
Comprehensive Transcriptome Analysis Provides Evidence of Local Thermal Adaptation in Three Loaches (Genus: Misgurnus)


Abstract
:
1. Introduction
2. Results
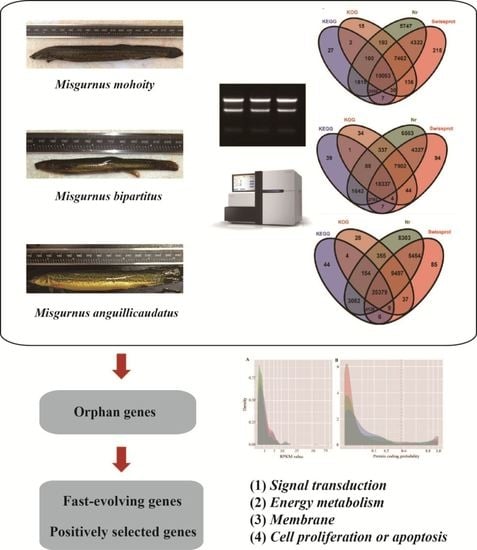
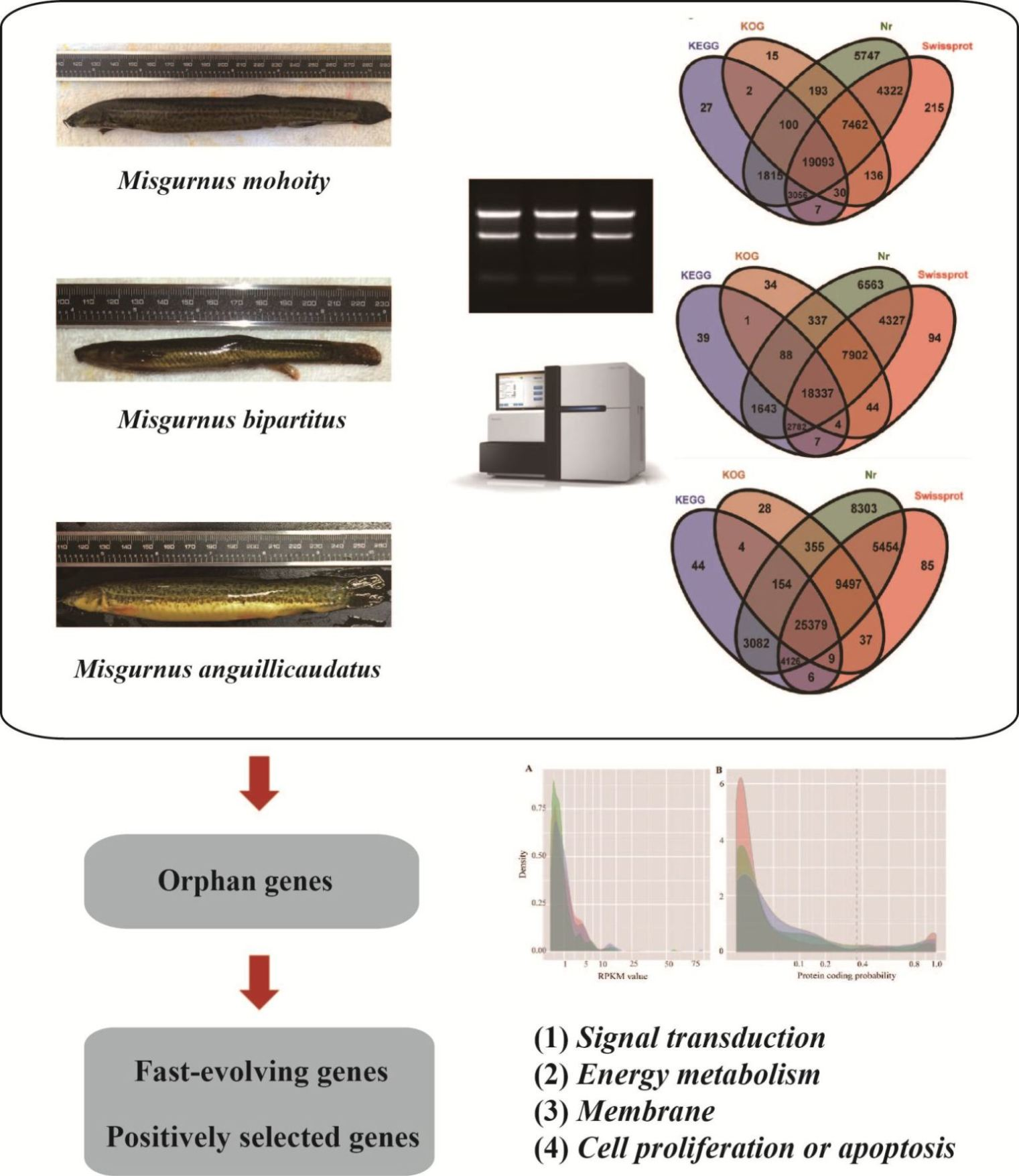
2.1. Transcriptome Assembly and Annotation
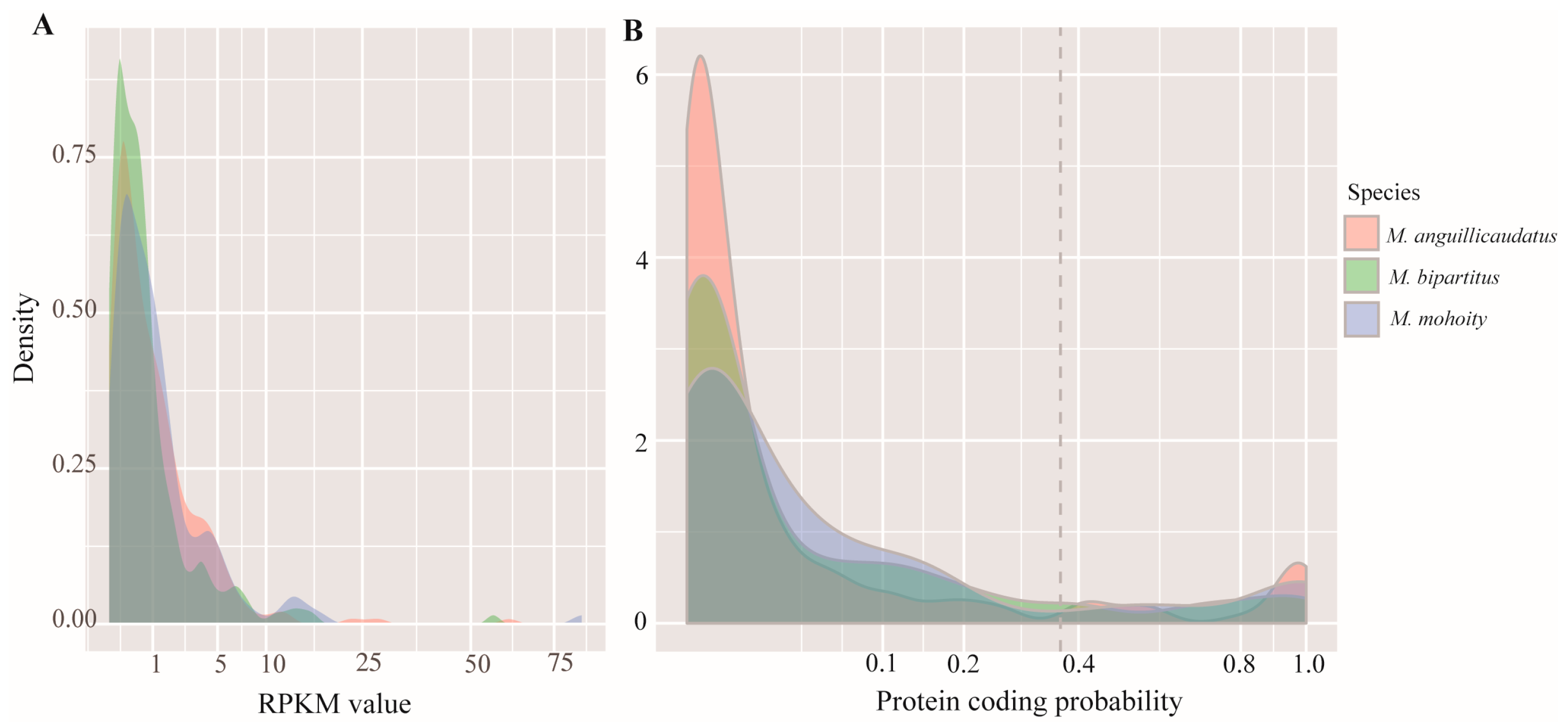
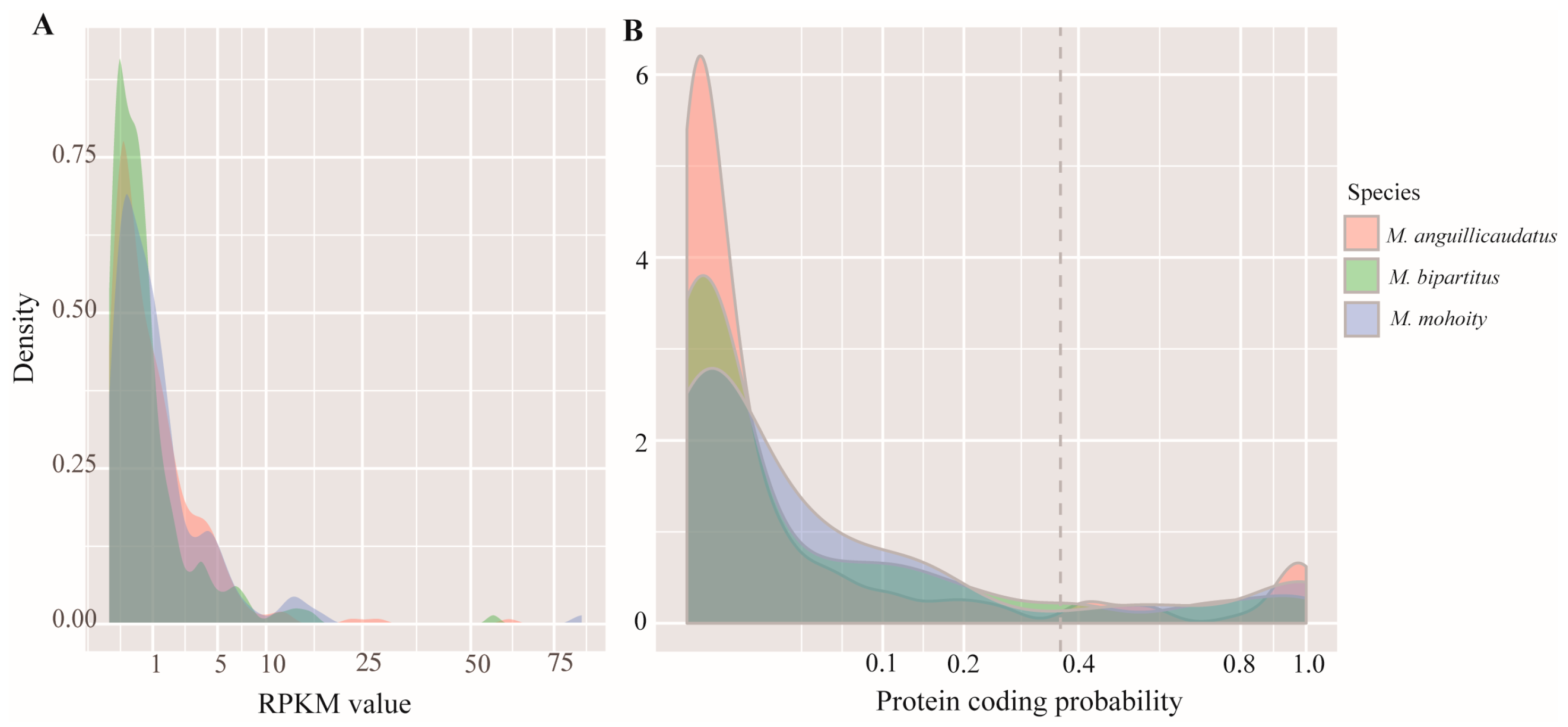
2.2. Species-Specific Orphan Genes
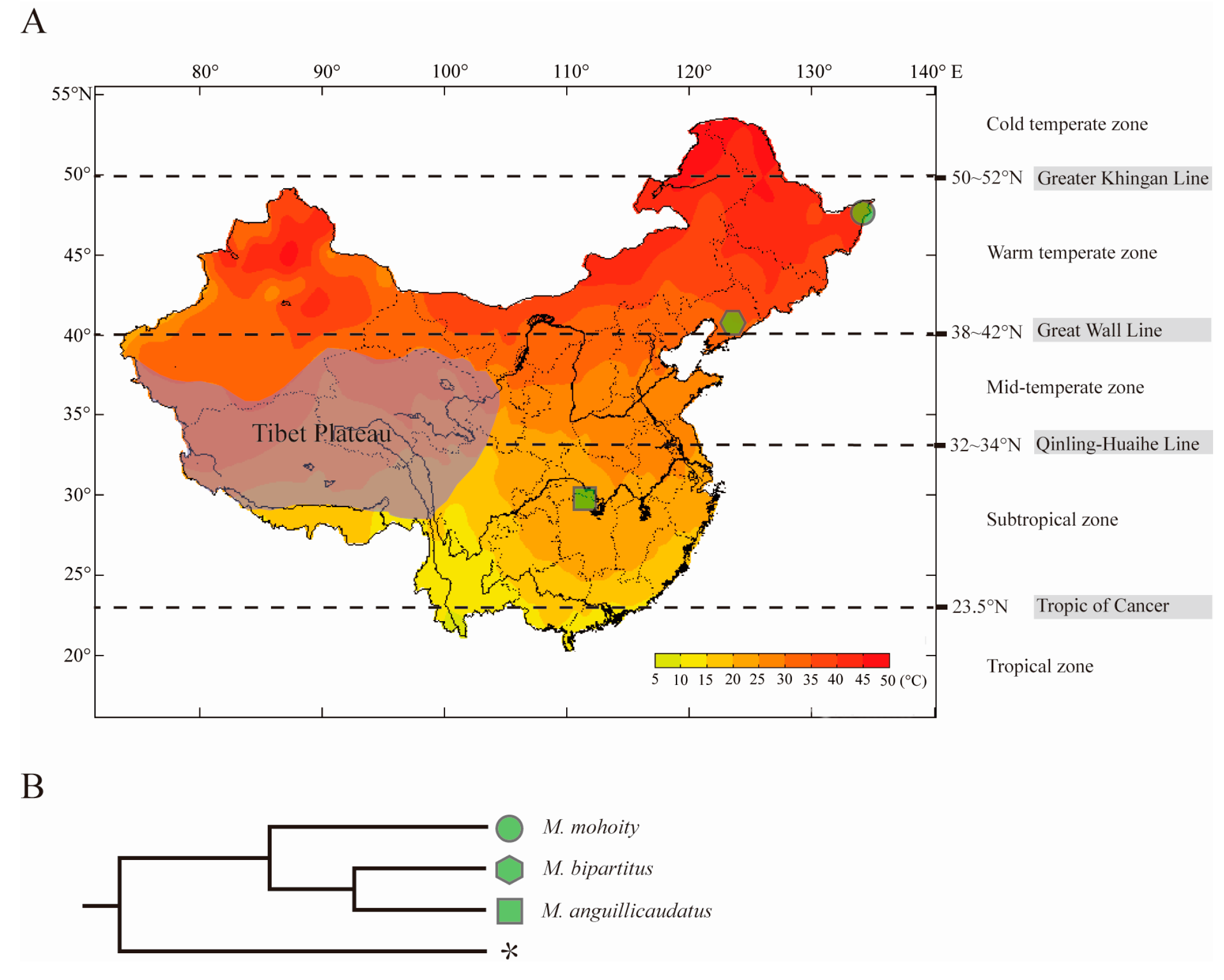
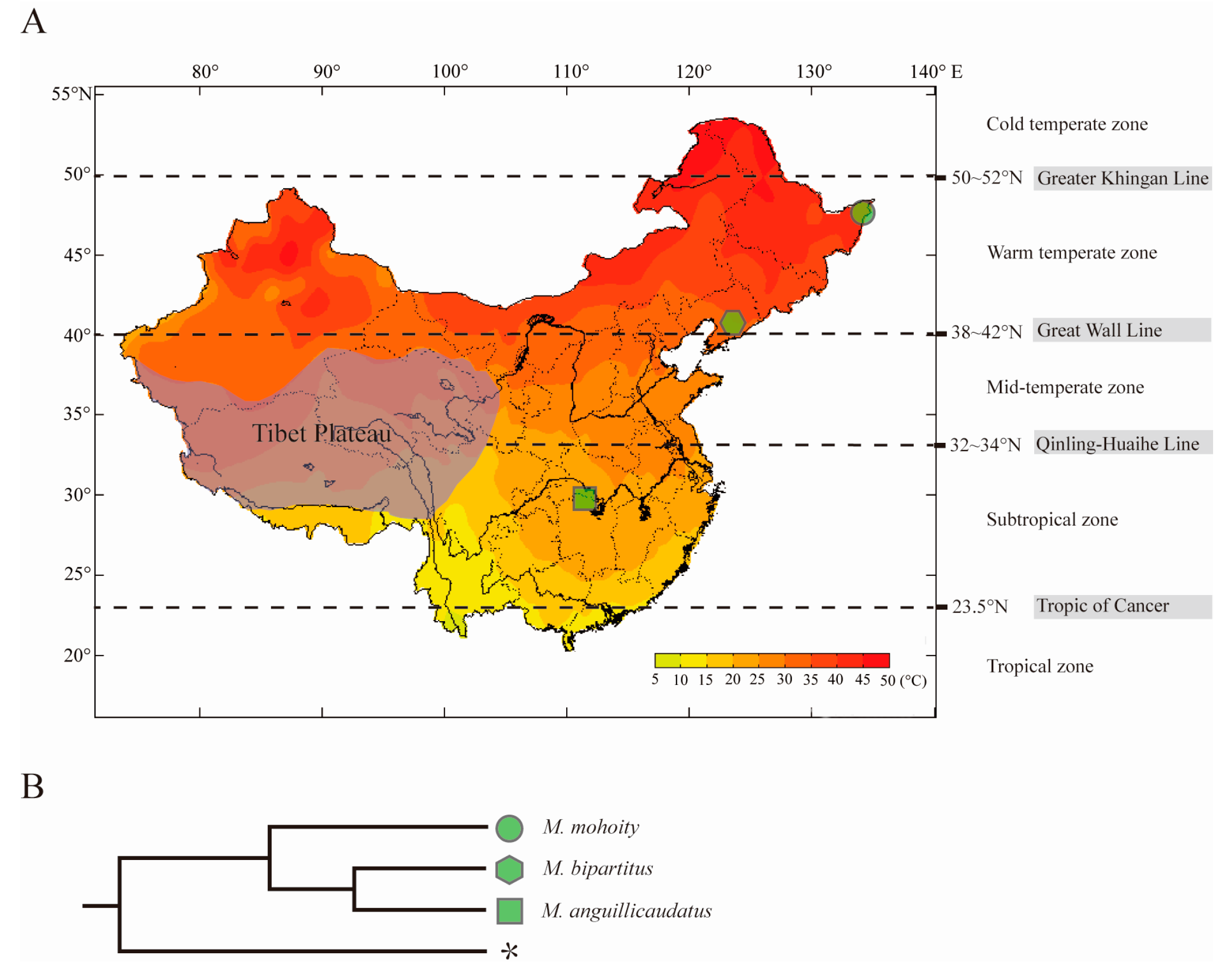
2.3. Orthologs Identification and Phylogenetic Relationship
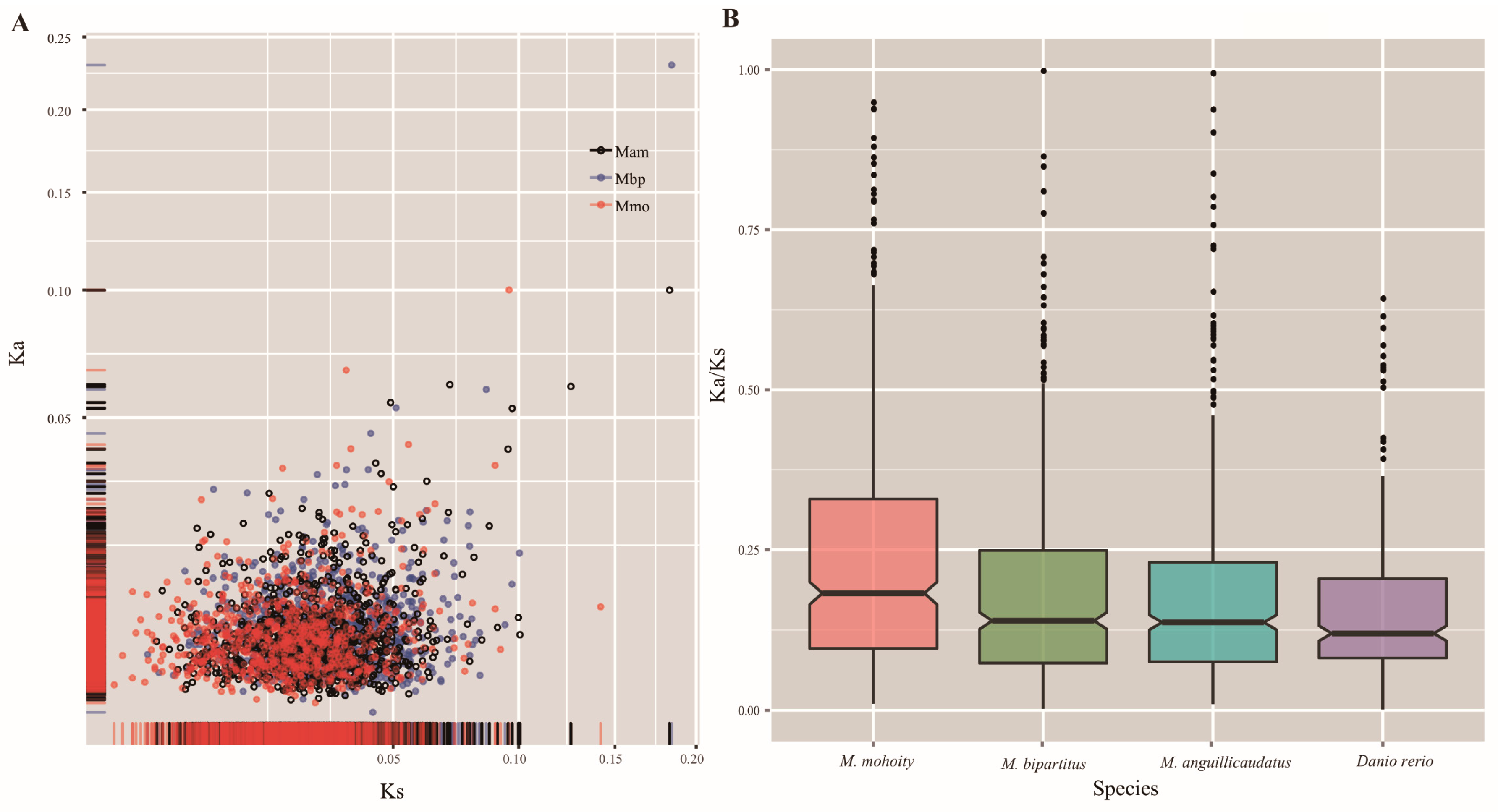
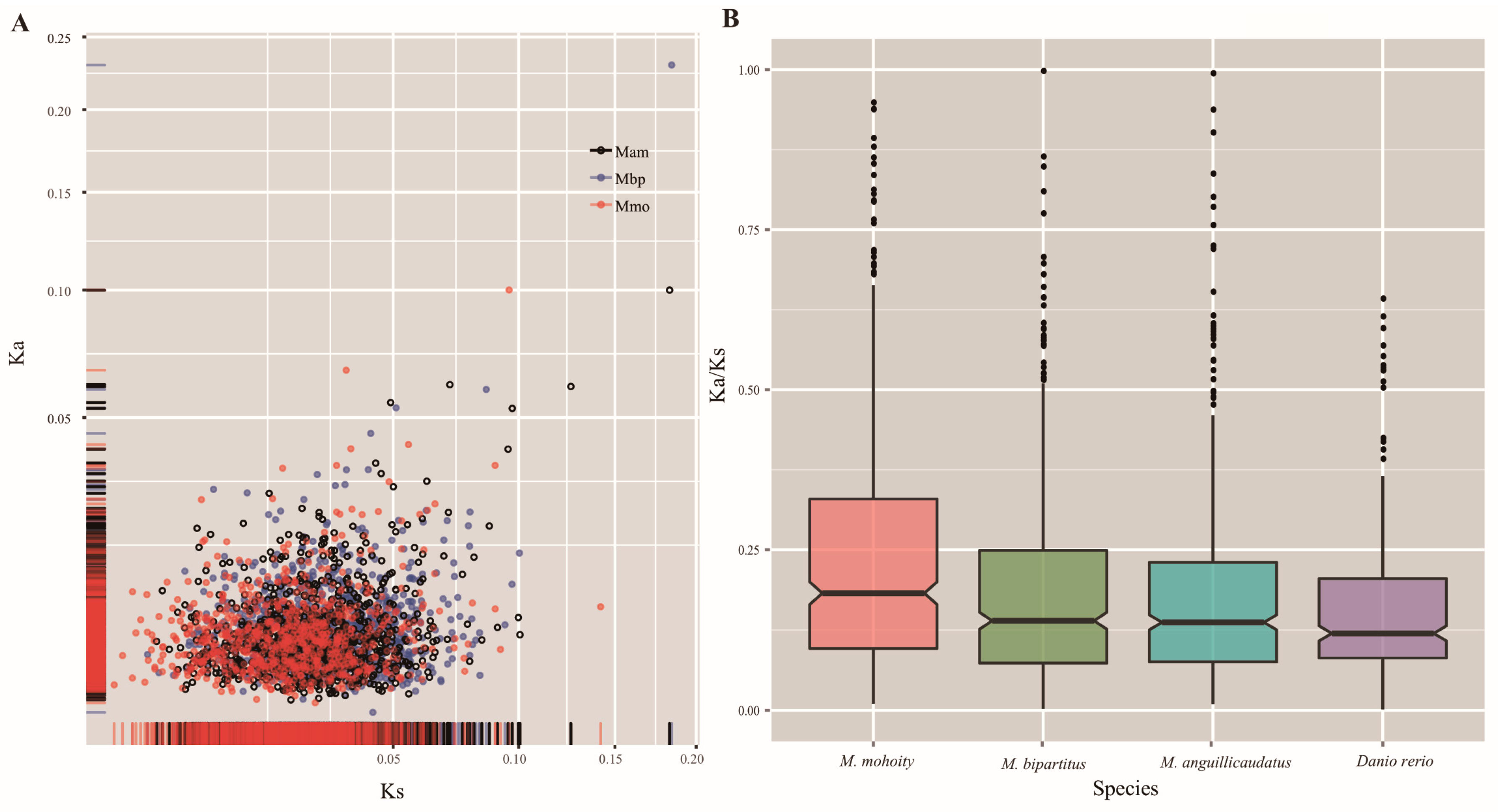
2.4. Accelerated Evolution in the Misgurnus Lineages
2.5. Fast Evolving Genes and Positive Selection Analysis
2.6. Lineage-Specific Positively Selected Sites
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Sample Collection
4.3. Sequencing, Assembly and Annotation
4.4. Identification of Orthologous Genes
4.5. Phylogenetic Analysis and Substitution Rate Estimation
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Guderley, H. Metabolic responses to low temperature in fish muscle. Biol. Rev. 2004, 79, 409–427. [Google Scholar] [CrossRef] [PubMed]
- Holopainen, I.J.; Tonn, W.M.; Paszkowski, C.A. Tales of two fish: The dichotomous biology of crucian carp (Carassius carassius (L.)) in northern Europe. Ann. Zool. Fenn. 1997, 34, 1–22. [Google Scholar]
- Tanck, M.W.T.; Booms, G.H.R.; Eding, E.H.; Wendellar Bonga, S.E.; Komen, J. Cold shocks: A stressor for common carp. J. Fish Biol. 2000, 57, 881–894. [Google Scholar] [CrossRef]
- Atwood, H.L.; Tomasso, J.R.; Webb, K.; Gatlin, D.M. Low-temperature tolerance of Nile tilapia, Oreochromis niloticus: Effects of environmental and dietary factors. Aquac. Res. 2003, 34, 241–251. [Google Scholar] [CrossRef]
- Sifa, L.; Chenhong, L.; Dey, M.; Gagalac, F.; Dunham, R. Cold tolerance of three strains of Nile tilapia, Oreochromis niloticus, in China. Aquaculture 2002, 213, 123–129. [Google Scholar] [CrossRef]
