
Tocotrienol Affects Oxidative Stress, Cholesterol Homeostasis and the Amyloidogenic Pathway in Neuroblastoma Cells: Consequences for Alzheimer’s Disease

 ,
,  ,
,  , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
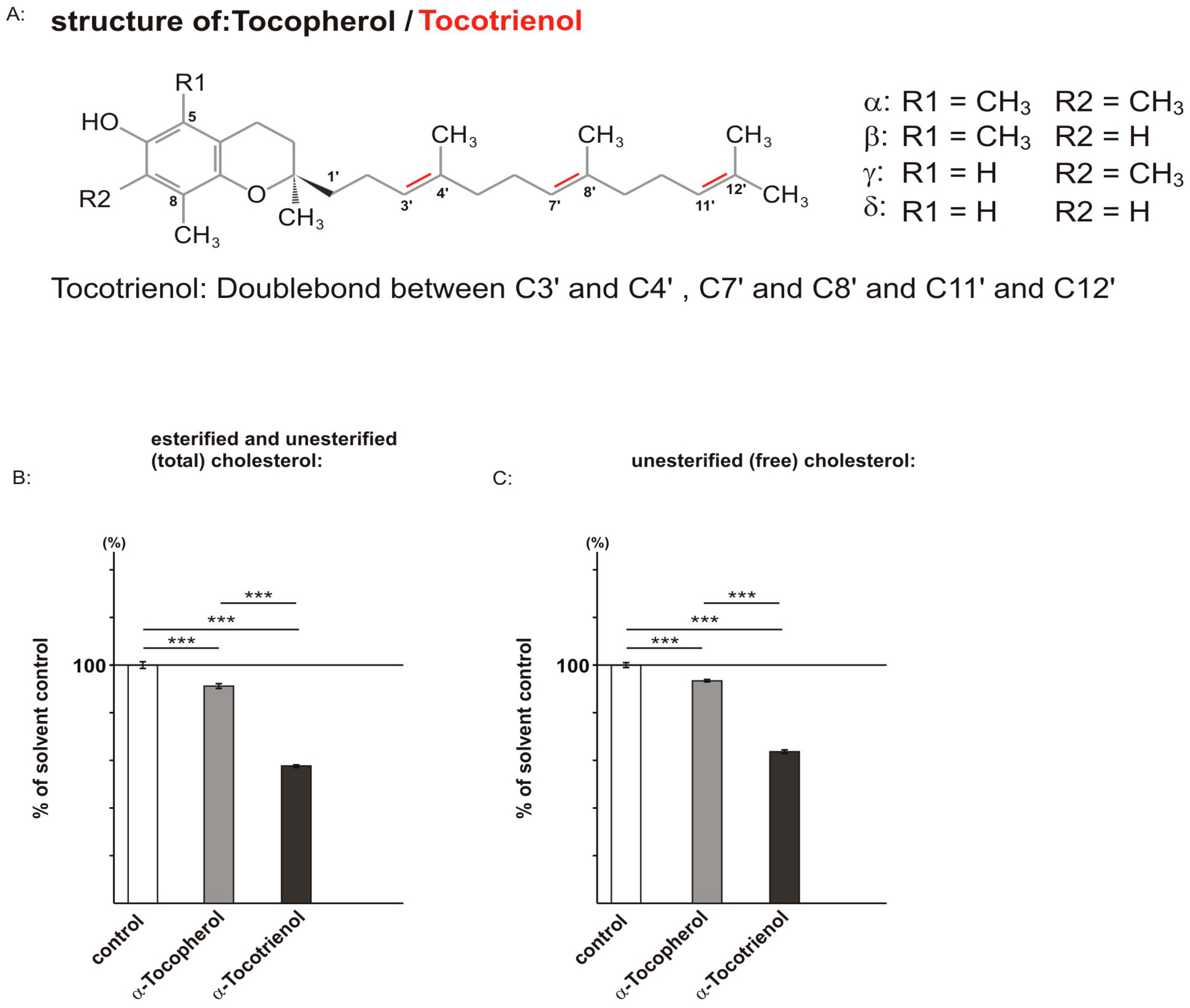
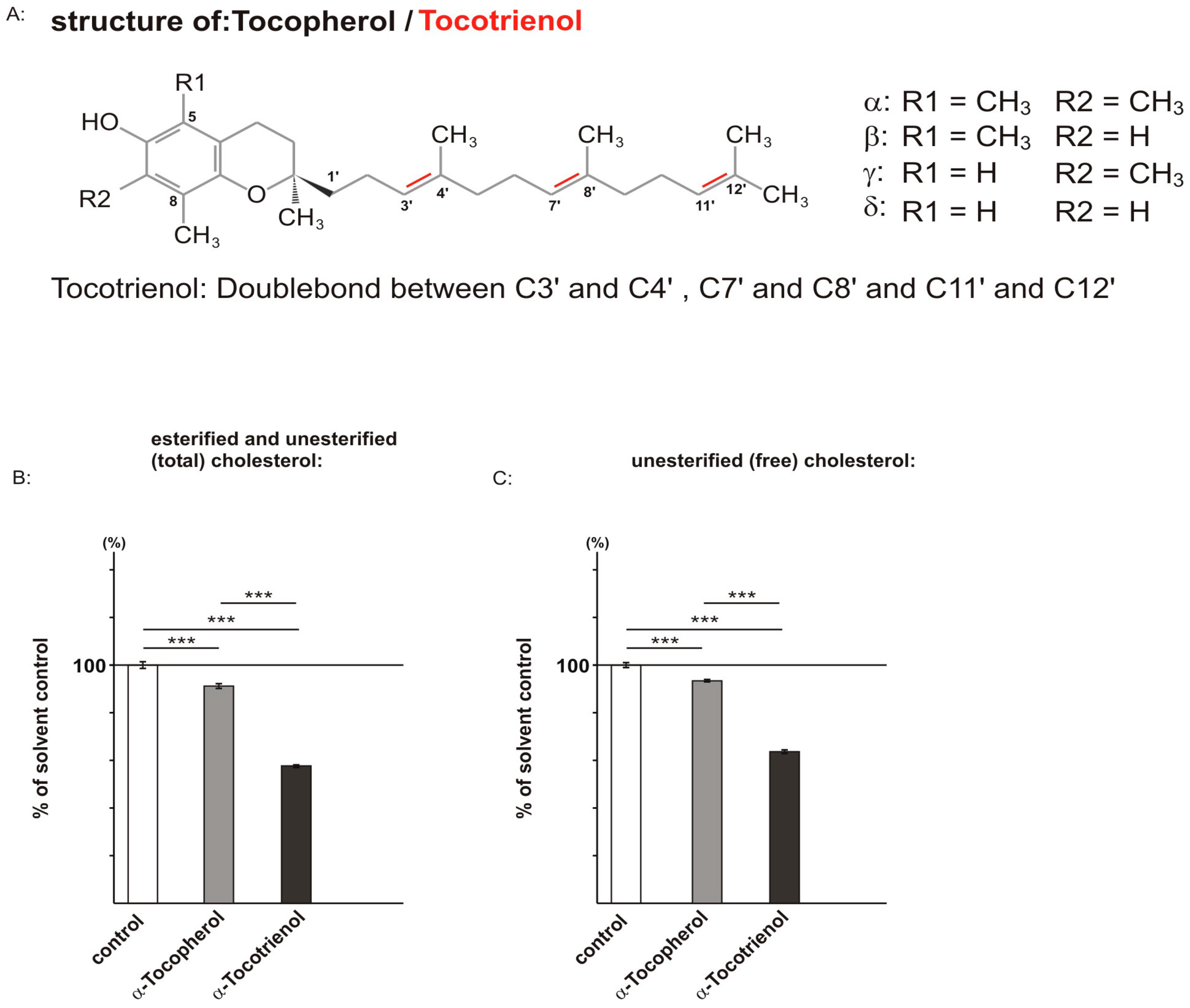
2.1. Effect of α-Tocopherol and α-Tocotrienol on Cholesterol Level
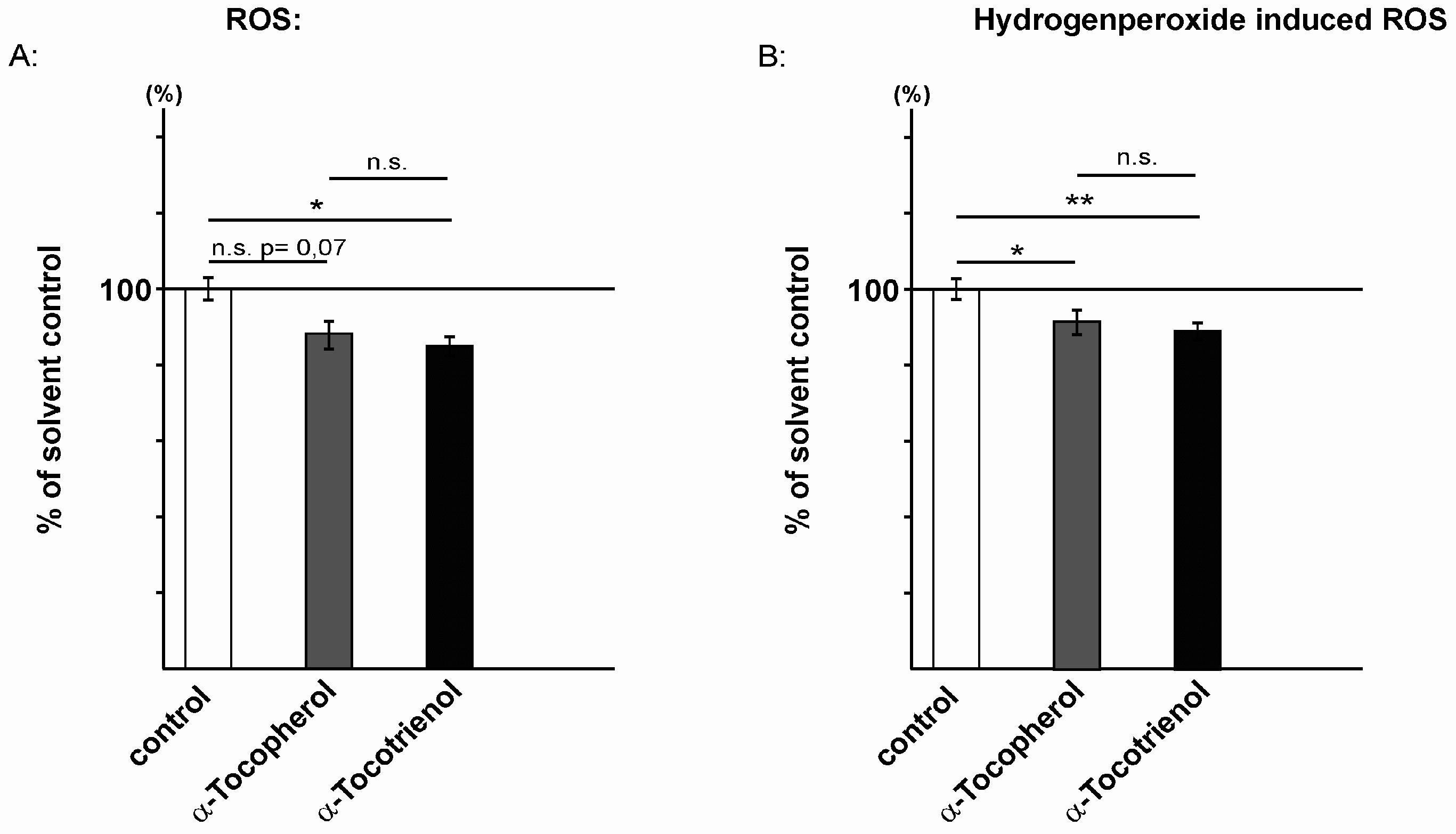
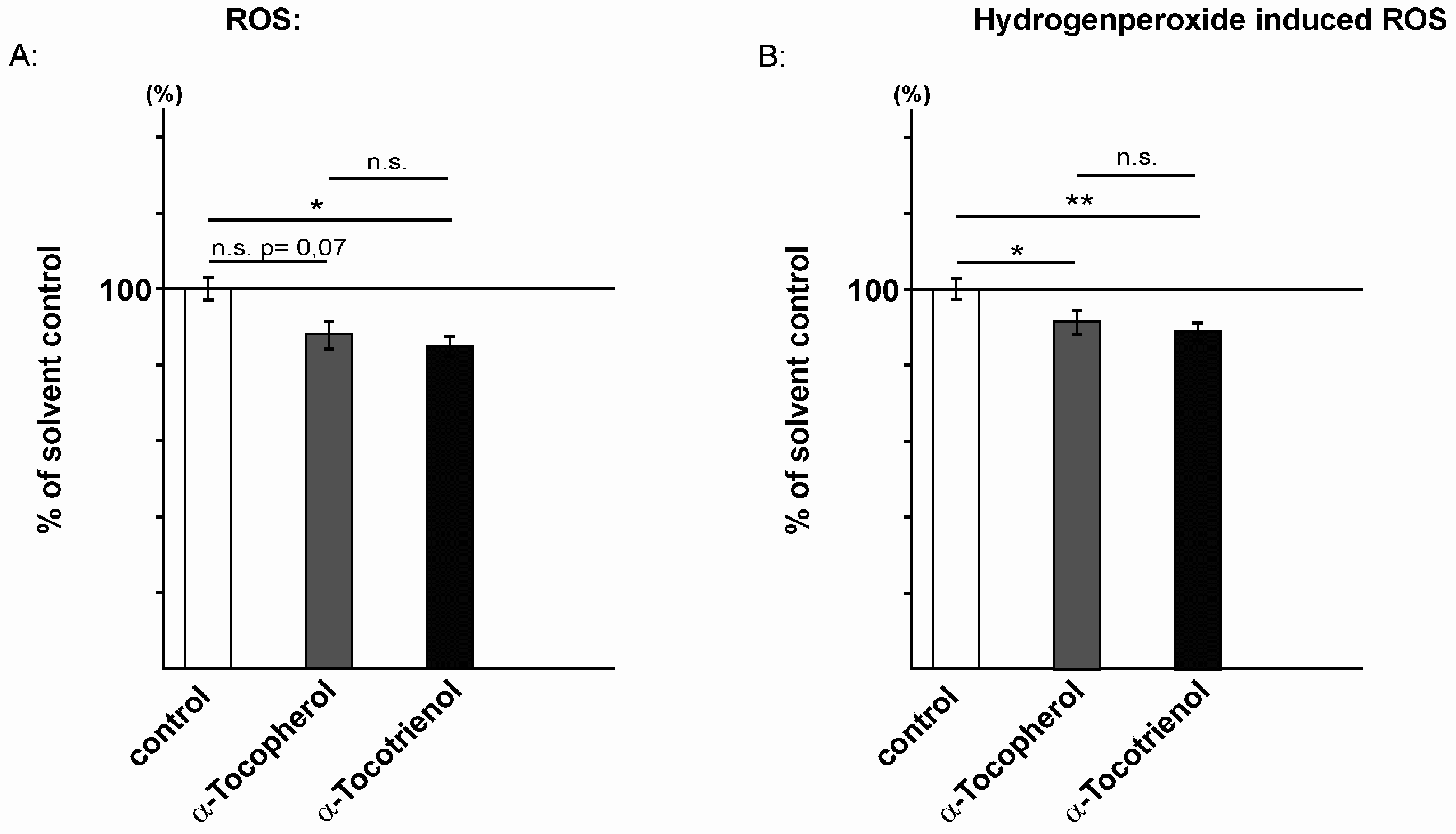
2.2. Reactive Oxidative Species Are Reduced in the Presence of α-Tocopherol and α-Tocotrienol
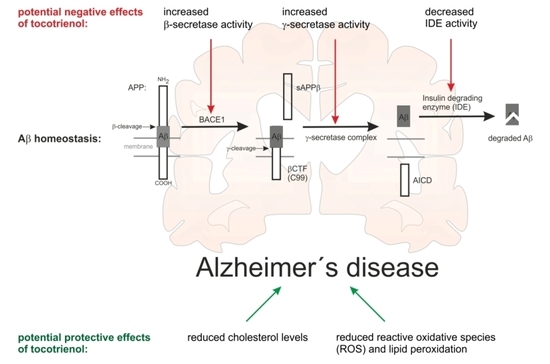
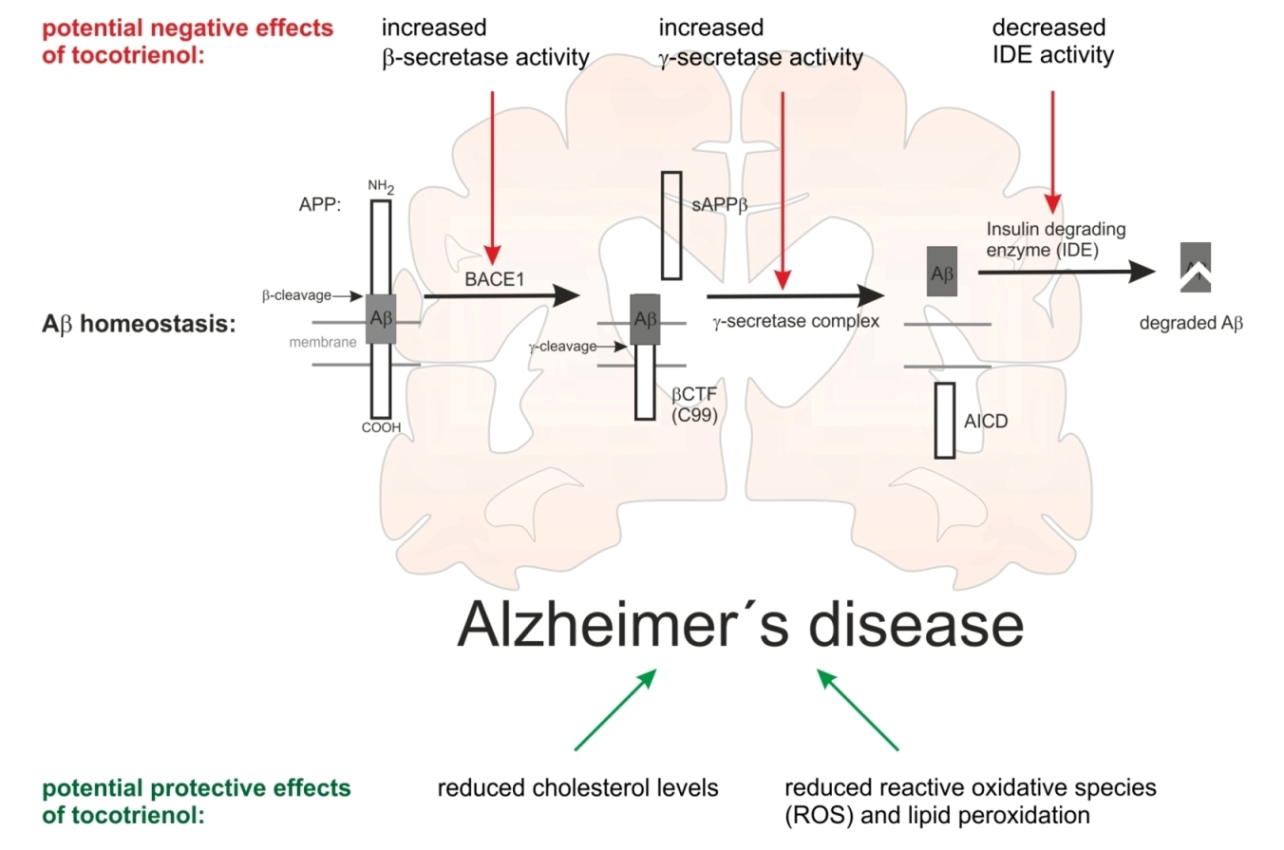
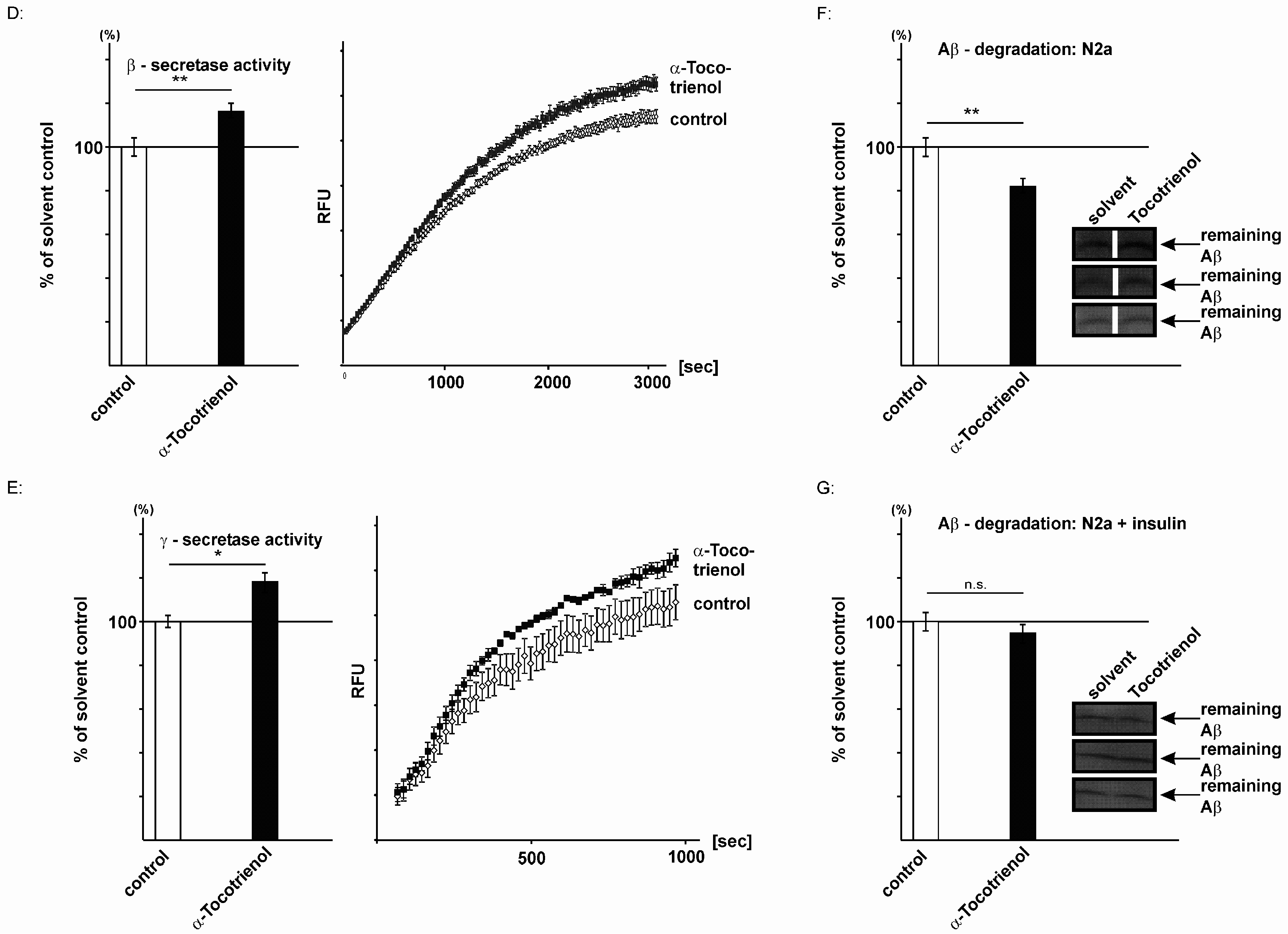
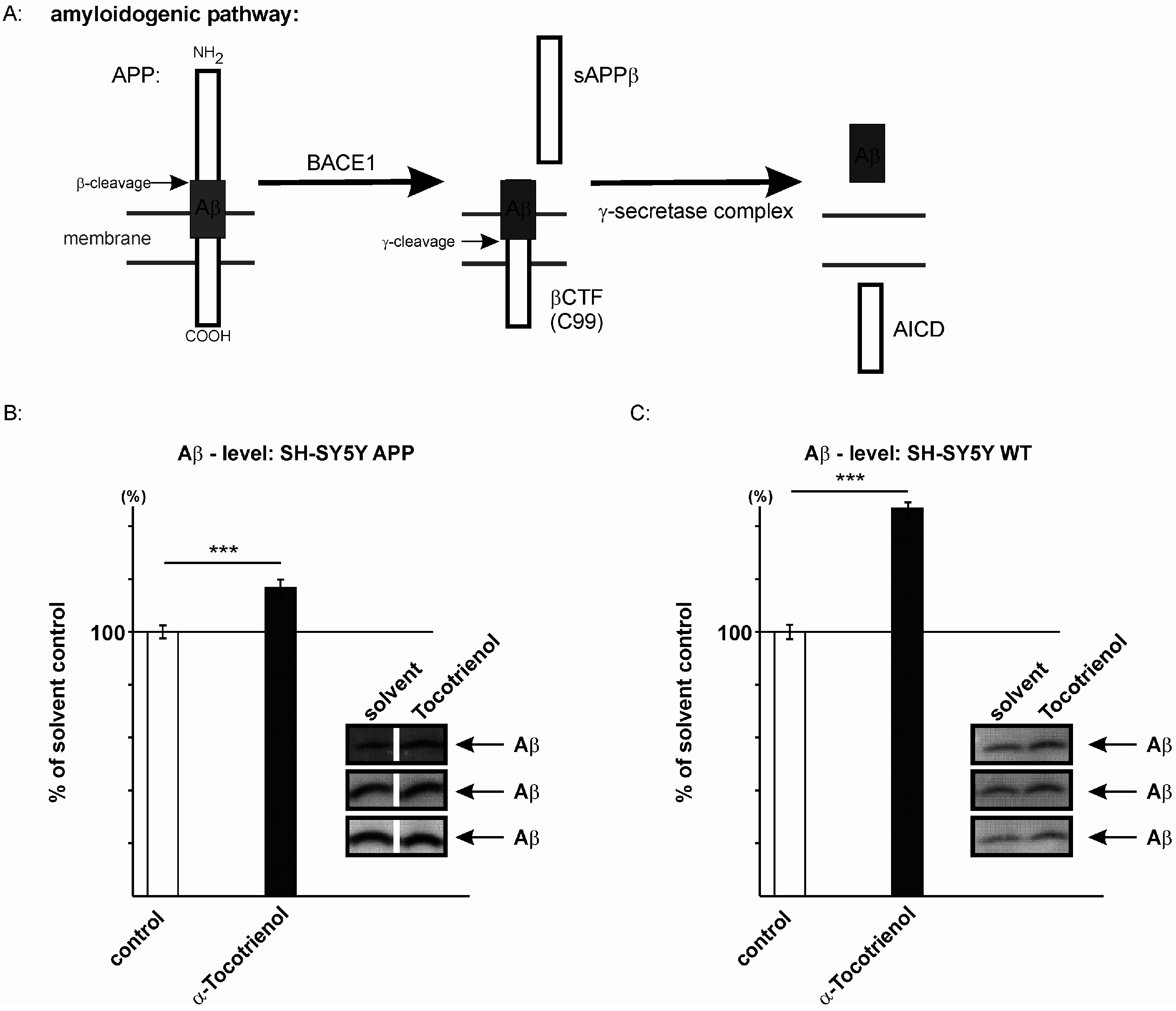
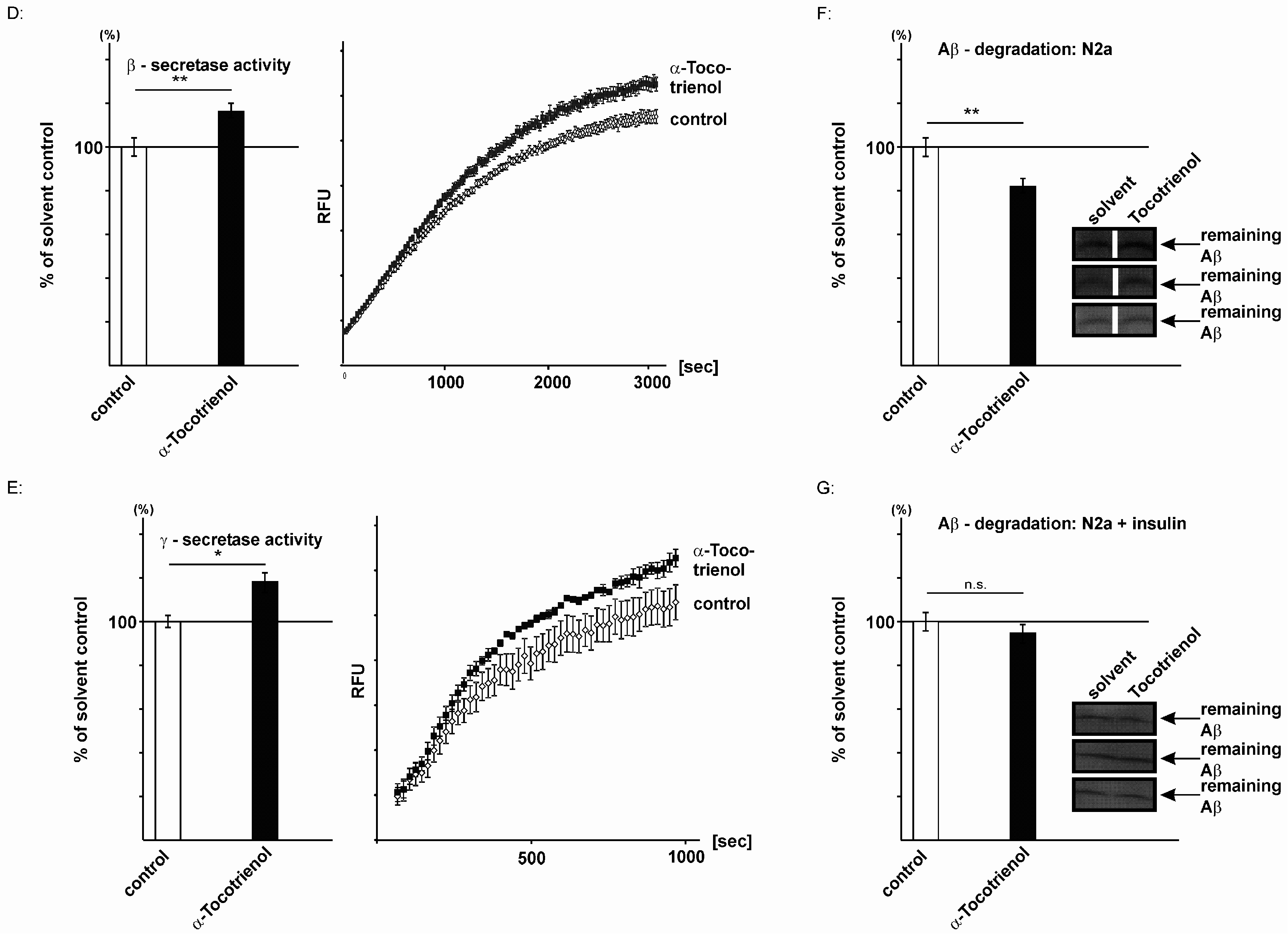
2.3. Effect of α-Tocotrienol on Aβ Generation and Aβ Degradation
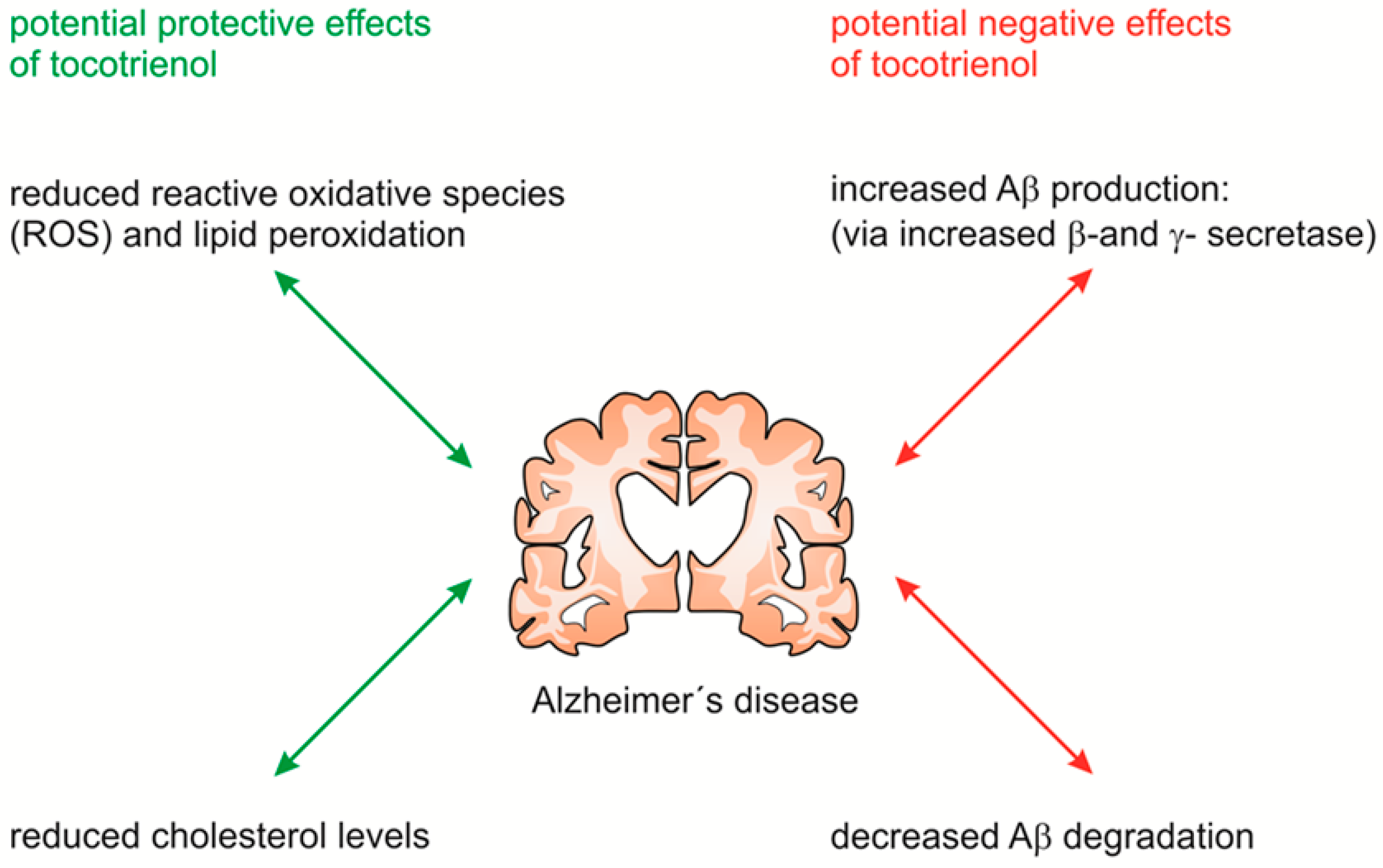
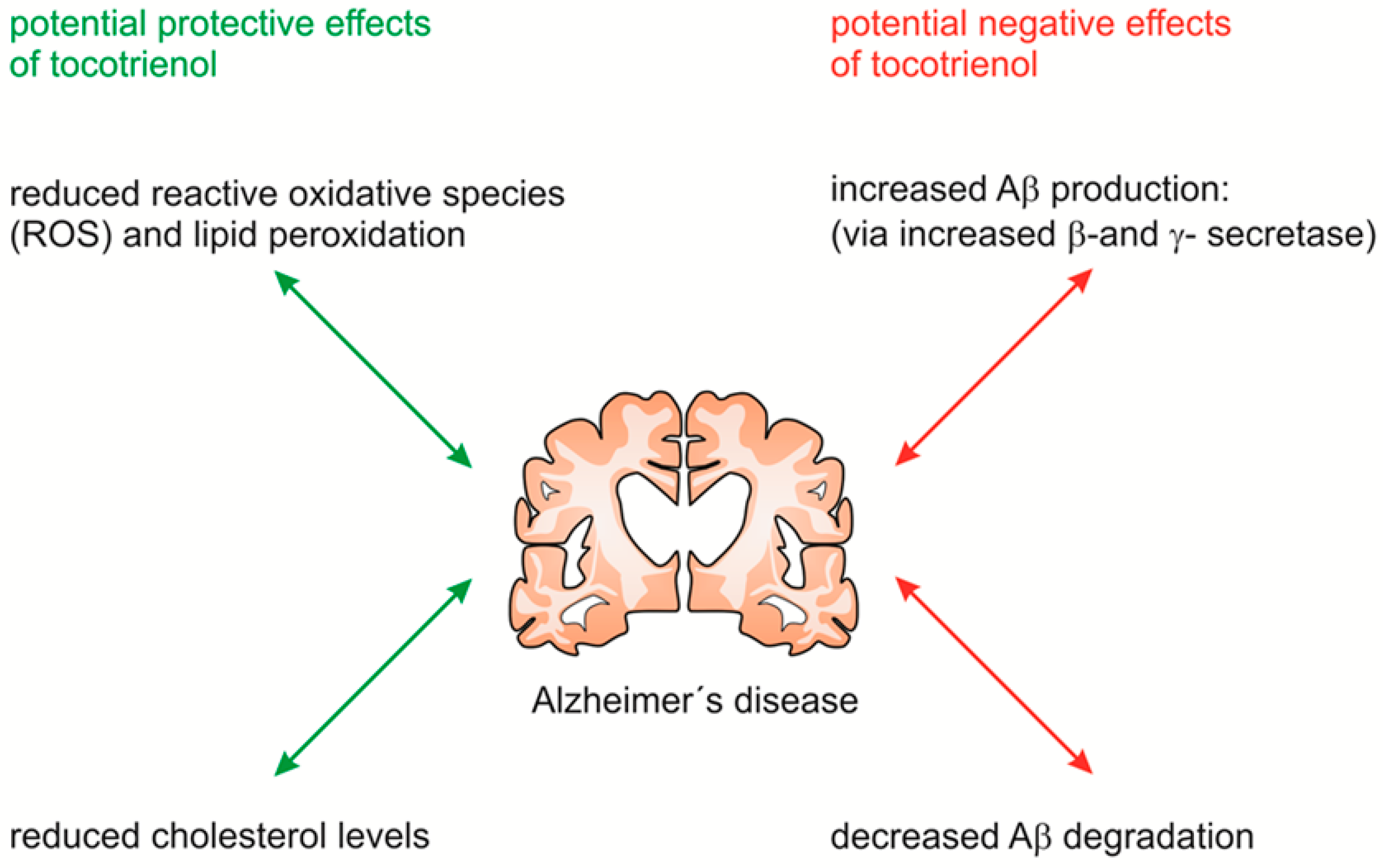
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. Cell Viability
4.4. Cholesterol Concentration
4.5. Detection of Reactive Oxygen Species
4.6. Determination of Protein Concentration
4.7. Western Blot Analysis
4.8. Preparation of Purified Membranes
4.9. Measurement of β- and γ-Secretase Activity
4.10. Measurement of Secretase Activity in Living Cells
4.11. Quantitative Real-Time Experiments
4.12. Determination of Total Aβ Degradation
4.13. Tocopherol and Tocotrienol Uptake
4.13.1. Sample Preparation
4.13.2. LC-MS/MS
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [PubMed]
- Haass, C. Take five—Bace and the γ-secretase quartet conduct Alzheimer’s amyloid β-peptide generation. EMBO J. 2004, 23, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Grundke-Iqbal, I.; Iqbal, K.; Quinlan, M.; Tung, Y.C.; Zaidi, M.S.; Wisniewski, H.M. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J. Biol. Chem. 1986, 261, 6084–6089. [Google Scholar] [PubMed]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Demuro, A.; Parker, I.; Stutzmann, G.E. Calcium signaling and amyloid toxicity in Alzheimer disease. J. Biol. Chem. 2010, 285, 12463–12468. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Vondrakova, D.; Lawson, M.; Valko, M. Metals, oxidative stress and neurodegenerative disorders. Mol. Cell. Biochem. 2010, 345, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Mandrekar-Colucci, S.; Landreth, G.E. Microglia and inflammation in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2010, 9, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Fedrizzi, L.; Carafoli, E. Ca2+ dysfunction in neurodegenerative disorders: Alzheimer’s disease. BioFactors 2011, 37, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Gotz, J.; Eckert, A.; Matamales, M.; Ittner, L.M.; Liu, X. Modes of abeta toxicity in Alzheimer’s disease. Cell. Mol. Life Sci. 2011, 68, 3359–3375. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Kivipelto, M.; Helkala, E.L.; Laakso, M.P.; Hanninen, T.; Hallikainen, M.; Alhainen, K.; Soininen, H.; Tuomilehto, J.; Nissinen, A. Midlife vascular risk factors and Alzheimer’s disease in later life: Longitudinal, population based study. Br. Med. J. 2001, 322, 1447–1451. [Google Scholar] [CrossRef]
