
Amyloid β Peptide Enhances RANKL-Induced Osteoclast Activation through NF-κB, ERK, and Calcium Oscillation Signaling


Abstract
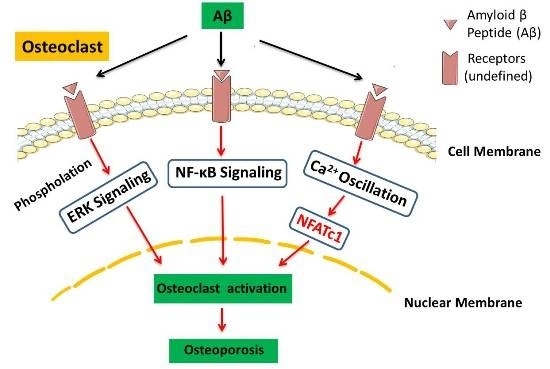
:
1. Introduction
2. Results
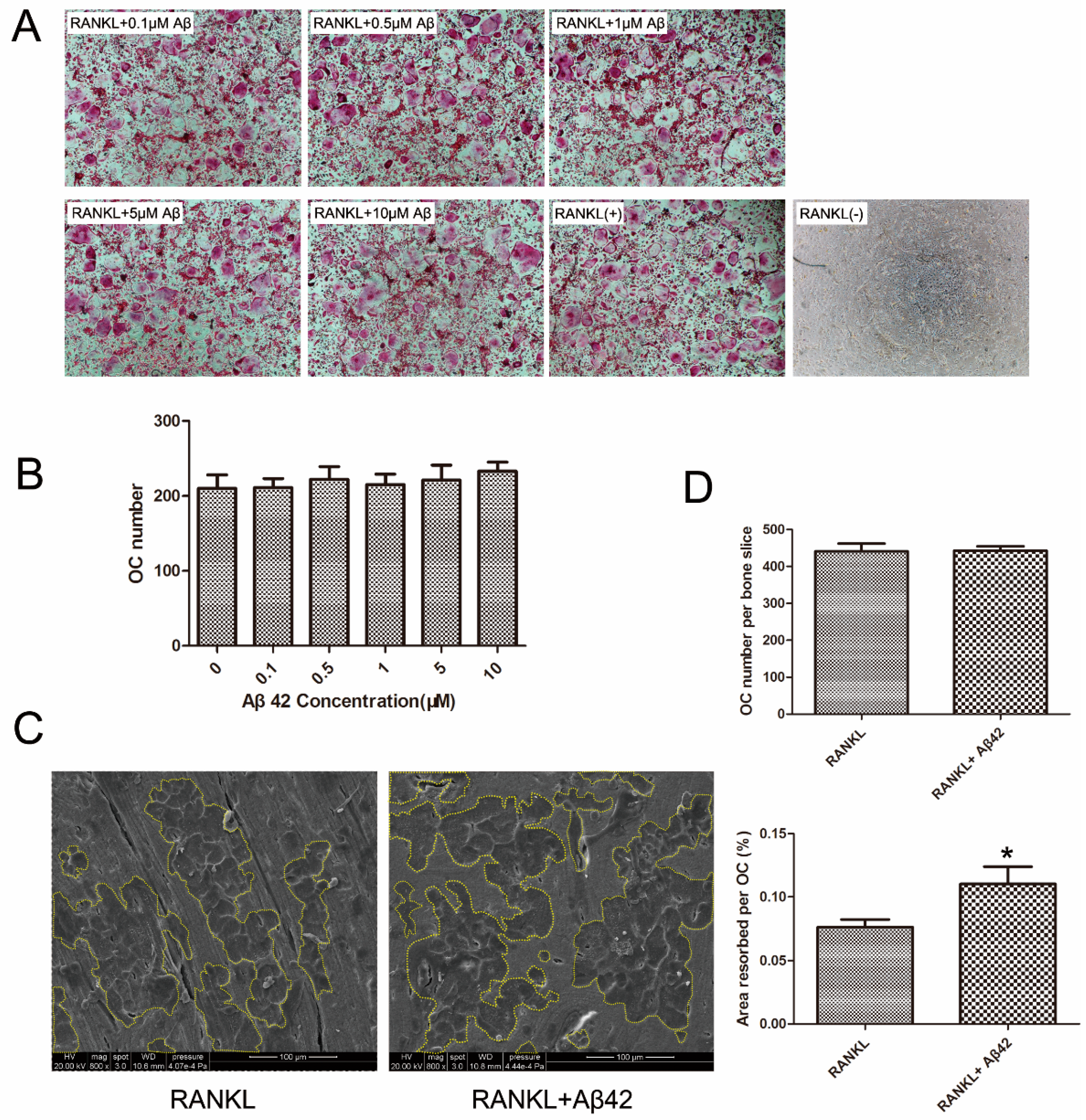
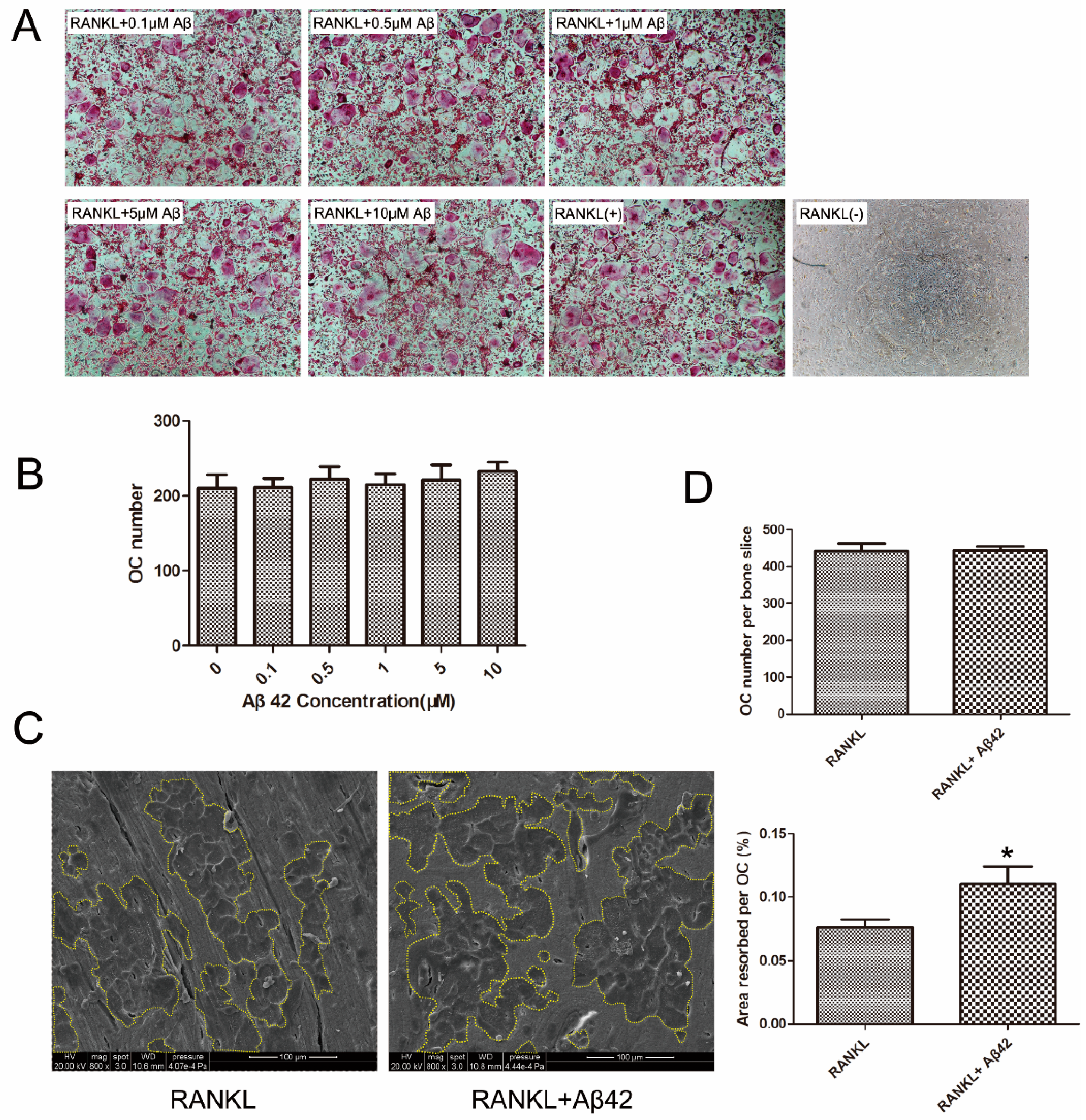
2.1. Aβ42 Exerts No Effect on RANKL-Induced Osteoclastogenesis but Enhances Osteoclastic Bone Resorption
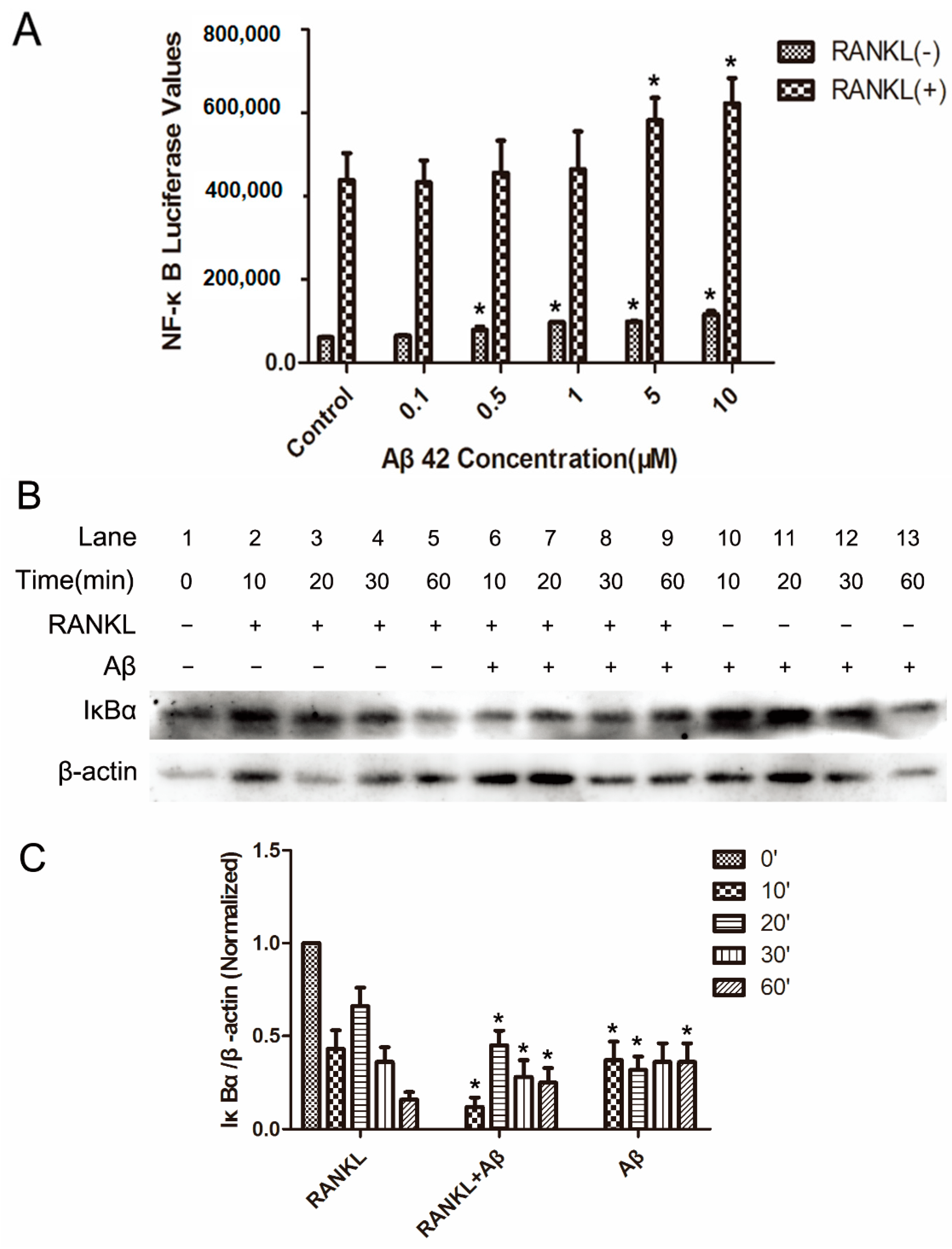
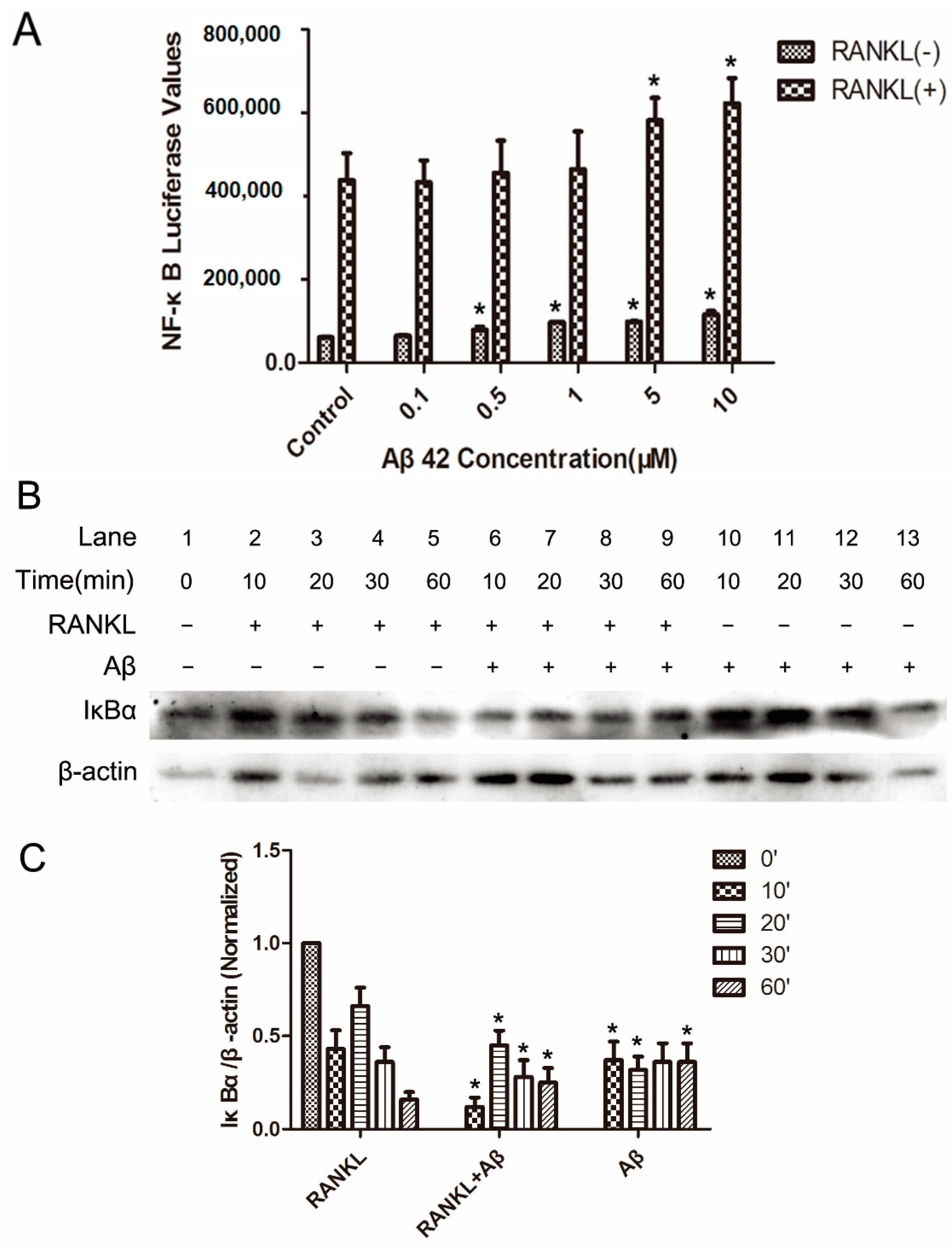
2.2. Aβ42 Enhances RANKL-Stimulated NF-κB Activity and IκB-α Degeneration
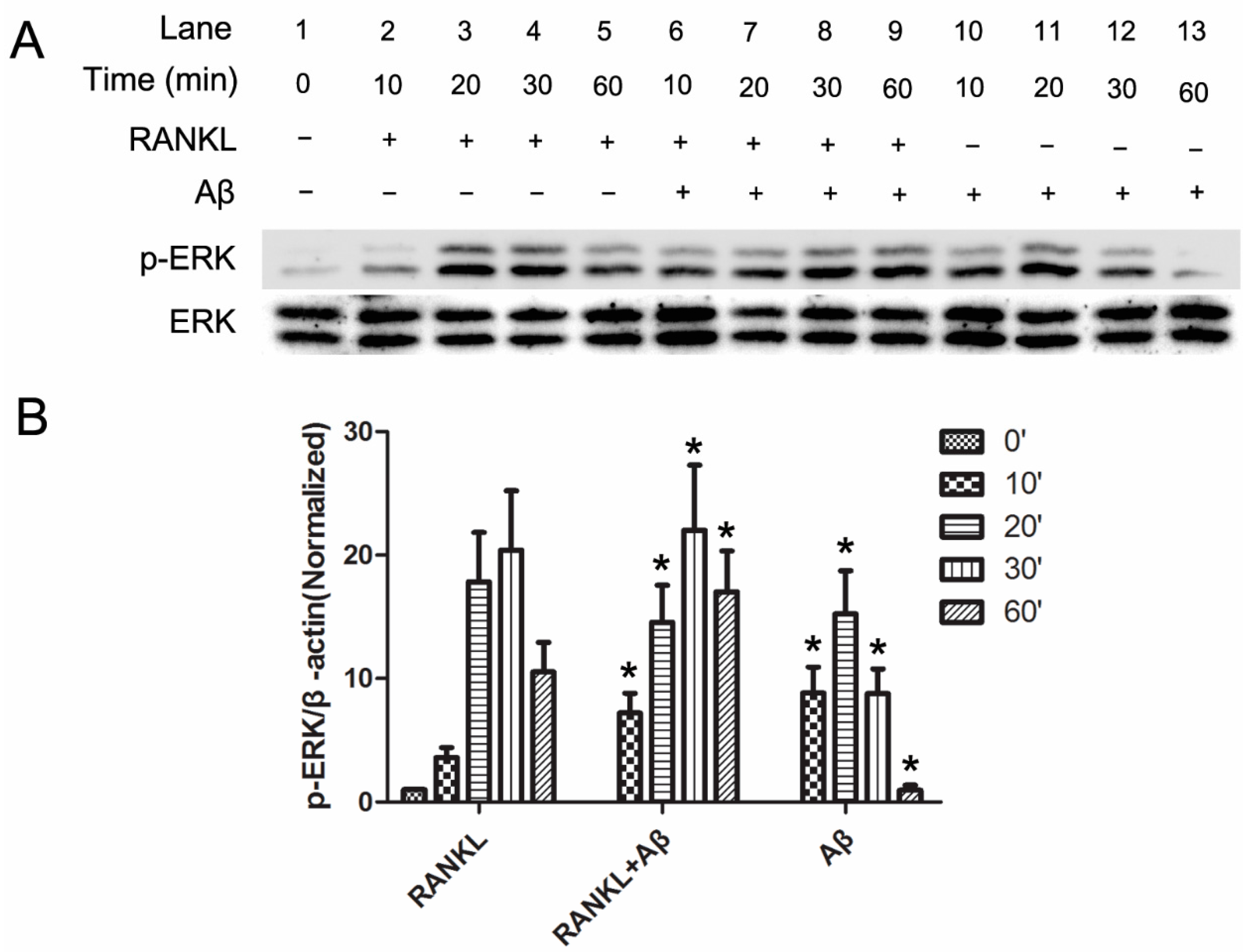
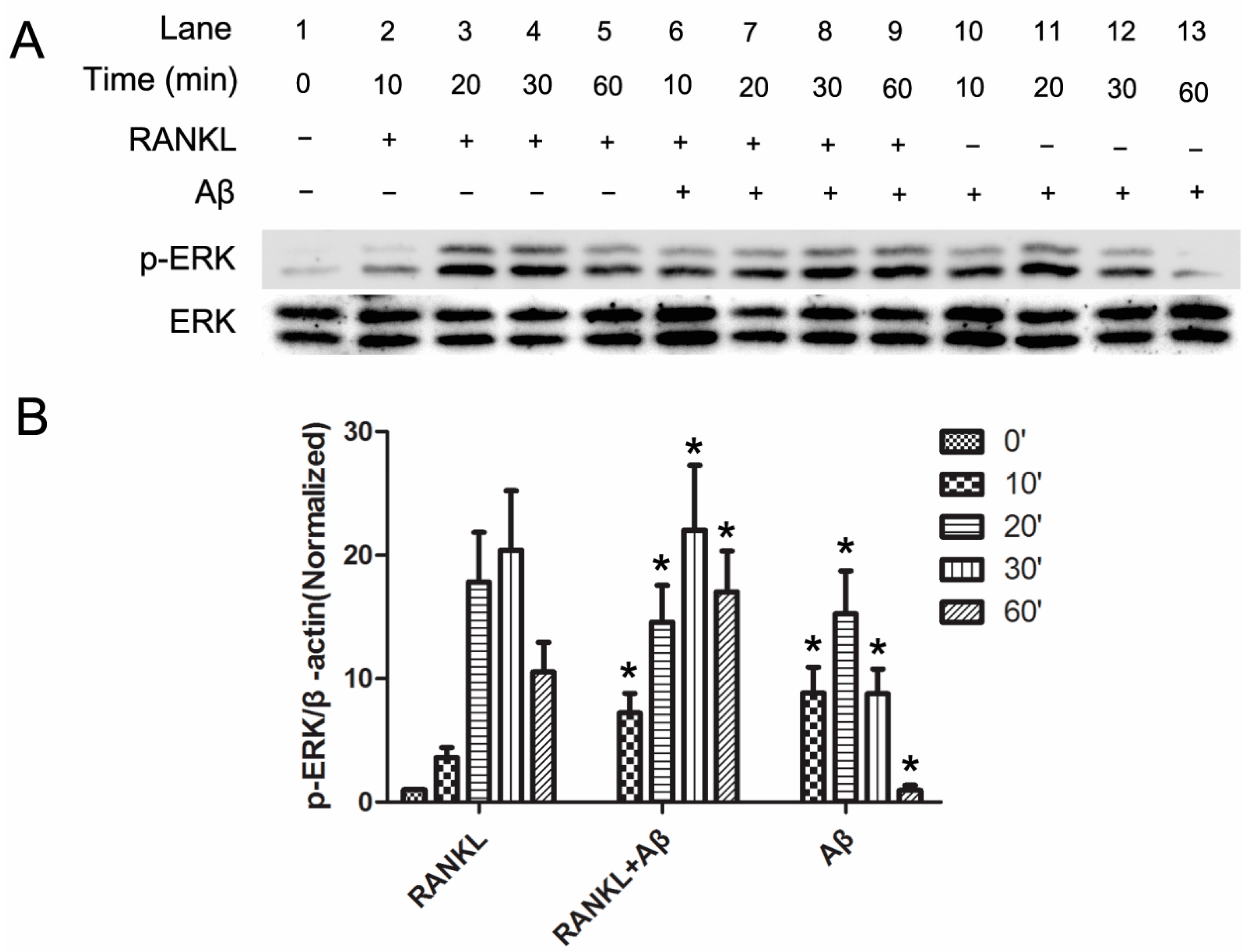
2.3. Aβ42 Promotes the Phosphorylation of ERK Induced by RANKL
2.4. Aβ42 Increases RANKL-Induced Calcium Oscillation during OC Differentiation
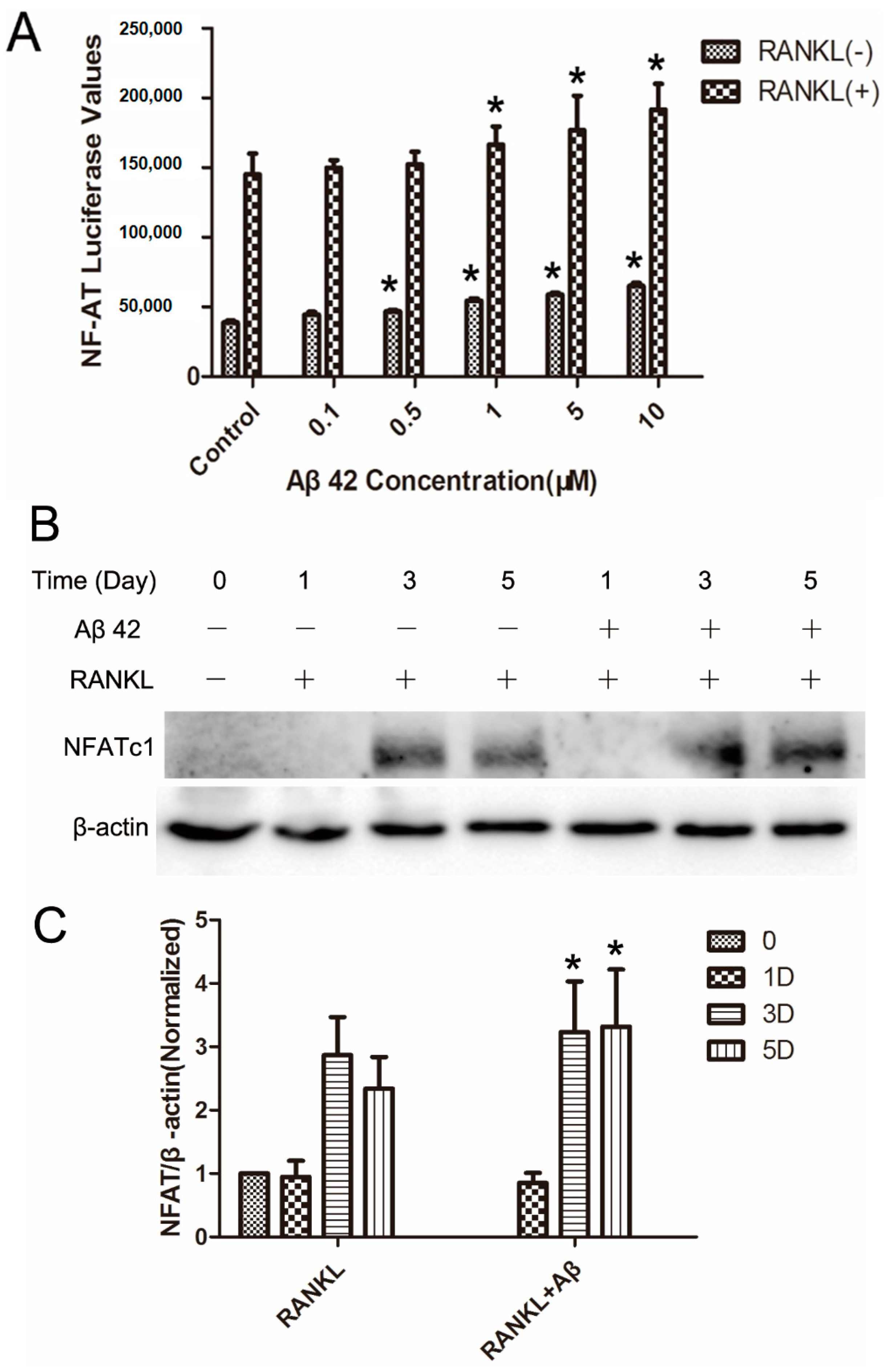
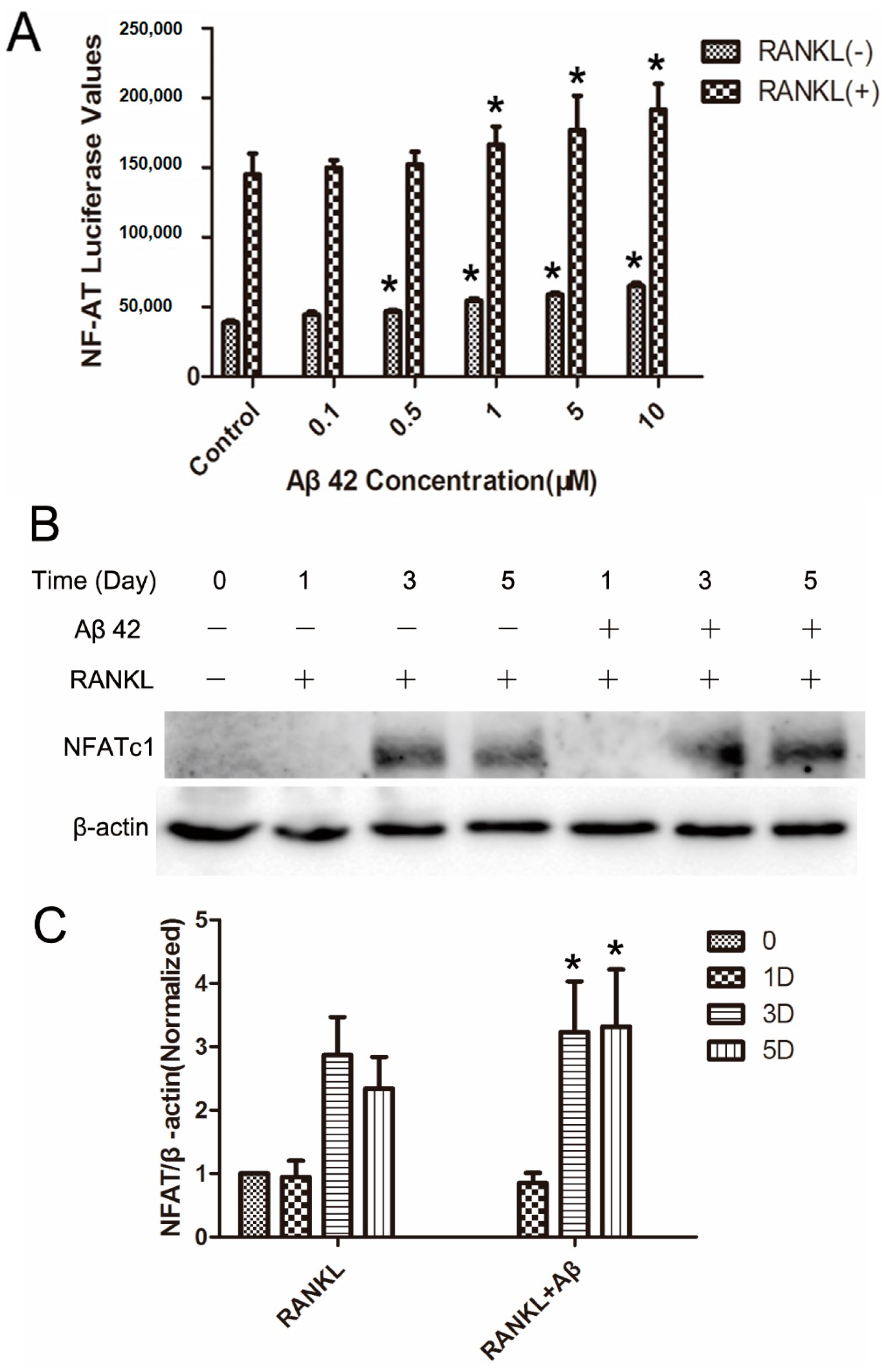
2.5. Aβ42 Enhances NFAT Activity and Protein Expression of NFATc1 during OC Differentiation
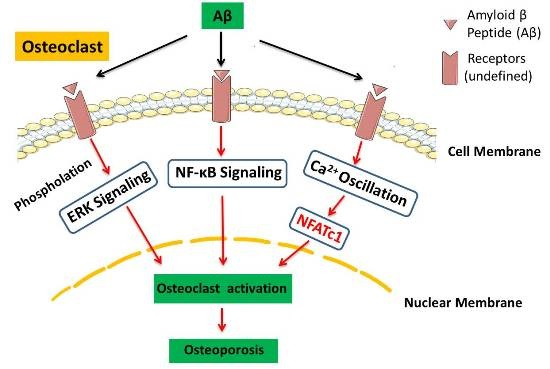
3. Discussion
4. Experimental Section
4.1. Reagents
4.2. Macrophage Isolation from Mouse Bone Marrow, Osteoclastogenesis Assay, and Tartrate Resistant Acid Phosphatase (TRAP) Staining
4.3. Functional Lacunar Resorption Pit Formation Assay