- Shaklee, J.B.; Christiansen, J.A.; Sidell, B.D.; Prosser, C.L.; Whitt, G.S. Molecular aspects of temperature acclimation in fish: Contributions of changes in enzyme activities and isozyme patterns to metabolic reorganization in the green sunfish. J. Exp. Zool. 1977, 201, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Gracey, A.Y.; Fraser, E.J.; Li, W.Z.; Fang, Y.X.; Taylor, R.R.; Rogers, J.; Brass, A.; Cossins, A.R. Coping with cold: An integrative, multitissue analysis of the transcriptome of a poikilothermic vertebrate. Proc. Natl. Acad. Sci. USA 2004, 101, 16970–16975. [Google Scholar] [CrossRef] [PubMed]
- Ju, Z.; Dunham, R.A.; Liu, Z. Differential gene expression in the brain of channel catfish (Ictalurus punctatus) in response to cold acclimation. Mol. Genet. Genom. 2002, 268, 87–95. [Google Scholar]
- Smith, S.; Bernatchez, L.; Beheregaray, L.B. RNA-seq analysis reveals extensive transcriptional plasticity to temperature stress in a freshwater fish species. BMC Genom. 2013, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Nichols, J.T. An analysis of Chinese loaches of the genus Misgurnus. Am. Mus. Novit. 1925, 169, 1–7. [Google Scholar]
- Chen, J.X.; Zhu, S.Q. Phylogenetic relationships of the subfamily in the loach family Cobitidae (Pisces). Acta Zootax. Sin. 1984, 9, 201–207. [Google Scholar]
- Perdices, A.; Bohlen, J.; Slechtova, V.; Doadrio, I. Molecular evidence for multiple origins of the European spined loaches (Teleostei, Cobitidae). PLoS ONE 2016, 11, e0151228. [Google Scholar] [CrossRef] [PubMed]
- Perdices, A.; Vasilev, V.; Vasil'eva, E. Molecular phylogeny and intraspecific structure of loaches (genera Cobitis and Misgurnus) from the Far East region of Russia and some conclusions on their systematics. Ichthyol. Res. 2012, 59, 113–123. [Google Scholar] [CrossRef]
- Vasil’eva, E.D.; Vasil’ev, V.P.; Skomorokhov, M.O. Loaches (Misgurnus, Cobitidae) of Russian Asia: II. Morphology, synonymy, diagnoses, karyology, biology, and distribution. J. Ichthyol. 2003, 7, 501. [Google Scholar]