- Refolo, L.M.; Malester, B.; LaFrancois, J.; Bryant-Thomas, T.; Wang, R.; Tint, G.S.; Sambamurti, K.; Duff, K.; Pappolla, M.A. Hypercholesterolemia accelerates the Alzheimer’s amyloid pathology in a transgenic mouse model. Neurobiol. Dis. 2000, 7, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Shie, F.S.; Jin, L.W.; Cook, D.G.; Leverenz, J.B.; LeBoeuf, R.C. Diet-induced hypercholesterolemia enhances brain Aβ accumulation in transgenic mice. Neuroreport 2002, 13, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Refolo, L.M.; Pappolla, M.A.; LaFrancois, J.; Malester, B.; Schmidt, S.D.; Thomas-Bryant, T.; Tint, G.S.; Wang, R.; Mercken, M.; Petanceska, S.S.; et al. A cholesterol-lowering drug reduces β-amyloid pathology in a transgenic mouse model of Alzheimer’s disease. Neurobiol. Dis. 2001, 8, 890–899. [Google Scholar] [CrossRef] [PubMed]
- Sparks, D.L.; Kuo, Y.M.; Roher, A.; Martin, T.; Lukas, R.J. Alterations of Alzheimer’s disease in the cholesterol-fed rabbit, including vascular inflammation. Preliminary observations. Ann. N. Y. Acad. Sci. 2000, 903, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Fassbender, K.; Simons, M.; Bergmann, C.; Stroick, M.; Lutjohann, D.; Keller, P.; Runz, H.; Kuhl, S.; Bertsch, T.; von Bergmann, K.; et al. Simvastatin strongly reduces levels of Alzheimer’s disease β-amyloid peptides Aβ 42 and Aβ 40 in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2001, 98, 5856–5861. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Keller, P.; De Strooper, B.; Beyreuther, K.; Dotti, C.G.; Simons, K. Cholesterol depletion inhibits the generation of β-amyloid in hippocampal neurons. Proc. Natl. Acad. Sci. USA 1998, 95, 6460–6464. [Google Scholar] [CrossRef] [PubMed]
- Wahrle, S.; Das, P.; Nyborg, A.C.; McLendon, C.; Shoji, M.; Kawarabayashi, T.; Younkin, L.H.; Younkin, S.G.; Golde, T.E. Cholesterol-dependent γ-secretase activity in buoyant cholesterol-rich membrane microdomains. Neurobiol. Dis. 2002, 9, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Grimm, H.S.; Tomic, I.; Beyreuther, K.; Hartmann, T.; Bergmann, C. Independent inhibition of Alzheimer disease β- and γ-secretase cleavage by lowered cholesterol levels. J. Biol. Chem. 2008, 283, 11302–11311. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Callaghan, D.; Jones, A.; Walker, D.G.; Lue, L.F.; Beach, T.G.; Sue, L.I.; Woulfe, J.; Xu, H.; Stanimirovic, D.B.; et al. Cholesterol retention in Alzheimer’s brain is responsible for high β- and γ-secretase activities and Aβ production. Neurobiol. Dis. 2008, 29, 422–437. [Google Scholar] [CrossRef] [PubMed]
