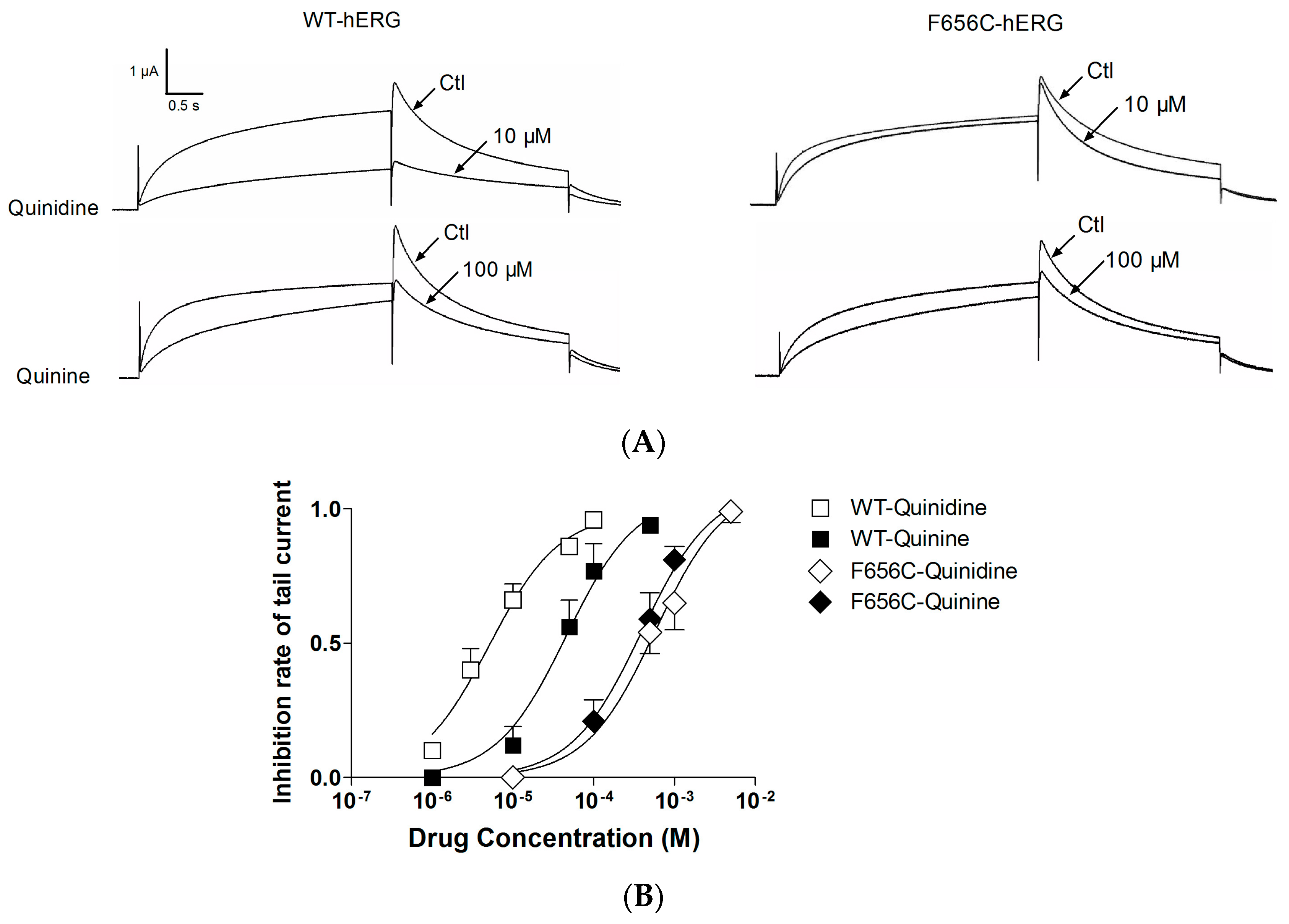
2.1. Stereoselective Difference in hERG Blockage by Quinidine and Quinine
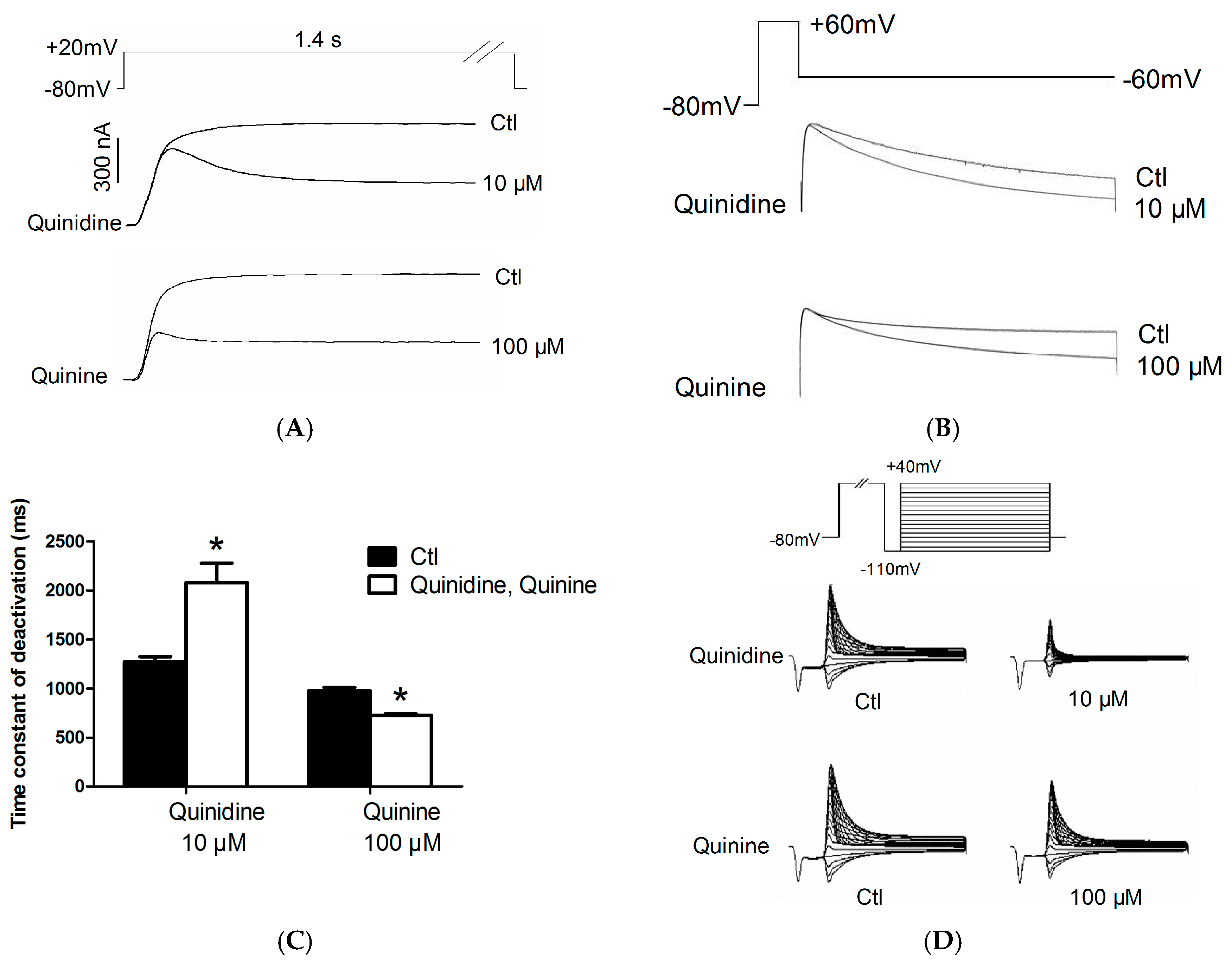
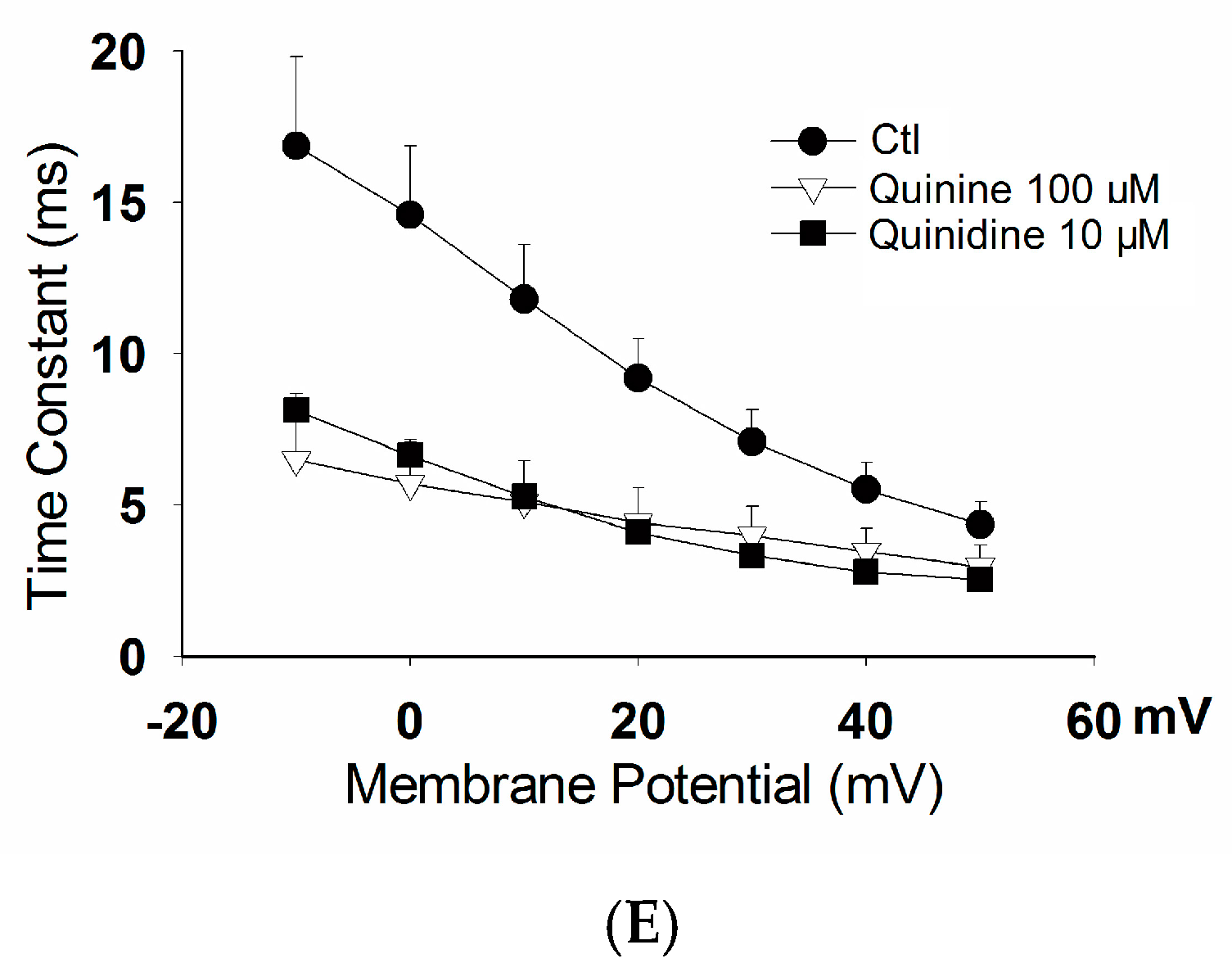
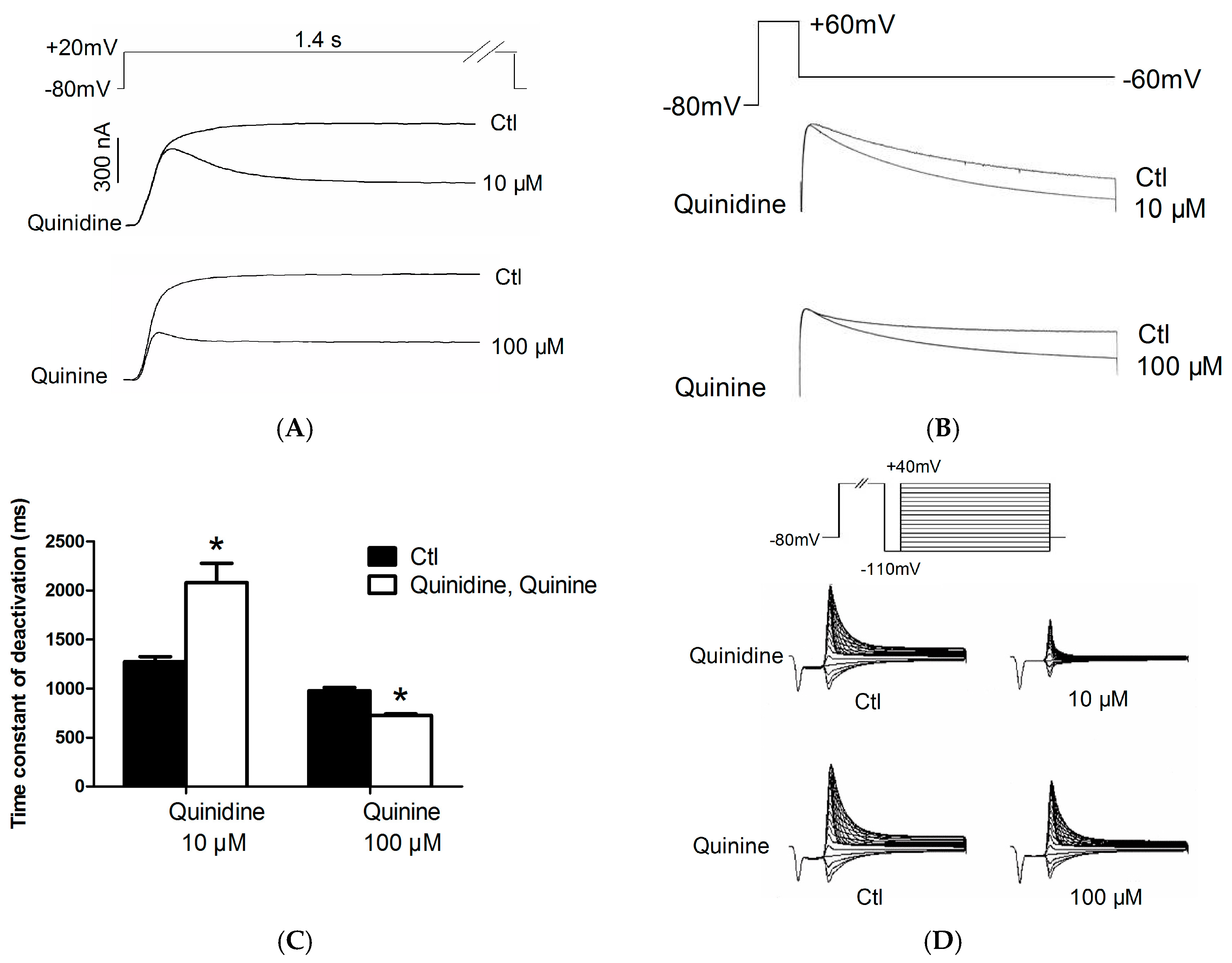
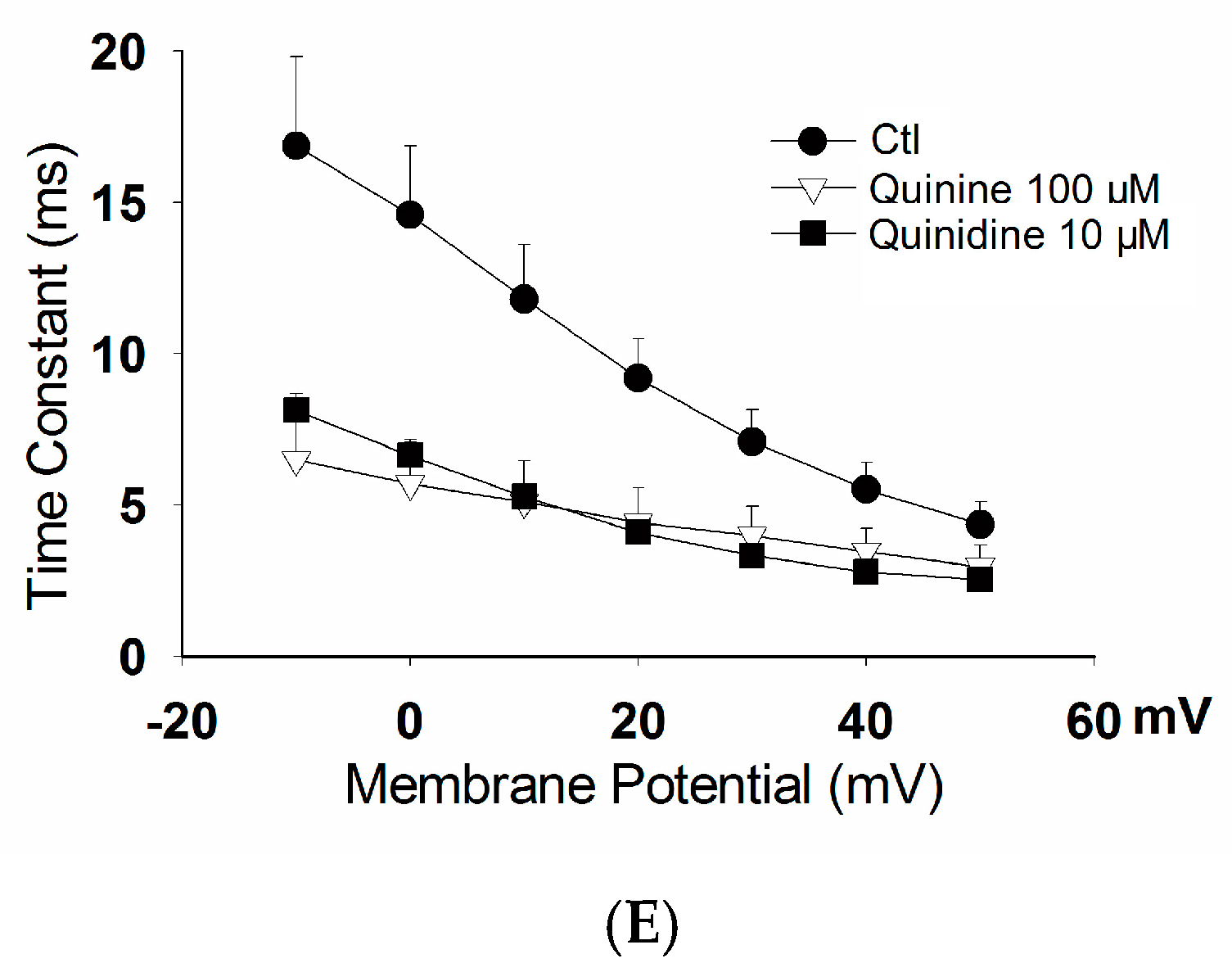
2.2. Effects of Quinidine and Quinine on Gating Properties of the hERG Channel
2.3. F656C Abrogates the Stereospecific Differences between Quinidine and Quinine
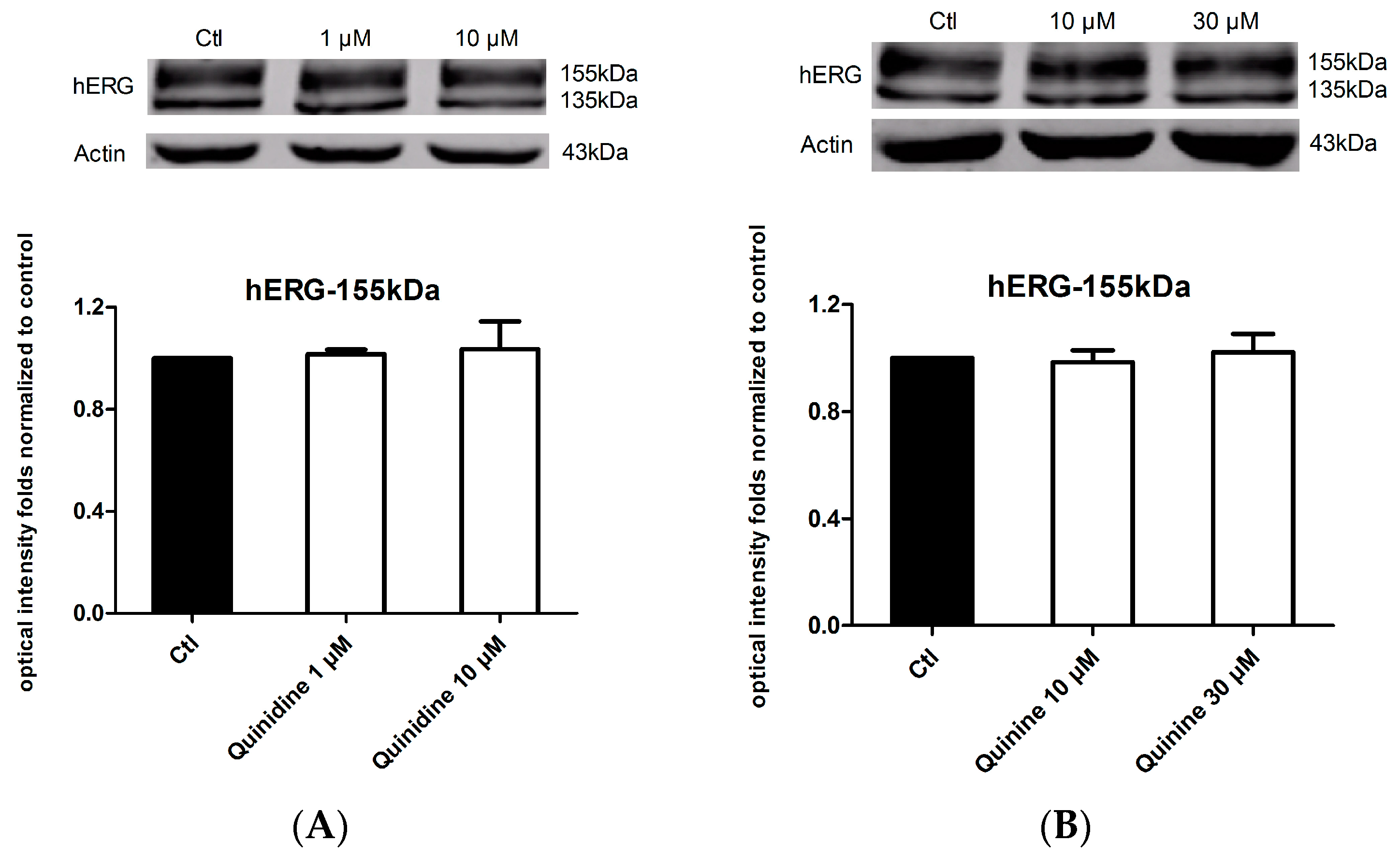
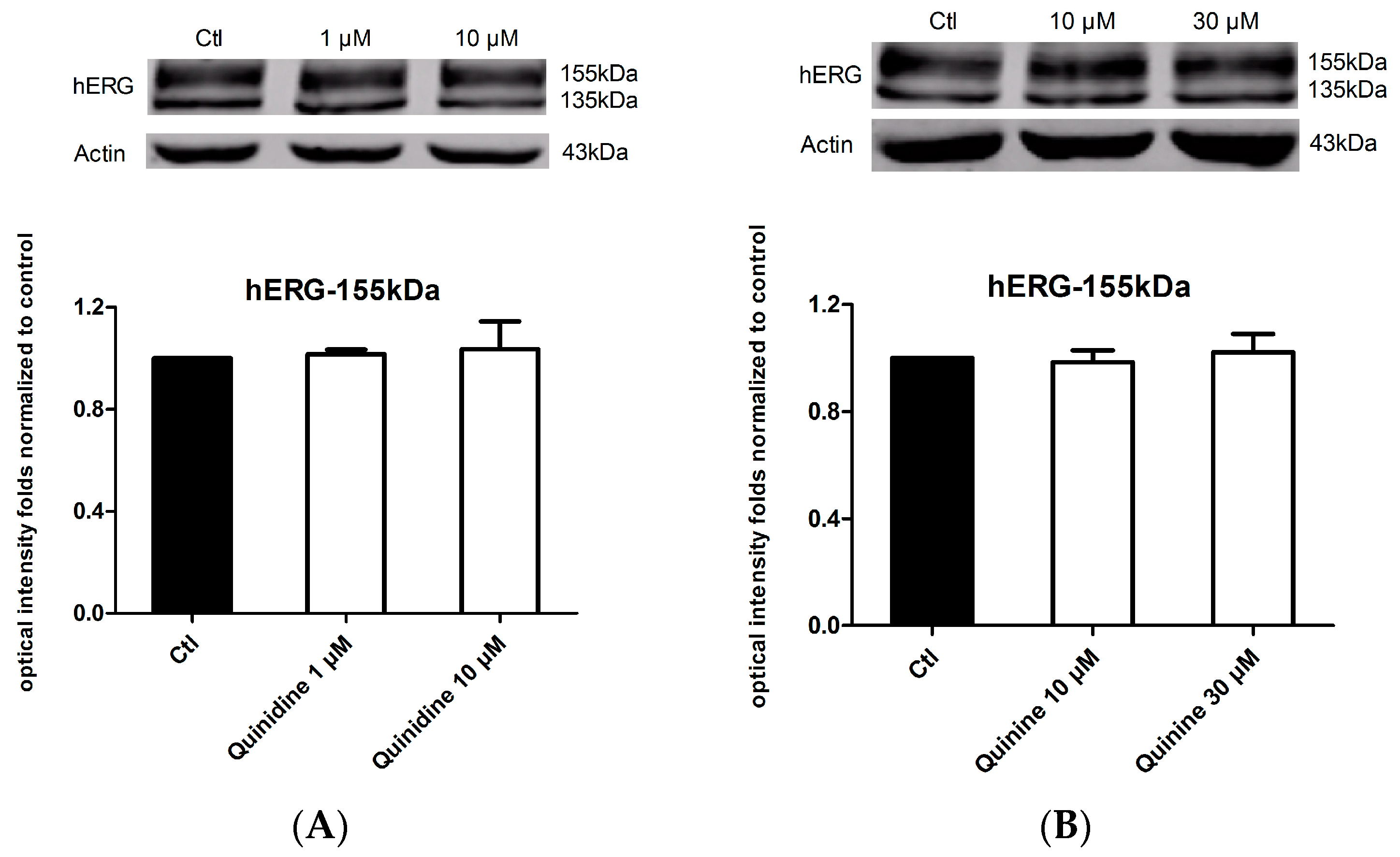
2.4. Effects of Quinidine and Quinine on the Expression of the hERG Channel
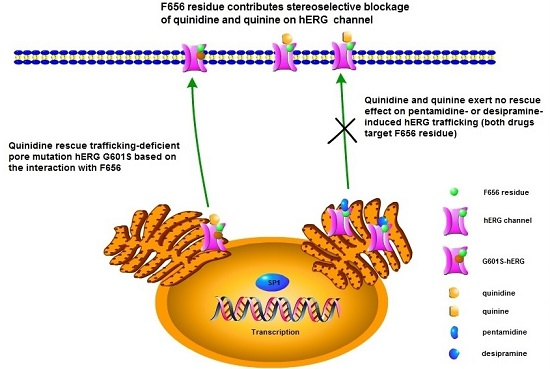
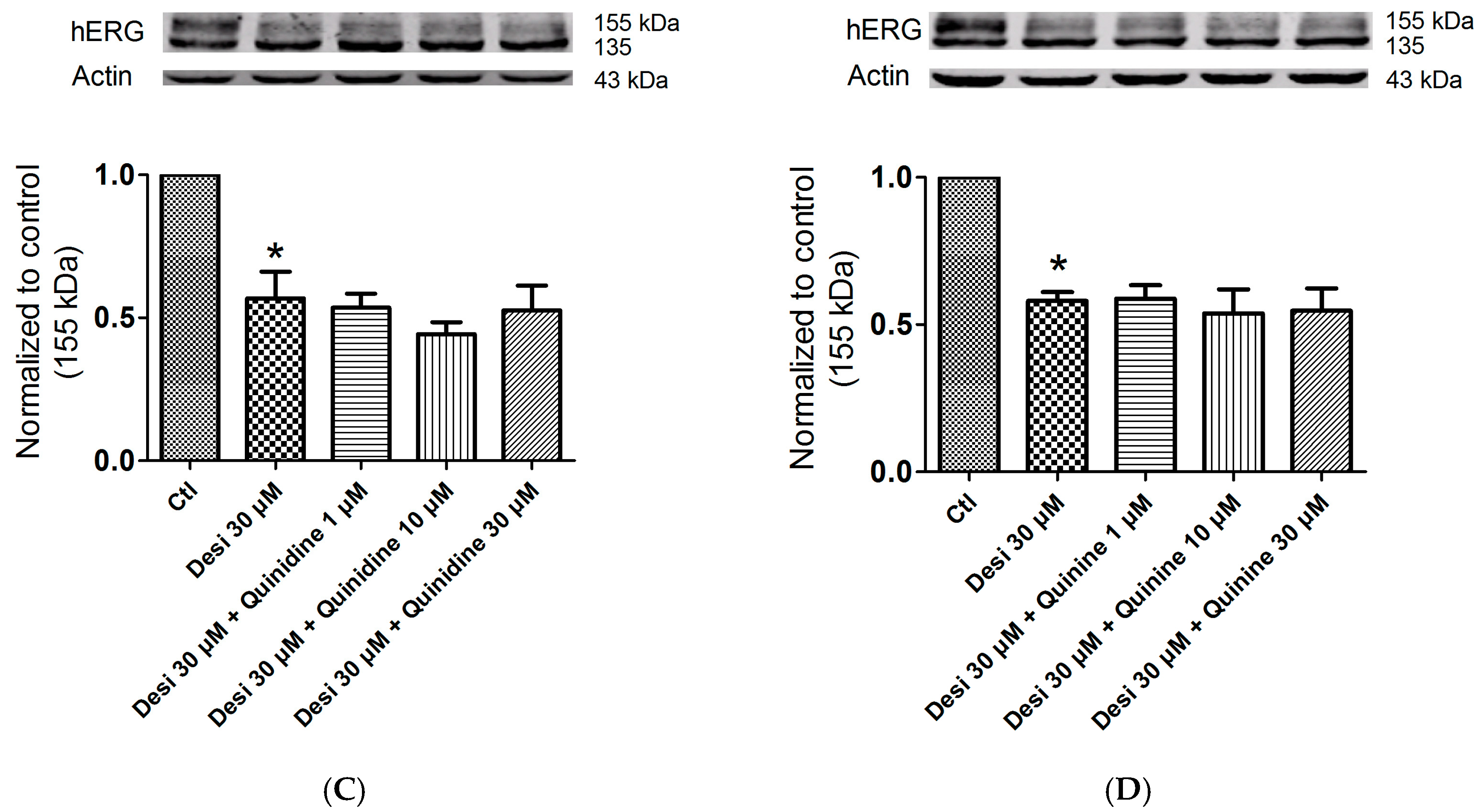
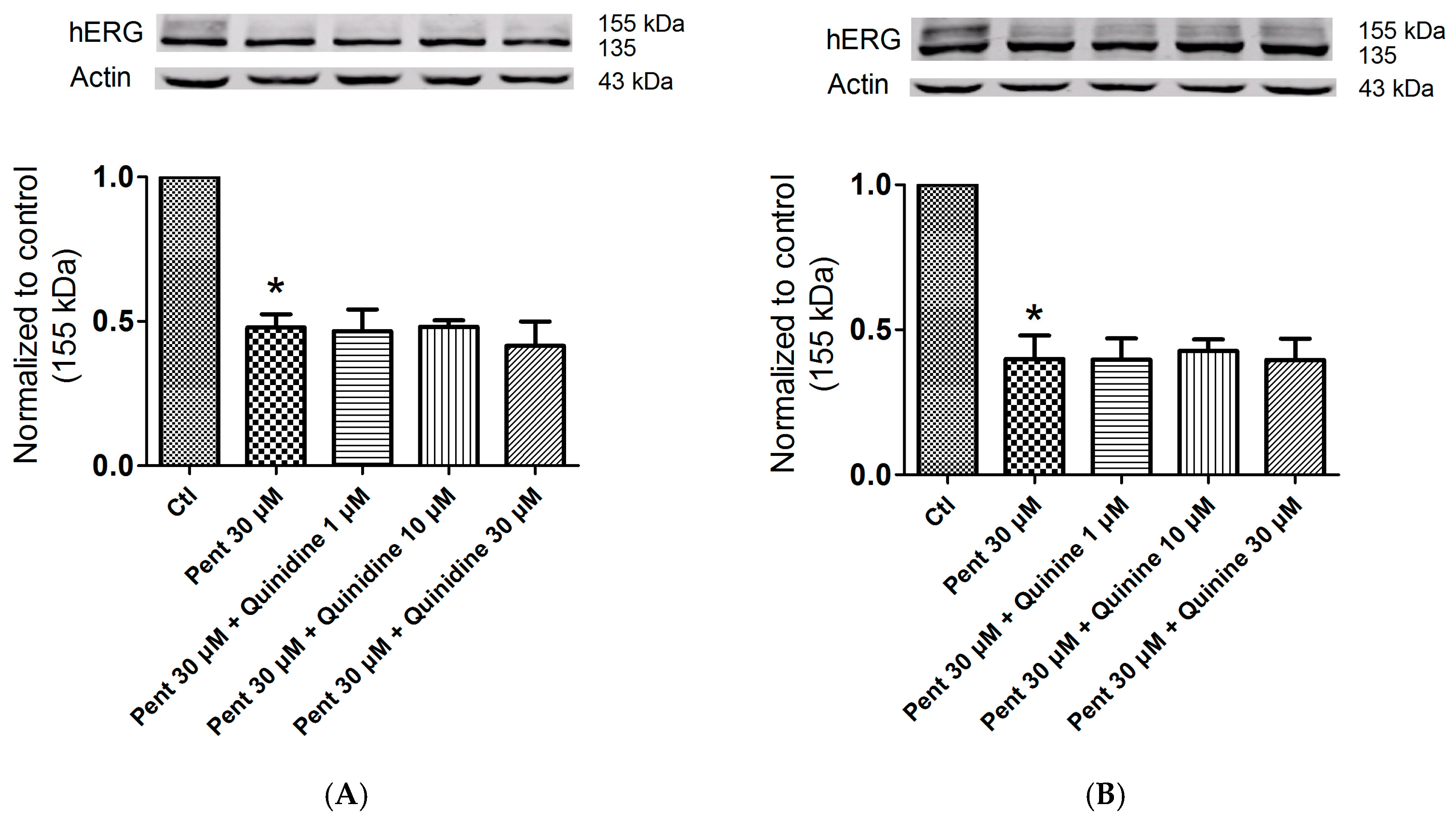
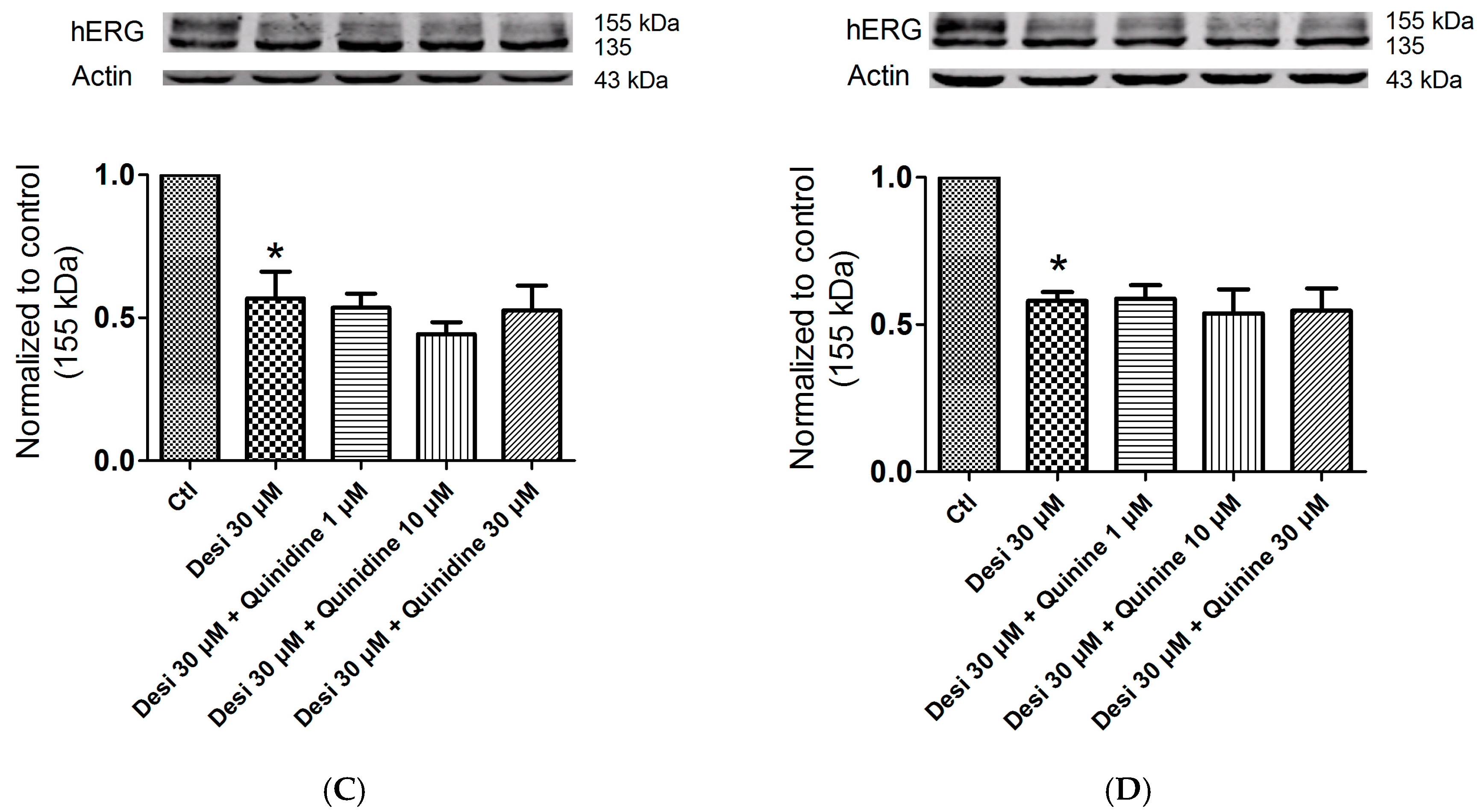
2.5. Effects of Quinidine and Quinine on Drug-Induced hERG Trafficking Deficiency
3. Discussion
4. Materials and Methods
4.1. Isolation and Maintenance of Xenopus Oocytes
4.2. Site-Directed Mutagenesis and cRNA Injection
4.3. Electrophysiology in Xenopus Oocytes
4.4. Expression of hERG in Ltk− Cells
4.5. Electrophysiology in Ltk− Cells
4.6. Western Blot Analysis
