
The Molecular Pathway of Argon-Mediated Neuroprotection


Abstract
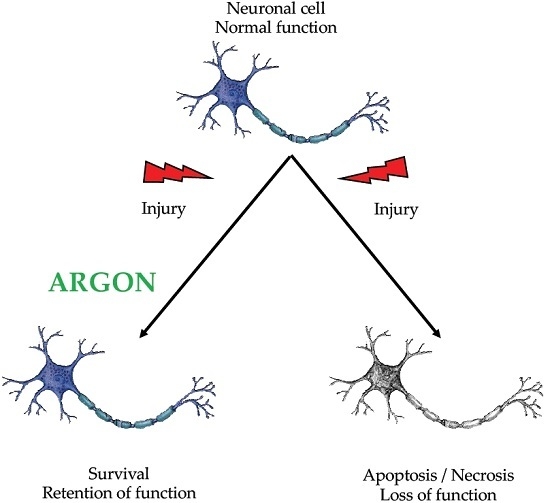
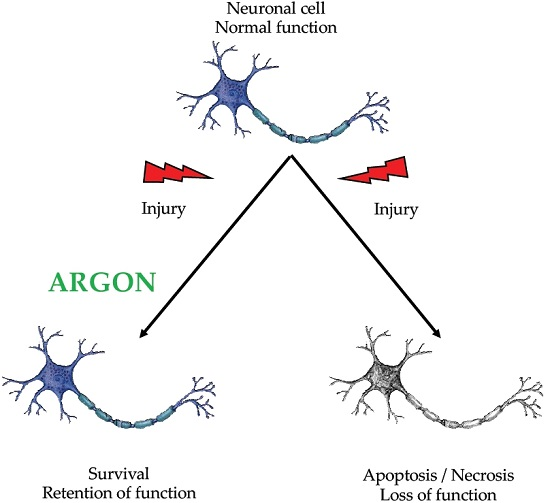
:
1. Introduction
2. Neuroprotection
3. Argon as a Neuroprotective Gaseous Molecule in Various Models of Injury
4. Receptor Mediated Neuroprotection
5. Intracellular Pathways Displaying Argon-Mediated Neuroprotection
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ballentine, C.J. Geochemistry: Earth holds its breath. Nature 2007, 449, 294–296. [Google Scholar] [CrossRef] [PubMed]
- Cavandish, H. Experiments on air. Philos. Trans. R. Soc. Lond. 1784, 119–153. [Google Scholar]
- Raleigh, L.; Ramsay, W. Argon, a new constituent of the atmosphere. Proc. R. Soc. Lond. 1984, 55, 340–346. [Google Scholar]
- Ruzicka, J.; Benes, J.; Bolek, L.; Markvartova, V. Biological effects of noble gases. Physiol. Res. Acad. Sci. Bohemoslov. 2007, 56, S39–S44. [Google Scholar]
- Trudell, J.R.; Koblin, D.D.; Eger, E.I., II. A molecular description of how noble gases and nitrogen bind to a model site of anesthetic action. Anesth. Analg. 1998, 87, 411–418. [Google Scholar] [PubMed]
- Quillin, M.L.; Breyer, W.A.; Griswold, I.J.; Matthews, B.W. Size versus polarizability in protein-ligand interactions: Binding of noble gases within engineered cavities in phage T4 lysozyme. J. Mol. Biol. 2000, 302, 955–977. [Google Scholar] [CrossRef] [PubMed]
- Behnke, A.R.; Yarbrough, O.D. Respiratory resistance, oil-water solubility and mental effects of argon compared with helium and nitrogen. Am. J. Physiol. 1939, 126, 409–415. [Google Scholar]
- Bertaccini, E.J.; Trudell, J.R.; Franks, N.P. The common chemical motifs within anesthetic binding sites. Anesth. Analg. 2007, 104, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Wang, R. Two’s company, three’s a crowd: Can H2S be the third endogenous gaseous transmitter? FASEB J. 2002, 16, 1792–1798. [Google Scholar] [CrossRef] [PubMed]
- Moretti, A.; Ferrari, F.; Villa, R.F. Neuroprotection for ischaemic stroke: Current status and challenges. Pharmacol. Ther. 2015, 146, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Gill, R.; Foster, A.C.; Woodruff, G.N. Systemic administration of MK-801 protects against ischemia-induced hippocampal neurodegeneration in the gerbil. J. Neurosci. 1987, 7, 3343–3349. [Google Scholar] [PubMed]
- Seidl, S.E.; Potashkin, J.A. The promise of neuroprotective agents in parkinson’s disease. Front. Neurol. 2011, 2, 68. [Google Scholar] [CrossRef] [PubMed]
