
TP53/MicroRNA Interplay in Hepatocellular Carcinoma


Abstract
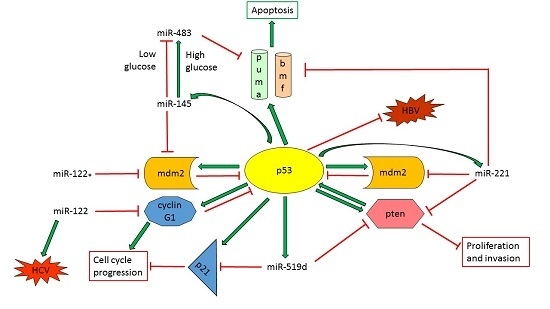
:
1. Introduction
2. TP53 and MicroRNAs
2.1. TP53-Effector MicroRNAs in Hepatocellular Carcinoma (HCC)
2.1.1. miR-34a Paradigm in HCC: Tumor Suppressor or Oncogene?
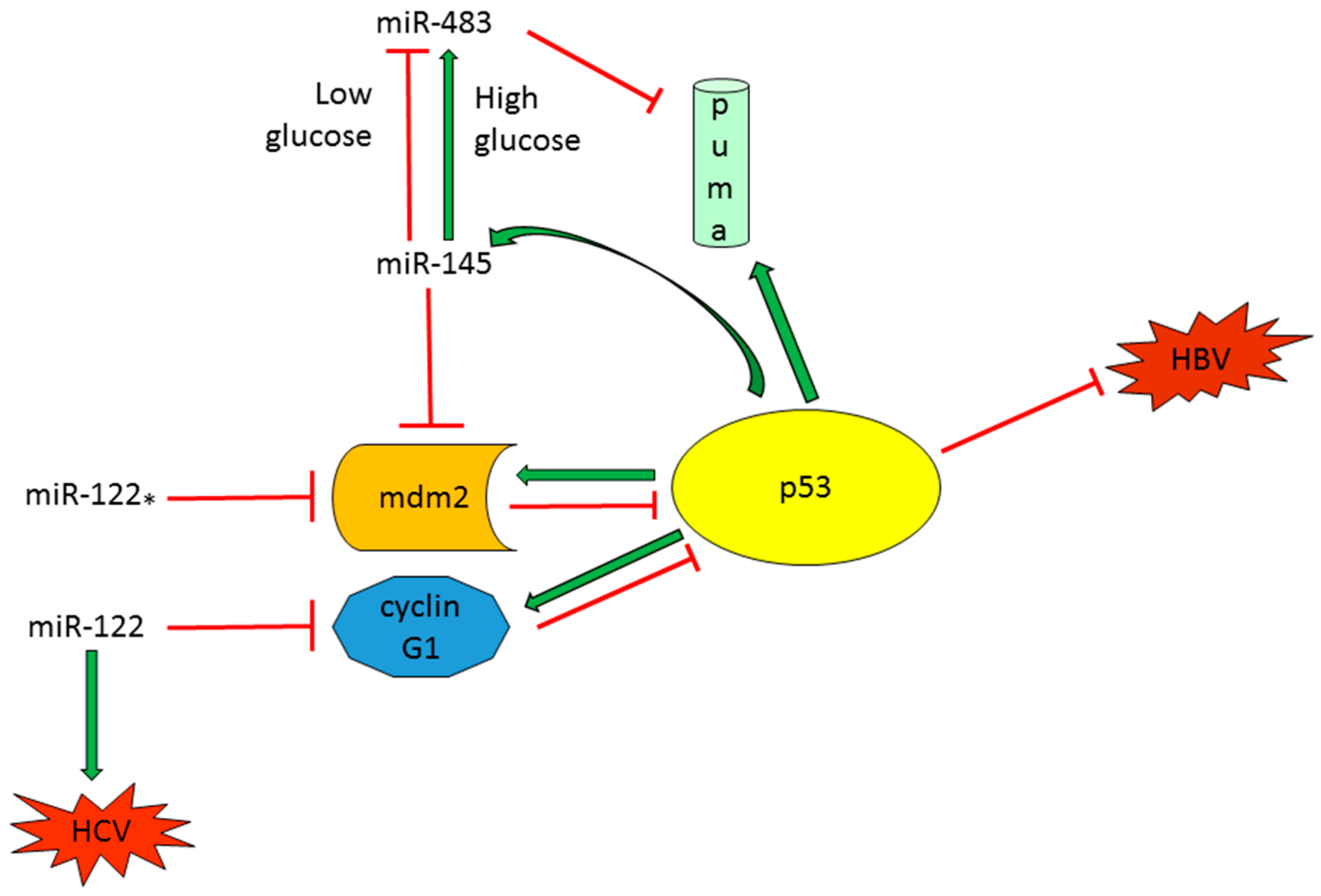
2.1.2. TP53-Regulated miRNAs with a Role in Proliferation, Tumor Growth and Apoptosis
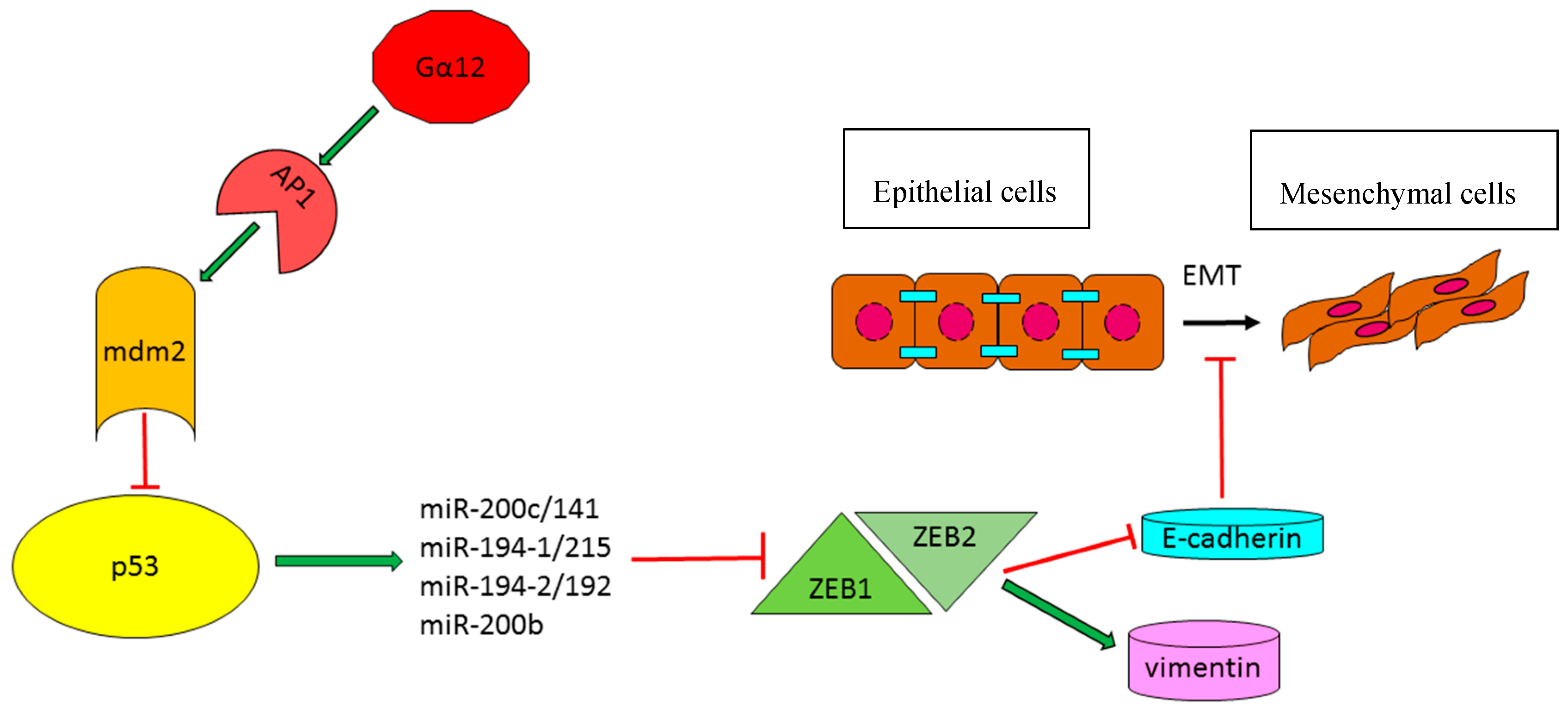
2.1.3. TP53-Effector miRNAs with a Role in the Modulation of Epithelial–Mesenchymal Transition
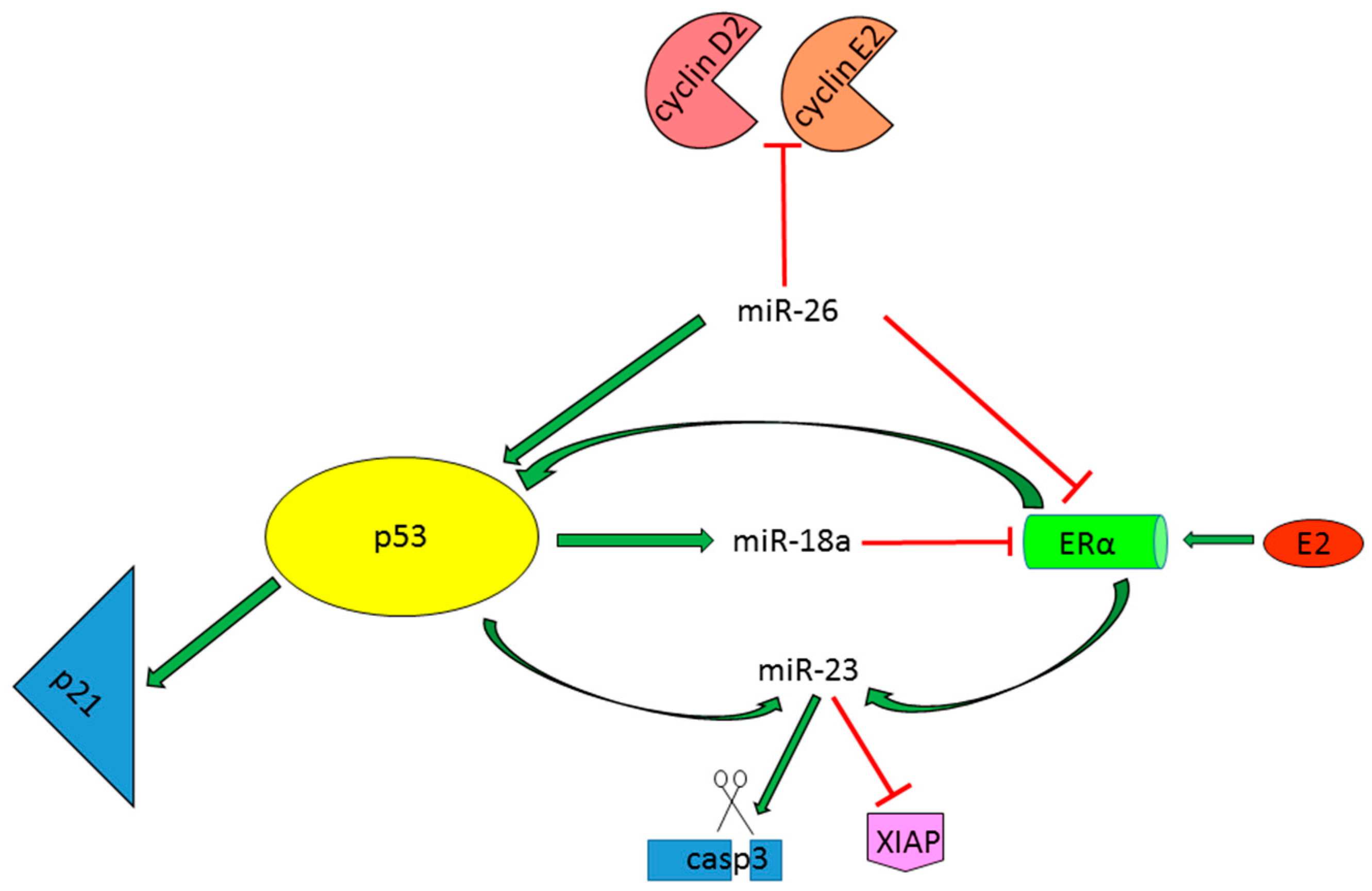
2.1.4. miRNAs as p53-Dependent Players in the Regulation of Estrogen Protective Activity
2.2. TP53-Modulator miRNAs in HCC
2.2.1. Tumor Suppressor miRNAs Regulate p53 Activity in HCC
2.2.2. Oncogenic miRNAs Involved in p53 Regulation in HCC
3. DNA Hypomethylation Contributes to the Maintenance of TP53/miRNA Loops in HCC
4. TP53-Effector and Modulator miRNAs as Therapeutic Targets in HCC
5. Conclusions and Clinical Challenges
Acknowledgments
Author Contributions
Conflicts of Interest
References
- GBD 2013 Mortality and Causes of Death Collaborators. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: A systematic analysis for the global burden of disease study 2013. Lancet 2015, 385, 117–171. [Google Scholar]
- Forner, A.; Llovet, J.M.; Bruix, J. Hepatocellular carcinoma. Lancet 2012, 379, 1245–1255. [Google Scholar] [CrossRef]
- Laurent-Puig, P.; Legoix, P.; Bluteau, O.; Belghiti, J.; Franco, D.; Binot, F.; Monges, G.; Thomas, G.; Bioulac-Sage, P.; Zucman-Rossi, J. Genetic alterations associated with hepatocellular carcinomas define distinct pathways of hepatocarcinogenesis. Gastroenterology 2001, 120, 1763–1773. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2016, 2, 16018. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Hu, W.; Feng, Z. The p53 pathway: What questions remain to be explored? Cell Death Differ. 2006, 13, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Jeon, K.; Lee, J.T.; Kim, S.; Kim, V.N. MicroRNA maturation: Stepwise processing and subcellular localization. EMBO J. 2002, 21, 4663–4670. [Google Scholar] [CrossRef] [PubMed]
