
The Double Role of p53 in Cancer and Autoimmunity and Its Potential as Therapeutic Target



Abstract
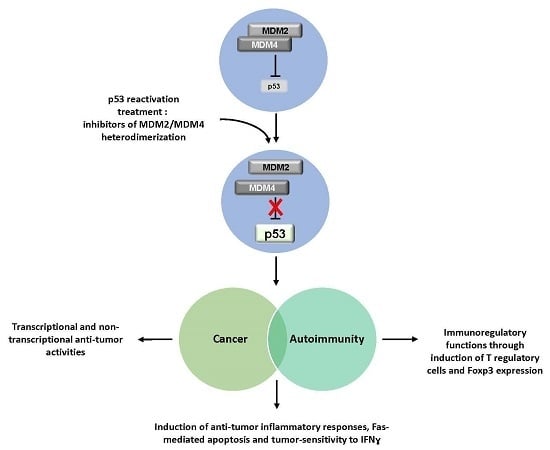
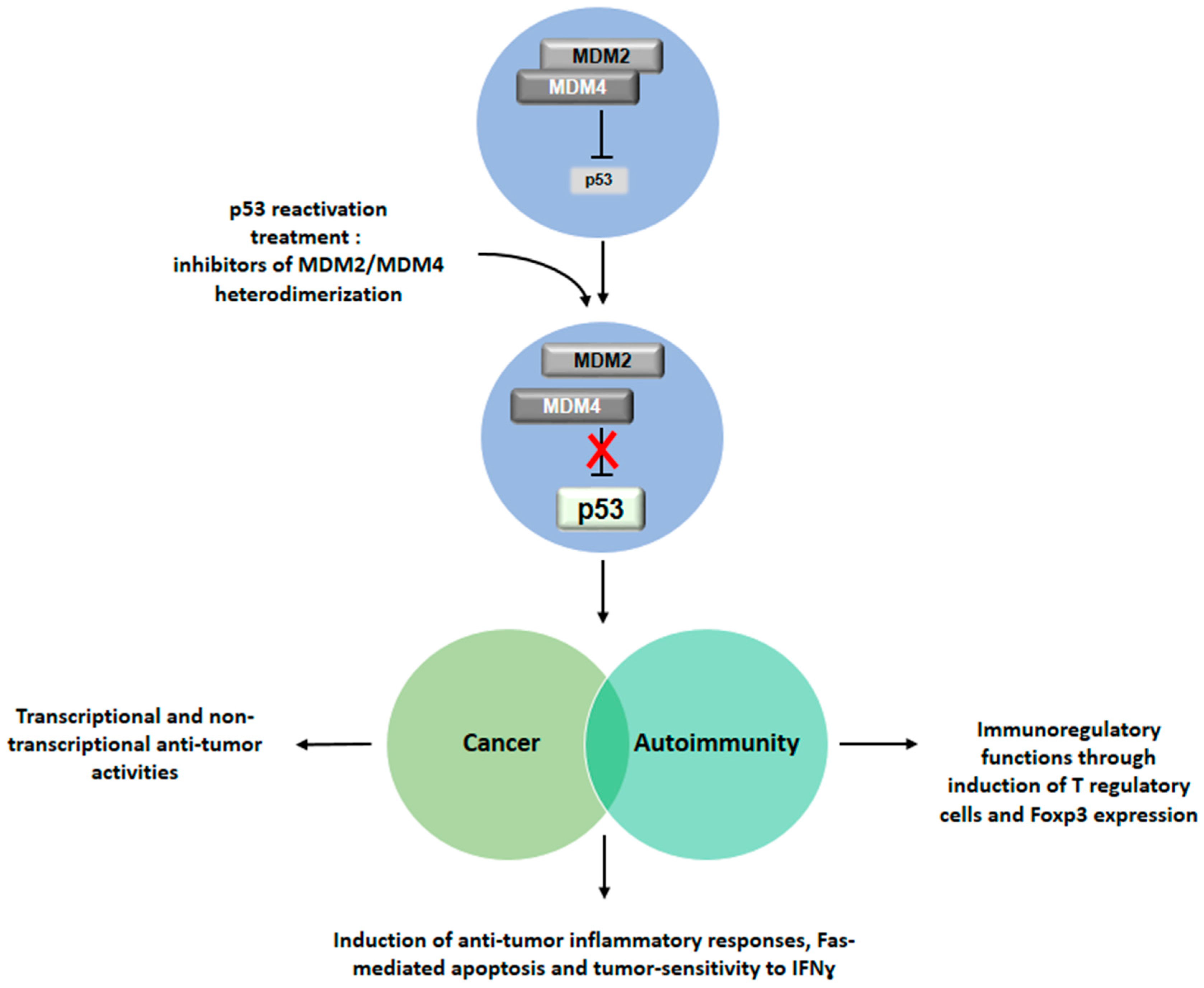
:
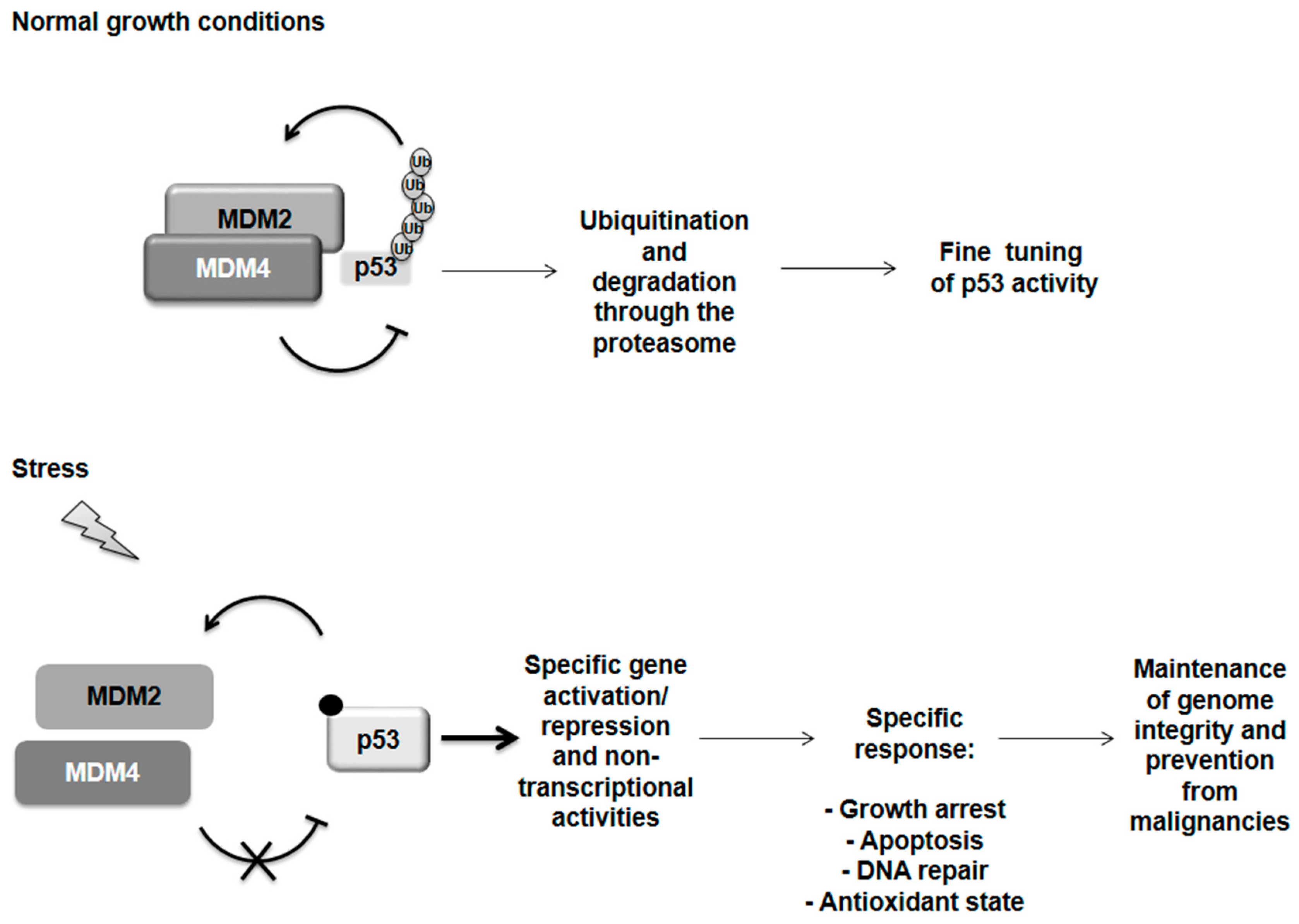
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. p53 in Human Tumors
3. p53 in Inflammatory and Autoimmune Conditions
4. Cancer and Autoimmunity: p53 Probing the Link
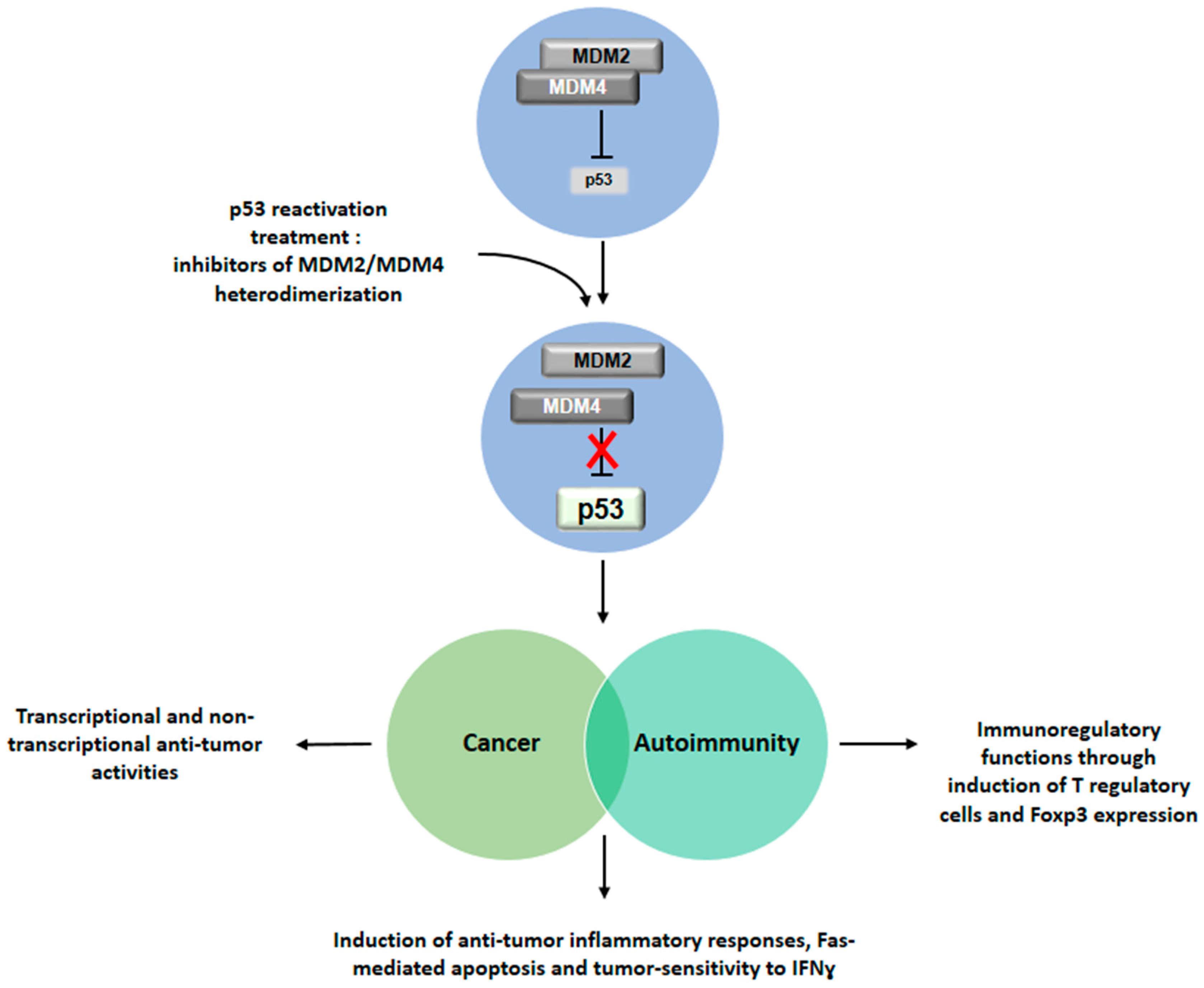
5. Conclusive Remarks: Therapeutic Perspectives Based on p53 Reactivation
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Levrero, M.; de Laurenzi, V.; Costanzo, A.; Gong, J.; Wang, J.Y.; Melino, G.J. The p53/p63/p73 family of transcription factors: Overlapping and distinct functions. Cell Sci. 2000, 113, 1661–1670. [Google Scholar]
- Arrowsmith, C.H. Structure and function in the p53 family. Cell Death Differ. 1999, 6, 1169–1173. [Google Scholar] [CrossRef] [PubMed]
- Yamanishi, Y.; Boyle, D.L.; Pinkoski, M.J.; Mahboubi, A.; Lin, T.; Han, Z.; Zvaifler, N.J.; Green, D.R.; Firestein, G.S. Regulation of joint destruction and inflammation by p53 in collagen-induced arthritis. Am. J. Pathol. 2002, 160, 123–130. [Google Scholar] [CrossRef]
- Sakhi, S.; Bruce, A.; Sun, N.; Tocco, G.; Baudry, M.; Schreiber, S.S. p53 induction is associated with neuronal damage in the central nervous system. Proc. Natl. Acad. Sci. USA 1994, 91, 7525–7529. [Google Scholar] [CrossRef] [PubMed]
- Moon, C.; Kim, S.; Wie, M.; Kim, H.; Cheong, J.; Park, J.; Jee, Y.; Tanuma, N.; Matsumoto, Y.; Shin, T. Increased expression of p53 and Bax in the spinal cords of rats with experimental autoimmune encephalomyelitis. Neurosci. Lett. 2000, 289, 41–44. [Google Scholar] [CrossRef]
- Green, D.R.; Kroemer, G. Cytoplasmic functions of the tumour suppressor p53. Nature 2009, 458, 1127–1130. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.S.; Burns, T.F.; McDonald, E.R., 3rd; Jiang, W.; Meng, R.; Krantz, I.D.; Kao, G.; Gan, D.D.; Zhou, J.Y.; Muschel, R.; et al. KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat. Genet. 1997, 17, 141–143. [Google Scholar] [CrossRef] [PubMed]
- Guan, B.; Yue, P.; Clayman, G.L.; Sun, S.Y. Evidence that the death receptor DR4 is a DNA damage-inducible, p53-regulated gene. J. Cell Physiol. 2001, 188, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Wilder, S.; Bannasch, D.; Israeli, D.; Lehlbach, K.; Li-Weber, M.; Friedman, S.L.; Galle, P.R.; Stremmel, W.; Oren, M.; et al. p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J. Exp. Med. 1998, 188, 2033–2045. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.; Macdonald, K.; Chan, S.W.; Luzio, J.P.; Simari, R.; Weissberg, P. Cell surface trafficking of Fas: A rapid mechanism of p53-mediated apoptosis. Science 1998, 282, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Xia, Y.; Zweier, J.L.; Kinzler, K.W.; Vogelstein, B. A model for p53-induced apoptosis. Nature 1997, 389, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Buckbinder, L.; Talbott, R.; Velasco-Miguel, S.; Takenaka, I.; Faha, B.; Seizinger, B.R.; Kley, N. Induction of the growth inhibitor IGF-binding protein 3 by p53. Nature 1995, 377, 646–649. [Google Scholar] [CrossRef] [PubMed]
- El-Deiry, W.S.; Harper, J.W.; O’Connor, P.M.; Velculescu, V.E.; Canman, C.E.; Jackman, J.; Pietenpol, J.A.; Burrell, M.; Hill, D.E.; Wang, Y.; et al. WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res. 1994, 5, 1169–1174. [Google Scholar]