- Shabab, T.; Khanabdali, R.; Moghadamtousi, S.Z.; Kadir, H.A.; Mohan, G. Neuroinflammation pathways: A general review. Int. J. Neurosci. 2016, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Witherspoon, J.D.; Wiebers, J.E.; Hiestand, W.A.; Heimlich, A.H. Decompression of mice in atmosphere containing helium or argon in place of nitrogen. Aerosp. Med. 1964, 35, 529–532. [Google Scholar] [PubMed]
- Bennett, P.B. Prevention in rats of narcosis produced by inert gases at high pressures. Am. J. Physiol. 1963, 205, 1013–1018. [Google Scholar] [PubMed]
- Antonov, A.A.; Ershova, T.A. Retention of the skill to perform adaptive bio-control of bioelectrical activity synchronization in the human brain cortex in an argon-nitrogen-oxygen atmosphere with various oxygen content. Aviakosm. Ekolog. Med. 2009, 43, 27–31. [Google Scholar] [PubMed]
- Soldatov, P.E.; D’Iachenko, A.I.; Pavlov, B.N.; Fedotov, A.P.; Chuguev, A.P. Survival of laboratory animals in argon-containing hypoxic gaseous environments. Aviakosm. Ekolog. Med. 1998, 32, 33–37. [Google Scholar] [PubMed]
- Ulbrich, F.; Goebel, U. Argon: A novel therapeutic option to treat neuronal ischemia and reperfusion injuries? Neural Regen. Res. 2015, 10, 1043–1044. [Google Scholar] [PubMed]
- Jawad, N.; Rizvi, M.; Gu, J.; Adeyi, O.; Tao, G.; Maze, M.; Ma, D. Neuroprotection (and lack of neuroprotection) afforded by a series of noble gases in an in vitro model of neuronal injury. Neurosci. Lett. 2009, 460, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Loetscher, P.D.; Rossaint, J.; Rossaint, R.; Weis, J.; Fries, M.; Fahlenkamp, A.; Ryang, Y.M.; Grottke, O.; Coburn, M. Argon: Neuroprotection in in vitro models of cerebral ischemia and traumatic brain injury. Crit. Care 2009, 13, R206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, H.N.; Haelewyn, B.; Degoulet, M.; Colomb, D.G., Jr.; Risso, J.J.; Abraini, J.H. Ex vivo and in vivo neuroprotection induced by argon when given after an excitotoxic or ischemic insult. PLoS ONE 2012, 7, e30934. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Mitchell, S.; Ciechanowicz, S.; Savage, S.; Wang, T.; Ji, X.; Ma, D. Argon protects against hypoxic-ischemic brain injury in neonatal rats through activation of nuclear factor (erythroid-derived 2)-like 2. Oncotarget 2016, 7, 25640–25651. [Google Scholar] [CrossRef] [PubMed]