4.4. Luciferase Assay
4.5. Western Blotting Assay
4.6. Measurement of Intracellular Ca2+ Oscillation
4.7. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Roos, P.M. Osteoporosis in neurodegeneration. J. Trace Elem. Med. Biol. 2014, 28, 418–421. [Google Scholar] [CrossRef] [PubMed]
- Tysiewicz-Dudek, M.; Pietraszkiewicz, F.; Drozdzowska, B. Alzheimer’s disease and osteoporosis: Common risk factors or one condition predisposing to the other? Ortop. Traumatol. Rehabil. 2008, 10, 315–323. [Google Scholar] [PubMed]
- Chang, K.H.; Chung, C.J.; Lin, C.L.; Sung, F.C.; Wu, T.N.; Kao, C.H. Increased risk of dementia in patients with osteoporosis: A population-based retrospective cohort analysis. Age 2014, 36, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Weller, I.; Schatzker, J. Hip fractures and Alzheimer’s disease in elderly institutionalized Canadians. Ann. Epidemiol. 2004, 14, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Zhou, H.; Rui, L.; Xu, J. Bone loss and osteoporosis are associated with conversion from mild cognitive impairment to Alzheimer’s disease. Curr. Alzheimer Res. 2014, 11, 706–713. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Deng, J.; Zhang, M.; Zhou, H.D.; Wang, Y.J. Association between bone mineral density and the risk of Alzheimer’s disease. J. Alzheimer’s Dis. 2011, 24, 101–108. [Google Scholar]