- Sun, W.; Zhao, X.W.; Zhang, Z. Identification and evolution of the orphan genes in the domestic silkworm, Bombyx mori. FEBS Lett. 2015, 589, 2731–2738. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.Q.; Kocher, J.P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef] [PubMed]
- Weikard, R.; Hadlich, F.; Kuehn, C. Identification of novel transcripts and noncoding RNAs in bovine skin by deep next generation sequencing. BMC Genom. 2013, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Yi, S.; Zhong, J.; Wang, S.; Huang, S.; Wang, W. Mitochondrial DNA reveals evolutionary status and population genetics of two closely related fish ( Misgurnus bipartitus and Misgurnus mohoity ) in northeast China. Biochem. Syst. Ecol. 2016, 68, 192–199. [Google Scholar] [CrossRef]
- Yang, Z.; Nielsen, R. Synonymous and nonsynonymous rate variation in nuclear genes of mammals. J. Mol. Evol. 1998, 46, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Breuker, C.; Moreau, A.; Lakhal, L.; Tamasi, V.; Parmentier, Y.; Meyer, U.; Maurel, P.; Lumbroso, S.; Vilarem, M.J.; Pascussi, J.M. Hepatic expression of thyroid hormone-responsive spot 14 protein is regulated by constitutive androstane receptor (NR1I3). Endocrinology 2010, 151, 1653–1661. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.-B.; Shao, Y.-M.; Miao, S.; Wang, L. The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell. Mol. Life Sci. 2006, 63, 2560–2570. [Google Scholar] [CrossRef] [PubMed]
- Sanders, B.M. Stress proteins in aquatic organisms: An environmental perspective. Crit. Rev. Toxicol. 1993, 23, 49–75. [Google Scholar] [CrossRef] [PubMed]
- Currie, S. Heat Shock Proteins and Temperature. In Encyclopedia of Fish Physiology; Academic Press: San Diego, CA, USA, 2011; pp. 1732–1737. [Google Scholar]
- Wojtasik, B.; Kuczynska-Wisnik, D. Temperature shock tolerance and heat shock proteins in Arctic freshwater ostracod Candona rectangulata—Preliminary results. Pol. Polar Res. 2012, 33, 199–206. [Google Scholar] [CrossRef]
