
Expression of Carbonic Anhydrase I in Motor Neurons and Alterations in ALS


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
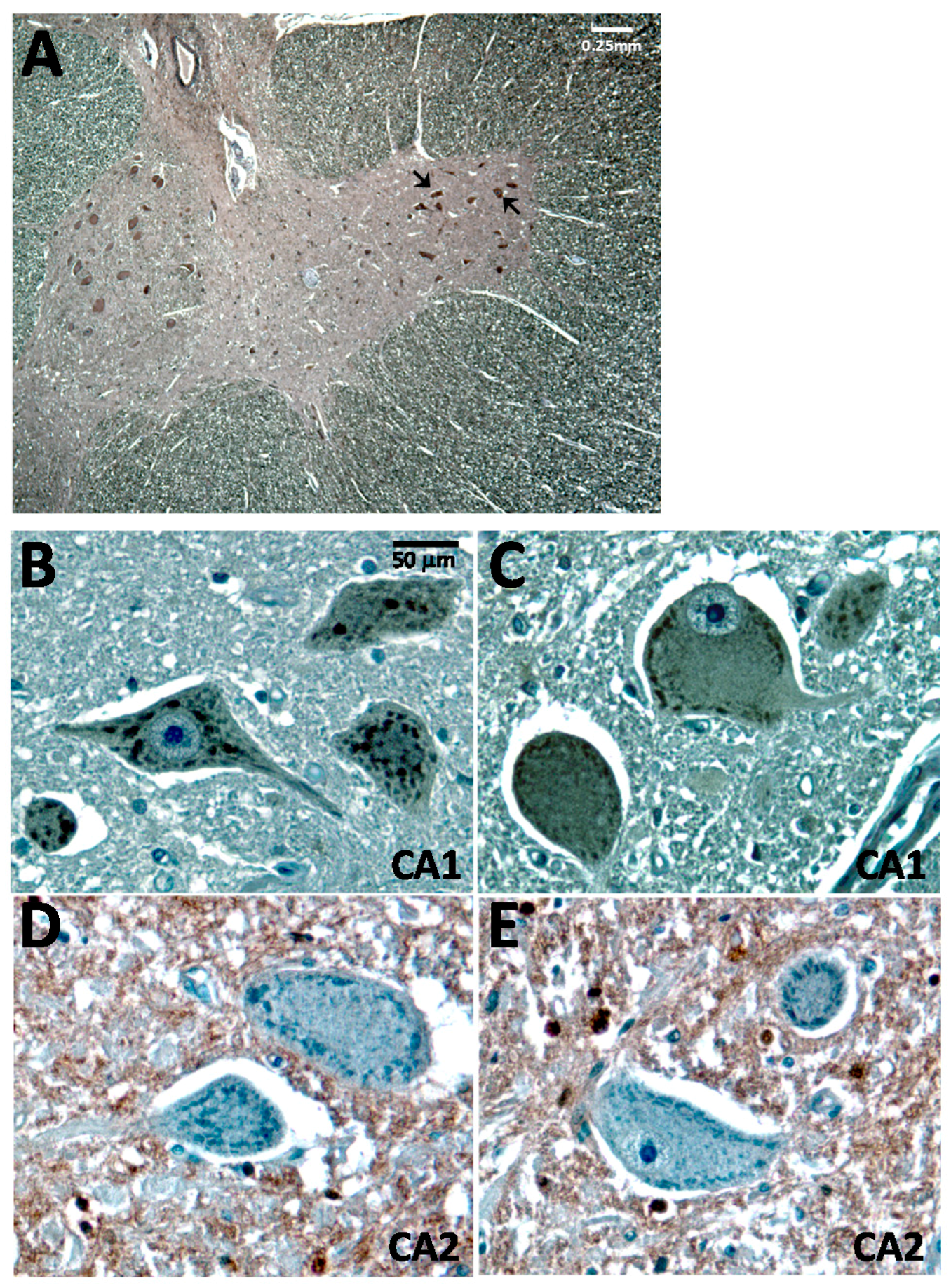
2.1. Carbonic Anhydrase I (CA1) Is Expressed in Human Spinal Cord Motor Neurons
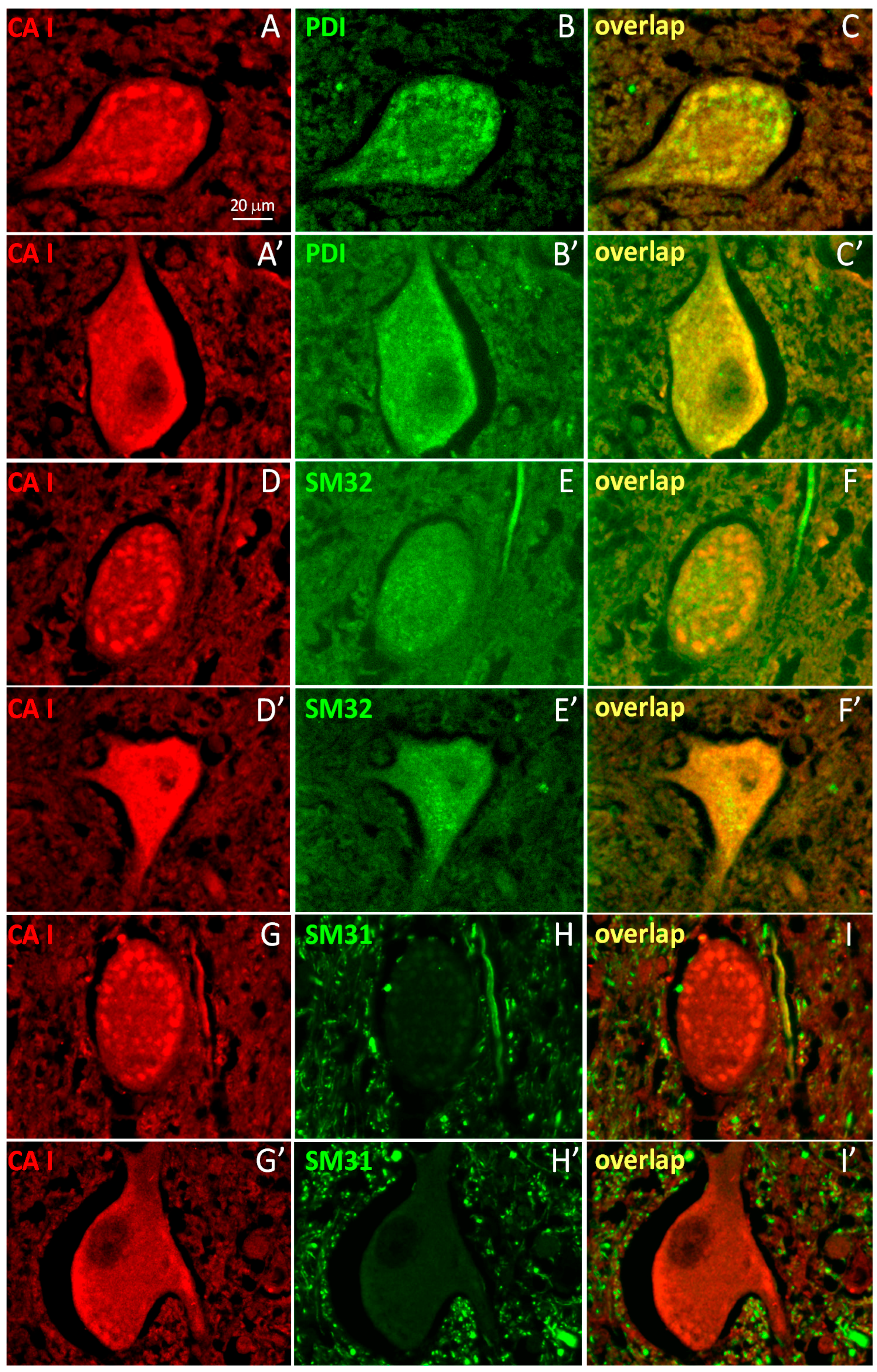
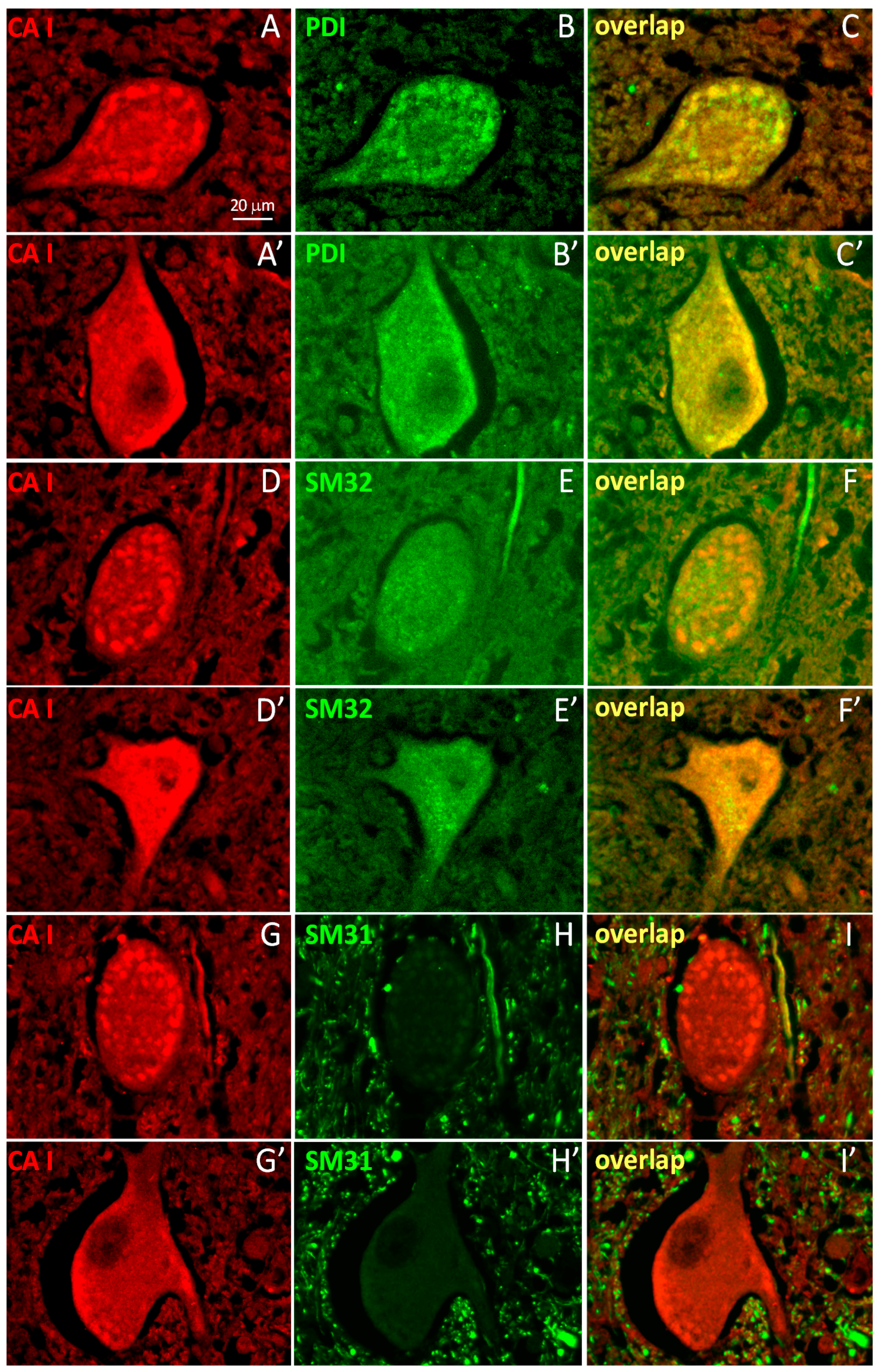
2.2. A Subpopulation of Neuronal CA1 Appears to Be Associated with the Endoplasmic Reticulum Subcellular Structure
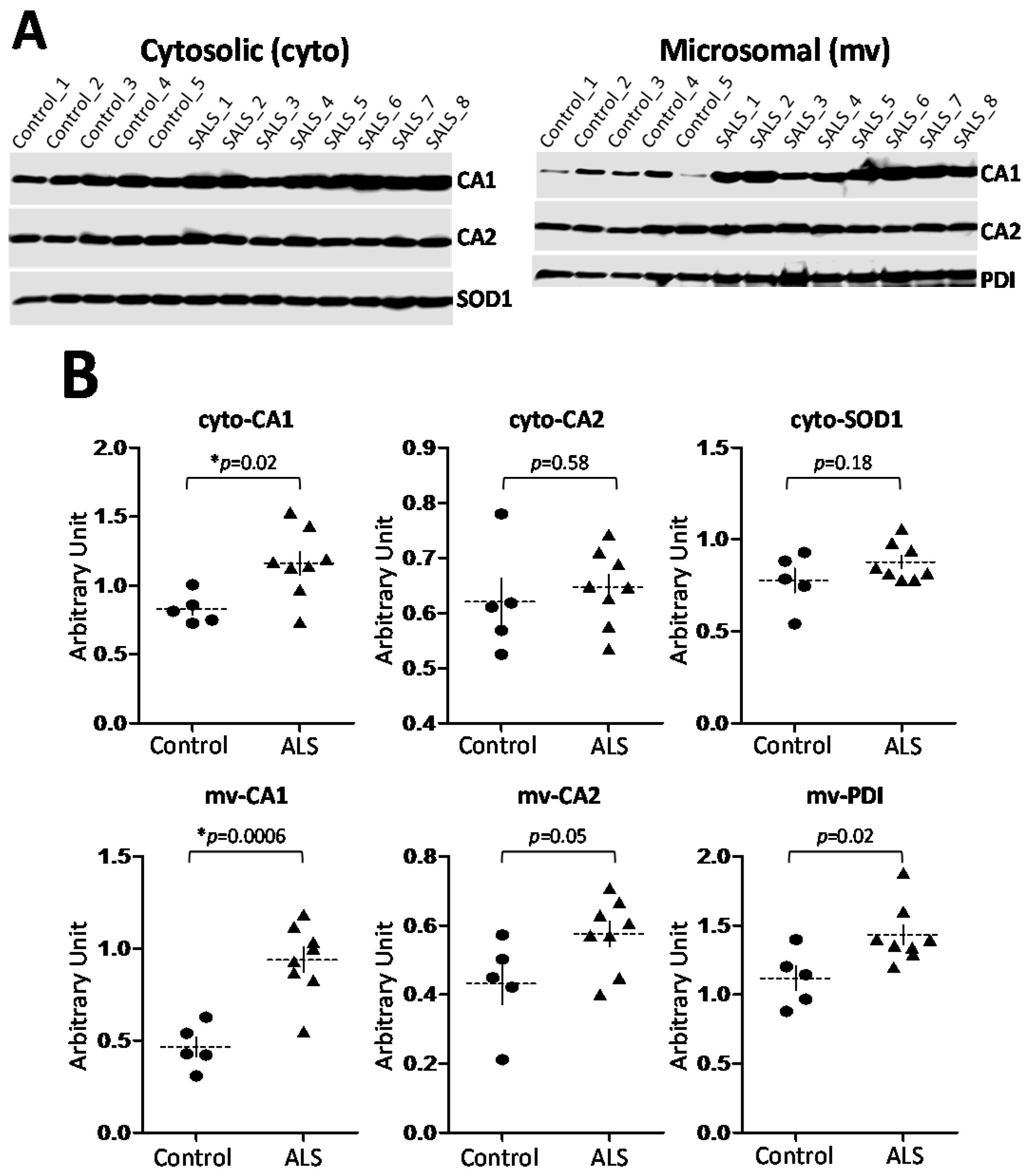
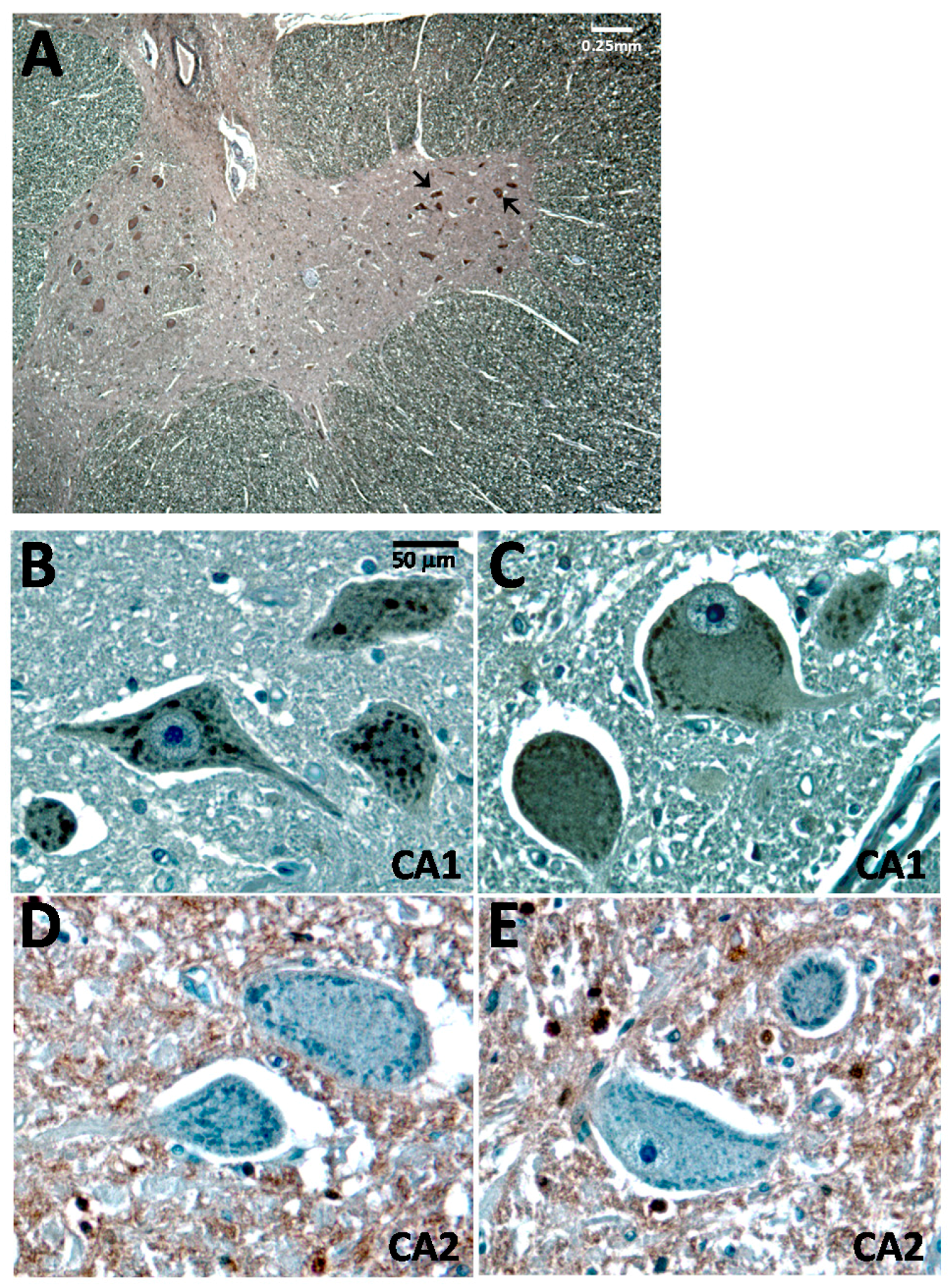
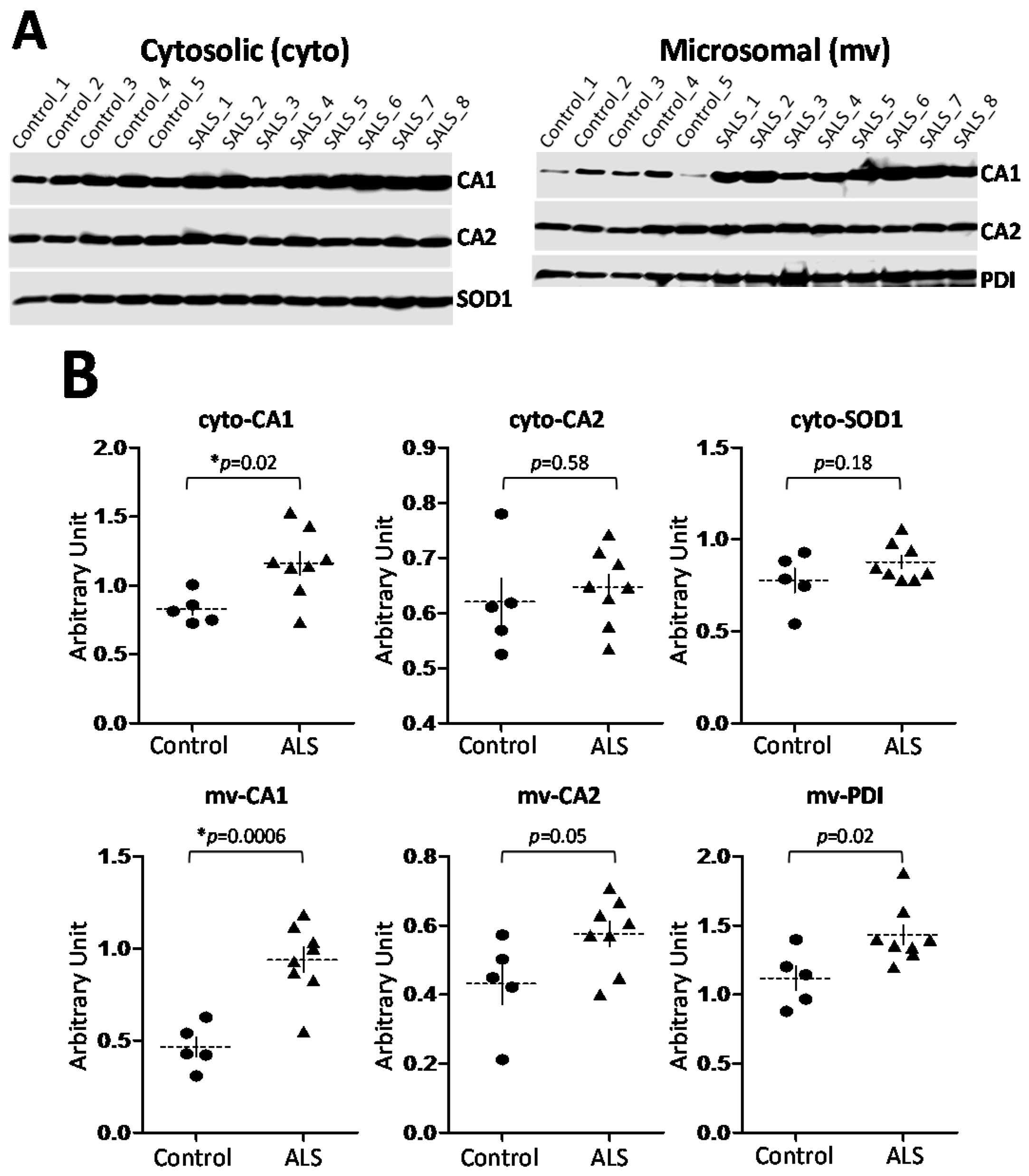
2.3. CA1 Is Preferentially Elevated in ALS Spinal Cord
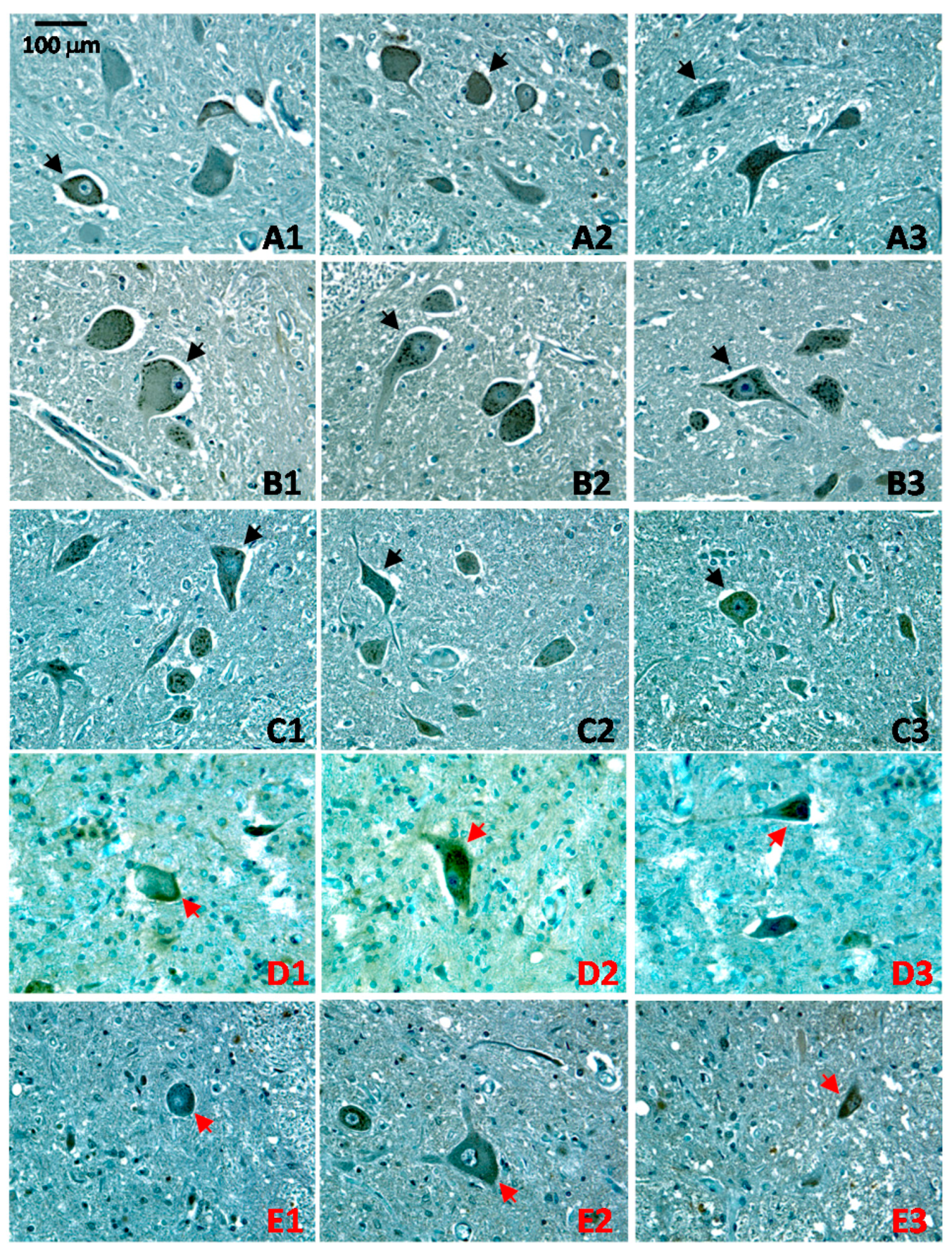
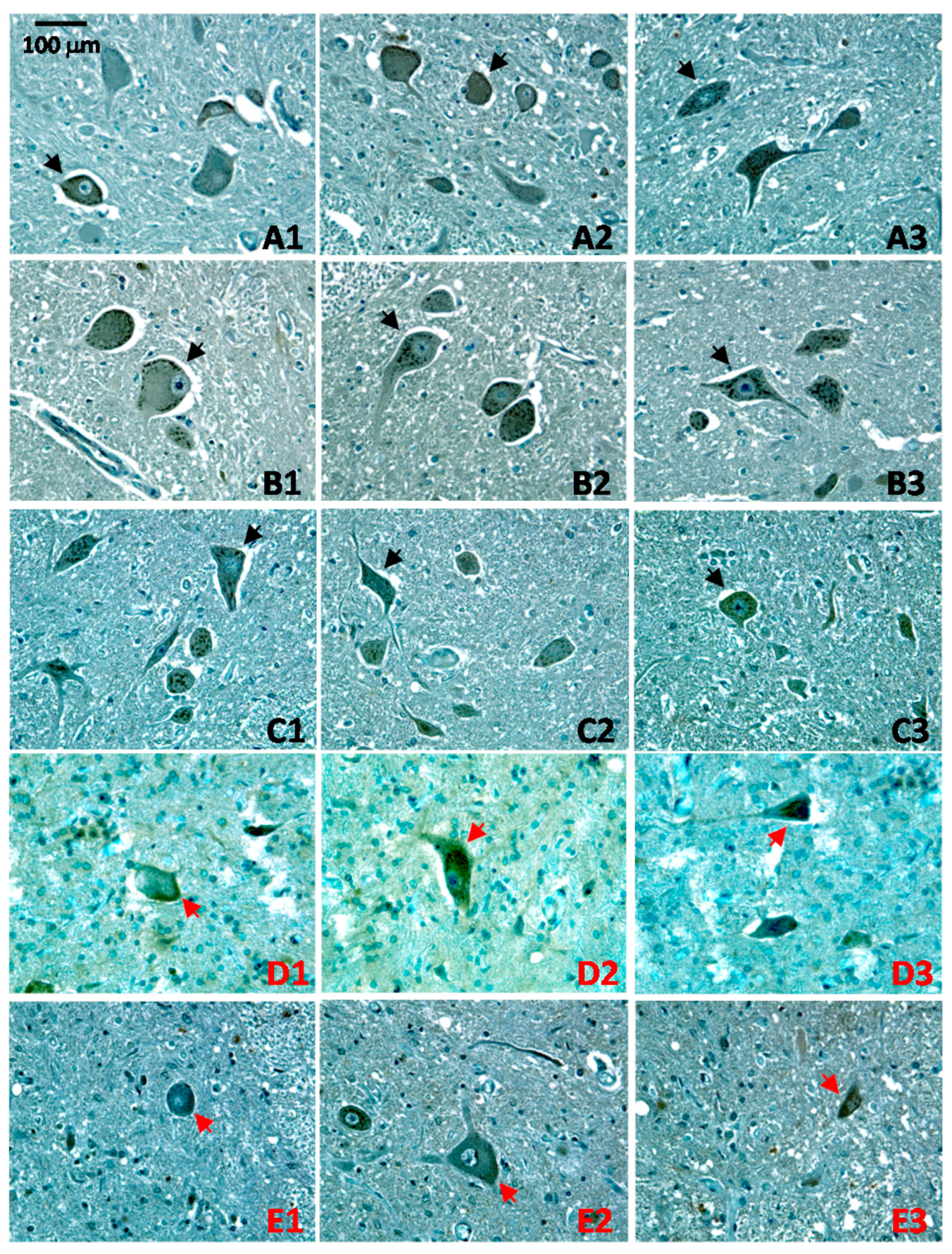
2.4. Altered Patterns of CA1 Expression in ALS Pathology
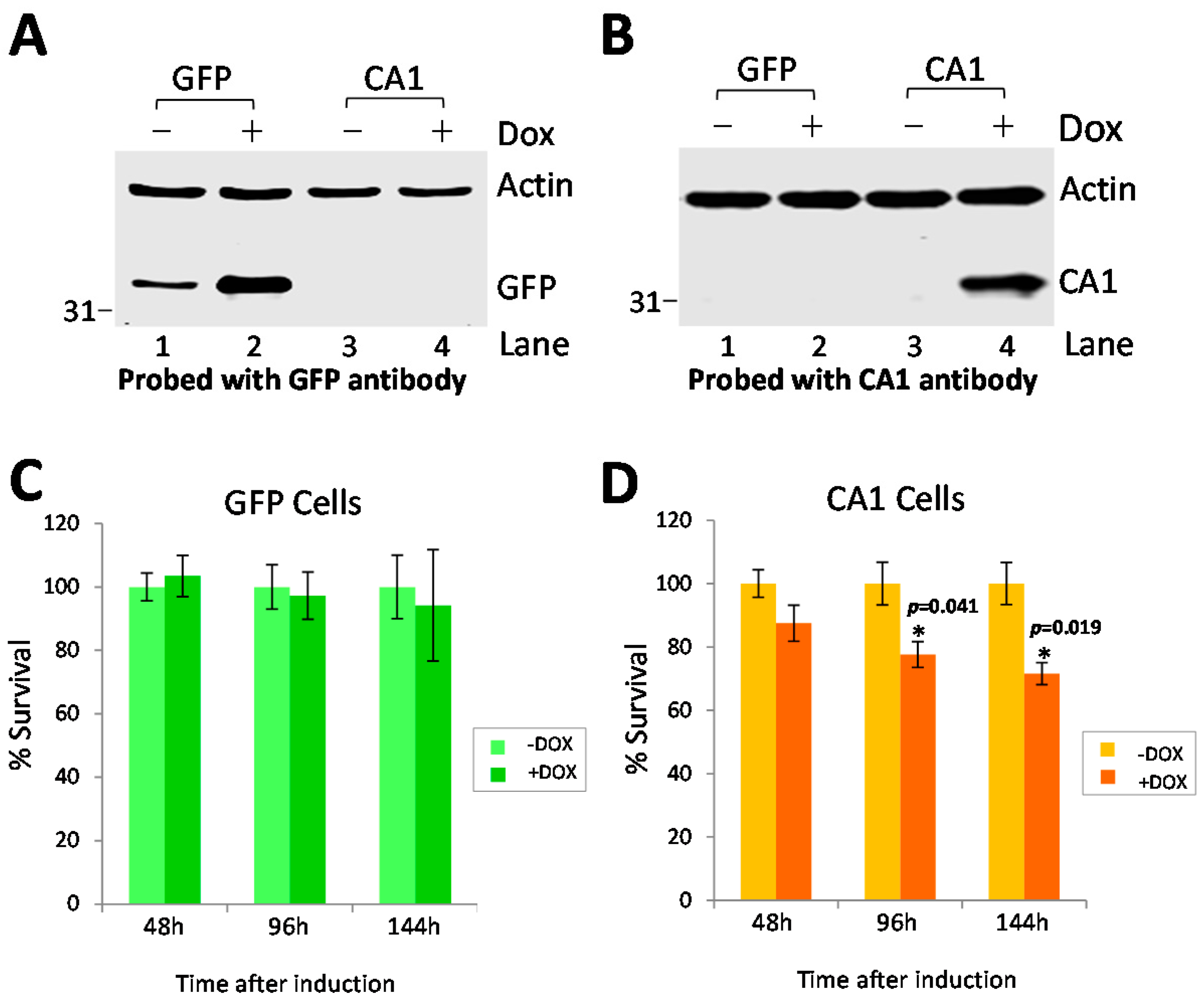
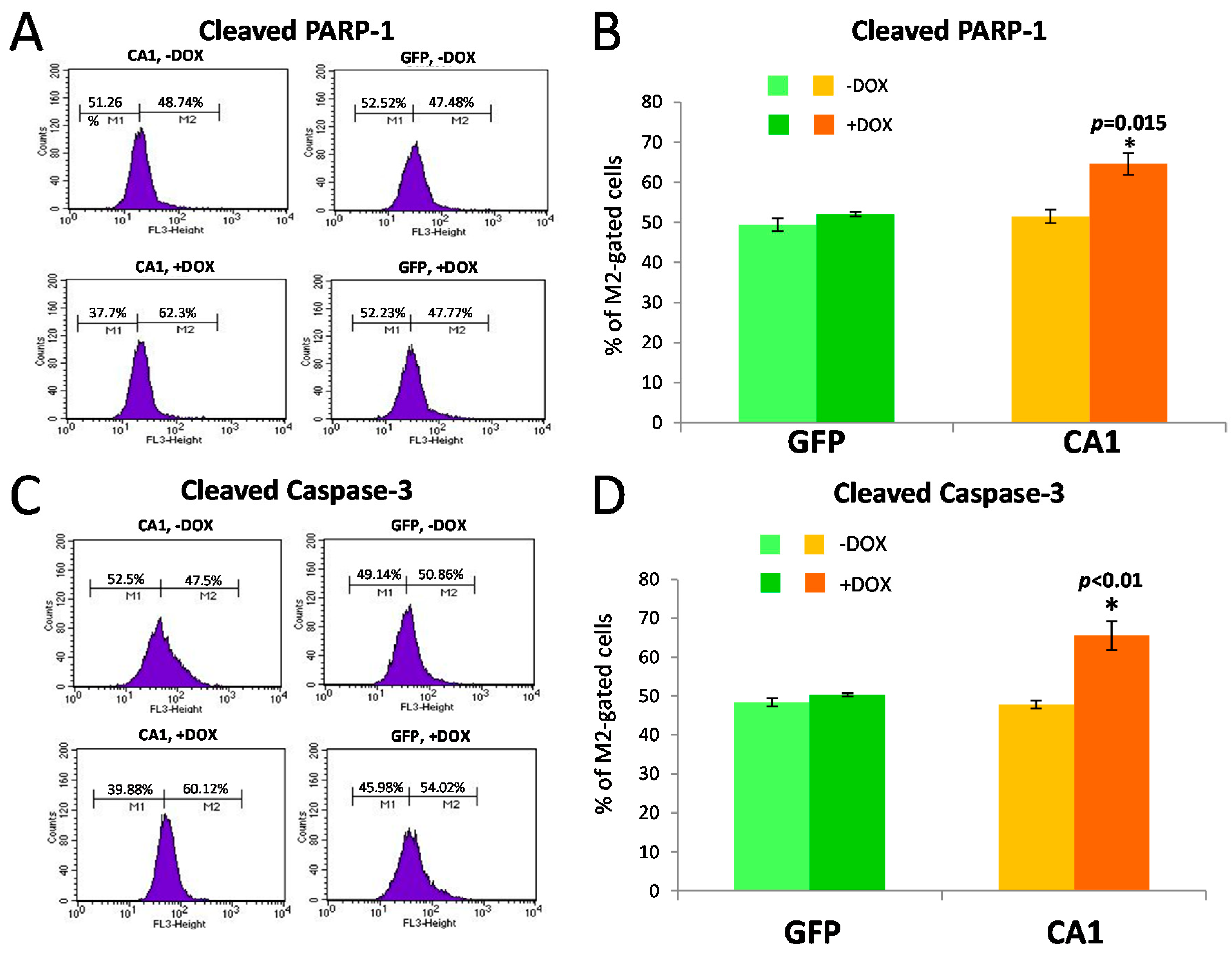
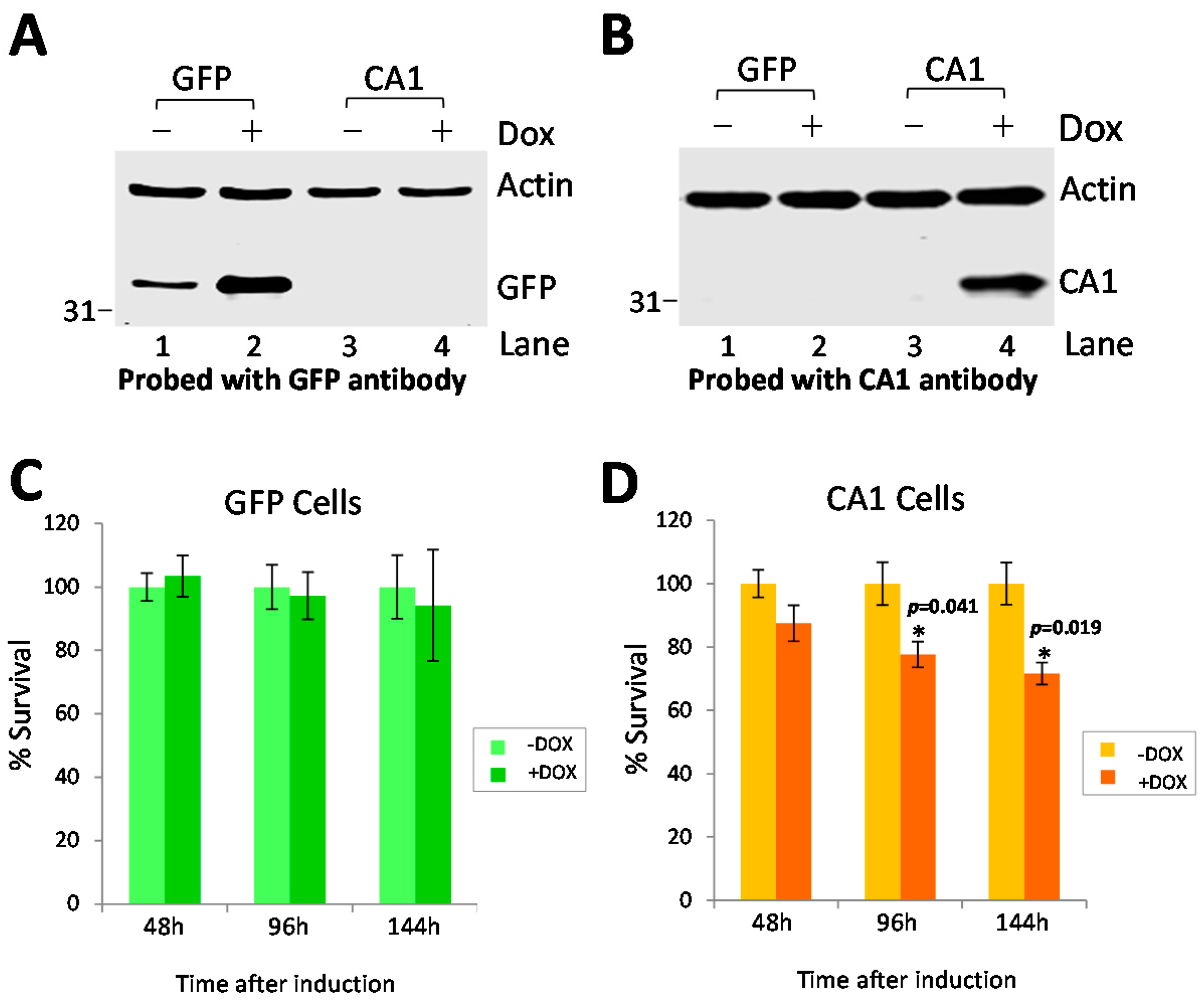
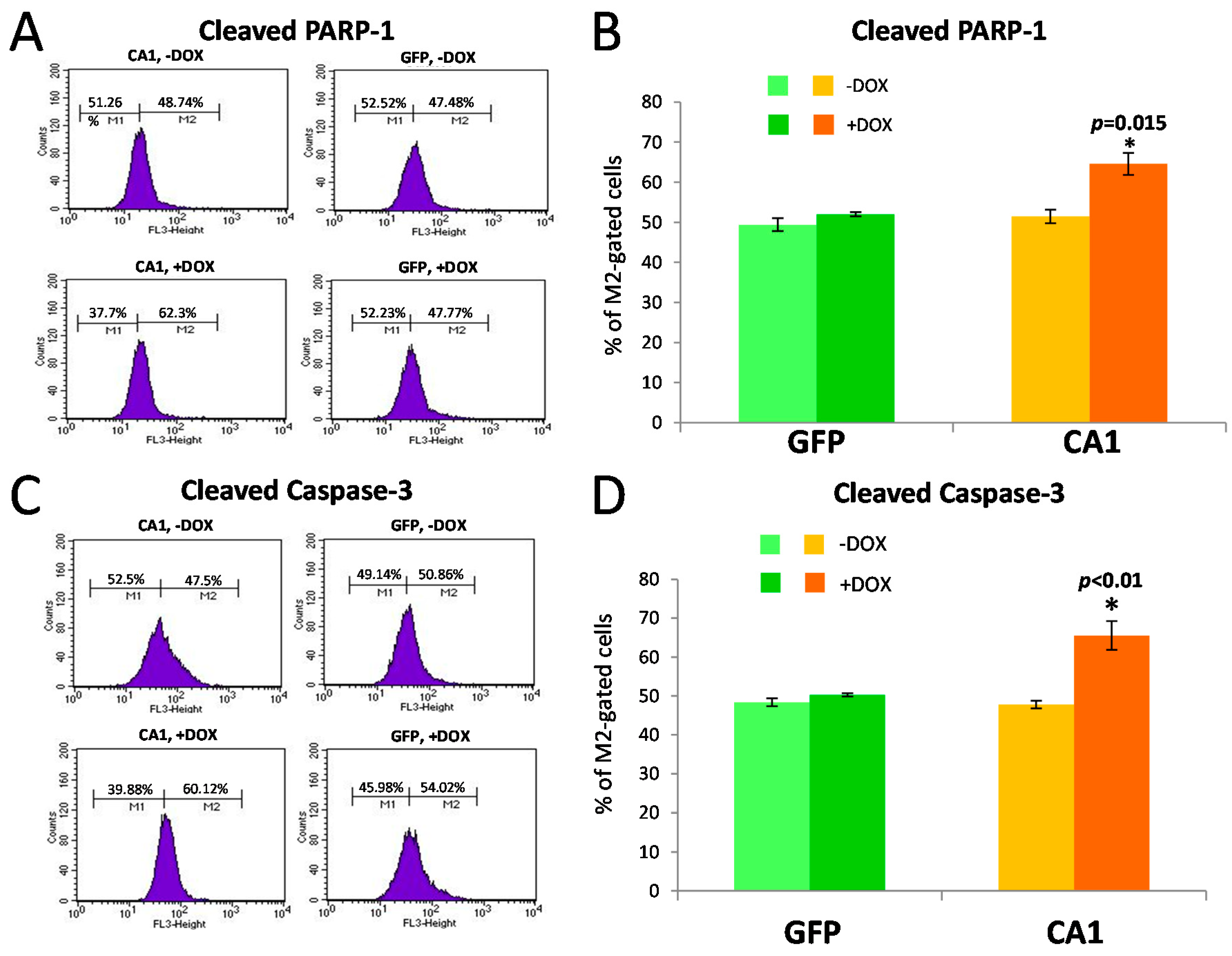
2.5. Long-Term CA1 Expression Induces Apoptosis in HEK293 Cells
3. Discussion
4. Experimental Section
4.1. Human Tissues
4.2. Antibodies and Other Reagents
4.3. Cell Cultures and Establishing Inducible Stable Cell Lines
4.4. Immunohistochemistry and Immunofluorescent Labeling
4.5. Western Blot Analysis
4.6. Cell Survival Assay
4.7. Flow Cytometry Analysis
4.8. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Supuran, C.T. Structure and function of carbonic anhydrases. Biochem. J. 2016, 473, 2023–2032. [Google Scholar] [CrossRef] [PubMed]
- Tolvanen, M.E.; Ortutay, C.; Barker, H.R.; Aspatwar, A.; Patrikainen, M.; Parkkila, S. Analysis of evolution of carbonic anhydrases IV and XV reveals a rich history of gene duplications and a new group of isozymes. Bioorg. Med. Chem. 2013, 21, 1503–1510. [Google Scholar] [CrossRef] [PubMed]
- McKenna, R.; Frost, S.C. Overview of the carbonic anhydrase family. Subcell. Biochem. 2014, 75, 3–5. [Google Scholar] [PubMed]
- Boron, W.F. Regulation of intracellular pH. Adv. Physiol. Educ. 2004, 28, 160–179. [Google Scholar] [CrossRef] [PubMed]