- Haag, M.D.; Hofman, A.; Koudstaal, P.J.; Stricker, B.H.; Breteler, M.M. Statins are associated with a reduced risk of Alzheimer disease regardless of lipophilicity. The rotterdam study. J. Neurol. Neurosurg. Psychiatry 2009, 80, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Wolozin, B.; Kellman, W.; Ruosseau, P.; Celesia, G.G.; Siegel, G. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme a reductase inhibitors. Arch. Neurol. 2000, 57, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Wolozin, B.; Wang, S.W.; Li, N.C.; Lee, A.; Lee, T.A.; Kazis, L.E. Simvastatin is associated with a reduced incidence of dementia and Parkinson’s disease. BMC Med. 2007, 5, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Rea, T.D.; Breitner, J.C.; Psaty, B.M.; Fitzpatrick, A.L.; Lopez, O.L.; Newman, A.B.; Hazzard, W.R.; Zandi, P.P.; Burke, G.L.; Lyketsos, C.G.; et al. Statin use and the risk of incident dementia: The cardiovascular health study. Arch. Neurol. 2005, 62, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Feldman, H.H.; Doody, R.S.; Kivipelto, M.; Sparks, D.L.; Waters, D.D.; Jones, R.W.; Schwam, E.; Schindler, R.; Hey-Hadavi, J.; DeMicco, D.A.; et al. Randomized controlled trial of atorvastatin in mild to moderate Alzheimer disease: Leade. Neurology 2010, 74, 956–964. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Bell, K.L.; Galasko, D.; Galvin, J.E.; Thomas, R.G.; van Dyck, C.H.; Aisen, P.S. A randomized, double-blind, placebo-controlled trial of simvastatin to treat Alzheimer disease. Neurology 2011, 77, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Pearce, B.C.; Parker, R.A.; Deason, M.E.; Qureshi, A.A.; Wright, J.J. Hypocholesterolemic activity of synthetic and natural tocotrienols. J. Med. Chem. 1992, 35, 3595–3606. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, A.A.; Bradlow, B.A.; Brace, L.; Manganello, J.; Peterson, D.M.; Pearce, B.C.; Wright, J.J.; Gapor, A.; Elson, C.E. Response of hypercholesterolemic subjects to administration of tocotrienols. Lipids 1995, 30, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Parker, R.A.; Pearce, B.C.; Clark, R.W.; Gordon, D.A.; Wright, J.J. Tocotrienols regulate cholesterol production in mammalian cells by post-transcriptional suppression of 3-hydroxy-3-methylglutaryl-coenzyme a reductase. J. Biol. Chem. 1993, 268, 11230–11238. [Google Scholar] [PubMed]
- Qureshi, A.A.; Qureshi, N.; Hasler-Rapacz, J.O.; Weber, F.E.; Chaudhary, V.; Crenshaw, T.D.; Gapor, A.; Ong, A.S.; Chong, Y.H.; Peterson, D.; et al. Dietary tocotrienols reduce concentrations of plasma cholesterol, apolipoprotein B, thromboxane B2, and platelet factor 4 in pigs with inherited hyperlipidemias. Am. J. Clin. Nutr. 1991, 53, 1042–1046. [Google Scholar]
- Xia, W.; Mo, H. Potential of tocotrienols in the prevention and therapy of Alzheimer’s disease. J. Nutr. Biochem. 2016, 31, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohe, R.; Traber, M.G. Vitamin E: Function and metabolism. FASEB J. 1999, 13, 1145–1155. [Google Scholar] [PubMed]