- Hermeking, H.; Lengauer, C.; Polyak, K.; He, T.C.; Zhang, L.; Thiagalingam, S.; Kinzler, K.W.; Vogelstein, B. 14-3-3sigma is a p53-regulated inhibitor of G2/M progression. Mol. Cell 1997, 1, 3–11. [Google Scholar] [CrossRef]
- Zhan, Q.; Fan, S.; Bae, I.; Guillouf, C.; Liebermann, D.A.; O’Connor, P.M.; Fornace, A.J., Jr. Induction of bax by genotoxic stress in human cells correlates with normal p53 status and apoptosis. Oncogene 1994, 9, 3743–3751. [Google Scholar] [PubMed]
- Utrera, R.; Collavin, L.; Lazarević, D.; Delia, D.; Schneider, C. A novel p53-inducible gene coding for a microtubule-localized protein with G2-phase-specific expression. EMBO J. 1998, 17, 5015–5025. [Google Scholar] [CrossRef] [PubMed]
- Gudkov, A.V.; Gurova, K.V.; Komarova, E.A. Inflammation and p53: A tale of two stresses. Genes Cancer 2011, 2, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, A.; Sun, X.; Li, Y.; Zhang, Y.; Eckner, R.; Doi, T.S.; Takahashi, T.; Obata, Y.; Yoshioka, K.; Yamamoto, K. p300/CBP-dependent and -independent transcriptional interference between NF-κB RelA and p53. Biochem. Biophys. Res. Commun. 2000, 272, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Zhu, N.; Findley, H.W.; Woods, W.G.; Zhou, M. Identification and characterization of the IKKα promoter: Positive and negative regulation by ETS-1 and p53, respectively. J. Biol. Chem. 2004, 279, 52141–52149. [Google Scholar] [CrossRef] [PubMed]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. Activated p53 induces NF-κB DNA binding but suppresses its transcriptional activation. Biochem. Biophys. Res. Commun. 2008, 372, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.M.; Ernst, M.K.; Rice, N.R.; Vousden, K.H. Role of NF-κB in p53-mediated programmed cell death. Nature 2000, 404, 892–897. [Google Scholar] [CrossRef] [PubMed]
- Marine, J.C.; Francoz, S.; Maetens, M.; Wahl, G.; Toledo, F.; Lozano, G. Keeping p53 in check: Essential and synergistic functions of MDM2 and MDM4. Cell Death Differ. 2006, 13, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Parant, J.; Chavez-Reyes, A.; Little, N.A.; Yan, W.; Reinke, V.; Jochemsen, A.G.; Lozano, G. Rescue of embryonic lethality in MDM4-null mice by loss of Trp53 suggests a non-overlapping pathway with MDM2 to regulate p53. Nat. Genet. 2001, 29, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Migliorini, D.; Danovi, D.; Colombo, E.; Carbone, R.; Pelicci, P.G.; Marine, J.C. Hdmx recruitment into the nucleus by HDM2 is essential for its ability to regulate p53 stability and transactivation. J. Biol. Chem. 2002, 277, 7318–7323. [Google Scholar] [CrossRef] [PubMed]
- Finch, R.A.; Donoviel, D.B.; Potter, D.; Shi, M.; Fan, A.; Freed, D.D.; Wang, C.Y.; Zambrowicz, B.P.; Ramirez-Solis, R.; Sands, A.T.; et al. MDMX is a negative regulator of p53 activity in vivo. Cancer Res. 2002, 62, 3221–3225. [Google Scholar] [PubMed]
- Barak, Y.; Juven, T.; Haffner, R.; Oren, M. MDM2 expression is induced by wild type p53 activity. EMBO J. 1993, 12, 461–468. [Google Scholar] [PubMed]
- Wu, X.; Bayle, J.H.; Olson, D.; Levine, A.J. The p53-MDM-2 auto-regulatory feedback loop. Genes Dev. 1993, 7, 1126–1132. [Google Scholar] [CrossRef] [PubMed]
