
Cinnamide Derivatives as Mammalian Arginase Inhibitors: Synthesis, Biological Evaluation and Molecular Docking


Abstract
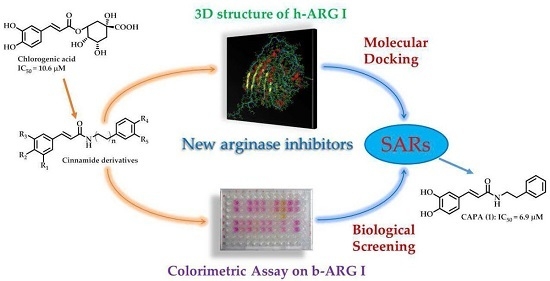
:
1. Introduction
2. Results and Discussion
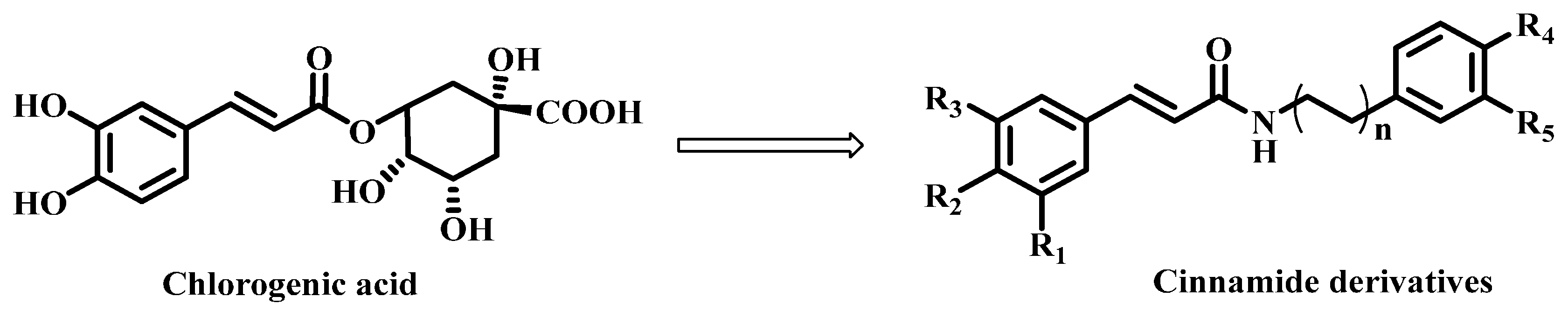
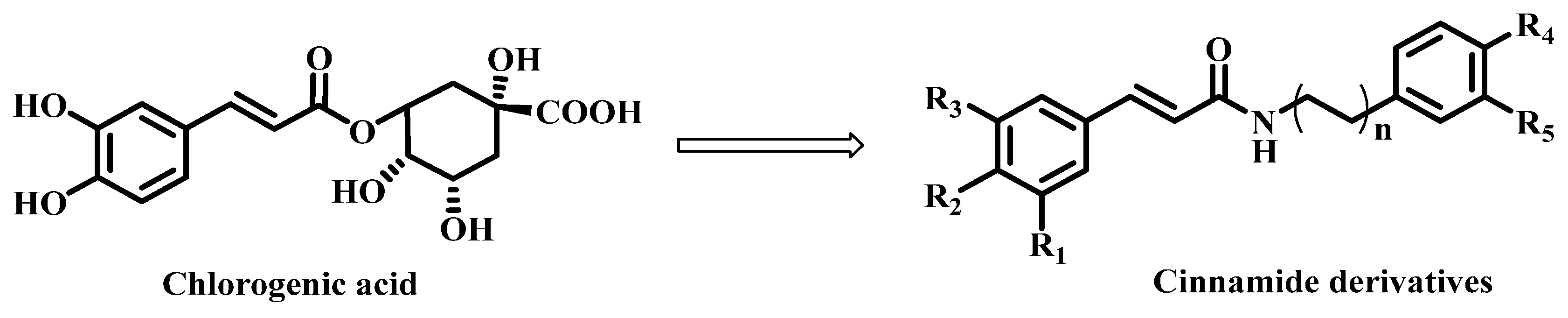
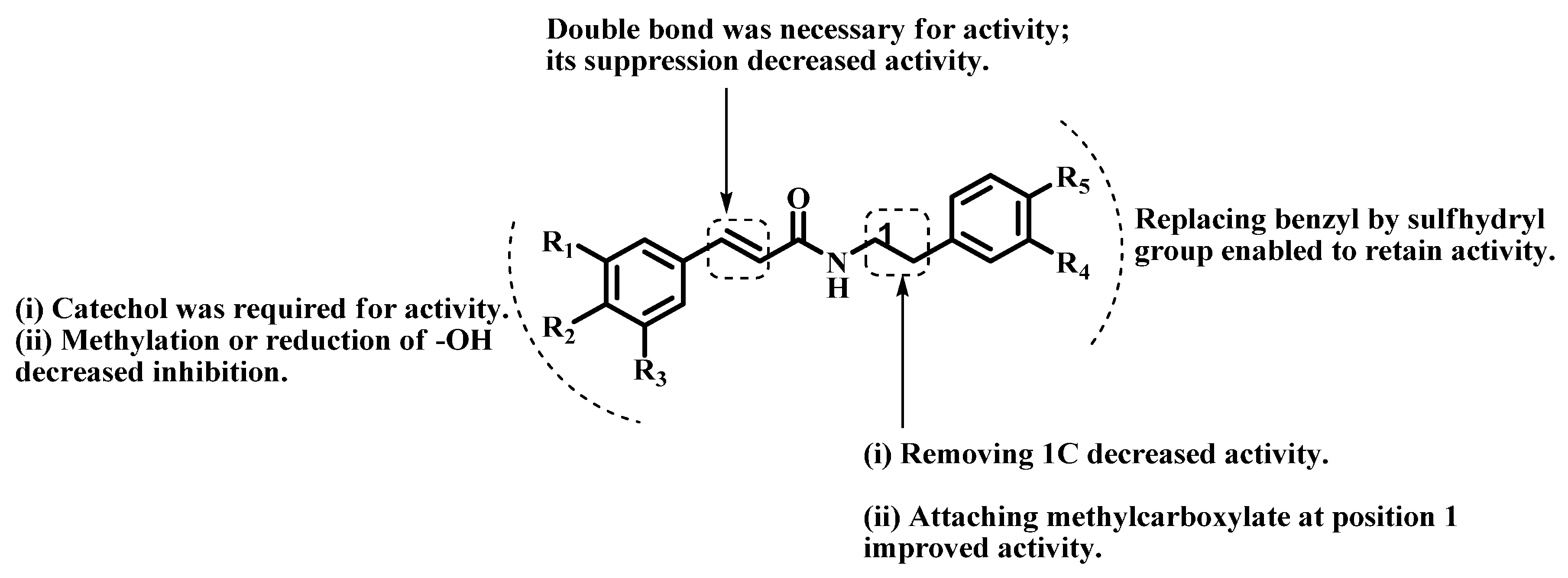
2.1. Chemistry
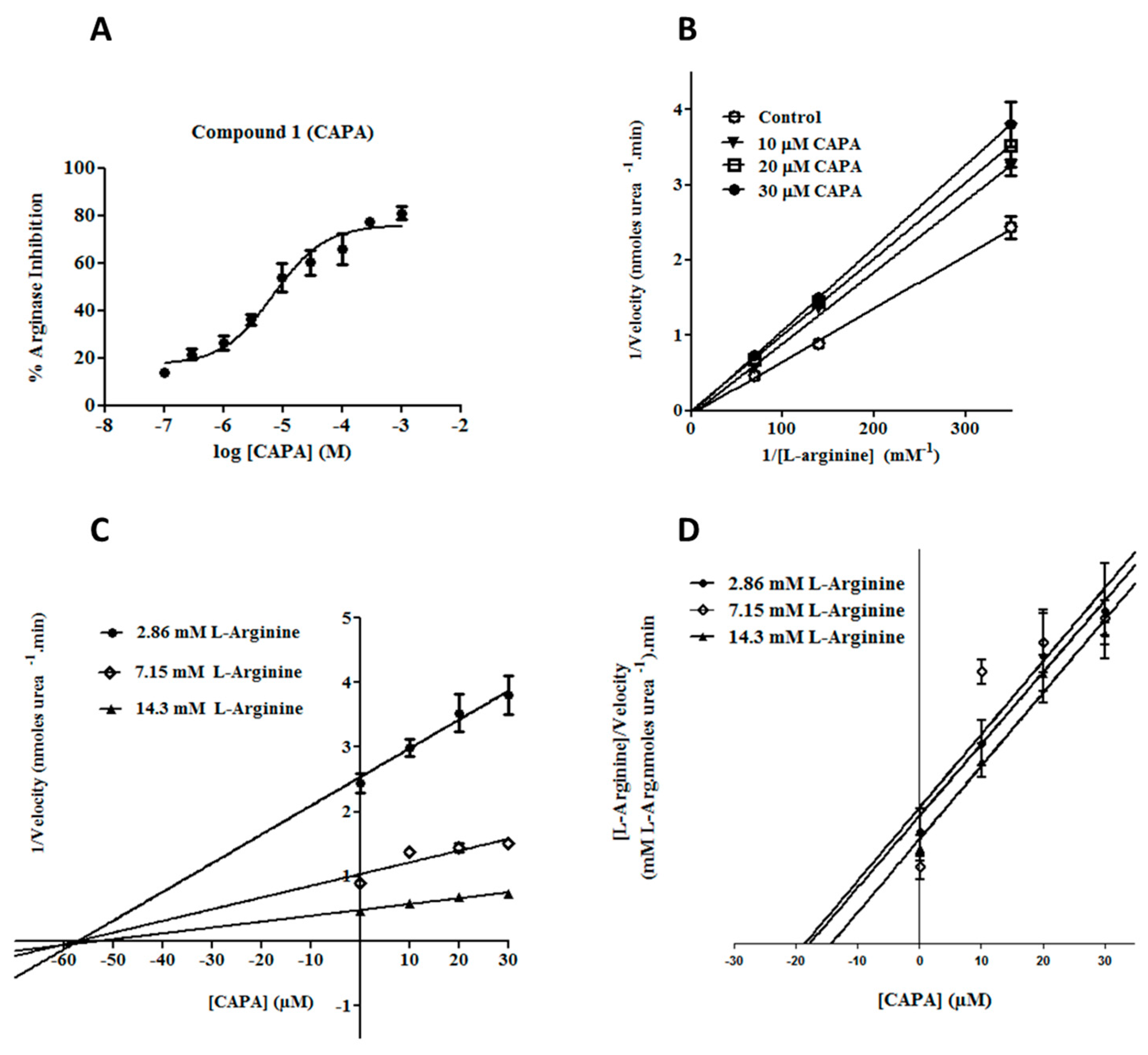
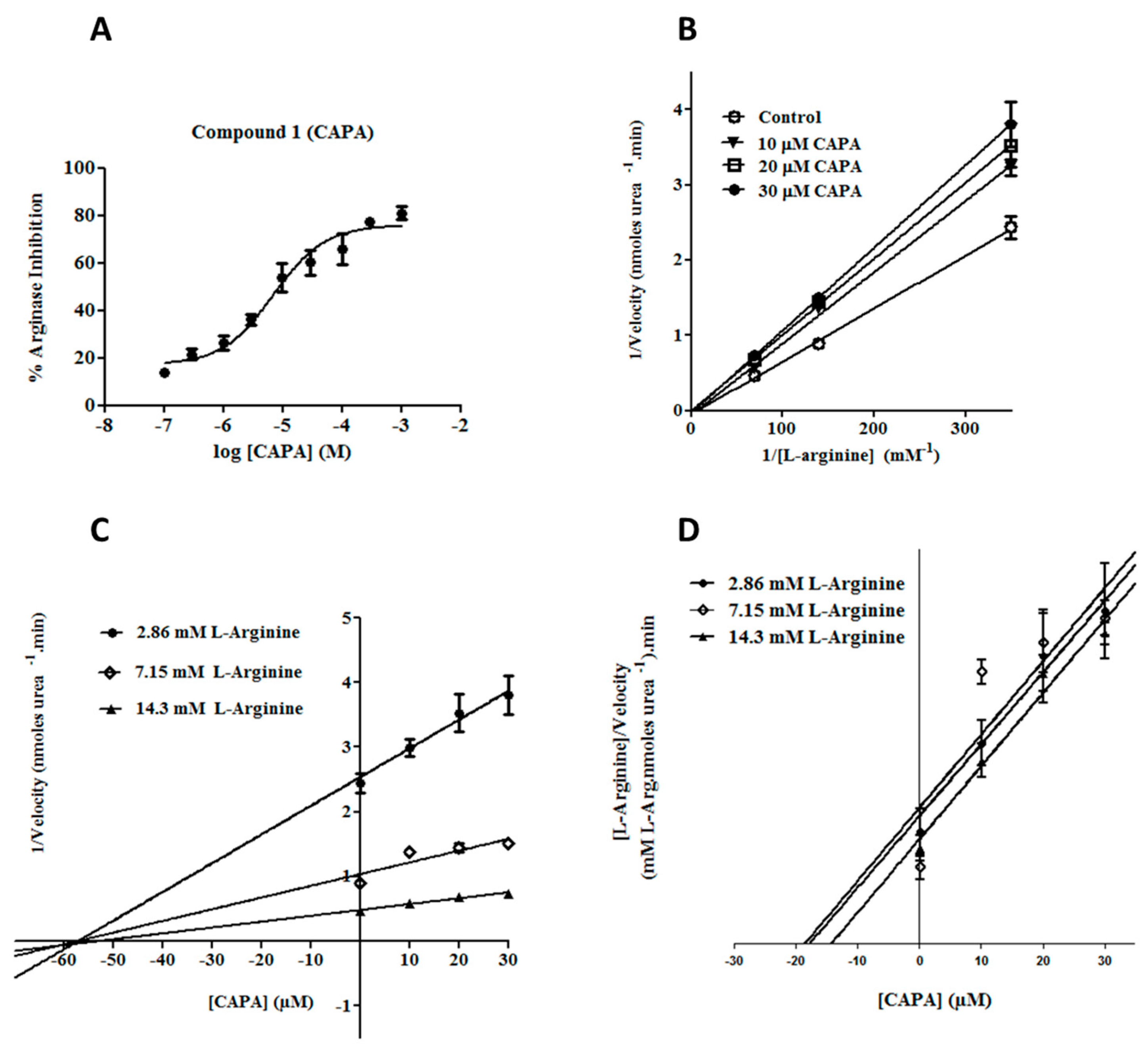
2.2. Arginase Inhibitory Activity
2.3. Enzyme Kinetic Studies for CAPA (1)
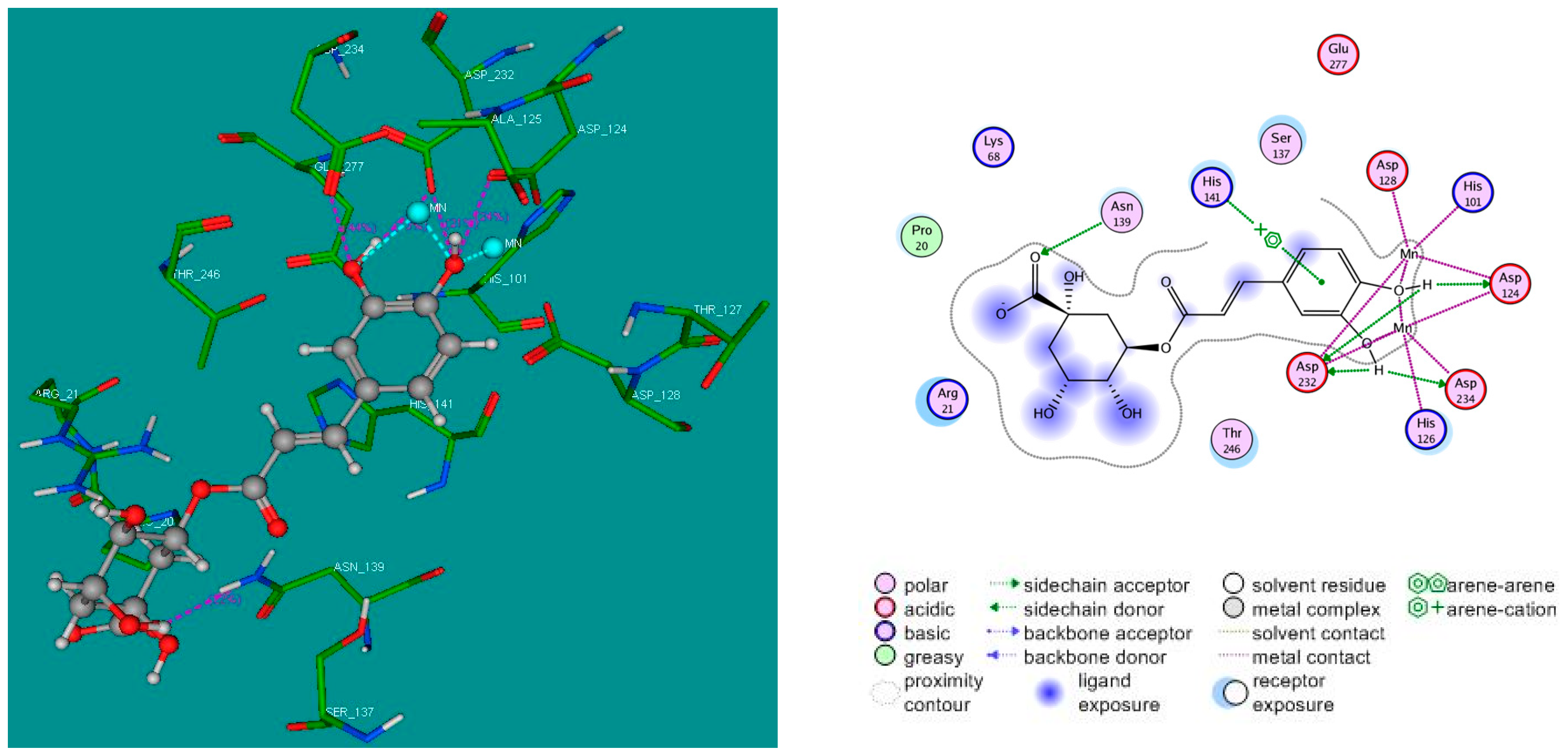
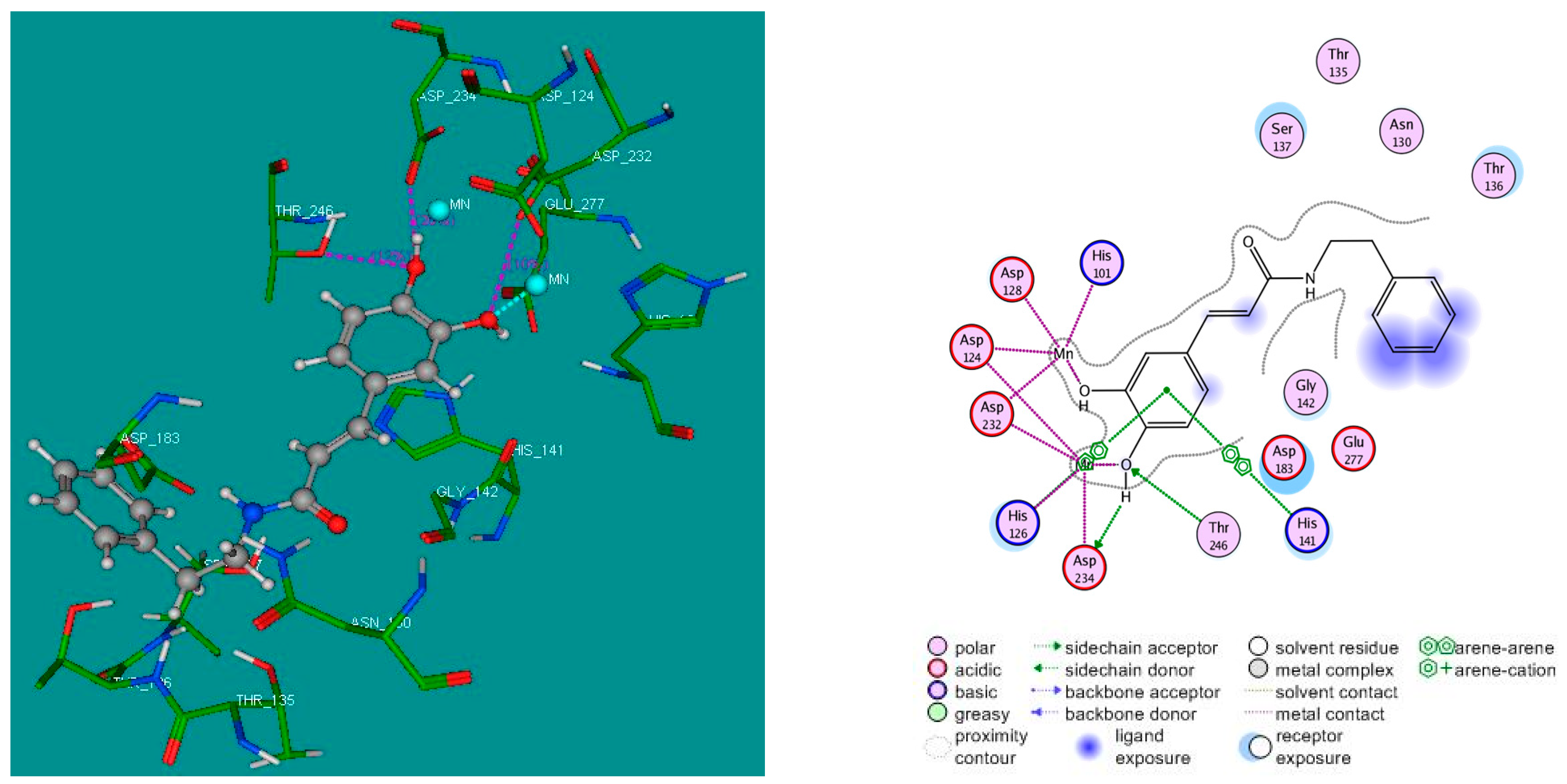
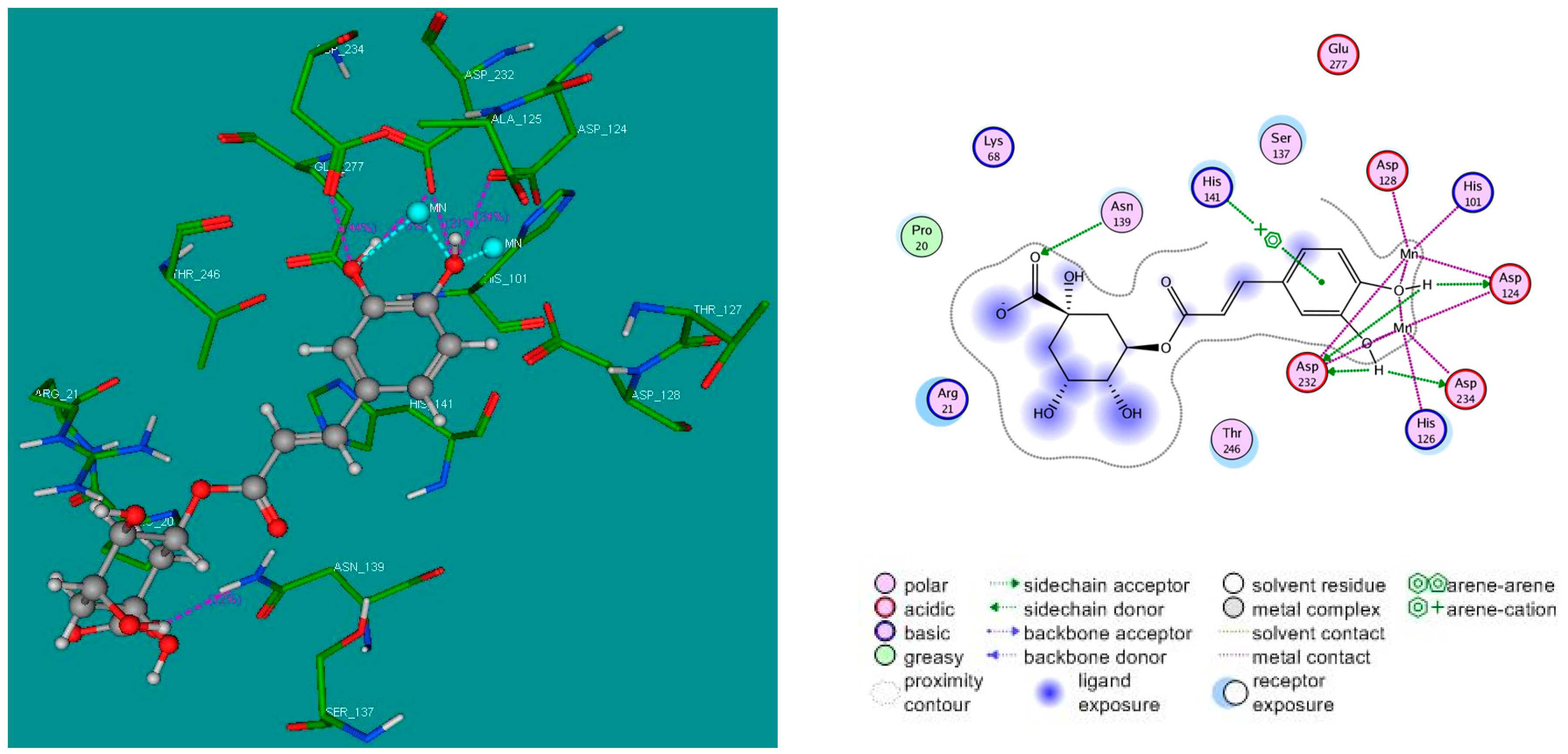
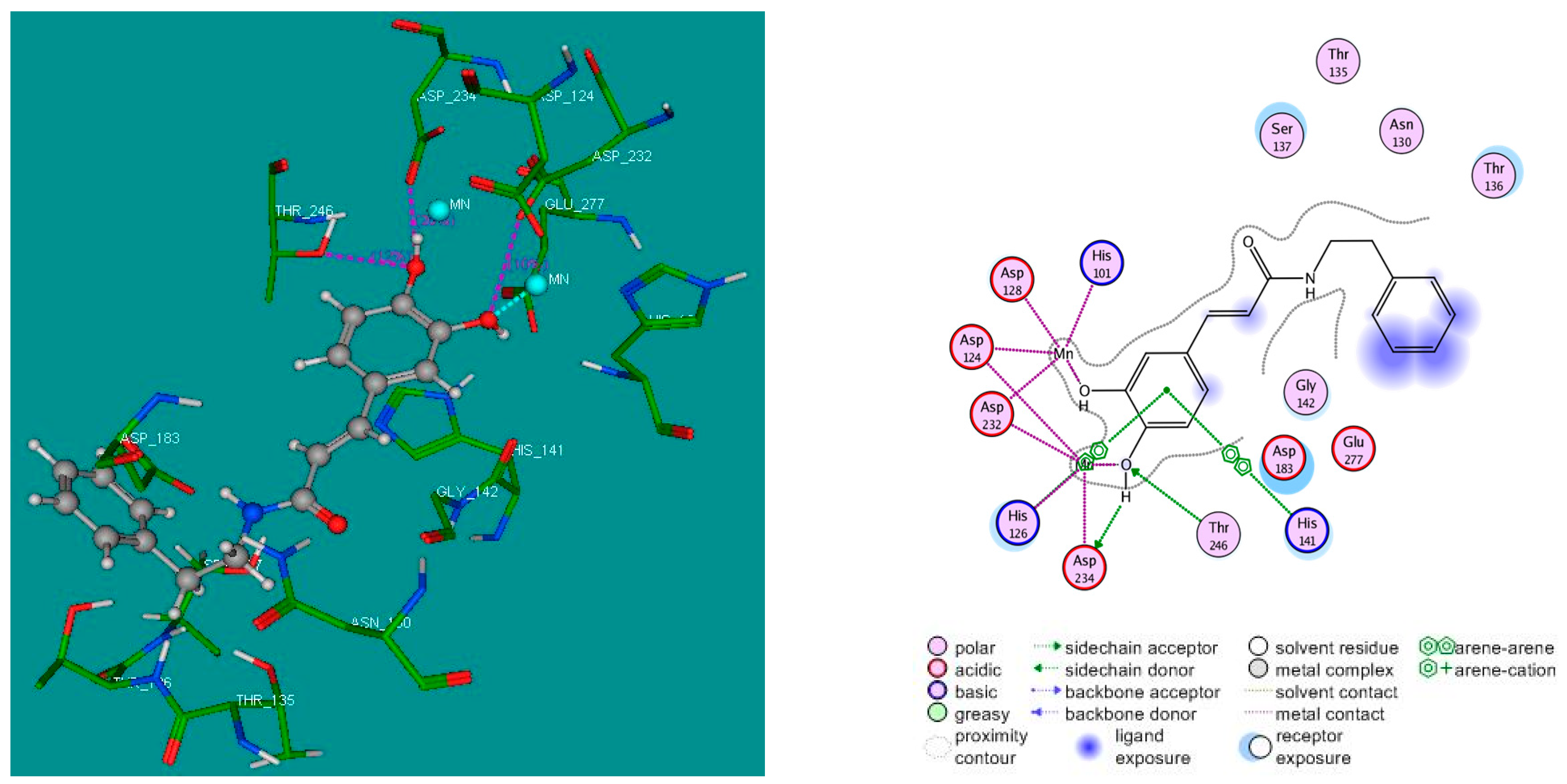
2.4. Molecular Docking Studies
3. Materials and Methods
3.1. Chemistry
3.2. General Procedure for the Synthesis of Amide Derivatives 1−19
3.3. Biological Evaluation
3.3.1. In Vitro Enzymatic Assay
3.3.2. Determination of IC50 Values and Percentages of Arginase Inhibition
3.3.3. Enzyme Kinetic Study and Determination of Ki Values
3.4. Molecular Docking Study
3.4.1. Preparation of Target Protein
3.4.2. Preparation of Ligands
3.4.3. Docking Simulation
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Maccallini, C.; Fantacuzzi, M.; Amoroso, R. Amidine-based bioactive compounds for the regulation of arginine metabolism. Mini Rev. Med. Chem. 2013, 13, 1305–1310. [Google Scholar] [CrossRef] [PubMed]
- Dimski, D.S. Ammonia metabolism and the urea cycle: Function and clinical implications. J. Vet. Intern. Med. Am. Coll. Vet. Int. Med. 1994, 8, 73–78. [Google Scholar] [CrossRef]
- Morris, S.M. Regulation of enzymes of the urea cycle and arginine metabolism. Annu. Rev. Nutr. 2002, 22, 87–105. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, R.B.; Toque, H.A.; Narayanan, S.P.; Caldwell, R.W. Arginase: An old enzyme with new tricks. Trends Pharmacol. Sci. 2015, 36, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Jenkinson, C.P.; Grody, W.W.; Cederbaum, S.D. Comparative properties of arginases. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1996, 114, 107–132. [Google Scholar] [CrossRef]