- Volinia, S.; Calin, G.A.; Liu, C.G.; Ambs, S.; Cimmino, A.; Petrocca, F.; Visone, R.; Iorio, M.; Roldo, C.; Ferracin, M.; et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. USA 2006, 103, 2257–2261. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Croce, C.M. MicroRNA-Cancer connection: The beginning of a new tale. Cancer Res. 2006, 66, 7390–7394. [Google Scholar] [CrossRef] [PubMed]
- Hermeking, H. MicroRNAs in the p53 network: Micromanagement of tumour suppression. Nat. Rev. Cancer 2012, 12, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Callegari, E.; Gramantieri, L.; Domenicali, M.; D’Abundo, L.; Sabbioni, S.; Negrini, M. MicroRNAs in liver cancer: A model for investigating pathogenesis and novel therapeutic approaches. Cell Death Differ. 2015, 22, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.I.; Yamagata, K.; Sugimoto, K.; Iwamoto, T.; Kato, S.; Miyazono, K. Modulation of microRNA processing by p53. Nature 2009, 460, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, W.; Ding, R.; Xiong, L.; Dou, R.; Zhang, Y.; Guo, Z. Comprehensive expression profiling and functional network analysis of p53-regulated microRNAs in HEPG2 cells treated with doxorubicin. PLoS ONE 2016, 11, e0149227. [Google Scholar] [CrossRef] [PubMed]
- He, L.; He, X.; Lim, L.P.; de Stanchina, E.; Xuan, Z.; Liang, Y.; Xue, W.; Zender, L.; Magnus, J.; Ridzon, D.; et al. A microRNA component of the p53 tumour suppressor network. Nature 2007, 447, 1130–1134. [Google Scholar] [CrossRef] [PubMed]