- Shvarts, A.; Steegenga, W.T.; Riteco, N.; van Laar, T.; Dekker, P.; Bazuine, M.; van Ham, R.C.; van der Houven van Oordt, W.; Hateboer, G.; van der Eb, A.J.; et al. MDMX: A novel p53-binding protein with some functional properties of MDM2. EMBO J. 1996, 15, 5349–5357. [Google Scholar] [PubMed]
- Linares, L.K.; Hengstermann, A.; Ciechanover, A.; Müller, S.; Scheffner, M. HDMX stimulates HDM2-mediated ubiquitination and degradation of p53. Proc. Natl. Acad. Sci. USA 2003, 100, 12009–12014. [Google Scholar] [CrossRef] [PubMed]
- Linke, K.; Mace, P.D.; Smith, C.A.; Vaux, D.L.; Silke, J.; Day, C.L. Structure of the MDM2/MDMX RING domain heterodimer reveals dimerization is required for their ubiquitylation in trans. Cell Death Differ. 2008, 15, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Taya, Y.; Nakagama, H. MDMX enhances p53 ubiquitination by altering the substrate preference of the MDM2 ubiquitin ligase. FEBS Lett. 2003, 583, 2710–2714. [Google Scholar] [CrossRef] [PubMed]
- Shmueli, A.; Oren, M. MDM2: p53’s lifesaver? Mol. Cell 2007, 25, 794–796. [Google Scholar] [CrossRef] [PubMed]
- Mancini, F.; Di Conza, G.; Pellegrino, M.; Rinaldo, C.; Prodosmo, A.; Giglio, S.; D’Agnano, I.; Florenzano, F.; Felicioni, L.; Buttitta, F.; et al. MDM4 (MDMX) localizes at the mitochondria and facilitates the p53-mediated intrinsic-apoptotic pathway. EMBO J. 2009, 28, 1926–1939. [Google Scholar] [CrossRef] [PubMed]
- Mancini, F.; Pieroni, L.; Monteleone, V.; Lucà, R.; Fici, L.; Luca, E.; Urbani, A.; Xiong, S.; Soddu, S.; Masetti, R.; et al. MDM4/HIPK2/p53 cytoplasmic assembly uncovers coordinated repression of molecules with anti-apoptotic activity during early DNA damage response. Oncogene 2016, 35, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Meek, D.W. Tumour suppression by p53: A role for the DNA damage response. Nat. Rev. Cancer 2009, 9, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Fontela, C.; Mandinova, A.; Aaronson, S.A.; Lee, S.W. Emerging roles of p53 and other tumour-suppressor genes in immune regulation. Nat. Rev. Immunol. 2016, 16, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Galluzzi, L.; Morselli, E.; Kepp, O.; Malik, S.A.; Kroemer, G. Autophagy regulation by p53. Curr. Opin. Cell Biol. 2010, 22, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Berkers, C.R.; Maddocks, O.D.; Cheung, E.C.; Mor, I.; Vousden, K.H. Metabolic regulation by p53 family members. Cell Metab. 2013, 18, 617–633. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Tomasini, R.; McKeon, F.D.; Mak, T.W.; Melino, G. The p53 family: Guardians of maternal reproduction. Nat. Rev. Mol. Cell Biol. 2011, 12, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Chen, X. Senescence regulation by the p53 protein family. Methods Mol. Biol. 2013, 965, 37–61. [Google Scholar] [PubMed]
- Insinga, A.; Cicalese, A.; Pelicci, P.G. DNA damage response in adult stem cells. Blood Cells Mol. Dis. 2014, 52, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; Evan, G.I. p53—A Jack of all trades but master of none. Nat. Rev. Cancer 2009, 9, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Takatori, H.; Kawashima, H.; Suzuki, K.; Nakajima, H. Role of p53 in systemic autoimmune diseases. Crit. Rev. Immunol. 2014, 34, 509–516. [Google Scholar] [PubMed]