- Bagnost, T.; Ma, L.; da Silva, R.F.; Rezakhaniha, R.; Houdayer, C.; Stergiopulos, N.; André, C.; Guillaume, Y.; Berthelot, A.; Demougeot, C. Cardiovascular effects of arginase inhibition in spontaneously hypertensive rats with fully developed hypertension. Cardiovasc. Res. 2010, 87, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Segal, R.; Hannan, J.L.; Liu, X.; Kutlu, O.; Burnett, A.L.; Champion, H.C.; Kim, J.H.; Steppan, J.; Berkowitz, D.E.; Bivalacqua, T.J. Chronic oral administration of the arginase inhibitor 2(S)-amino-6-boronohexanoic acid (ABH) improves erectile function in aged rats. J. Androl. 2012, 33, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Grasemann, H.; Dhaliwal, R.; Ivanovska, J.; Kantores, C.; McNamara, P.J.; Scott, J.A.; Belik, J.; Jankov, R.P. Arginase inhibition prevents bleomycin-induced pulmonary hypertension, vascular remodeling, and collagen deposition in neonatal rat lungs. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L503–L510. [Google Scholar] [CrossRef] [PubMed]
- Olivon, V.C.; Fraga-Silva, R.A.; Segers, D.; Demougeot, C.; de Oliveira, A.M.; Savergnini, S.S.; Berthelot, A.; de Crom, R.; Krams, R.; Stergiopulos, N.; et al. Arginase inhibition prevents the low shear stress-induced development of vulnerable atherosclerotic plaques in ApoE−/− mice. Atherosclerosis 2013, 227, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.M.; Gao, T.; Cooper, T.K.; Kepka-Lenhart, D.; Awad, A.S. Arginase-2 mediates diabetic renal injury. Diabetes 2011, 60, 3015–3022. [Google Scholar] [CrossRef] [PubMed]
- Meurs, H.; Zaagsma, J.; Maarsingh, H.; van Duin, M. Use of Arginase Inhibitors in the Treatment of Asthma and Allergic Rhinitis. US 20150164930 A1, 4 March 2010. [Google Scholar]
- Van Zandt, M.C.; Whitehouse, D.L.; Golebiowski, A.; Ji, M.K.; Zhang, M.; Beckett, R.P.; Jagdmann, G.E.; Ryder, T.R.; Sheeler, R.; Andreoli, M.; et al. Discovery of (R)-2-Amino-6-borono-2-(2-(piperidin-1-yl)ethyl)hexanoic acid and congeners as highly potent inhibitors of human arginases I and II for treatment of myocardial reperfusion injury. J. Med. Chem. 2013, 56, 2568–2580. [Google Scholar] [CrossRef] [PubMed]
- Kavalukas, S.L.; Uzgare, A.R.; Bivalacqua, T.J.; Barbul, A. Arginase inhibition promotes wound healing in mice. Surgery 2012, 151, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Pervin, S.; Karimi, A.; Cederbaum, S.; Chaudhuri, G. Arginase Activity in human breast cancer cell lines: Nω-Hydroxy-l-arginine selectively inhibits cell proliferation and induces apoptosis in MDA-MB-468 cells. Cancer Res. 2000, 60, 3305–3312. [Google Scholar] [PubMed]