- Ryang, Y.M.; Fahlenkamp, A.V.; Rossaint, R.; Wesp, D.; Loetscher, P.D.; Beyer, C.; Coburn, M. Neuroprotective effects of argon in an in vivo model of transient middle cerebral artery occlusion in rats. Crit. Care Med. 2011, 39, 1448–1453. [Google Scholar] [CrossRef] [PubMed]
- Fahlenkamp, A.V.; Coburn, M.; de Prada, A.; Gereitzig, N.; Beyer, C.; Haase, H.; Rossaint, R.; Gempt, J.; Ryang, Y.M. Expression analysis following argon treatment in an in vivo model of transient middle cerebral artery occlusion in rats. Med. Gas Res. 2014, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Yang, T.; Zhao, H.; Fidalgo, A.R.; Vizcaychipi, M.P.; Sanders, R.D.; Yu, B.; Takata, M.; Johnson, M.R.; Ma, D. The protective profile of argon, helium, and xenon in a model of neonatal asphyxia in rats. Crit. Care Med. 2012, 40, 1724–1730. [Google Scholar] [CrossRef] [PubMed]
- Brucken, A.; Cizen, A.; Fera, C.; Meinhardt, A.; Weis, J.; Nolte, K.; Rossaint, R.; Pufe, T.; Marx, G.; Fries, M. Argon reduces neurohistopathological damage and preserves functional recovery after cardiac arrest in rats. Br. J. Anaesth. 2013, 110, i106–i112. [Google Scholar] [CrossRef] [PubMed]
- Ulbrich, F.; Schallner, N.; Coburn, M.; Loop, T.; Lagreze, W.A.; Biermann, J.; Goebel, U. Argon inhalation attenuates retinal apoptosis after ischemia/reperfusion injury in a time- and dose-dependent manner in rats. PLoS ONE 2014, 9, e115984. [Google Scholar] [CrossRef] [PubMed]
- Hollig, A.; Weinandy, A.; Liu, J.; Clusmann, H.; Rossaint, R.; Coburn, M. Beneficial properties of argon after experimental subarachnoid hemorrhage: Early treatment reduces mortality and influences hippocampal protein expression. Crit. Care Med. 2016, 44, e520–e529. [Google Scholar] [CrossRef] [PubMed]
- Rostain, J.C.; Balon, N. Recent neurochemical basis of inert gas narcosis and pressure effects. Undersea Hyperb. Med. 2006, 33, 197–204. [Google Scholar] [PubMed]
- David, H.N.; Haelewyn, B.; Rouillon, C.; Lecoq, M.; Chazalviel, L.; Apiou, G.; Risso, J.J.; Lemaire, M.; Abraini, J.H. Neuroprotective effects of xenon: A therapeutic window of opportunity in rats subjected to transient cerebral ischemia. FASEB J. 2008, 22, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.; Ma, D.; Maze, M.; Franks, N.P. Effects of xenon on in vitro and in vivo models of neuronal injury. Anesthesiology 2002, 96, 1485–1491. [Google Scholar] [CrossRef] [PubMed]
- Homi, H.M.; Yokoo, N.; Ma, D.; Warner, D.S.; Franks, N.P.; Maze, M.; Grocott, H.P. The neuroprotective effect of xenon administration during transient middle cerebral artery occlusion in mice. Anesthesiology 2003, 99, 876–881. [Google Scholar] [CrossRef] [PubMed]