- Li, S.; Liu, B.; Zhang, L.; Rong, L. Amyloid β peptide is elevated in osteoporotic bone tissues and enhances osteoclast function. Bone 2014, 61, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Immel, D.; Xi, C.X.; Bierhaus, A.; Feng, X.; Mei, L.; Nawroth, P.; Stern, D.M.; Xiong, W.C. Regulation of osteoclast function and bone mass by rage. J. Exp. Med. 2006, 203, 1067–1080. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Xiong, F.; Hong, Y.; Jung, J.U.; Li, X.S.; Liu, J.Z.; Yan, R.; Mei, L.; Feng, X.; Xiong, W.C. APPswe/Aβ regulation of osteoclast activation and rage expression in an age-dependent manner. J. Bone Miner. Res. 2011, 26, 1084–1098. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.R.; Mundy, G.R. Advances in osteoclast biology: Old findings and new insights from mouse models. Nat. Rev. Rheumatol. 2011, 7, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Deng, H.W.; Shen, H. Circulating monocytes: An appropriate model for bone-related study. Osteoporos. Int. 2015, 26, 2561–2572. [Google Scholar] [CrossRef] [PubMed]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Asagiri, M.; Takayanagi, H. The molecular understanding of osteoclast differentiation. Bone 2007, 40, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Abu-Amer, Y. NF-κB signaling and bone resorption. Osteoporos. Int. 2013, 24, 2377–2386. [Google Scholar] [CrossRef] [PubMed]
- Kajiya, H. Calcium signaling in osteoclast differentiation and bone resorption. Adv. Exp. Med. Biol. 2012, 740, 917–932. [Google Scholar] [PubMed]
- Nakamura, I.; Takahashi, N.; Jimi, E.; Udagawa, N.; Suda, T. Regulation of osteoclast function. Mod. Rheumatol. Jpn. Rheum. Assoc. 2012, 22, 167–177. [Google Scholar] [CrossRef]
- Sitara, D.; Aliprantis, A.O. Transcriptional regulation of bone and joint remodeling by NFAT. Immunol. Rev. 2010, 233, 286–300. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, T.; Takayanagi, H. New regulation mechanisms of osteoclast differentiation. Ann. N. Y. Acad. Sci. 2011, 1240, E13–E18. [Google Scholar] [CrossRef] [PubMed]