- Alper, S.L. Genetic diseases of acid-base transporters. Annu. Rev. Physiol. 2002, 64, 899–923. [Google Scholar] [CrossRef] [PubMed]
- Frost, S.C. Physiological functions of the alpha class of carbonic anhydrases. Subcell. Biochem. 2014, 75, 9–30. [Google Scholar] [PubMed]
- McMurtrie, H.L.; Cleary, H.J.; Alvarez, B.V.; Loiselle, F.B.; Sterling, D.; Morgan, P.E.; Johnson, D.E.; Casey, J.R. The bicarbonate transport metabolon. J. Enzym. Inhib. Med. Chem. 2004, 19, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Tresguerres, M.; Buck, J.; Levin, L.R. Physiological carbon dioxide, bicarbonate, and pH sensing. Pflug. Arch. 2010, 460, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Purkerson, J.M.; Schwartz, G.J. The role of carbonic anhydrases in renal physiology. Kidney Int. 2007, 71, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Harju, A.K.; Bootorabi, F.; Kuuslahti, M.; Supuran, C.T.; Parkkila, S. Carbonic anhydrase III: A neglected isozyme is stepping into the limelight. J. Enzym. Inhib. Med. Chem. 2013, 28, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Cammer, W.; Bieler, L.; Fredman, T.; Norton, W.T. Quantitation of myelin carbonic anhydrase-development and subfractionation of rat brain myelin and comparison with myelin from other species. Brain Res. 1977, 138, 17–28. [Google Scholar] [CrossRef]
- Kumpulainen, T.; Korhonen, L.K. Immunohistochemical localization of carbonic anhydrase isoenzyme C in the central and peripheral nervous system of the mouse. J. Histochem. Cytochem. 1982, 30, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Kumpulainen, T.; Nystrom, S.H. Immunohistochemical localization of carbonic anhydrase isoenzyme C in human brain. Brain Res. 1981, 220, 220–225. [Google Scholar] [CrossRef]
- Sapirstein, V.S.; Strocchi, P.; Wesolowski, M.; Gilbert, J.M. Characterization and biosynthesis of soluble and membrane-bound carbonic anhydrase in brain. J. Neurochem. 1983, 40, 1251–1261. [Google Scholar] [CrossRef] [PubMed]