- Khoo, K.H.; Verma, C.S.; Lane, D.P. Drugging the p53 pathway: Understanding the route to clinical efficacy. Nat. Rev. Drug Discov. 2014, 13, 217–236. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Cheok, C.F.; Verma, C.S.; Lane, D.P. Reactivation of p53: From peptides to small molecules. Trends Pharmacol. Sci. 2010, 32, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Danovi, D.; Meulmeester, E.; Pasini, D.; Migliorini, D.; Capra, M.; Frenk, R.; de Graaf, P.; Francoz, S.; Gasparini, P.; Gobbi, A.; et al. Amplification of MDMX (or MDM4) directly contributes to tumor formation by inhibiting p53 tumor suppressor activity. Mol. Cell. Biol. 2004, 24, 5835–5843. [Google Scholar] [CrossRef] [PubMed]
- Laurie, N.A.; Donovan, S.L.; Shih, C.S.; Zhang, J.; Mills, N.; Fuller, C.; Teunisse, A.; Lam, S.; Ramos, Y.; Mohan, A.; et al. Inactivation of the p53 pathway in retinoblastoma. Nature 2006, 444, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Riemenschneider, M.J.; Büschges, R.; Wolter, M.; Reifenberger, J.; Boström, J.; Kraus, J.A.; Schlegel, U.; Reifenberger, G. Amplification and overexpression of the MDM4 (MDMX) gene from 1q32 in a subset of malignant gliomas without TP53 mutation or MDM2 amplification. Cancer Res. 1999, 59, 6091–6096. [Google Scholar] [PubMed]
- Murray-Zmijewski, F.; Slee, E.A.; Lu, X. A complex barcode underlies the heterogeneous response of p53 to stress. Nat. Rev. Mol. Cell Biol. 2008, 9, 702–712. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M.; Cordon-Cardo, C.; Corn, P.G.; Meltzer, S.J.; Pohar, K.S.; Watkins, D.N.; Capella, G.; Peinado, M.A.; Matias-Guiu, X.; Prat, J.; et al. p14ARF silencing by promoter hypermethylation mediates abnormal intracellular localization of MDM2. Cancer Res. 2001, 61, 2816–2821. [Google Scholar] [PubMed]
- Sherr, C.J.; Weber, J.D. The ARF/p53 pathway. Curr. Opin. Genet. Dev. 2000, 10, 94–99. [Google Scholar] [CrossRef]
- Milner, J.; Medcalf, E.A. Cotranslation of activated mutant p53 with wild-type p53 protein into the mutant conformation. Cell 1991, 65, 765–774. [Google Scholar] [CrossRef]
- Sigal, A.; Rotter, V. Oncogenic mutations of the p53 tumor suppressor: The demons of the guardian of the genome. Cancer Res. 2000, 60, 6788–6793. [Google Scholar]
- Levine, A.J.; Wu, M.C.; Chang, A.; Silver, A.; Attiyeh, E.F.; Lin, J.; Epstein, C.B. The spectrum of mutations at the p53 locus. Evidence for tissue-specific mutagenesis, selection of mutant alleles, and a “gain of function” phenotype. Ann. N. Y. Acad. Sci. 1995, 768, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.J.; Rodrigues, C.M.; Moreira, R.; Santos, M.M. Chemical variations on the p53 reactivation theme. Pharmaceuticals 2016, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Issaeva, N.; Bozko, P.; Enge, M.; Protopopova, M.; Verhoef, L.G.; Masucci, M.; Pramanik, A.; Selivanova, G. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat. Med. 2004, 10, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Reed, D.; Shen, Y.; Shelat, A.A.; Arnold, L.A.; Ferreira, A.M.; Zhu, F.; Mills, N.; Smithson, D.C.; Regni, C.A.; Bashford, D.; et al. Identification and characterization of the first small molecule inhibitor of MDMX. J. Biol. Chem. 2010, 285, 10786–10796. [Google Scholar] [CrossRef] [PubMed]