4.7. Data Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Weinreb, S.M. Chemistry. Synthetic lessons from quinine. Nature 2001, 411, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Salerno, D.M. Quinidine: Is it a good drug or a bad drug? Postgrad. Med. 1992, 92, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Achan, J.; Talisuna, A.O.; Erhart, A.; Yeka, A.; Tibenderana, J.K.; Baliraine, F.N.; Rosenthal, P.J.; D’Alessandro, U. Quinine, an old anti-malarial drug in a modern world: Role in the treatment of malaria. Malar. J. 2011, 10, 144. [Google Scholar] [CrossRef] [PubMed]
- Bonington, A.; Davidson, R.N.; Winstanley, P.A.; Pasvol, G. Fatal quinine cardiotoxicity in the treatment of falciparum malaria. Trans. R. Soc. Trop. Med. Hyg. 1996, 90, 305–307. [Google Scholar] [CrossRef]
- Wroblewski, H.A.; Kovacs, R.J.; Kingery, J.R.; Overholser, B.R.; Tisdale, J.E. High risk of QT interval prolongation and torsades de pointes associated with intravenous quinidine used for treatment of resistant malaria or babesiosis. Antimicrob. Agents Chemother. 2012, 56, 4495–4499. [Google Scholar] [CrossRef] [PubMed]