- Raver-Shapira, N.; Marciano, E.; Meiri, E.; Spector, Y.; Rosenfeld, N.; Moskovits, N.; Bentwich, Z.; Oren, M. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol. Cell 2007, 26, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Son, M.S.; Jang, M.J.; Jeon, Y.J.; Kim, W.H.; Kwon, C.I.; Ko, K.H.; Park, P.W.; Hong, S.P.; Rim, K.S.; Kwon, S.W.; et al. Promoter polymorphisms of pri-miR-34b/c are associated with hepatocellular carcinoma. Gene 2013, 524, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Fu, H.; Tie, Y.; Hu, Z.; Kong, W.; Wu, Y.; Zheng, X. miR-34a inhibits migration and invasion by down-regulation of C-met expression in human hepatocellular carcinoma cells. Cancer Lett. 2009, 275, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Tryndyak, V.P.; Ross, S.A.; Beland, F.A.; Pogribny, I.P. Down-regulation of the microRNAs miR-34a, miR-127, and miR-200b in rat liver during hepatocarcinogenesis induced by a methyl-deficient diet. Mol. Carcinog. 2009, 48, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Pineau, P.; Volinia, S.; McJunkin, K.; Marchio, A.; Battiston, C.; Terris, B.; Mazzaferro, V.; Lowe, S.W.; Croce, C.M.; Dejean, A. miR-221 overexpression contributes to liver tumorigenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Gougelet, A.; Sartor, C.; Bachelot, L.; Godard, C.; Marchiol, C.; Renault, G.; Tores, F.; Nitschke, P.; Cavard, C.; Terris, B.; et al. Antitumour activity of an inhibitor of miR-34a in liver cancer with beta-catenin-mutations. Gut 2016, 65, 1024–1034. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.I.; Matoso, A.; Corney, D.C.; Flesken-Nikitin, A.; Korner, S.; Wang, W.; Boccaccio, C.; Thorgeirsson, S.S.; Comoglio, P.M.; Hermeking, H.; et al. Wild-Type p53 controls cell motility and invasion by dual regulation of met expression. Proc. Natl. Acad. Sci. USA 2011, 108, 14240–14245. [Google Scholar] [CrossRef] [PubMed]