- Wade, M.; Wahl, G.M. Targeting MDM2 and MDMX in cancer therapy: Better living through medicinal chemistry? Mol. Cancer Res. 2009, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, M.; Mancini, F.; Lucà, R.; Coletti, A.; Giacchè, N.; Manni, I.; Arisi, I.; Florenzano, F.; Teveroni, E.; Buttarelli, M.; et al. Targeting the MDM2/MDM4 interaction interface as a promising approach for p53 reactivation therapy. Cancer Res. 2015, 75, 4560–4572. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Nikolova, P.V.; Wong, K.B.; DeDecker, B.; Henckel, J.; Fersht, A.R. Mechanism of rescue of common p53 cancer mutations by second-site suppressor mutations. EMBO J. 2000, 19, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Puca, R.; Nardinocchi, L.; Porru, M.; Simon, A.J.; Rechavi, G.; Leonetti, C.; Givol, D.; D’Orazi, G. Restoring p53 active conformation by zinc increases the response of mutant p53 tumor cells to anticancer drugs. Cell Cycle 2011, 10, 1679–1689. [Google Scholar] [CrossRef] [PubMed]
- Petitjean, A.; Mathe, E.; Kato, S.; Ishioka, C.; Tavtigian, S.V.; Hainaut, P.; Olivier, M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: Lessons from recent developments in the IARC TP53 database. Hum. Mutat. 2007, 28, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.K.; Chan, F.S.; O’Connor, D.J.; Mittnacht, S.; Zhong, S.; Lu, X. RB regulates the stability and the apoptotic function of p53 via MDM2. Mol. Cell 1999, 3, 181–193. [Google Scholar] [CrossRef]
- Saha, S.; Bhattacharjee, P.; Guha, D.; Kajal, K.; Khan, P.; Chakraborty, S.; Mukherjee, S.; Paul, S.; Manchanda, R.; Khurana, A.; et al. Sulphur alters NFκB-p300 cross-talk in favor of p53–p300 to induce apoptosis in non-small cell lung carcinoma. Int. J. Oncol. 2015, 47, 573–582. [Google Scholar] [PubMed]
- Kawashima, H.; Takatori, H.; Suzuki, K.; Iwata, A.; Yokota, M.; Suto, A.; Minamino, T.; Hirose, K.; Nakajima, H. Tumor suppressor p53 inhibits systemic autoimmune diseases by inducing regulatory T cells. J. Immunol. 2013, 191, 3614–3623. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Lim, M.A.; Cho, M.L.; Ryu, J.G.; Moon, Y.M.; Jhun, J.Y.; Byun, J.K.; Kim, E.K.; Hwang, S.Y.; Ju, J.H.; et al. p53 controls autoimmune arthritis via STAT-mediated regulation of the Th17 cell/Treg cell balance in mice. Arthritis Rheum. 2013, 65, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Thomasova, D.; Mulay, S.R.; Bruns, H.; Anders, H.J. p53-independent roles of MDM2 in NF-κB signaling: Implications for cancer therapy, wound healing, and autoimmune diseases. Neoplasia 2012, 14, 1097–1101. [Google Scholar] [CrossRef] [PubMed]
- Simelyte, E.; Rosengren, S.; Boyle, D.L.; Corr, M.; Green, D.R.; Firestein, G.S. Regulation of arthritis by p53: Critical role of adaptive immunity. Arthritis Rheum. 2005, 52, 1876–1884. [Google Scholar] [CrossRef] [PubMed]
- Leech, M.; Xue, J.R.; Dacumos, A.; Hall, P.; Santos, L.; Yang, Y.; Li, M.; Kitching, A.R.; Morand, E.F. The tumour suppressor gene p53 modulates the severity of antigen-induced arthritis and the systemic immune response. Clin. Exp. Immunol. 2008, 152, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Okuda, Y.; Okuda, M.; Bernard, C.C. Regulatory role of p53 in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2003, 135, 29–37. [Google Scholar] [CrossRef]
- Zheng, S.J.; Lamhamedi-Cherradi, S.E.; Wang, P.; Xu, L.; Chen, Y.H. Tumor suppressor p53 inhibits autoimmune inflammation and macrophage function. Diabetes 2005, 54, 1423–1428. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zheng, M.; Kibe, R.; Huang, Y.; Marrero, L.; Warren, S.; Zieske, A.W.; Iwakuma, T.; Kolls, J.K.; Cui, Y. Trp53 negatively regulates autoimmunity via the STAT3-Th17 axis. FASEB J. 2011, 25, 2387–2398. [Google Scholar] [CrossRef] [PubMed]