- Mirro, M.J.; Watanabe, A.M.; Bailey, J.C. Electrophysiological effects of the optical isomers of disopyramide and quinidine in the dog. Dependence on stereochemistry. Circ. Res. 1981, 48, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.S.; Rahmberg, M.; Duff, H.J. Quinidine/quinine: Stereospecific electrophysiologic and antiarrhythmic effects in a canine model of ventricular tachycardia. J. Cardiovasc. Pharmacol. 1990, 16, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, A.; Yamada, H.; Kotaki, H.; Kimura, M. Effects of anti-malarial drugs on the electrocardiographic QT interval modelled in the isolated perfused guinea pig heart system. Malar. J. 2010, 9, 318. [Google Scholar] [CrossRef] [PubMed]
- Touze, J.E.; Heno, P.; Fourcade, L.; Deharo, J.C.; Thomas, G.; Bohan, S.; Paule, P.; Riviere, P.; Kouassi, E.; Buguet, A. The effects of antimalarial drugs on ventricular repolarization. Am. J. Trop. Med. Hyg. 2002, 67, 54–60. [Google Scholar] [PubMed]
- Yang, T.; Snyders, D.; Roden, D.M. Drug block of I Kr: Model systems and relevance to human arrhythmias. J. Cardiovasc. Pharmacol. 