- Feder, M.E.; Hofmann, G.E. Heat-shock proteins, molecular chaperones, and the stress response: Evolutionary and Ecological Physiology. Annu. Rev. Physiol. 2003, 61, 243. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.H.; Dai, W.; Kang, J.L.; Yang, L.D.; He, S.P. Comprehensive Transcriptome Analysis of Six Catfish Species from an Altitude Gradient Reveals Adaptive Evolution in Tibetan Fishes. G3-Genes Genom. Genet. 2016, 6, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, L.D.; Zhou, K.; Zhang, Y.P.; Song, Z.B.; He, S.P. Evidence for Adaptation to the Tibetan Plateau Inferred from Tibetan Loach Transcriptomes. Genome Biol. Evol. 2015, 7, 2970–2982. [Google Scholar] [CrossRef] [PubMed]
- Backstrom, N.; Zhang, Q.; Edwards, S.V. Evidence from a House Finch (Haemorhous mexicanus) Spleen Transcriptome for Adaptive Evolution and Biased Gene Conversion in Passerine Birds. Mol. Biol. Evol. 2013, 30, 1046–1050. [Google Scholar] [CrossRef] [PubMed]
- Vasilev, V.P.; Vasileva, E.D. Comparative karyological analysis of mud loach and spined loach species (genera Misgurnus and Cobitis) from the Far East region of Russia. Folia Zool. 2008, 57, 51–59. [Google Scholar]
- Li, Y.J.; Zhang, M.Z.; Zhuo, Y.U.; Pang, Y.M.; Qian, C.; Katsutoshi, A. Morphological variations in amur weatherfish Misgurnus mohoity, northern weatherfish Misgurnus bipartitus and oriental weatherfish Misgurnus anguillicaudatus in China. J. Dalian Ocean Univ. 2010, 25, 397–401. [Google Scholar]
- Grandaubert, J.; Bhattacharyya, A.; Stukenbrock, E.H. RNA-seq-Based Gene Annotation and Comparative Genomics of Four Fungal Grass Pathogens in the Genus Zymoseptoria Identify Novel Orphan Genes and Species-Specific Invasions of Transposable Elements. G3-Genes Genom. Genet. 2015, 5, 1323–1333. [Google Scholar]