- Ricciarelli, R.; Argellati, F.; Pronzato, M.A.; Domenicotti, C. Vitamin E and neurodegenerative diseases. Mol. Asp. Med. 2007, 28, 591–606. [Google Scholar] [CrossRef] [PubMed]
- La Fata, G.; Weber, P.; Mohajeri, M.H. Effects of vitamin E on cognitive performance during ageing and in Alzheimer’s disease. Nutrients 2014, 6, 5453–5472. [Google Scholar] [CrossRef] [PubMed]
- Mangialasche, F.; Polidori, M.C.; Monastero, R.; Ercolani, S.; Camarda, C.; Cecchetti, R.; Mecocci, P. Biomarkers of oxidative and nitrosative damage in Alzheimer’s disease and mild cognitive impairment. Ageing Res. Rev. 2009, 8, 285–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muid, S.; Froemming, G.R.; Rahman, T.; Ali, A.M.; Nawawi, H.M. δ- and γ-tocotrienol isomers are potent in inhibiting inflammation and endothelial activation in stimulated human endothelial cells. Food Nutr. Res. 2016, 60, 31526–31537. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.Y.; Ward, L.C.; Fong, C.W.; Yap, W.N.; Brown, L. Anti-inflammatory γ- and δ-tocotrienols improve cardiovascular, liver and metabolic function in diet-induced obese rats. Eur. J. Nutr. 2015. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.J.; Liu, P.L.; Ng, L.T. Tocotrienol-rich fraction of palm oil exhibits anti-inflammatory property by suppressing the expression of inflammatory mediators in human monocytic cells. Mol. Nutr. Food Res. 2008, 52, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Reiter, E.; Jiang, Q.; Christen, S. Anti-inflammatory properties of α- and γ-tocopherol. Mol. Asp. Med. 2007, 28, 668–691. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q. Natural forms of vitamin E: Metabolism, antioxidant, and anti-inflammatory activities and their role in disease prevention and therapy. Free Radic. Biol. Med. 2014, 72, 76–90. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Stahlmann, C.P.; Mett, J.; Haupenthal, V.J.; Zimmer, V.C.; Lehmann, J.; Hundsdorfer, B.; Endres, K.; Grimm, H.S.; Hartmann, T. Vitamin E: Curse or benefit in Alzheimer’s disease? A systematic investigation of the impact of α-, γ- and δ-tocopherol on ass generation and degradation in neuroblastoma cells. J. Nutr. Health Aging 2015, 19, 646–656. [Google Scholar] [CrossRef] [PubMed]
- Fairus, S.; Nor, R.M.; Cheng, H.M.; Sundram, K. α-tocotrienol is the most abundant tocotrienol isomer circulated in plasma and lipoproteins after postprandial tocotrienol-rich vitamin E supplementation. Nutr. J. 2012, 11, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Song, B.L.; DeBose-Boyd, R.A. Insig-dependent ubiquitination and degradation of 3-hydroxy-3-methylglutaryl coenzyme a reductase stimulated by δ- and γ-tocotrienols. J. Biol. Chem. 2006, 281, 25054–25061. [Google Scholar] [CrossRef] [PubMed]