2001, 38, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Chapula, J.A.; Ferrer, T.; Navarro-Polanco, R.A.; Sanguinetti, M.C. Voltage-dependent profile of human Ether-a-go-go-related gene channel block is influenced by a single residue in the S6 transmembrane domain. Mol. Pharmacol. 2003, 63, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Villoutreix, B.O.; Taboureau, O. Computational investigations of herg channel blockers: New insights and current predictive models. Adv. Drug Deliv. Rev. 2015, 86, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Vonderlin, N.; Fischer, F.; Zitron, E.; Seyler, C.; Scherer, D.; Thomas, D.; Katus, H.A.; Scholz, E.P. Anesthetic drug midazolam inhibits cardiac human ether-a-go-go-related gene channels: Mode of action. Drug Des. Dev. Ther. 2015, 9, 867–877. [Google Scholar]
- Zhang, K.; Zhi, D.; Huang, T.; Gong, Y.; Yan, M.; Liu, C.; Wei, T.; Dong, Z.; Li, B.; Yang, B. Berberine induces herg channel deficiency through trafficking inhibition. Cell. Physiol. Biochem. 2014, 34, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.Q.; Yan, C.C.; Zhang, X.; Yan, M.; Liu, L.R.; Geng, H.Z.; Lv, L.; Li, B.X. Mechanisms underlying probucol-induced HERG-channel deficiency. Drug Des. Dev. Ther. 2015, 9, 3695–3704. [Google Scholar]