- Dingley, J.; Tooley, J.; Porter, H.; Thoresen, M. Xenon provides short-term neuroprotection in neonatal rats when administered after hypoxia-ischemia. Stroke 2006, 37, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Hossain, M.; Chow, A.; Arshad, M.; Battson, R.M.; Sanders, R.D.; Mehmet, H.; Edwards, A.D.; Franks, N.P.; Maze, M. Xenon and hypothermia combine to provide neuroprotection from neonatal asphyxia. Ann. Neurol. 2005, 58, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Abraini, J.H.; Kriem, B.; Balon, N.; Rostain, J.C.; Risso, J.J. Gamma-aminobutyric acid neuropharmacological investigations on narcosis produced by nitrogen, argon, or nitrous oxide. Anesth. Analg. 2003, 96, 746–749. [Google Scholar] [CrossRef]
- Harris, K.; Armstrong, S.P.; Campos-Pires, R.; Kiru, L.; Franks, N.P.; Dickinson, R. Neuroprotection against traumatic brain injury by xenon, but not argon, is mediated by inhibition at the N-methyl-d-aspartate receptor glycine site. Anesthesiology 2013, 119, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Brucken, A.; Kurnaz, P.; Bleilevens, C.; Derwall, M.; Weis, J.; Nolte, K.; Rossaint, R.; Fries, M. Dose dependent neuroprotection of the noble gas argon after cardiac arrest in rats is not mediated by KATP-channel opening. Resuscitation 2014, 85, 826–832. [Google Scholar] [CrossRef] [PubMed]
- Ulbrich, F.; Kaufmann, K.; Roesslein, M.; Wellner, F.; Auwarter, V.; Kempf, J.; Loop, T.; Buerkle, H.; Goebel, U. Argon mediates anti-apoptotic signaling and neuroprotection via inhibition of toll-like receptor 2 and 4. PLoS ONE 2015, 10, e0143887. [Google Scholar] [CrossRef] [PubMed]
- Ulbrich, F.; Lerach, T.; Biermann, J.; Kaufmann, K.B.; Lagreze, W.A.; Buerkle, H.; Loop, T.; Goebel, U. Argon mediates protection by interleukin-8 suppression via a TLR2/TLR4/STAT3/NF-κB pathway in a model of apoptosis in neuroblastoma cells in vitro and following ischemia-reperfusion injury in rat retina in vivo. J. Neurochem. 2016, 138, 859–873. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, M.; Jawad, N.; Li, Y.; Vizcaychipi, M.P.; Maze, M.; Ma, D. Effect of noble gases on oxygen and glucose deprived injury in human tubular kidney cells. Exp. Biol. Med. 2010, 235, 886–891. [Google Scholar] [CrossRef] [PubMed]