- Valastyan, S.; Thakur, V.; Johnson, A.; Kumar, K.; Manor, D. Novel transcriptional activities of vitamin E: Inhibition of cholesterol biosynthesis. Biochemistry 2008, 47, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Krycer, J.R.; Phan, L.; Brown, A.J. A key regulator of cholesterol homoeostasis, SREBP-2, can be targeted in prostate cancer cells with natural products. Biochem. J. 2012, 446, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.Y.; Chang, C.C.; Bryleva, E.; Rogers, M.A.; Murphy, S.R. Neuronal cholesterol esterification by acat1 in Alzheimer’s disease. IUBMB Life 2010, 62, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Craft, N.E.; Haitema, T.B.; Garnett, K.M.; Fitch, K.A.; Dorey, C.K. Carotenoid, tocopherol, and retinol concentrations in elderly human brain. J. Nutr. Health Aging 2004, 8, 156–162. [Google Scholar] [PubMed]
- Setsukinai, K.; Urano, Y.; Kakinuma, K.; Majima, H.J.; Nagano, T. Development of novel fluorescence probes that can reliably detect reactive oxygen species and distinguish specific species. J. Biol. Chem. 2003, 278, 3170–3175. [Google Scholar] [CrossRef] [PubMed]
- Shirotani, K.; Tsubuki, S.; Iwata, N.; Takaki, Y.; Harigaya, W.; Maruyama, K.; Kiryu-Seo, S.; Kiyama, H.; Iwata, H.; Tomita, T.; et al. Neprilysin degrades both amyloid β peptides 1–40 and 1–42 most rapidly and efficiently among thiorphan- and phosphoramidon-sensitive endopeptidases. J. Biol. Chem. 2001, 276, 21895–21901. [Google Scholar] [CrossRef] [PubMed]
- Takaki, Y.; Iwata, N.; Tsubuki, S.; Taniguchi, S.; Toyoshima, S.; Lu, B.; Gerard, N.P.; Gerard, C.; Lee, H.J.; Shirotani, K.; et al. Biochemical identification of the neutral endopeptidase family member responsible for the catabolism of amyloid β peptide in the brain. J. Biochem. 2000, 128, 897–902. [Google Scholar] [CrossRef] [PubMed]
- Vekrellis, K.; Ye, Z.; Qiu, W.Q.; Walsh, D.; Hartley, D.; Chesneau, V.; Rosner, M.R.; Selkoe, D.J. Neurons regulate extracellular levels of amyloid beta-protein via proteolysis by insulin-degrading enzyme. J. Neurosci. 2000, 20, 1657–1665. [Google Scholar] [PubMed]
- Qiu, W.Q.; Walsh, D.M.; Ye, Z.; Vekrellis, K.; Zhang, J.; Podlisny, M.B.; Rosner, M.R.; Safavi, A.; Hersh, L.B.; Selkoe, D.J. Insulin-degrading enzyme regulates extracellular levels of amyloid β-protein by degradation. J. Biol. Chem. 1998, 273, 32730–32738. [Google Scholar] [CrossRef] [PubMed]
- Evin, G.; Weidemann, A. Biogenesis and metabolism of Alzheimer’s disease Aβ amyloid peptides. Peptides 2002, 23, 1285–1297. [Google Scholar] [CrossRef]
- Rohan de Silva, H.A.; Jen, A.; Wickenden, C.; Jen, L.S.; Wilkinson, S.L.; Patel, A.J. Cell-specific expression of β-amyloid precursor protein isoform mrnas and proteins in neurons and astrocytes. Brain Res. Mol. Brain Res. 1997, 47, 147–156. [Google Scholar] [CrossRef]
- Duering, M.; Grimm, M.O.; Grimm, H.S.; Schroder, J.; Hartmann, T. Mean age of onset in familial Alzheimer’s disease is determined by amyloid β42. Neurobiol. Aging 2005, 26, 785–788. [Google Scholar] [CrossRef] [PubMed]