- Dennis, A.T.; Nassal, D.; Deschenes, I.; Thomas, D.; Ficker, E. Antidepressant-induced ubiquitination and degradation of the cardiac potassium channel herg. J. Biol. Chem. 2011, 286, 34413–34425. [Google Scholar] [CrossRef] [PubMed]
- Ficker, E.; Obejero-Paz, C.A.; Zhao, S.; Brown, A.M. The binding site for channel blockers that rescue misprocessed human long qt syndrome type 2 ether-a-gogo-related gene (HERG) mutations. J. Biol. Chem. 2002, 277, 4989–4998. [Google Scholar] [CrossRef] [PubMed]
- Rajamani, S.; Anderson, C.L.; Anson, B.D.; January, C.T. Pharmacological rescue of human K+ channel long-QT2 mutations: Human ether-a-go-go-related gene rescue without block. Circulation 2002, 105, 2830–2835. [Google Scholar] [CrossRef] [PubMed]
- Dennis, A.T.; Wang, L.; Wan, H.; Nassal, D.; Deschenes, I.; Ficker, E. Molecular determinants of pentamidine-induced herg trafficking inhibition. Mol. Pharmacol. 2012, 81, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.K.; Park, M.H.; Lee, B.H.; Jo, S.H. Block of the human ether-a-go-go-related gene (HERG) K+ channel by the antidepressant desipramine. Biochem. Biophys. Res. Commun. 