- Casey, J.R.; Sly, W.S.; Shah, G.N.; Alvarez, B.V. Bicarbonate homeostasis in excitable tissues: Role of AE3 Cl-/HCO3-exchanger and carbonic anhydrase XIV interaction. Am. J. Physiol. Cell Physiol. 2009, 297, C1091–C1102. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa-Adachi, K.; Nishimori, I.; Taguchi, T.; Yuri, K.; Onishi, S. cDNA sequence, mRNA expression, and chromosomal localization of human carbonic anhydrase-related protein, CA-RP XI. Biochim. Biophys. Acta 1999, 1431, 518–524. [Google Scholar] [CrossRef]
- Ghandour, M.S.; Parkkila, A.K.; Parkkila, S.; Waheed, A.; Sly, W.S. Mitochondrial carbonic anhydrase in the nervous system: Expression in neuronal and glial cells. J. Neurochem. 2000, 75, 2212–2220. [Google Scholar] [CrossRef] [PubMed]
- Halmi, P.; Parkkila, S.; Honkaniemi, J. Expression of carbonic anhydrases II, IV, VII, VIII and XII in rat brain after kainic acid induced status epilepticus. Neurochem. Int. 2006, 48, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Kaya, N.; Aldhalaan, H.; Al-Younes, B.; Colak, D.; Shuaib, T.; Al-Mohaileb, F.; Al-Sugair, A.; Nester, M.; Al-Yamani, S.; Al-Bakheet, A.; et al. Phenotypical spectrum of cerebellar ataxia associated with a novel mutation in the CA8 gene, encoding carbonic anhydrase (CA) VIII. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2011, 156, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Parkkila, S.; Parkkila, A.K.; Rajaniemi, H.; Shah, G.N.; Grubb, J.H.; Waheed, A.; Sly, W.S. Expression of membrane-associated carbonic anhydrase XIV on neurons and axons in mouse and human brain. Proc. Natl. Acad. Sci. USA 2001, 98, 1918–1923. [Google Scholar] [CrossRef] [PubMed]
- Shah, G.N.; Ulmasov, B.; Waheed, A.; Becker, T.; Makani, S.; Svichar, N.; Chesler, M.; Sly, W.S. Carbonic anhydrase IV and XIV knockout mice: Roles of the respective carbonic anhydrases in buffering the extracellular space in brain. Proc. Natl. Acad. Sci. USA 2005, 102, 16771–16776. [Google Scholar] [CrossRef] [PubMed]