- Boucher, J.L.; Custot, J.; Vadon, S.; Delaforge, M.; Lepoivre, M.; Tenu, J.P.; Yapo, A.; Mansuy, D. Nω-Hydroxy-l-arginine, an intermediate in the l-arginine to nitric oxide pathway, is a strong inhibitor of liver and macrophage arginase. Biochem. Biophys. Res. Commun. 1994, 203, 1614–1621. [Google Scholar] [CrossRef] [PubMed]
- Custot, J.; Boucher, J.-L.; Vadon, S.; Guedes, C.; Dijols, S.; Delaforge, M.; Mansuy, D. Nω-Hydroxyamino-α-amino acids as a new class of very strong inhibitors of arginases. JBIC J. Biol. Inorg. Chem. 1996, 1, 73–82. [Google Scholar] [CrossRef]
- Custot, J.; Moali, C.; Brollo, M.; Boucher, J.L.; Delaforge, M.; Mansuy, D.; Tenu, J.P.; Zimmermann, J.L. The new α-amino acid Nω-hydroxy-nor-l-arginine: A high-affinity inhibitor of arginase well adapted to bind to its manganese cluster. J. Am. Chem. Soc. 1997, 119, 4086–4087. [Google Scholar] [CrossRef]
- Kim, N.N.; Cox, J.D.; Baggio, R.F.; Emig, F.A.; Mistry, S.K.; Harper, S.L.; Speicher, D.W.; Morris, S.M.; Ash, D.E.; Traish, A.; et al. Probing erectile function: S-(2-Boronoethyl)-l-cysteine binds to arginase as a transition state analogue and enhances smooth muscle relaxation in human penile corpus cavernosum. Biochemistry 2001, 40, 2678–2688. [Google Scholar] [CrossRef] [PubMed]
- Baggio, R.; Emig, F.A.; Christianson, D.W.; Ash, D.E.; Chakder, S.; Rattan, S. Biochemical and functional profile of a newly developed potent and isozyme-selective arginase inhibitor. J. Pharmacol. Exp. Ther. 1999, 290, 1409–1416. [Google Scholar] [PubMed]
- Ivanenkov, Y.A.; Chufarova, N. V. Small-molecule arginase inhibitors. Pharm. Pat. Anal. 2013, 3, 65–85. [Google Scholar] [CrossRef] [PubMed]
- Havlinova, Z.; Babicova, A.; Hroch, M.; Chladek, J. Comparative pharmacokinetics of Nω-hydroxy-nor-l-arginine, an arginase inhibitor, after single-dose intravenous, intraperitoneal and intratracheal administration to brown Norway rats. Xenobiotica 2013, 43, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.M., Jr. Recent advances in arginine metabolism: Roles and regulation of the arginases. Br. J. Pharmacol. 2009, 157, 922–930. [Google Scholar] [CrossRef] [PubMed]