2010, 394, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.; Duff, H.; Koshman, M.L. Antiarrhythmic activity of quinine in humans. Circulation 1995, 92, 2944–2950. [Google Scholar] [CrossRef] [PubMed]
- White, N.J. Cardiotoxicity of antimalarial drugs. Lancet Infect. Dis. 2007, 7, 549–558. [Google Scholar] [CrossRef]
- Kong, L.Y.; Tan, R.X. Artemisinin, a miracle of traditional chinese medicine. Nat. Prod. Rep. 2015, 32, 1617–1621. [Google Scholar] [CrossRef] [PubMed]
- Sanders, N.G.; Meyers, D.J.; Sullivan, D.J. Antimalarial efficacy of hydroxyethylapoquinine (SN-119) and its derivatives. Antimicrob. Agents Chemother. 2014, 58, 820–827. [Google Scholar] [CrossRef] [PubMed]
- White, N.J.; Looareesuwan, S.; Warrell, D.A. Quinine and quinidine: A comparison of EKG effects during the treatment of malaria. J. Cardiovasc. Pharmacol. 1983, 5, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Snyders, D.J.; Yeola, S.W. Determinants of antiarrhythmic drug action. Electrostatic and hydrophobic components of block of the human cardiac HKV1.5 channel. Circ. Res. 1995, 77, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Mitcheson, J.S.; Chen, J.; Lin, M.; Culberson, C.; Sanguinetti, M.C. A structural basis for drug-induced long qt syndrome. Proc. Natl. Acad. Sci. USA 2000, 97, 12329–12333. [Google Scholar] [CrossRef] [PubMed]
- Saxena, P.; Zangerl-Plessl, E.M.; Linder, T.; Windisch, A.; Hohaus, A.; Timin, E.; Hering, S.; Stary-Weinzinger, A. New potential binding determinant for herg channel inhibitors. Sci. Rep. 2016, 6, 24182. [Google Scholar] [CrossRef] [PubMed]
- Duff, H.J.; Feng, Z.P.; Sheldon, R.S. High- and low-affinity sites for [3H]dofetilide binding to guinea pig myocytes. Circ. Res. 1995, 77, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Gong, Q.; January, C.T. Correction of defective protein trafficking of a mutant HERG potassium channel in human long QT syndrome. Pharmacological and temperature effects. J. Biol. Chem. 1999, 274, 31123–31126. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Zhang, K.; Shi, Y.; Feng, L.; Lv, L.; Li, B. Mechanism and pharmacological rescue of berberine-induced HERG channel deficiency. Drug Des. Dev. Ther. 2015, 9, 5737–5747. [Google Scholar]