- Dang, Y.; Luo, D.; Rong, M.; Chen, G. Underexpression of miR-34a in hepatocellular carcinoma and its contribution towards enhancement of proliferating inhibitory effects of agents targeting C-met. PLoS ONE 2013, 8, e61054. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Li, Q.J.; Feng, Y.; Zhang, Y.; Markowitz, G.J.; Ning, S.; Deng, Y.; Zhao, J.; Jiang, S.; Yuan, Y.; et al. TGF-β-miR-34a-CCL22 signaling-induced Treg cell recruitment promotes venous metastases of hbv-positive hepatocellular carcinoma. Cancer Cell 2012, 22, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.I.; Katsura, A.; Matsuyama, H.; Miyazono, K. MicroRNA regulons in tumor microenvironment. Oncogene 2015, 34, 3085–3094. [Google Scholar] [CrossRef] [PubMed]
- Bi, Q.; Tang, S.; Xia, L.; Du, R.; Fan, R.; Gao, L.; Jin, J.; Liang, S.; Chen, Z.; Xu, G.; et al. Ectopic expression of miR-125a inhibits the proliferation and metastasis of hepatocellular carcinoma by targeting mmp11 and VEGF. PLoS ONE 2012, 7, e40169. [Google Scholar] [CrossRef] [PubMed]
- Le, M.T.; Teh, C.; Shyh-Chang, N.; Xie, H.; Zhou, B.; Korzh, V.; Lodish, H.F.; Lim, B. MicroRNA-125b is a novel negative regulator of p53. Genes Dev. 2009, 23, 862–876. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Noh, J.H.; Jung, K.H.; Eun, J.W.; Bae, H.J.; Kim, M.G.; Chang, Y.G.; Shen, Q.; Park, W.S.; Lee, J.Y.; et al. Sirtuin7 oncogenic potential in human hepatocellular carcinoma and its regulation by the tumor suppressors miR-125a-5p and miR-125b. Hepatology 2013, 57, 1055–1067. [Google Scholar] [CrossRef] [PubMed]
- Pollutri, D.; Gramantieri, L.; Bolondi, L.; Fornari, F. miR-30e/p53 positive feedback loop in hepatocellular carcinoma. MCR 2017. in preparation. [Google Scholar]
- Kim, T.; Veronese, A.; Pichiorri, F.; Lee, T.J.; Jeon, Y.J.; Volinia, S.; Pineau, P.; Marchio, A.; Palatini, J.; Suh, S.S.; et al. P53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J. Exp. Med. 2011, 208, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.M.; Lee, W.H.; Lee, C.G.; An, J.; Kim, E.S.; Kim, S.H.; Lee, S.K.; Lee, C.H.; Dhanasekaran, D.N.; Moon, A.; et al. Gα12 GEP oncogene deregulation of p53-responsive microRNAs promotes epithelial-mesenchymal transition of hepatocellular carcinoma. Oncogene 2015, 34, 2910–2921. [Google Scholar] [CrossRef] [PubMed]