- Fierabracci, A. Recent insights into the role and molecular mechanisms of the autoimmune regulator (AIRE) gene in autoimmunity. Autoimmun. Rev. 2011, 10, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Cools, N.; van Tendeloo, V.F.; Smits, E.L.; Lenjou, M.; Nijs, G.; van Bockstaele, D.R.; Berneman, Z.N.; Ponsaerts, P. Immunosuppression induced by immature dendritic cells is mediated by TGF-β/IL-10 double-positive CD4+ regulatory T cells. J. Cell Mol. Med. 2008, 2, 690–700. [Google Scholar] [CrossRef] [PubMed]
- Collison, L.W.; Workman, C.J.; Kuo, T.T.; Boyd, K.; Wang, Y.; Vignali, K.M.; Cross, R.; Sehy, D.; Blumberg, R.S.; Vignali, D.A. The inhibitory cytokine IL-35 contributes to regulatory T cell function. Nature 2007, 450, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Pandiyan, P.; Zheng, L.; Ishihara, S.; Reed, J.; Lenardo, M.J. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat. Immunol. 2007, 8, 1353–1362. [Google Scholar] [CrossRef] [PubMed]
- Onishi, Y.; Fehervari, Z.; Yamaguchi, T.; Sakaguchi, S. Foxp3+ natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc. Natl. Acad. Sci. USA 2008, 105, 10113–10118. [Google Scholar] [CrossRef] [PubMed]
- Krammer, P.H.; Arnold, R.; Lavrik, I.N. Life and death in peripheral T cells. Nat. Rev. Immunol. 2007, 7, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Droin, N.; Pinkoski, M. Activation-induced cell death in T cells. Immunol. Rev. 2003, 193, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Lenardo, M.J. Interleukin-2 programs mouse alpha beta T lymphocytes for apoptosis. Nature 1991, 353, 858–861. [Google Scholar] [CrossRef] [PubMed]
- Boehme, S.A.; Lenardo, M.J. TCR-mediated death of mature T lymphocytes occurs in the absence of p53. J. Immunol. 1996, 156, 4075–4078. [Google Scholar] [PubMed]
- Singh, N.; Yamamoto, M.; Takami, M.; Seki, Y.; Takezaki, M.; Mellor, A.L.; Iwashima, M. CD4(+)CD25(+) regulatory T cells resist a novel form of CD28- and Fas-dependent p53-induced T cell apoptosis. J. Immunol. 2010, 184, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Moon, K.D.; Vacchio, M.S.; Hathcock, K.S.; Hodes, R.J. Downmodulation of tumor suppressor p53 by T cell receptor signaling is critical for antigen-specific CD4+ T cell responses. Immunity 2014, 40, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Minton, K. p53 controls the crowd. Nat. Rev. Immunol. 2014, 14, 358–359. [Google Scholar] [CrossRef] [PubMed]
- Lowe, J.M.; Menendez, D.; Bushel, P.R.; Shatz, M.; Kirk, E.L.; Troester, M.A.; Garantziotis, S.; Fessler, M.B.; Resnick, M.A. p53 and NF-κB coregulate proinflammatory gene responses in human macrophages. Cancer Res. 2014, 74, 2182–2192. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Fontela, C.; Macip, S.; Martinez-Sobrido, L.; Brown, L.; Ashour, J.; Garcia-Sastre, A.; Lee, S.W.; Aaronson, S.A. Transcriptional role of p53 in interferon-medaited antiviral immunity. J. Exp. Med. 2008, 205, 1929–1938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taura, M.; Eguma, A.; Suico, M.A.; Shuto, T.; Koga, T.; Komatsu, K.; Komune, T.; Sato, T.; Saya, H.; Li, J.-D.; et al. p53 regulates Toll-like receptor 3 expression and function in huamn epithelial cell lines. Mol. Cell. Biol. 2008, 28, 6557–6567. [Google Scholar] [CrossRef] [PubMed]