- Schade, D.; Kotthaus, J.; Klein, N.; Kotthaus, J.; Clement, B. Prodrug design for the potent cardiovascular agent Nω-hydroxy-l-arginine (NOHA): Synthetic approaches and physicochemical characterization. Org. Biomol. Chem. 2011, 9, 5249–5259. [Google Scholar] [CrossRef] [PubMed]
- Ilies, M.; di Costanzo, L.; Dowling, D.P.; Thorn, K.J.; Christianson, D.W. Binding of α,α-disubstituted amino acids to arginase suggests new avenues for inhibitor design. J. Med. Chem. 2011, 54, 5432–5443. [Google Scholar] [CrossRef] [PubMed]
- Golebiowski, A.; Beckett, R.P.; van Zandt, M.; Ji, M.K.; Whitehouse, D.; Ryder, T.R.; Jagdmann, E.; Andreoli, M.; Mazur, A.; Padmanilayam, M.; et al. 2-Substituted-2-amino-6-boronohexanoic acids as arginase inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 2027–2030. [Google Scholar] [CrossRef] [PubMed]
- Golebiowski, A.; Whitehouse, D.; Beckett, R.P.; van Zandt, M.; Ji, M.K.; Ryder, T.R.; Jagdmann, E.; Andreoli, M.; Lee, Y.; Sheeler, R.; et al. Synthesis of quaternary α-amino acid-based arginase inhibitors via the Ugi reaction. Bioorg. Med. Chem. Lett. 2013, 23, 4837–4841. [Google Scholar] [CrossRef] [PubMed]
- Cama, E.; Shin, H.; Christianson, D.W. Design of amino acid sulfonamides as transition-state analogue inhibitors of arginase. J. Am. Chem. Soc. 2003, 125, 13052–13057. [Google Scholar] [CrossRef] [PubMed]
- Ilies, M.; di Costanzo, L.; North, M.L.; Scott, J.A.; Christianson, D.W. 2-Aminoimidazole amino acids as inhibitors of the binuclear manganese metalloenzyme human arginase I. J. Med. Chem. 2010, 53, 4266–4276. [Google Scholar] [CrossRef] [PubMed]
- Zakharian, T.Y.; di Costanzo, L.; Christianson, D.W. S-2-Amino-6-nitrohexanoic acid binds to human arginase I through multiple nitro−metal coordination interactions in the binuclear manganese cluster. J. Am. Chem. Soc. 2008, 130, 17254–17255. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Cama, E.; Christianson, D.W. Design of amino acid aldehydes as transition-state analogue inhibitors of arginase. J. Am. Chem. Soc. 2004, 126, 10278–10284. [Google Scholar] [CrossRef] [PubMed]
- Zakharian, T.Y.; di Costanzo, L.; Christianson, D.W. Synthesis of (2S)-2-amino-7,8-epoxyoctanoic acid and structure of its metal-bridging complex with human arginase I. Org. Biomol. Chem. 2008, 6, 3240–3243. [Google Scholar] [CrossRef] [PubMed]