- Fornari, F.; Gramantieri, L.; Giovannini, C.; Veronese, A.; Ferracin, M.; Sabbioni, S.; Calin, G.A.; Grazi, G.L.; Croce, C.M.; Tavolari, S.; et al. miR-122/cyclin G1 interaction modulates p53 activity and affects doxorubicin sensitivity of human hepatocarcinoma cells. Cancer Res. 2009, 69, 5761–5767. [Google Scholar] [CrossRef] [PubMed]
- Bosch, F.X.; Ribes, J.; Diaz, M.; Cleries, R. Primary liver cancer: Worldwide incidence and trends. Gastroenterology 2004, 127, S5–S16. [Google Scholar] [CrossRef] [PubMed]
- Naugler, W.E.; Sakurai, T.; Kim, S.; Maeda, S.; Kim, K.; Elsharkawy, A.M.; Karin, M. Gender disparity in liver cancer due to sex differences in MYD88-dependent IL-6 production. Science 2007, 317, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Shi, J.; Budhu, A.; Yu, Z.; Forgues, M.; Roessler, S.; Ambs, S.; Chen, Y.; Meltzer, P.S.; Croce, C.M.; et al. MicroRNA expression, survival, and response to interferon in liver cancer. N. Engl. J. Med. 2009, 361, 1437–1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kota, J.; Chivukula, R.R.; O’Donnell, K.A.; Wentzel, E.A.; Montgomery, C.L.; Hwang, H.W.; Chang, T.C.; Vivekanandan, P.; Torbenson, M.; Clark, K.R.; et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell 2009, 137, 1005–1017. [Google Scholar] [CrossRef] [PubMed]
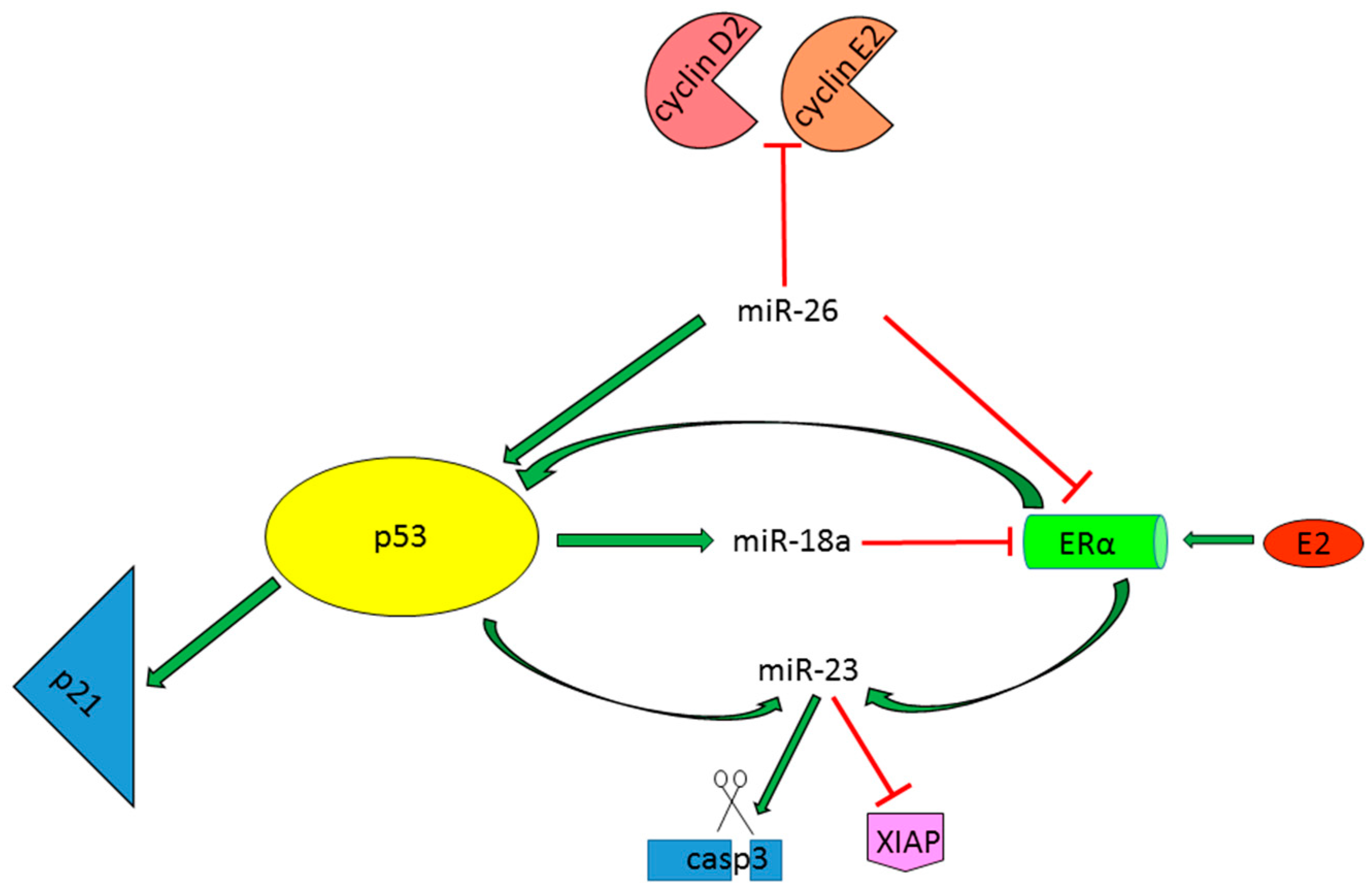
- Chen, L.; Zheng, J.; Zhang, Y.; Yang, L.; Wang, J.; Ni, J.; Cui, D.; Yu, C.; Cai, Z. Tumor-Specific expression of microRNA-26a suppresses human hepatocellular carcinoma growth via cyclin-dependent and -independent pathways. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 1521–1528. [Google Scholar] [CrossRef] [PubMed]