- Rossano, A.J.; Chouhan, A.K.; Macleod, G.T. Genetically encoded pH-indicators reveal activity-dependent cytosolic acidification of Drosophila motor nerve termini in vivo. J. Physiol. 2013, 591, 1691–1706. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Nguyen, K.T.; Barrett, E.F.; David, G. Vesicular ATPase inserted into the plasma membrane of motor terminals by exocytosis alkalinizes cytosolic pH and facilitates endocytosis. Neuron 2010, 68, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Klier, M.; Schuler, C.; Halestrap, A.P.; Sly, W.S.; Deitmer, J.W.; Becker, H.M. Transport activity of the high-affinity monocarboxylate transporter MCT2 is enhanced by extracellular carbonic anhydrase IV but not by intracellular carbonic anhydrase II. J. Biol. Chem. 2011, 286, 27781–27791. [Google Scholar] [CrossRef] [PubMed]
- Obara, M.; Szeliga, M.; Albrecht, J. Regulation of pH in the mammalian central nervous system under normal and pathological conditions: Facts and hypotheses. Neurochem. Int. 2008, 52, 905–919. [Google Scholar] [CrossRef] [PubMed]
- Stridh, M.H.; Alt, M.D.; Wittmann, S.; Heidtmann, H.; Aggarwal, M.; Riederer, B.; Seidler, U.; Wennemuth, G.; McKenna, R.; Deitmer, J.W.; et al. Lactate flux in astrocytes is enhanced by a non-catalytic action of carbonic anhydrase II. J. Physiol. 2012, 590, 2333–2351. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Dahl, D.; Alkon, D.L. Heterosynaptic transformation of GABAergic gating in the hippocampus and effects of carbonic anhydrase inhibition. J. Pharmacol. Exp. Ther. 2001, 296, 811–817. [Google Scholar] [PubMed]
- Sun, M.K.; Zhao, W.Q.; Nelson, T.J.; Alkon, D.L. Theta rhythm of hippocampal CA1 neuron activity: Gating by GABAergic synaptic depolarization. J. Neurophysiol. 2001, 85, 269–279. [Google Scholar] [PubMed]
- Theparambil, S.M.; Deitmer, J.W. High effective cytosolic H+ buffering in mouse cortical astrocytes attributable to fast bicarbonate transport. Glia 2015, 63, 1581–1594. [Google Scholar] [CrossRef] [PubMed]
- Shah, G.N.; Bonapace, G.; Hu, P.Y.; Strisciuglio, P.; Sly, W.S. Carbonic anhydrase II deficiency syndrome (osteopetrosis with renal tubular acidosis and brain calcification): Novel mutations in CA2 identified by direct sequencing expand the opportunity for genotype-phenotype correlation. Hum. Mutat. 2004. [Google Scholar] [CrossRef] [PubMed]