- Fahlenkamp, A.V.; Rossaint, R.; Haase, H.; Al Kassam, H.; Ryang, Y.M.; Beyer, C.; Coburn, M. The noble gas argon modifies extracellular signal-regulated kinase 1/2 signaling in neurons and glial cells. Eur. J. Pharmacol. 2012, 674, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Hetman, M.; Xia, Z. Signaling pathways mediating anti-apoptotic action of neurotrophins. Acta Neurobiol. Exp. 2000, 60, 531–545. [Google Scholar]
- Subramaniam, S.; Unsicker, K. ERK and cell death: ERK1/2 in neuronal death. FEBS J. 2010, 277, 22–29. [Google Scholar] [CrossRef] [PubMed]
- De Rossi, L.W.; Brueckmann, M.; Rex, S.; Barderschneider, M.; Buhre, W.; Rossaint, R. Xenon and isoflurane differentially modulate lipopolysaccharide-induced activation of the nuclear transcription factor KB and production of tumor necrosis factor-α and interleukin-6 in monocytes. Anesth. Analg. 2004, 98, 1007–1012. [Google Scholar] [CrossRef]
- Ulbrich, F.; Kaufmann, K.B.; Coburn, M.; Lagreze, W.A.; Roesslein, M.; Biermann, J.; Buerkle, H.; Loop, T.; Goebel, U. Neuroprotective effects of argon are mediated via an ERK-1/2 dependent regulation of heme-oxygenase-1 in retinal ganglion cells. J. Neurochem. 2015, 134, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Weber, N.C.; Stursberg, J.; Wirthle, N.M.; Toma, O.; Schlack, W.; Preckel, B. Xenon preconditioning differently regulates p44/42 MAPK (ERK 1/2) and p46/54 MAPK (JNK 1/2 and 3) in vivo. Br. J. Anaesth. 2006, 97, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Mitchell, S.; Koumpa, S.; Cui, Y.T.; Lian, Q.; Hagberg, H.; Johnson, M.R.; Takata, M.; Ma, D. Heme oxygenase-1 mediates neuroprotection conferred by argon in combination with hypothermia in neonatal hypoxia-ischemia brain injury. Anesthesiology 2016, 125, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Arancibia, S.A.; Beltran, C.J.; Aguirre, I.M.; Silva, P.; Peralta, A.L.; Malinarich, F.; Hermoso, M.A. Toll-like receptors are key participants in innate immune responses. Biol. Res. 2007, 40, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Jennings, P.; Limonciel, A.; Felice, L.; Leonard, M.O. An overview of transcriptional regulation in response to toxicological insult. Arch. Toxicol. 2013, 87, 49–72. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Feng, J.; He, G. Hypoxia increases Nrf2-induced HO-1 expression via the PI3K/Akt pathway. Front. Biosci. 2016, 21, 385–396. [Google Scholar]
- Gupta, V.; Garg, R.K.; Khattri, S. Levels of IL-8 and TNF-α decrease in parkinson’s disease. Neurol. Res. 2016, 38, 98–102. [Google Scholar] [CrossRef] [PubMed]