- Li, C.L.; Yeh, K.H.; Liu, W.H.; Chen, C.L.; Chen, D.S.; Chen, P.J.; Yeh, S.H. Elevated p53 promotes the processing of miR-18a to decrease estrogen receptor-α in female hepatocellular carcinoma. Int. J. Cancer 2015, 136, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.Y.; Wong, D.K.; Seto, W.K.; Lai, C.L.; Yuen, M.F. Estradiol induces apoptosis via activation of miRNA-23a and p53: Implication for gender difference in liver cancer development. Oncotarget 2015, 6, 34941–34952. [Google Scholar] [PubMed]
- Gramantieri, L.; Ferracin, M.; Fornari, F.; Veronese, A.; Sabbioni, S.; Liu, C.G.; Calin, G.A.; Giovannini, C.; Ferrazzi, E.; Grazi, G.L.; et al. Cyclin G1 is a target of miR-122a, a microRNA frequently down-regulated in human hepatocellular carcinoma. Cancer Res. 2007, 67, 6092–6099. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Yasuda, T.; Saigo, K.; Urashima, T.; Toyoda, H.; Okanoue, T.; Shimotohno, K. Comprehensive analysis of microRNA expression patterns in hepatocellular carcinoma and non-tumorous tissues. Oncogene 2006, 25, 2537–2545. [Google Scholar] [CrossRef] [PubMed]
- Petrelli, A.; Perra, A.; Cora, D.; Sulas, P.; Menegon, S.; Manca, C.; Migliore, C.; Kowalik, M.A.; Ledda-Columbano, G.M.; Giordano, S.; et al. MicroRNA/gene profiling unveils early molecular changes and nuclear factor erythroid related factor 2 (Nrf2) activation in a rat model recapitulating human hepatocellular carcinoma (HCC). Hepatology 2014, 59, 228–241. [Google Scholar] [CrossRef] [PubMed]
- Kutay, H.; Bai, S.; Datta, J.; Motiwala, T.; Pogribny, I.; Frankel, W.; Jacob, S.T.; Ghoshal, K. Downregulation of miR-122 in the rodent and human hepatocellular carcinomas. J. Cell. Biochem. 2006, 99, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Budhu, A.; Jia, H.L.; Forgues, M.; Liu, C.G.; Goldstein, D.; Lam, A.; Zanetti, K.A.; Ye, Q.H.; Qin, L.X.; Croce, C.M.; et al. Identification of metastasis-related microRNAs in hepatocellular carcinoma. Hepatology 2008, 47, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Li, H.; Jensen, M.R.; Zhang, T.; Taya, Y.; Thorgeirsson, S.S.; Prives, C. Cyclin G recruits PP2A to dephosphorylate MDM2. Mol. Cell 2002, 9, 761–771. [Google Scholar] [CrossRef]