- Dyrks, T.; Dyrks, E.; Monning, U.; Urmoneit, B.; Turner, J.; Beyreuther, K. Generation of β A4 from the amyloid protein precursor and fragments thereof. FEBS Lett. 1993, 335, 89–93. [Google Scholar] [CrossRef]
- Selkoe, D.J.; American College of Physicians; American Physiological Society. Alzheimer disease: Mechanistic understanding predicts novel therapies. Ann. Intern. Med. 2004, 140, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Sen, C.K.; Khanna, S.; Roy, S. Tocotrienols: Vitamin E beyond tocopherols. Life Sci. 2006, 78, 2088–2098. [Google Scholar] [CrossRef] [PubMed]
- Rondanelli, M.; Faliva, M.A.; Peroni, G.; Moncaglieri, F.; Infantino, V.; Naso, M.; Perna, S. Focus on pivotal role of dietary intake (diet and supplement) and blood levels of tocopherols and tocotrienols in obtaining successful aging. Int. J. Mol. Sci. 2015, 16, 23227–23249. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.W.; Hartmann, T. Recent understanding of the molecular mechanisms of Alzheimer’s disease. J. Addict. Res. Ther. 2012, 5, 1–27. [Google Scholar]
- Nohturfft, A.; Yabe, D.; Goldstein, J.L.; Brown, M.S.; Espenshade, P.J. Regulated step in cholesterol feedback localized to budding of scap from er membranes. Cell 2000, 102, 315–323. [Google Scholar] [CrossRef]
- Sakai, J.; Duncan, E.A.; Rawson, R.B.; Hua, X.; Brown, M.S.; Goldstein, J.L. Sterol-regulated release of SREBP-2 from cell membranes requires two sequential cleavages, one within a transmembrane segment. Cell 1996, 85, 1037–1046. [Google Scholar] [CrossRef]
- Mangialasche, F.; Solomon, A.; Kareholt, I.; Hooshmand, B.; Cecchetti, R.; Fratiglioni, L.; Soininen, H.; Laatikainen, T.; Mecocci, P.; Kivipelto, M. Serum levels of vitamin E forms and risk of cognitive impairment in a finnish cohort of older adults. Exp. Gerontol. 2013, 48, 1428–1435. [Google Scholar] [CrossRef] [PubMed]
- Mangialasche, F.; Westman, E.; Kivipelto, M.; Muehlboeck, J.S.; Cecchetti, R.; Baglioni, M.; Tarducci, R.; Gobbi, G.; Floridi, P.; Soininen, H.; et al. Classification and prediction of clinical diagnosis of Alzheimer’s disease based on mri and plasma measures of α-/γ-tocotrienols and γ-tocopherol. J. Intern. Med. 2013, 273, 602–621. [Google Scholar] [CrossRef] [PubMed]
- Mangialasche, F.; Kivipelto, M.; Mecocci, P.; Rizzuto, D.; Palmer, K.; Winblad, B.; Fratiglioni, L. High plasma levels of vitamin E forms and reduced Alzheimer’s disease risk in advanced age. J. Alzheimers Dis. 2010, 20, 1029–1037. [Google Scholar] [PubMed]
- Mangialasche, F.; Xu, W.; Kivipelto, M.; Costanzi, E.; Ercolani, S.; Pigliautile, M.; Cecchetti, R.; Baglioni, M.; Simmons, A.; Soininen, H.; et al. Tocopherols and tocotrienols plasma levels are associated with cognitive impairment. Neurobiol. Aging 2012, 33, 2282–2290. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Evans, D.A.; Tangney, C.C.; Bienias, J.L.; Wilson, R.S.; Aggarwal, N.T.; Scherr, P.A. Relation of the tocopherol forms to incident Alzheimer disease and to cognitive change. Am. J. Clin. Nutr. 2005, 81, 508–514. [Google Scholar] [PubMed]