- Colbourne, J.K.; Pfrender, M.E.; Gilbert, D.; Thomas, W.K.; Tucker, A.; Oakley, T.H.; Tokishita, S.; Aerts, A.; Arnold, G.J.; Basu, M.K.; et al. The Ecoresponsive Genome of Daphnia pulex. Science 2011, 331, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Donoghue, M.T.A.; Keshavaiah, C.; Swamidatta, S.H.; Spillane, C. Evolutionary origins of Brassicaceae specific genes in Arabidopsis thaliana. BMC Evol. Biol. 2011, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.D.; Wang, Y.; Zhang, Z.L.; He, S.P. Comprehensive Transcriptome Analysis Reveals Accelerated Genic Evolution in a Tibet Fish, Gymnodiptychus pachycheilus. Genome Biol. Evol. 2015, 7, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Song, G.L.; Yan, J.J.; He, X.Z.; Li, Q.; Cui, Z.B. Transcriptomic characterization of cold acclimation in larval zebrafish. BMC Genom. 2013, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ferguson, S.S.G.; Barak, L.S.; Bodduluri, S.R.; Laporte, S.A.; Caron, M.G. Role for G protein-coupled receptor kinase in agonist-specific regulation of μ-opioid receptor responsiveness. Proc. Natl. Acad. Sci. USA 1998, 95, 7157. [Google Scholar] [CrossRef] [PubMed]
- Egami, Y. Molecular imaging analysis of Rab GTPases in the regulation of phagocytosis and macropinocytosis. Anat. Sci. Int. 2016, 91, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Samet, J.M.; Dewar, B.J.; Wu, W.; Graves, L.M. Mechanisms of Zn2+-induced signal initiation through the epidermal growth factor receptor. Toxicol. Appl. Pharm. 2003, 191, 86–93. [Google Scholar] [CrossRef]
- Hu, J.W.; You, F.; Wang, Q.; Weng, S.D.; Liu, H.; Wang, L.J.; Zhang, P.J.; Tan, X.G. Transcriptional Responses of Olive Flounder (Paralichthys olivaceus) to Low Temperature. PLoS ONE 2014, 9, e108582. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Wu, S.B.; Hong, C.H.; Yu, H.S.; Wei, Y.H. Molecular Mechanisms of UV-Induced Apoptosis and Its Effects on Skin Residential Cells: The Implication in UV-Based Phototherapy. Int. J. Mol. Sci. 2013, 14, 6414–6435. [Google Scholar] [CrossRef] [PubMed]