- Wang, S.; Qiu, L.; Yan, X.; Jin, W.; Wang, Y.; Chen, L.; Wu, E.; Ye, X.; Gao, G.F.; Wang, F.; et al. Loss of microRNA 122 expression in patients with hepatitis b enhances hepatitis B virus replication through cyclin G(1)-modulated p53 activity. Hepatology 2012, 55, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Krutzfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of microRNAs in vivo with “antagomirs”. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Janssen, H.L.; Kauppinen, S.; Hodges, M.R. Hcv infection and miravirsen. N. Engl. J. Med. 2013, 369, 878. [Google Scholar] [PubMed]
- Janssen, H.L.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; van der Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatment of HCV infection by targeting microRNA. N. Engl. J. Med. 2013, 368, 1685–1694. [Google Scholar] [CrossRef] [PubMed]
- Simerzin, A.; Zorde-Khvalevsky, E.; Rivkin, M.; Adar, R.; Zucman-Rossi, J.; Couchy, G.; Roskams, T.; Govaere, O.; Oren, M.; Giladi, H.; et al. The liver-specific microRNA-122*, the complementary strand of microRNA-122, acts as a tumor suppressor by modulating the p53/mouse double minute 2 homolog circuitry. Hepatology 2016, 64, 1623–1636. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.I.; Katsura, A.; Yasuda, T.; Ueno, T.; Mano, H.; Sugimoto, K.; Miyazono, K. Small-RNA asymmetry is directly driven by mammalian argonautes. Nat. Struct. Mol. Biol. 2015, 22, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Bandres, E.; Cubedo, E.; Agirre, X.; Malumbres, R.; Zarate, R.; Ramirez, N.; Abajo, A.; Navarro, A.; Moreno, I.; Monzo, M.; et al. Identification by real-time PCR of 13 mature microRNAs differentially expressed in