- Asagiri, M.; Sato, K.; Usami, T.; Ochi, S.; Nishina, H.; Yoshida, H.; Morita, I.; Wagner, E.F.; Mak, T.W.; Serfling, E.; et al. Autoamplification of nfatc1 expression determines its essential role in bone homeostasis. J. Exp. Med. 2005, 202, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Rojas, A.; Delgado-Lopez, F.; Gonzalez, I.; Perez-Castro, R.; Romero, J.; Rojas, I. The receptor for advanced glycation end-products: A complex signaling scenario for a promiscuous receptor. Cell. Signal. 2013, 25, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Son, S.M.; Jin, S.M.; Hong, H.S.; Shin, D.H.; Kim, S.J.; Huh, K.; Mook-Jung, I. Rage regulates bace1 and Aβ generation via NFAT1 activation in alzheimer’s disease animal model. FASEB J. 2009, 23, 2639–2649. [Google Scholar] [CrossRef] [PubMed]
- Cianferotti, L.; Gomes, A.R.; Fabbri, S.; Tanini, A.; Brandi, M.L. The calcium-sensing receptor in bone metabolism: From bench to bedside and back. Osteoporos. Int. 2015, 26, 2055–2071. [Google Scholar] [CrossRef] [PubMed]
- Demuro, A.; Parker, I.; Stutzmann, G.E. Calcium signaling and amyloid toxicity in Alzheimer’s disease. J. Biol. Chem. 2010, 285, 12463–12468. [Google Scholar] [CrossRef] [PubMed]