- Moon, K.; Zhou, S.Y.; Lee, S.H.; Owyang, C.; DiMagno, M.J. Hsp70 and XIAP are potential key molecular mechanisms causing impaired apoptosis and severe acute pancreatitis (AP) in CF mice. Pancreas 2007, 35, 417. [Google Scholar] [CrossRef]
- Zucchini, N.; de Sousa, G.; Pizzol, J.; Rahmani, R. Molecular mechanisms underlying the hepatotoxicity of lindane in rat hepatocytes: Apoptosis regulation and oxidative stress. Drug Metab. Rev. 2005, 37, 23. [Google Scholar]
- Gorman, A.M.; Healy, S.J.M.; Jager, R.; Samali, A. Stress management at the ER: Regulators of ER stress-induced apoptosis. Pharmacol. Ther. 2012, 134, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Li, W.Z.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [PubMed]
- Iseli, C.; Jongeneel, C.V.; Bucher, P. ESTScan: A program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1999, 99, 138–148. [Google Scholar]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Loytynoja, A.; Goldman, N. An algorithm for progressive multiple alignment of sequences with insertions. Proc. Natl. Acad. Sci. USA 2005, 102, 10557–10562. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Roure, B.; Rodriguez-Ezpeleta, N.; Philippe, H. SCaFoS: A tool for selection, concatenation and fusion of sequences for phylogenomics. BMC Evol. Biol. 2007, 7, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.H. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Goodman, M.; Sterner, K.N.; Islam, M.; Uddin, M.; Sherwood, C.C.; Hof, P.R.; Hou, Z.C.; Lipovich, L.; Jia, H.; Grossman, L.I.; et al. Phylogenomic analyses reveal convergent patterns of adaptive evolution in elephant and human ancestries. Proc. Natl. Acad. Sci. USA 2009, 106, 20824–20829. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Ks | Ka/Ks | GC3 | FEGs | PSGs | Overlapping |
|---|---|---|---|---|---|---|
| M. anguillicaudatus | 0.0127 | 0.1827 | 0.5099 | 205 | 63 | 28 |
| M. bipartitus | 0.0111 | 0.1994 | 0.5091 | 193 | 53 | 18 |
| M. mohoity | 0.0092 | 0.2271 | 0.5095 | 191 | 50 | 15 |
| Species | Gene | Function Terms | GO Number |