- Sanguinetti, M.C.; Xu, Q.P. Mutations of the S4–S5 linker alter activation properties of HERG potassium channels expressed in Xenopus oocytes. J. Physiol. 1999, 514, 667–675. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, M.; Fan, P.; Shi, Y.; Feng, L.; Wang, J.; Zhan, G.; Li, B. Stereoselective Blockage of Quinidine and Quinine in the hERG Channel and the Effect of Their Rescue Potency on Drug-Induced hERG Trafficking Defect. Int. J. Mol. Sci. 2016, 17, 1648. https://doi.org/10.3390/ijms17101648
Yan M, Fan P, Shi Y, Feng L, Wang J, Zhan G, Li B. Stereoselective Blockage of Quinidine and Quinine in the hERG Channel and the Effect of Their Rescue Potency on Drug-Induced hERG Trafficking Defect. International Journal of Molecular Sciences. 2016; 17(10):1648. https://doi.org/10.3390/ijms17101648
Chicago/Turabian StyleYan, Meng, Pan Fan, Yanhui Shi, Lifang Feng, Junnan Wang, Ge Zhan, and Baoxin Li. 2016. "Stereoselective Blockage of Quinidine and Quinine in the hERG Channel and the Effect of Their Rescue Potency on Drug-Induced hERG Trafficking Defect" International Journal of Molecular Sciences 17, no. 10: 1648. https://doi.org/10.3390/ijms17101648