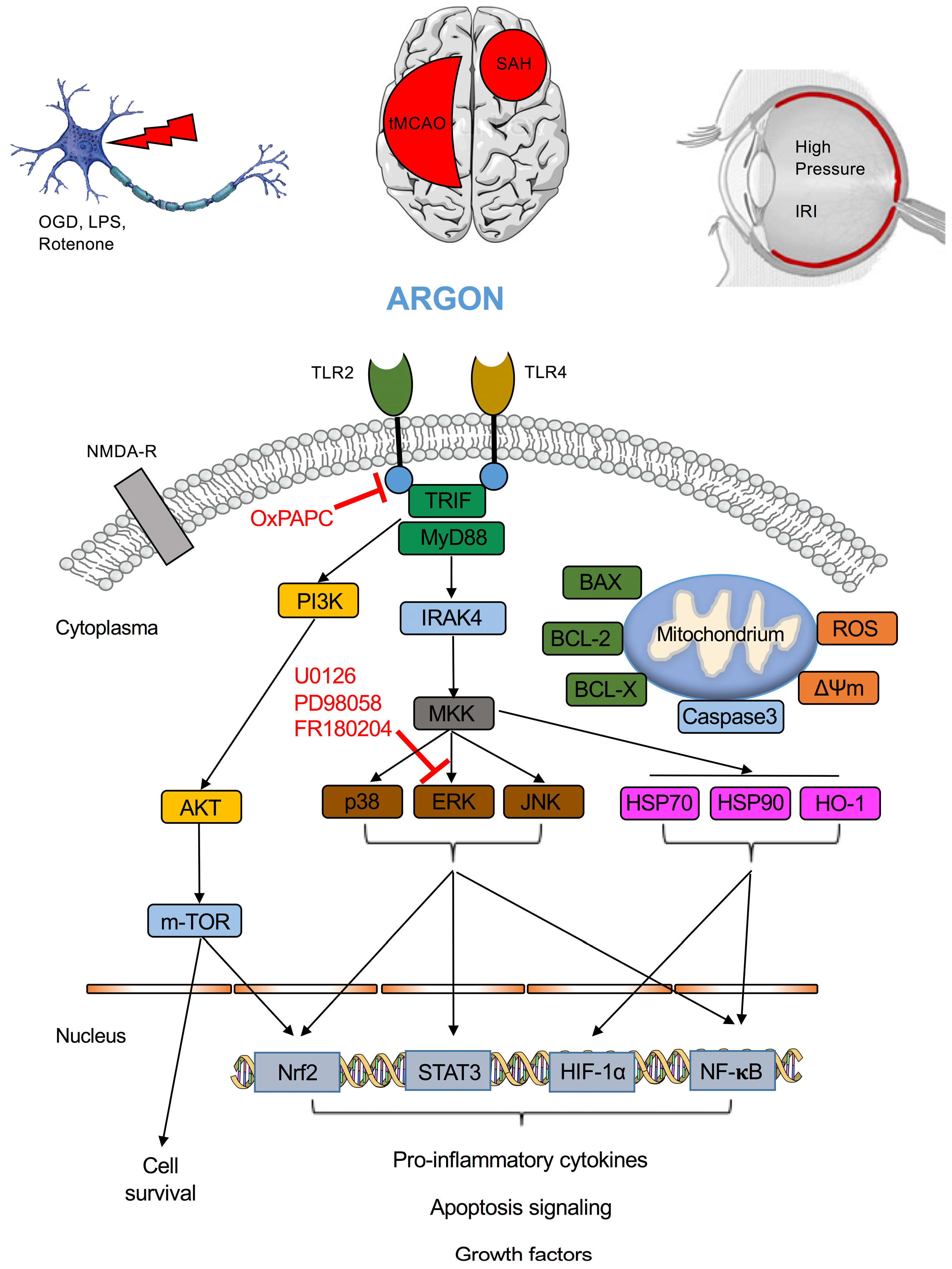

{kind=link}
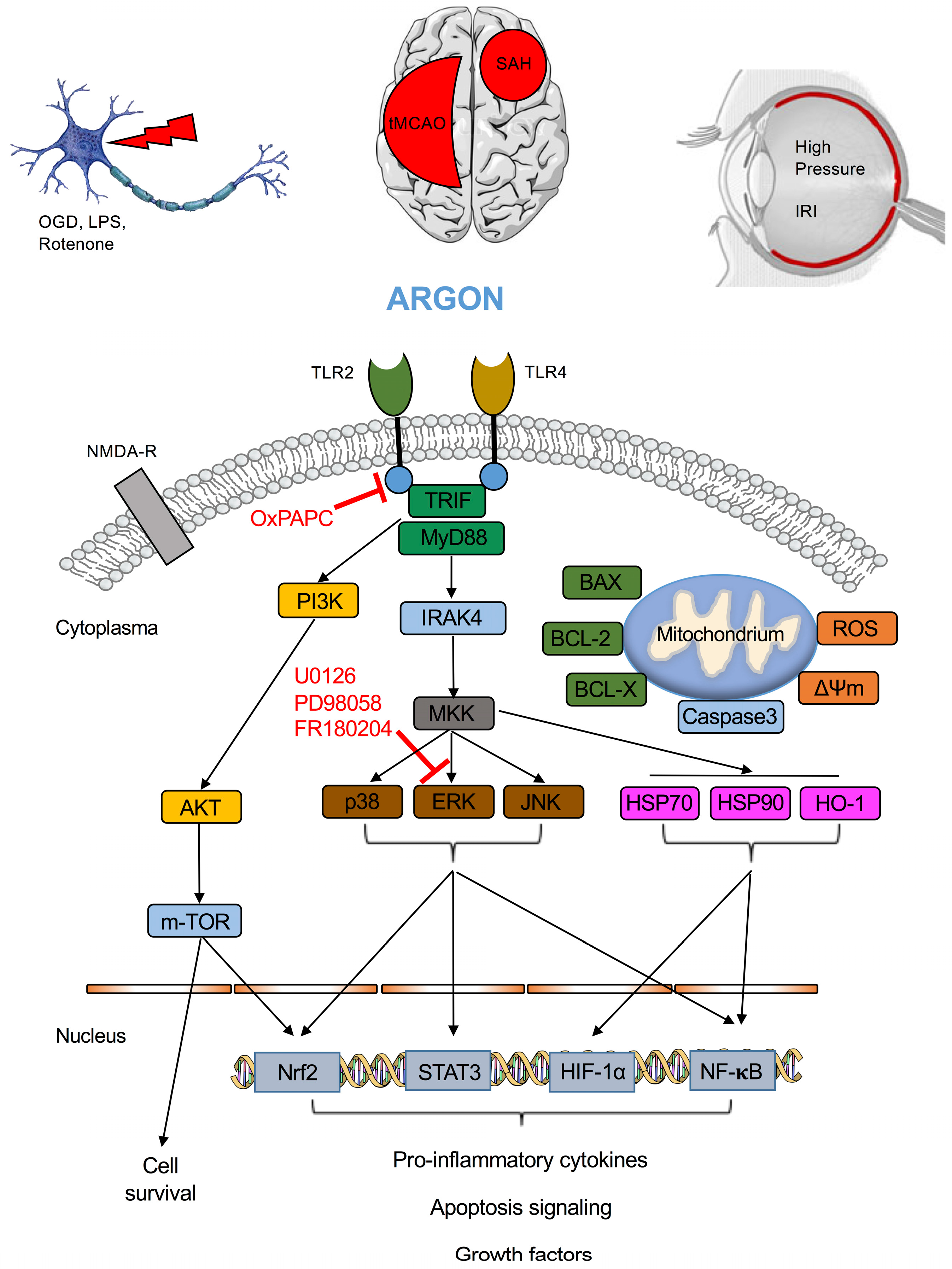{kind=link}
| Ref. | Model/Setup | Cell Culture Animals | Groups (n = x) | Concentration Duration | Primary Outcome Parameter | General Results | Proposed Mechanism |
|---|---|---|---|---|---|---|---|
| Abraini 2003 [35] | Ar anesthesia under hyperbaric conditions | Adult Sprague Dawley rats | n = 6 n = 4 | unknown | Loss of righting reflexes | gabazine and flumazenil reduced anesthetic action of Ar | GABAA receptor |
| Harris 2013 [36] | 1. Measurement of receptor currents 2. TBI to hippocampal brain slices | (a) HEK-293 cells (b) six day old C57/BL6 pups | n = 10 (NMDA) n = 5 (TREK) n = 105 (SHAM) n = 141 (TBI) n = 44 (Ar) n = 37 (Glycin + Ar) | Ar 80 Vol % 0.5 atm for 30 min up to 24 h | Currents via NMDA or TREK-1 receptor Quantification of cell inury | Ar does not affect NMDA or TREK currents Ar provides neuroprotection Pretreatment with Glycin did not alter Ar effects | NMDA receptor TREK-1 receptor |
| Brücken 2014 [37] | Cardiac arrest model | Adult Sprague Dawley rats | n = 47 | Ar 40 Vol % and Ar 70 Vol %, each for 1 h | NDS, reduction of neuronal damage | Improvement of NDS after Ar inhalation KATP channel blocking had no effect | KATP channel |
| Fahlenkamp 2012 [41] | Ar with or without LPS exposure | BV-2 microglia primary neurons primary astrocytes | n = 3 | Ar 50 Vol % 15, 30, 60 and 120 min | ERK1/2 phosphorylation cytokine expression | Ar activates ERK No effect on LPS mediated cytokine expression (IL-1β, TNF-α, IL-6) | ERK1/2 phosphorylation |
| Zhuang 2012 [25] | Hypoxic and ischemic brain injury | Sprague Dawley rat pups | 5 groups n = 5–7 | Ar 70 Vol % for 90 min, administration 2 h after injury | Infarct size and neurological function | Ar reduced infarction size and improved neurological function Ar increased BCL-2 expression | n.n. |
| Fahlenkamp 2014 [24] | tMCAO | Adult Sprague Dawley rats | 4 groups n = 12–15 | Ar 50 Vol % for 60 min administration 2 h after injury | Expression of growth factors and inflammatory cytokines | Ar increased: TGF-β, NGF, IL-6 and iNOS No effect on: HIF-1α, MMP-9, CNP, GFAP, VEGF-α, IGF-1 and microglia | n.n. |
| Zhao 2016 [47] | OGD Right common artery ligation and 90 min hypoxia | Cortical neuronal cell culture (SDR) Neonatal Sprague Dawley rats | n = 8 | Ar 70/75 Vol % 2 h after hypothermia | Neuronal injury | Ar after hypothermia increased p-AKT, HO-1 and decreased Cytochrome C, Caspase-3, NF-κB and infarct size | n.n. |