- Youlyouz-Marfak, I.; Gachard, N.; le Clorennec, C.; Najjar, I.; Baran-Marszak, F.; Reminieras, L.; May, E.; Bornkamm, G.W.; Fagard, R.; Feuillard, J. Identification of a novel p53-dependent activation pathway of STAT1 by antitumour genotoxic agents. Cell Death Differ. 2008, 15, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Dirisina, R.; Katzman, R.B.; Goretsky, T.; Managlia, E.; Mittal, N.; Williams, D.B.; Qiu, W.; Yu, J.; Chandel, N.S.; Zhang, L.; et al. p53 and PUMA independently regulate apoptosis of intestinal epithelial cells in patients and mice with colitis. Gastroenterology 2011, 141, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Herkel, J.; Kam, N.; Erez, N.; Mimran, A.; Heifetz, A.; Eisenstein, M.; Rotter, V.; Cohen, I.R. Monoclonal antibody to a DNA-binding domain of p53 mimics charge structure of DNA: Anti-idiotypes to the anti-p53 antibody are anti-DNA. Eur. J. Immunol. 2004, 34, 3623–3632. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Ogawa, F.; Muroi, E.; Komura, K.; Takenaka, M.; Hasegawa, M.; Fujimoto, M.; Sato, S. Anti-p53 autoantibody in systemic sclerosis: Association with limited cutaneous systemic sclerosis. J. Rheumatol. 2008, 35, 451–457. [Google Scholar] [PubMed]
- Kovacs, B.; Patel, A.; Hershey, J.N.; Dennis, G.J.; Kirschfink, M.; Tsokos, G.C. Antibodies against p53 in sera from patients with systemic lupus erythematosus and other rheumatic diseases. Arthritis Rheum. 1997, 40, 980–985. [Google Scholar] [CrossRef] [PubMed]
- Shiau, M.Y.; Kuo, T.M.; Tsay, G.J.; Chiou, H.L.; Lee, Y.L.; Chang, Y.H. Absence of anti-p53 antibodies in Chinese patients with rheumatoid arthritis and systemic lupus erythematosus: Comment on the concise communication by Kovacs et al. Arthritis Rheum. 2002, 46, 276–277. [Google Scholar] [CrossRef]
- Chauhan, R.; Handai, R.; Das, T.P.; Pati, U. Over-expression of TATA binding protein (TBP) and p53 and autoantibodies to these antigens are features of systemic sclerosis, systemic lupus erythematosus and overlap syndromes. Clin. Exp. Immunol. 2004, 136, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Mimura, Y.; Yazawa, N.; Tamaki, Z.; Ashida, R.; Jinnin, M.; Asano, Y.; Tada, Y.; Kubo, M.; Ihn, H.; Tamaki, K. Anti-p53 antibodies in patients with dermatomyositis/polymyositis. Clin. Rheumatol. 2007, 26, 1328–1331. [Google Scholar] [CrossRef] [PubMed]
- Herkel, J.; Mimran, A.; Erez, N.; Kam, N.; Lohse, A.W.; Märker-Hermann, E.; Rotter, V.; Cohen, I.R. Autoimmunity to the p53 protein is a feature of systemic lupus erythematosus (SLE) related to anti-DNA antibodies. J. Autoimmun. 2001, 17, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, H.M.; Kromminga, A.; Flammann, H.T.; Frey, M.; Layer, P.; Arndt, R. p53 autoantibodies in patients with autoimmune diseases: A quantitative approach. Autoimmunity 1999, 31, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Achiron, A.; Feldman, A.; Magalashvili, D.; Dolev, M.; Gurevich, M. Suppressed RNA-polymerase 1 pathway is associated with benign multiple sclerosis. PLoS ONE 2012, 7, e4687. [Google Scholar] [CrossRef] [PubMed]
- Firestein, G.S.; Echeverri, F.; Yeo, M.; Zvaifler, N.J.; Green, D.R. Somatic mutations in the p53 tumor suppressor gene in rheumatoid arthritis synovium. Proc. Natl. Acad. Sci. USA 1997, 94, 10895–10900. [Google Scholar] [CrossRef] [PubMed]