- Girard-Thernier, C.; Pham, T.-N.; Demougeot, C. The promise of plant-derived substances as inhibitors of arginase. Mini Rev. Med. Chem. 2015, 15, 798–808. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.; Guglielmetti, A.; Fimbel, S.; Demougeot, C.; Girard-Thernier, C. Arginase inhibitory activity of several natural polyphenols using a novel in vitro test on purified bovine arginase. Planta Med. 2014, 80, P1L9. [Google Scholar] [CrossRef]
- Bordage, S.; Pham, T.-N.; Zedet, A.; Gugglielmetti, A.-S.; Nappey, M.; Demougeot, C.; Girard-Thernier, C. Investigation of mammal arginase inhibitory properties of natural ubiquitous polyphenols by using an optimized colorimetric microplate assay. Planta Med. 2016. accepted. [Google Scholar]
- Wang, L.-N.; Wang, W.; Hattori, M.; Daneshtalab, M.; Ma, C.-M. Synthesis, Anti-HCV, antioxidant and reduction of intracellular reactive oxygen species generation of a chlorogenic acid analogue with an amide bond replacing the ester bond. Molecules 2016, 21, 737. [Google Scholar] [CrossRef] [PubMed]
- Le Mellay-Hamon, V.; Criton, M. Phenylethylamide and phenylmethylamide derivatives as new tyrosinase inhibitors. Biol. Pharm. Bull. 2009, 32, 301–303. [Google Scholar] [CrossRef] [PubMed]
- Okombi, S.; Rival, D.; Boumendjel, A.; Mariotte, A.-M.; Perrier, E. Para-Coumaric Acid or Para-Hydroxycinnamic Acid Derivatives and Their Use in Cosmetic or Dermatological Compositions. US 8481593 B2, 9 August 2007. [Google Scholar]
- Coste, J.; Le-Nguyen, D.; Castro, B. PyBOP: A new peptide coupling reagent devoid of toxic by-product. Tetrahedron Lett. 1990, 31, 205–208. [Google Scholar] [CrossRef]
- Berndt, M.; Hölemann, A.; Niermann, A.; Bentz, C.; Zimmer, R.; Reissig, H.-U. Replacement of HMPA in Samarium Diiodide Promoted Cyclizations and Reactions of Organolithium Compounds. Eur. J. Org. Chem. 2012, 2012, 1299–1302. [Google Scholar] [CrossRef]
- Corraliza, I.M.; Campo, M.L.; Soler, G.; Modolell, M. Determination of arginase activity in macrophages: A micromethod. J. Immunol. Methods 1994, 174, 231–235. [Google Scholar] [CrossRef]