- Gopalan, Y.; Shuaib, I.L.; Magosso, E.; Ansari, M.A.; Abu Bakar, M.R.; Wong, J.W.; Khan, N.A.; Liong, W.C.; Sundram, K.; Ng, B.H.; et al. Clinical investigation of the protective effects of palm vitamin E tocotrienols on brain white matter. Stroke 2014, 45, 1422–1428. [Google Scholar] [CrossRef] [PubMed]
- Farina, N.; Isaac, M.G.; Clark, A.R.; Rusted, J.; Tabet, N. Vitamin E for Alzheimer’s dementia and mild cognitive impairment. Cochrane Database Syst. Rev. 2012, 11. [Google Scholar] [CrossRef] [Green Version]
- Barnes, D.E.; Yaffe, K. Vitamin E and donepezil for the treatment of mild cognitive impairment. N. Engl. J. Med. 2005, 353, 951–952. [Google Scholar] [PubMed]
- Usoro, O.B.; Mousa, S.A. Vitamin E forms in Alzheimer’s disease: A review of controversial and clinical experiences. Crit. Rev. Food Sci. Nutr. 2010, 50, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.R., 3rd; Pastor-Barriuso, R.; Dalal, D.; Riemersma, R.A.; Appel, L.J.; Guallar, E. Meta-analysis: High-dosage vitamin E supplementation may increase all-cause mortality. Ann. Intern. Med. 2005, 142, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Behl, C. Amyloid β-protein toxicity and oxidative stress in Alzheimer’s disease. Cell Tissue Res. 1997, 290, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Ito, S.; Ohtsuki, S.; Yamamoto, N.; Takahashi, T.; Iwata, N.; Jishage, K.; Yamada, H.; Sasaguri, H.; Yokota, S.; et al. Depletion of vitamin E increases amyloid β accumulation by decreasing its clearances from brain and blood in a mouse model of Alzheimer disease. J. Biol. Chem. 2009, 284, 33400–33408. [Google Scholar] [CrossRef] [PubMed]
- Sinha, M.; Bir, A.; Banerjee, A.; Bhowmick, P.; Chakrabarti, S. Multiple mechanisms of age-dependent accumulation of amyloid β protein in rat brain: Prevention by dietary supplementation with N-acetylcysteine, α-lipoic acid and α-tocopherol. Neurochem. Int. 2016, 95, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Thakurta, I.G.; Banerjee, P.; Bagh, M.B.; Ghosh, A.; Sahoo, A.; Chattopadhyay, S.; Chakrabarti, S. Combination of N-acetylcysteine, α-lipoic acid and α-tocopherol substantially prevents the brain synaptosomal alterations and memory and learning deficits of aged rats. Exp. Gerontol. 2014, 50, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Grimm, H.S.; Beher, D.; Lichtenthaler, S.F.; Shearman, M.S.; Beyreuther, K.; Hartmann, T. γ-secretase cleavage site specificity differs for intracellular and secretory amyloid β. J. Biol. Chem. 2003, 278, 13077–13085. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Ida, N.; Hartmann, T.; Pantel, J.; Schroder, J.; Zerfass, R.; Forstl, H.; Sandbrink, R.; Masters, C.L.; Beyreuther, K. Analysis of heterogeneous A4 peptides in human cerebrospinal fluid and blood by a newly developed sensitive western blot assay. J. Biol. Chem. 1996, 271, 22908–22914. [Google Scholar] [PubMed]