- Yamanishi, Y.; Boyle, D.L.; Rosengren, S.; Green, D.R.; Zvaifler, N.J.; Firestein, G.S. Regional analysis of p53 mutations in rheumatoid arthritis synovium. Proc. Natl. Acad. Sci. USA 2002, 99, 10025–10030. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Boyle, D.L.; Shi, Y.; Green, D.R.; Firestein, G.S. Dominant-negative p53 mutations in rheumatoid arthritis. Arthritis Rheum. 1999, 42, 1088–1092. [Google Scholar] [CrossRef]
- Volodko, N.; Salla, M.; Eksteen, B.; Fedorak, R.N.; Huynh, H.Q.; Baksh, S. TP53 codon 72 Arg/Arg polymorphism is associated with a higher risk for inflammatory bowel disease development. World J. Gastroenterol. 2015, 21, 10358–10366. [Google Scholar] [CrossRef] [PubMed]
- Egiziano, G.; Bernatsky, S.; Shah, A.A. Cancer and autoimmunity: Harnessing longitudinal cohorts to probe the link. Best Pract. Res. Clin. Rheumatol. 2016, 30, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Giani, C.; Fierabracci, P.; Bonacci, R.; Gigliotti, A.; Campani, D.; de Negri, F.; Cecchetti, D.; Martino, E.; Pinchera, A. Relationship between breast cancer and thyroid disease: Relevance of autoimmune thyroid disorders in breast malignancy. J. Clin. Endocrinol. Metab. 1996, 81, 990–994. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.A.; Casciola-Rosen, L. Cancer and scleroderma: A paraneoplastic disease with implications for malignancy screening. Curr. Opin. Rheumatol. 2015, 27, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.J.; Jin, D.H.; Hong, S.W.; Kim, J.E.; Shin, J.S.; Kim, D.; Cho, B.J.; Hwang, Y.I.; Kang, J.S.; Lee, W.J. Foxp3 expression in p53-dependent DNA damage responses. J. Biol. Chem. 2010, 285, 7995–8002. [Google Scholar] [CrossRef] [PubMed]
- Katchman, B.A.; Barderas, R.; Alam, R.; Chowell, D.; Field, M.S.; Esserman, L.J.; Wallstrom, G.; LaBaer, J.; Cramer, D.W.; Hollingsworth, M.A.; et al. Proteomic mapping of p53 immunogenicity in pancreatic, ovarian, and breast cancers. Proteom. Clin. Appl. 2016, 10, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Garziera, M.; Montico, M.; Bidoli, E.; Scalone, S.; Sorio, R.; Giorda, G.; Lucia, E.; Toffoli, G. Prognostic value of serum antibody immunity to p53 oncogenic protein in ovarian cancer: A systematic review and a meta-analysis. PLoS ONE 2015, 10, e0140351. [Google Scholar] [CrossRef] [PubMed]
- Chai, Y.; Peng, B.; Dai, L.; Qian, W.; Zhang, Y.; Zhang, J.Y. Autoantibodies response to MDM2 and p53 in the immunodiagnosis of esophageal squamous cell carcinoma. Scand. J. Immunol. 2014, 80, 362–368. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fierabracci, A.; Pellegrino, M. The Double Role of p53 in Cancer and Autoimmunity and Its Potential as Therapeutic Target. Int. J. Mol. Sci. 2016, 17, 1975. https://doi.org/10.3390/ijms17121975
Fierabracci A, Pellegrino M. The Double Role of p53 in Cancer and Autoimmunity and Its Potential as Therapeutic Target. International Journal of Molecular Sciences. 2016; 17(12):1975. https://doi.org/10.3390/ijms17121975
Chicago/Turabian StyleFierabracci, Alessandra, and Marsha Pellegrino. 2016. "The Double Role of p53 in Cancer and Autoimmunity and Its Potential as Therapeutic Target" International Journal of Molecular Sciences 17, no. 12: 1975. https://doi.org/10.3390/ijms17121975