- Kular, J.; Tickner, J.; Chim, S.M.; Xu, J. An overview of the regulation of bone remodelling at the cellular level. Clin. Biochem. 2012, 45, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Xiu, Y.; Boyce, B.F. Osteoclast fusion and regulation by RANKl-dependent and independent factors. World J. Orthop. 2012, 3, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, H. Rankl, a necessary chance for clinical application to osteoporosis and cancer-related bone diseases. World J. Orthop. 2013, 4, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wu, H.F.; Ang, E.S.; Yip, K.; Woloszyn, M.; Zheng, M.H.; Tan, R.X. NF-κB modulators in osteolytic bone diseases. Cytokine Growth Factor Rev. 2009, 20, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, N. Regulation of NFATC1 in osteoclast differentiation. J. Bone Metab. 2014, 21, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Negishi-Koga, T.; Takayanagi, H. Ca2+-NFATC1 signaling is an essential axis of osteoclast differentiation. Immunol. Rev. 2009, 231, 241–256. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Teitelbaum, S.L. Osteoclasts: New insights. Bone Res. 2013, 1, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, E.; Mukohyama, H.; Aoki, K.; Duarte, W.R.; Lerner, U.H.; Ohya, K.; Omura, K.; Kasugai, S. High extracellular calcium affects osteoclastogenesis in mouse bone marrow cell culture. J. Med. Dent. Sci. 2002, 49, 109–120. [Google Scholar] [PubMed]