|---|---|---|---|
| Signal transduction | |||
| M. anguillicaudatus (n = 6) | stk17b | Intracellular signal transduction | GO: 0035556 |
| trim35 | Zinc ion binding | GO: 0008270 | |
| gatad1 | Zinc ion binding | GO: 0008270 | |
| oprm1 | G-protein coupled receptor | GO: 0001664 | |
| selenbp1 | Selenium binding | GO: 0008430 | |
| card19 | NF-κB signaling | GO: 0043124 | |
| M. bipartitus (n = 3) | rab20 | GTP binding | GO: 0005525 |
| mhc1laa | Antigen binding | GO: 0006955 | |
| spred1 | Inactivation of MAPK activity | GO: 0000188 | |
| M. mohoity (n = 2) | efcc1 | Calcium ion binding | GO: 0005509 |
| trim63b | Zinc ion binding | GO: 0008270 | |
| Membrane | |||
| M. anguillicaudatus (n = 2) | sgcb | Membrane organization | GO: 0061024 |
| cldn15lb | Tight junction | GO: 0030054 | |
| M. bipartitus (n = 2) | tmem101 | Transmembrane | GO: 0016021 |
| rhbdd2 | Serine-type endopeptidase activity | GO: 0004252 | |
| M. mohoity (n = 4) | tmem88b | Integral component of membrane | GO: 0016021 |
| tmem223 | Integral component of membrane | GO: 0016021 | |
| tm4sf5 | Integral component of membrane | GO: 0016021 | |
| tpbgb | Integral component of membrane | GO: 0016021 | |
| Energy metabolism | |||
| M. anguillicaudatus (n = 6) | ubiad1 | Antioxidant activity | GO: 0016209 |
| ndufb11 | Oxidation-reduction process | GO: 0055114 | |
| dnajc19 | Hsp70 protein binding | GO: 0030544 | |
| alg1 | Protein glycosylation | GO: 0006486 | |
| thrsp | Lipid metabolic process | GO: 0006629 | |
| impad1 | Myo-inositol biosynthesis | GO: 0006021 | |
| M. bipartitus (n = 4) | ndufs3 | Oxidoreductase activity, acting on NAD(P)H | GO: 0004128 |
| crot | Fatty acid beta-oxidation | GO: 0006631 | |
| pdxp | Heat shock protein binding | GO: 0031072 | |
| dnajb1a | Hsp70 protein binding | GO: 0030544 | |
| M. mohoity (n = 3) | slc50a1 | Sugar transmembrane transporter activity | GO: 0051119 |
| srd5a2a | Steroid metabolic process | GO: 0008202 | |
| dnajc1 | ATPase activator activity | GO: 0001671 | |