- Gut, M.O.; Parkkila, S.; Vernerova, Z.; Rohde, E.; Zavada, J.; Hocker, M.; Pastorek, J.; Karttunen, T.; Gibadulinova, A.; Zavadova, Z.; et al. Gastric hyperplasia in mice with targeted disruption of the carbonic anhydrase gene Car9. Gastroenterology 2002, 123, 1889–1903. [Google Scholar] [CrossRef] [PubMed]
- Pan, P.W.; Parkkila, A.K.; Autio, S.; Hilvo, M.; Sormunen, R.; Pastorekova, S.; Pastorek, J.; Haapasalo, H.; Parkkila, S. Brain phenotype of carbonic anhydrase IX-deficient mice. Transgenic Res. 2012, 21, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Leitans, J.; Kazaks, A.; Balode, A.; Ivanova, J.; Zalubovskis, R.; Supuran, C.T.; Tars, K. Efficient expression and crystallization system of cancer-associated carbonic anhydrase isoform IX. J. Med. Chem. 2015, 58, 9004–9009. [Google Scholar] [CrossRef] [PubMed]
- Feinstein, Y.; Yerushalmi, B.; Loewenthal, N.; Alkrinawi, S.; Birk, O.S.; Parvari, R.; Hershkovitz, E. Natural history and clinical manifestations of hyponatremia and hyperchlorhidrosis due to carbonic anhydrase XII deficiency. Horm. Res. Paediatr. 2014, 81, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Van Karnebeek, C.D.; Sly, W.S.; Ross, C.J.; Salvarinova, R.; Yaplito-Lee, J.; Santra, S.; Shyr, C.; Horvath, G.A.; Eydoux, P.; Lehman, A.M.; et al. Mitochondrial carbonic anhydrase VA deficiency resulting from CA5A alterations presents with hyperammonemia in early childhood. Am. J. Hum. Genet. 2014, 94, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Pan, P.W.; Kayra, K.; Leinonen, J.; Nissinen, M.; Parkkila, S.; Rajaniemi, H. Gene expression profiling in the submandibular gland, stomach, and duodenum of CAVI-deficient mice. Transgenic Res. 2011, 20, 675–698. [Google Scholar] [CrossRef] [PubMed]
- Patrikainen, M.; Pan, P.; Kulesskaya, N.; Voikar, V.; Parkkila, S. The role of carbonic anhydrase VI in bitter taste perception: Evidence from the Car6−/− mouse model. J. Biomed. Sci. 2014. [Google Scholar] [CrossRef] [PubMed]