- Xu, J.; Tan, J.W.; Huang, L.; Gao, X.H.; Laird, R.; Liu, D.; Wysocki, S.; Zheng, M.H. Cloning, sequencing, and functional characterization of the rat homologue of receptor activator of NF-κB ligand. J. Bone Miner. Res. 2000, 15, 2178–2186. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wu, H.; Chim, S.M.; Zhou, L.; Zhao, J.; Feng, H.; Wei, Q.; Wang, Q.; Zheng, M.H.; Tan, R.X.; et al. Sc-514, a selective inhibitor of IKKβ attenuates rankl-induced osteoclastogenesis and NF-κB activation. Biochem. Pharmacol. 2013, 86, 1775–1783. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Steer, J.H.; Joyce, D.A.; Yip, K.H.; Zheng, M.H.; Xu, J. 12-O-tetradecanoylphorbol-13-acetate (tPA) inhibits osteoclastogenesis by suppressing rankl-induced NF-κB activation. J. Bone Miner. Res. 2003, 18, 2159–2168. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.P.; van der Kraan, A.G.; Xu, J.; Gillespie, M.T.; Quinn, J.M. Membrane-bound receptor activator of NFκB ligand (rankl) activity displayed by osteoblasts is differentially regulated by osteolytic factors. Biochem. Biophys. Res. Commun. 2012, 422, 48–53. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Average Change in Oscillation Intensity (/cell) | Average Change in Oscillation Frequency (times/min) |