| Cell proliferation/apoptosis | |||
| M. anguillicaudatus (n = 9) | diabloa | Apoptotic process | GO: 0006915 |
| api5 | Apoptotic process | GO: 0006915 | |
| fam212aa | Cartilage development | GO: 0051216 | |
| yap1 | Cartilage development | GO: 0051216 | |
| thumpd2 | RNA binding | GO: 0003723 | |
| bmf1 | Positive regulation of apoptotic process | GO: 0043065 | |
| lcmt1 | Methyltransferase activity | GO: 0008168 | |
| ankrd1a | Sarcomere | GO: 0030017 | |
| krt222 | Structural molecule activity | GO: 0005198 | |
| M. bipartitus (n = 7) | gins2 | DNA replication | GO: 0006260 |
| cdc23 | Cell division | GO: 0051301 | |
| plekho1b | Regulation of myoblast fusion | GO: 1901739 | |
| cwc25 | Nucleoplasm | GO: 0005654 | |
| ctdspl3 | Protein dephosphorylation | GO: 0006470 | |
| ddx41 | Apoptotic process | GO: 0006915 | |
| ddx52 | rRNA processing | GO: 0006364 | |
| M. mohoity (n = 5) | ogfr | Opioid receptor activity | GO: 0004985 |
| eepd1 | DNA repair | GO: 0006281 | |
| dnph1 | Positive regulation of cell growth | GO: 0030307 | |
| rpz | Fin development | GO: 0033333 | |
| cited4b | Muscle cell differentiation | GO: 0042692 | |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yi, S.; Wang, S.; Zhong, J.; Wang, W. Comprehensive Transcriptome Analysis Provides Evidence of Local Thermal Adaptation in Three Loaches (Genus: Misgurnus). Int. J. Mol. Sci. 2016, 17, 1943. https://doi.org/10.3390/ijms17121943
Yi S, Wang S, Zhong J, Wang W. Comprehensive Transcriptome Analysis Provides Evidence of Local Thermal Adaptation in Three Loaches (Genus: Misgurnus). International Journal of Molecular Sciences. 2016; 17(12):1943. https://doi.org/10.3390/ijms17121943
Chicago/Turabian StyleYi, Shaokui, Sai Wang, Jia Zhong, and Weimin Wang. 2016. "Comprehensive Transcriptome Analysis Provides Evidence of Local Thermal Adaptation in Three Loaches (Genus: Misgurnus)" International Journal of Molecular Sciences 17, no. 12: 1943. https://doi.org/10.3390/ijms17121943