- Aspatwar, A.; Tolvanen, M.E.; Ortutay, C.; Parkkila, S. Carbonic anhydrase related proteins: Molecular biology and evolution. Subcell. Biochem. 2014, 75, 135–156. [Google Scholar] [PubMed]
- Lamont, M.G.; Weber, J.T. Mice deficient in carbonic anhydrase type 8 exhibit motor dysfunctions and abnormal calcium dynamics in the somatic region of cerebellar granule cells. Behav. Brain Res. 2015, 286, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Aspatwar, A.; Tolvanen, M.E.; Ojanen, M.J.; Barker, H.R.; Saralahti, A.K.; Bauerlein, C.A.; Ortutay, C.; Pan, P.; Kuuslahti, M.; Parikka, M.; et al. Inactivation of ca10a and ca10b genes leads to abnormal embryonic development and alters movement pattern in zebrafish. PLoS ONE 2015, 10, e0134263. [Google Scholar] [CrossRef] [PubMed]
- Villeval, J.L.; Testa, U.; Vinci, G.; Tonthat, H.; Bettaieb, A.; Titeux, M.; Cramer, P.; Edelman, L.; Rochant, H.; Breton-Gorius, J.; et al. Carbonic anhydrase I is an early specific marker of normal human erythroid differentiation. Blood 1985, 66, 1162–1170. [Google Scholar] [PubMed]
- Chang, X.; Han, J.; Zhao, Y.; Yan, X.; Sun, S.; Cui, Y. Increased expression of carbonic anhydrase I in the synovium of patients with ankylosing spondylitis. BMC Musculoskelet. Disord. 2010, 11, 279. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.B.; Clermont, A.; Rook, S.; Fonda, S.J.; Srinivasan, V.J.; Wojtkowski, M.; Fujimoto, J.G.; Avery, R.L.; Arrigg, P.G.; Bursell, S.E.; et al. Extracellular carbonic anhydrase mediates hemorrhagic retinal and cerebral vascular permeability through prekallikrein activation. Nat. Med. 2007, 13, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Lowe, N.; Brady, H.J.; Barlow, J.H.; Sowden, J.C.; Edwards, M.; Butterworth, P.H. Structure and methylation patterns of the gene encoding human carbonic anhydrase I. Gene 1990, 93, 277–283. [Google Scholar] [CrossRef]
- Spicer, S.S.; Stoward, P.J.; Tashian, R.E. The immunohistolocalization of carbonic anhydrase in rodent tissues. J. Histochem. Cytochem. 1979, 27, 820–831. [Google Scholar] [CrossRef] [PubMed]