| Zhao 2016 [50] | OGD Hypoxic and ischemic brain injury | Cortical neuronal cells of rat foetuses Sprague Dawley rat pups | 5 groups n = 8 | Ar 70 Vol % for 2 h after injury | Infarct size and protein expression | Ar reduced infarction size Different protein expression and inhibition of effects via U0126 (ERK inhibition) | Ar dependent activation of MAPK, p-mTOR, Nrf-2 and NQO1/SOD1 |
| Höllig 2016 [28] | tMCAO | Adult Sprague Dawley rats | 9 groups n = 9–11 | Ar 50 Vol % for 60 min one hour after SAH | Mortality after SAH, neurological testing, protein analysis, quantification of neurons | Ar increased: HO-1 and HIF-1α expression No difference in neuroscore due to treatment | Ar dependent HO-1 and HIF-1α regulation |
| Ulbrich 2015 [45] | Retinal IRI | Adult Sprague Dawley rats | 6 groups n = 8 | Ar 75 Vol % for 60 min either immediately after IRI or with a 1.5 or 3 h delay | Vital retinal ganglion cells | Ar reduced HSP-70, HSP-90 and HO-1 expression, while inducing p38 and ERK1/2 ERK inhibition abolished Ar effects | ERK1/2 |
| Ulbrich 2015 [38] | Rotenone induced apoptosis | SY5Y neuroblastoma cell line | n = 6 | Ar 25/50 and 75 Vol % for 2 or 4 h after rotenone induced apoptosis | Reduction of apoptosis | Ar inhibited TLR2 and TLR4 receptors and downstream signaling in vitro | TLR2 and TLR4 signaling via IRAK4 and ERK1/2 |
| Ulbrich 2016 [39] | Retinal IRI | SY5Y neuroblastoma cell line Adult Sprague Dawley rats | n = 6 6 groups n = 8 | Ar 75 Vol % for 2 h Ar 75 Vol % for 60 min after IRI | Transcription factor analysis, Cytokine expression TLR2/TLR4 signaling | Ar inhibited STAT3 and NF-κB, but not STAT5 Ar affects mitochondrial membrane potential | TLR2 and TLR4 signaling in vivo via STAT3 and NF-κB pathway, suppressing IL-8 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ulbrich, F.; Goebel, U. The Molecular Pathway of Argon-Mediated Neuroprotection. Int. J. Mol. Sci. 2016, 17, 1816. https://doi.org/10.3390/ijms17111816
Ulbrich F, Goebel U. The Molecular Pathway of Argon-Mediated Neuroprotection. International Journal of Molecular Sciences. 2016; 17(11):1816. https://doi.org/10.3390/ijms17111816
Chicago/Turabian StyleUlbrich, Felix, and Ulrich Goebel. 2016. "The Molecular Pathway of Argon-Mediated Neuroprotection" International Journal of Molecular Sciences 17, no. 11: 1816. https://doi.org/10.3390/ijms17111816