- Grimm, M.O.; Zinser, E.G.; Grosgen, S.; Hundsdorfer, B.; Rothhaar, T.L.; Burg, V.K.; Kaestner, L.; Bayer, T.A.; Lipp, P.; Muller, U.; et al. Amyloid precursor protein (APP) mediated regulation of ganglioside homeostasis linking Alzheimer’s disease pathology with ganglioside metabolism. PLoS ONE 2012, 7, e34095. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Kuchenbecker, J.; Grosgen, S.; Burg, V.K.; Hundsdorfer, B.; Rothhaar, T.L.; Friess, P.; de Wilde, M.C.; Broersen, L.M.; Penke, B.; et al. Docosahexaenoic acid reduces amyloid β production via multiple pleiotropic mechanisms. J. Biol. Chem. 2011, 286, 14028–14039. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Mett, J.; Stahlmann, C.P.; Grosgen, S.; Haupenthal, V.J.; Blumel, T.; Hundsdorfer, B.; Zimmer, V.C.; Mylonas, N.T.; Tanila, H.; et al. APP intracellular domain derived from amyloidogenic β- and γ-secretase cleavage regulates neprilysin expression. Front. Aging Neurosci. 2015, 7, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z. Comparison of extraction methods for quantifying vitamin E from animal tissues. Bioresour. Technol. 2008, 99, 8705–8709. [Google Scholar] [CrossRef] [PubMed]
- Lauridsen, C.; Leonard, S.W.; Griffin, D.A.; Liebler, D.C.; McClure, T.D.; Traber, M.G. Quantitative analysis by liquid chromatography-tandem mass spectrometry of deuterium-labeled and unlabeled vitamin E in biological samples. Anal. Biochem. 2001, 289, 89–95. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grimm, M.O.W.; Regner, L.; Mett, J.; Stahlmann, C.P.; Schorr, P.; Nelke, C.; Streidenberger, O.; Stoetzel, H.; Winkler, J.; Zaidan, S.R.; et al. Tocotrienol Affects Oxidative Stress, Cholesterol Homeostasis and the Amyloidogenic Pathway in Neuroblastoma Cells: Consequences for Alzheimer’s Disease. Int. J. Mol. Sci. 2016, 17, 1809. https://doi.org/10.3390/ijms17111809
Grimm MOW, Regner L, Mett J, Stahlmann CP, Schorr P, Nelke C, Streidenberger O, Stoetzel H, Winkler J, Zaidan SR, et al. Tocotrienol Affects Oxidative Stress, Cholesterol Homeostasis and the Amyloidogenic Pathway in Neuroblastoma Cells: Consequences for Alzheimer’s Disease. International Journal of Molecular Sciences. 2016; 17(11):1809. https://doi.org/10.3390/ijms17111809
Chicago/Turabian StyleGrimm, Marcus O. W., Liesa Regner, Janine Mett, Christoph P. Stahlmann, Pascal Schorr, Christopher Nelke, Olga Streidenberger, Hannah Stoetzel, Jakob Winkler, Shatha R. Zaidan, and et al. 2016. "Tocotrienol Affects Oxidative Stress, Cholesterol Homeostasis and the Amyloidogenic Pathway in Neuroblastoma Cells: Consequences for Alzheimer’s Disease" International Journal of Molecular Sciences 17, no. 11: 1809. https://doi.org/10.3390/ijms17111809