- Torella, D.; Ellison, G.M.; Torella, M.; Vicinanza, C.; Aquila, I.; Iaconetti, C.; Scalise, M.; Marino, F.; Henning, B.J.; Lewis, F.C.; et al. Carbonic anhydrase activation is associated with worsened pathological remodeling in human ischemic diabetic cardiomyopathy. J. Am. Heart Assoc. 2014, 3, e000434. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Fujimura, M.; Nishita, T.; Nishijima, K.; Atoji, Y.; Suzuki, Y. Immunohistochemical localization of carbonic anhydrase isozymes in the rat carotid body. J. Anat. 2003, 202, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Pan, P.; Leppilampi, M.; Pastorekova, S.; Pastorek, J.; Waheed, A.; Sly, W.S.; Parkkila, S. Carbonic anhydrase gene expression in CA II-deficient (Car2−/−) and CA IX-deficient (Car9−/−) mice. J. Physiol. 2006, 571, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Johnston-Wilson, N.L.; Sims, C.D.; Hofmann, J.P.; Anderson, L.; Shore, A.D.; Torrey, E.F.; Yolken, R.H. Disease-specific alterations in frontal cortex brain proteins in schizophrenia, bipolar disorder, and major depressive disorder. The Stanley Neuropathology Consortium. Mol. Psychiatry 2000, 5, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Ruiz Esparza-Garrido, R.; Velazquez-Flores, M.A.; Diegoperez-Ramirez, J.; Lopez-Aguilar, E.; Siordia-Reyes, G.; Hernandez-Ortiz, M.; Martinez-Batallar, A.G.; Encarnacion-Guevara, S.; Salamanca-Gomez, F.; Arenas-Aranda, D.J. A proteomic approach of pediatric astrocytomas: MiRNAs and network insight. J. Proteom. 2013, 94, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Visanji, N.P.; Wong, J.C.; Wang, S.X.; Cappel, B.; Kleinschmidt-Demasters, B.K.; Handler, M.H.; Ochi, A.; Otsubo, H.; Rutka, J.T.; Go, C.; et al. A proteomic analysis of pediatric seizure cases associated with astrocytic inclusions. Epilepsia 2012, 53, e50–e54. [Google Scholar] [CrossRef] [PubMed]