- Cornish-Bowden, A. A simple graphical method for determining the inhibition constants of mixed, uncompetitive and non-competitive inhibitors (Short Communication). Biochem. J. 1974, 137, 143–144. [Google Scholar] [CrossRef] [PubMed]
- Yung-Chi, C.; Prusoff, W.H. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]
- Di Costanzo, L.; Pique, M.E.; Christianson, D.W. Crystal structure of human arginase I complexed with thiosemicarbazide reveals an unusual thiocarbonyl μ-sulfide ligand in the binuclear manganese cluster. J. Am. Chem. Soc. 2007, 129, 6388–6389. [Google Scholar] [CrossRef] [PubMed]
- BioSolveIT–GmbH. FlexX (Protein–Ligand Docker) user & technical reference as part of LeadIT 2.0. Germany, 2012; p. 15. Available online: https://www.biosolveit.de/FlexX (accessed on 18 March 2016).
- Weng, Y.-C.; Chuang, C.-F.; Chuang, S.-T.; Chiu, H.-L.; Kuo, Y.-H.; Su, M.-J. KS370G, a synthetic caffeamide derivative, improves left ventricular hypertrophy and function in pressure-overload mice heart. Eur. J. Pharmacol. 2012, 684, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.-H.; Li, N.-G.; Shi, Q.-P.; Hao-Tang, B.S.P.; Tang, Y.-P.; Wei-Li, B.S.P.; Lian-Yin, B.S.P.; Yang, J.-P.; Duan, J.-A. Design, synthesis and biological evaluation of ferulic acid amides as selective matrix metalloproteinase inhibitors. Med. Chem. 2013, 9, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Kawano, Y.; Uebayasi, M. Induction of adiponectin by natural and synthetic phenolamides in mouse and human preadipocytes and its enhancement by docosahexaenoic acid. Life Sci. 2008, 82, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Tang, Y.; Yamamoto, K. Metal-free oxidative amide formation with N-hydroxysuccinimide and hypervalent iodine reagents. Tetrahedron Lett. 2012, 53, 5094–5098. [Google Scholar] [CrossRef]
- Hong, S.S.; Jeong, W.; Kwon, J.G.; Choi, Y.-H.; Ahn, E.-K.; Ko, H.-J.; Seo, D.-W.; Oh, J.S. Phenolic Amides from the Fruits of Tribulus terrestris and Their Inhibitory Effects on the Production of Nitric Oxide. Bull. Korean Chem. Soc. 2013, 34, 3105–3108. [Google Scholar] [CrossRef]
- Kan, S.; Chen, G.; Han, C.; Chen, Z.; Song, X.; Ren, M.; Jiang, H. Chemical constituents from the roots of Xanthium sibiricum. Nat. Prod. Res. 2011, 25, 1243–1249. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-F.; Lay, H.-L.; Ni, C.-L.; Chen, C.-C. Phenolic Components of Dendrobium antennatum. Chem. Nat. Compd. 2013, 49, 520–522. [Google Scholar] [CrossRef]