|---|---|---|
| Control | 8 ± 6 * | 1 ± 1 * |
| RANKL | 189 ± 45 | 6 ± 2 |
| Aβ (5 μM) | 98 ± 46 * | 2 ± 2 * |
| RANKL + Aβ (1 μM) | 205 ± 53 | 8 ± 3 * |
| RANKL + Aβ (5 μM) | 238 ± 47 * | 9 ± 2 * |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Yang, B.; Teguh, D.; Zhou, L.; Xu, J.; Rong, L. Amyloid β Peptide Enhances RANKL-Induced Osteoclast Activation through NF-κB, ERK, and Calcium Oscillation Signaling. Int. J. Mol. Sci. 2016, 17, 1683. https://doi.org/10.3390/ijms17101683
Li S, Yang B, Teguh D, Zhou L, Xu J, Rong L. Amyloid β Peptide Enhances RANKL-Induced Osteoclast Activation through NF-κB, ERK, and Calcium Oscillation Signaling. International Journal of Molecular Sciences. 2016; 17(10):1683. https://doi.org/10.3390/ijms17101683
Chicago/Turabian StyleLi, Shangfu, Bu Yang, Dian Teguh, Lin Zhou, Jiake Xu, and Limin Rong. 2016. "Amyloid β Peptide Enhances RANKL-Induced Osteoclast Activation through NF-κB, ERK, and Calcium Oscillation Signaling" International Journal of Molecular Sciences 17, no. 10: 1683. https://doi.org/10.3390/ijms17101683