- Yamanishi, H.; Murakami, H.; Ikeda, Y.; Abe, M.; Kumagi, T.; Hiasa, Y.; Matsuura, B.; Onji, M. Regulatory dendritic cells pulsed with carbonic anhydrase I protect mice from colitis induced by CD4+CD25− T cells. J. Immunol. 2012, 188, 2164–2172. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Hua, Y.; Wang, J.; Keep, R.F.; Xi, G. Inhibition of carbonic anhydrase reduces brain injury after intracerebral hemorrhage. Transl. Stroke Res. 2012, 3, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.B.; Lu, X.K.; Zhang, X.; Li, Z.G.; Li, C.X. Carbonic anhydrase 1 is a promising biomarker for early detection of non-small cell lung cancer. Tumour Biol. 2016, 37, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lillo, C.; Jonsson, P.A.; vande Velde, C.; Ward, C.M.; Miller, T.M.; Subramaniam, J.R.; Rothstein, J.D.; Marklund, S.; Andersen, P.M.; et al. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron 2004, 43, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Jaronen, M.; Goldsteins, G.; Koistinaho, J. ER stress and unfolded protein response in amyotrophic lateral sclerosis—A controversial role of protein disulphide isomerase. Front. Cell. Neurosci. 2014. [Google Scholar] [CrossRef] [PubMed]
- Okado-Matsumoto, A.; Fridovich, I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J. Biol. Chem. 2001, 276, 38388–38393. [Google Scholar] [CrossRef] [PubMed]
- Antinone, S.E.; Ghadge, G.D.; Lam, T.T.; Wang, L.; Roos, R.P.; Green, W.N. Palmitoylation of superoxide dismutase 1 (SOD1) is increased for familial amyotrophic lateral sclerosis-linked SOD1 mutants. J. Biol. Chem. 2013, 288, 21606–21617. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Lu, D.; Bowser, R.; Liu, J. Expression of Carbonic Anhydrase I in Motor Neurons and Alterations in ALS. Int. J. Mol. Sci. 2016, 17, 1820. https://doi.org/10.3390/ijms17111820
Liu X, Lu D, Bowser R, Liu J. Expression of Carbonic Anhydrase I in Motor Neurons and Alterations in ALS. International Journal of Molecular Sciences. 2016; 17(11):1820. https://doi.org/10.3390/ijms17111820
Chicago/Turabian StyleLiu, Xiaochen, Deyi Lu, Robert Bowser, and Jian Liu. 2016. "Expression of Carbonic Anhydrase I in Motor Neurons and Alterations in ALS" International Journal of Molecular Sciences 17, no. 11: 1820. https://doi.org/10.3390/ijms17111820