- Tamiz, A.P.; Cai, S.X.; Zhou, Z.-L.; Yuen, P.-W.; Schelkun, R.M.; Whittemore, E.R.; Weber, E.; Woodward, R.M.; Keana, J.F.W. Structure−Activity relationship of N-(Phenylalkyl)cinnamides as novel NR2B subtype-selective NMDA receptor antagonists. J. Med. Chem. 1999, 42, 3412–3420. [Google Scholar] [CrossRef] [PubMed]
- Son, S.; Lewis, B.A. Free radical scavenging and antioxidative activity of caffeic acid amide and ester analogues: Structure−activity relationship. J. Agric. Food Chem. 2001, 50, 468–472. [Google Scholar] [CrossRef]
- Wu, Z.; Zheng, L.; Li, Y.; Su, F.; Yue, X.; Tang, W.; Ma, X.; Nie, J.; Li, H. Synthesis and structure–activity relationships and effects of phenylpropanoid amides of octopamine and dopamine on tyrosinase inhibition and antioxidation. Food Chem. 2012, 134, 1128–1131. [Google Scholar] [CrossRef] [PubMed]
- Michalet, S.; Cartier, G.; David, B.; Mariotte, A.-M.; Dijoux-franca, M.-G.; Kaatz, G. W.; Stavri, M.; Gibbons, S. N-Caffeoylphenalkylamide derivatives as bacterial efflux pump inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 1755–1758. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.-H.; Li, N.-G.; Shi, Q.-P.; Tang, H.; Tang, Y.-P.; Li, W.; Yin, L.; Yang, J.-P.; Duan, J.-A. Design, synthesis and biological evaluation of caffeic acid amides as selective MMP-2 and MMP-9 inhibitors. Drug Dev. Res. 2012, 73, 343–351. [Google Scholar] [CrossRef]
- Chou, S.-C.; Su, C.-R.; Ku, Y.-C.; Wu, T.-S. The constituents and their bioactivities of Houttuynia cordata. Chem. Pharm. Bull. 2009, 57, 1227–1230. [Google Scholar] [CrossRef] [PubMed]
- Larsson, R.; Blanco, N.; Johansson, M.; Sterner, O. Synthesis of C-1 indol-3-yl substituted tetrahydroisoquinoline derivatives via a Pictet–Spengler approach. Tetrahedron Lett. 2012, 53, 4966–4970. [Google Scholar] [CrossRef]
- Lee, S.U.; Shin, C.-G.; Lee, C.-K.; Lee, Y.S. Caffeoylglycolic and caffeoylamino acid derivatives, halfmers of l-chicoric acid, as new HIV-1 integrase inhibitors. Eur. J. Med. Chem. 2007, 42, 1309–1315. [Google Scholar] [CrossRef] [PubMed]
- SYBYL-X, Tripos International. Available online: https://www.certara.com (accessed on 18 March 2016).
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar] [CrossRef] [PubMed]
- Chemical Computing Group Inc. Molecular Operating Environment (MOE). Available online: https://www.chemcomp.com (accessed on 18 March 2016).
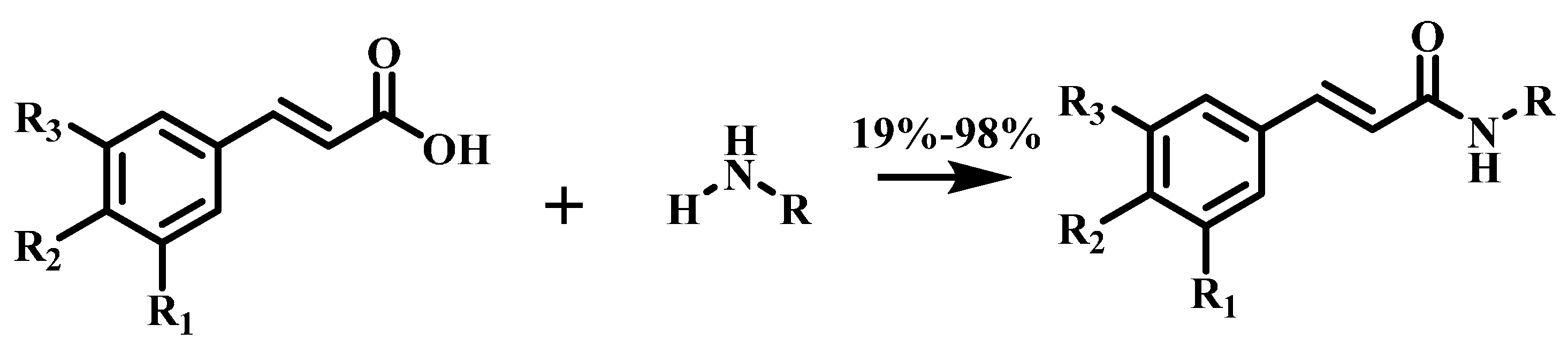


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 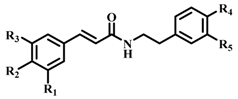 | b-ARG I Inhibition a (%) (100 μM) | IC50 b (µM) | ||||
|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | R4 | R5 | |||
| 1 (CAPA) | OH | OH | H | H | H | 64 ± 2 | 6.9 ± 1.3 |
| 2 | OCH3 | OH | H | H | H | 37 ± 4 | n.d c |
| 3 | H | OH | H | H | H | 31 ± 4 | n.d |
| 4 | OCH3 | OCH3 | OCH3 | H | H | 35 ± 2 | n.d |
| 5 | H | H | H | H | H | 34 ± 2 | n.d |
| 6 | OH | OH | H | OH | H | 58 ± 2 | 22.1 ± 1.6 |
| 7 | OCH3 | OH | H | OH | H | 24 ± 2 | n.d |
| 8 | H | OH | H | OH | H | 40 ± 2 | n.d |
| 9 | OCH3 | OCH3 | OCH3 | OH | H | 30 ± 2 | n.d |
| 10 | H | H | H | OH | H | 34 ± 3 | n.d |
| 11 | OH | OH | H | OH | OH | 62 ± 3 | 114.9 ± 1.3 |
| 12 | OCH3 | OH | H | OH | OH | 61 ± 3 | 198.7 ± 1.4 |
| 13 | H | OH | H | OH | OH | 54 ± 1 | 170.4 ± 1.7 |
| 14 | OCH3 | OCH3 | OCH3 | OH | OH | 53 ± 1 | 193.6 ± 1.4 |
| 15 | H | H | H | OH | OH | 68 ± 3 | 39.3 ± 1.4 |
| 16 |  | 59 ± 1 | 35.6 ± 1.3 | ||||
| 17 d | 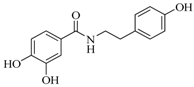 | 52 ± 2 | 175.3 ± 1.5 | ||||
| 18 |  | 62 ± 1 | 41.9 ± 1.3 | ||||
| 19 | 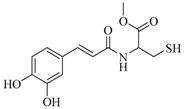 | 67 ± 1 | 37.0 ± 1.3 | ||||
| Caffeic acid | 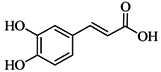 | 61 ± 4 | 86.7 ± 8.0 | ||||
| CGA |  | 71 ± 1 | 10.6 ± 1.6 | ||||
| nor-NOHA e | 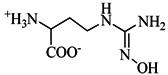 | 99 ± 1 | 1.7 ± 0.2 | ||||
| Inhibitor | Ki (µM) | Type of Inhibition |
|---|---|---|
| CAPA (1) | 5.5 ± 1.0 | competitive |
| CGA | 8.4 ± 1.2 | competitive |
| nor-NOHA | 1.3 ± 0.1 | competitive |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pham, T.-N.; Bordage, S.; Pudlo, M.; Demougeot, C.; Thai, K.-M.; Girard-Thernier, C. Cinnamide Derivatives as Mammalian Arginase Inhibitors: Synthesis, Biological Evaluation and Molecular Docking. Int. J. Mol. Sci. 2016, 17, 1656. https://doi.org/10.3390/ijms17101656
Pham T-N, Bordage S, Pudlo M, Demougeot C, Thai K-M, Girard-Thernier C. Cinnamide Derivatives as Mammalian Arginase Inhibitors: Synthesis, Biological Evaluation and Molecular Docking. International Journal of Molecular Sciences. 2016; 17(10):1656. https://doi.org/10.3390/ijms17101656
Chicago/Turabian StylePham, Thanh-Nhat, Simon Bordage, Marc Pudlo, Céline Demougeot, Khac-Minh Thai, and Corine Girard-Thernier. 2016. "Cinnamide Derivatives as Mammalian Arginase Inhibitors: Synthesis, Biological Evaluation and Molecular Docking" International Journal of Molecular Sciences 17, no. 10: 1656. https://doi.org/10.3390/ijms17101656