colorectal cancer and non-tumoral tissues. Mol. Cancer 2006, 5, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iorio, M.V.; Ferracin, M.; Liu, C.G.; Veronese, A.; Spizzo, R.; Sabbioni, S.; Magri, E.; Pedriali, M.; Fabbri, M.; Campiglio, M.; et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005, 65, 7065–7070. [Google Scholar] [CrossRef] [PubMed]
- Iorio, M.V.; Visone, R.; di Leva, G.; Donati, V.; Petrocca, F.; Casalini, P.; Taccioli, C.; Volinia, S.; Liu, C.G.; Alder, H.; et al. MicroRNA signatures in human ovarian cancer. Cancer Res. 2007, 67, 8699–8707. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sempere, L.F.; Galimberti, F.; Freemantle, S.J.; Black, C.; Dragnev, K.H.; Ma, Y.; Fiering, S.; Memoli, V.; Li, H.; et al. Uncovering growth-suppressive microRNAs in lung cancer. Clin. Cancer Res. 2009, 15, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Ozen, M.; Creighton, C.J.; Ozdemir, M.; Ittmann, M. Widespread deregulation of microRNA expression in human prostate cancer. Oncogene 2008, 27, 1788–1793. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sun, Q.; Zhang, Z.; Ge, S.; Han, Z.G.; Chen, W.T. Loss of microRNA-143/145 disturbs cellular growth and apoptosis of human epithelial cancers by impairing the MDM2-p53 feedback loop. Oncogene 2013, 32, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, M.; Zhu, S.; Wu, F.; Wu, H.; Walia, V.; Kumar, S.; Elble, R.; Watabe, K.; Mo, Y.Y. P53 represses C-MYC through induction of the tumor suppressor miR-145. Proc. Natl. Acad. Sci. USA 2009, 106, 3207–3212. [Google Scholar] [CrossRef] [PubMed]
- Spizzo, R.; Nicoloso, M.S.; Lupini, L.; Lu, Y.; Fogarty, J.; Rossi, S.; Zagatti, B.; Fabbri, M.; Veronese, A.; Liu, X.; et al. miR-145 participates with TP53 in a death-promoting regulatory loop and targets estrogen receptor-alpha in human breast cancer cells. Cell Death Differ. 2010, 17, 246–254. [Google Scholar] [CrossRef] [PubMed]
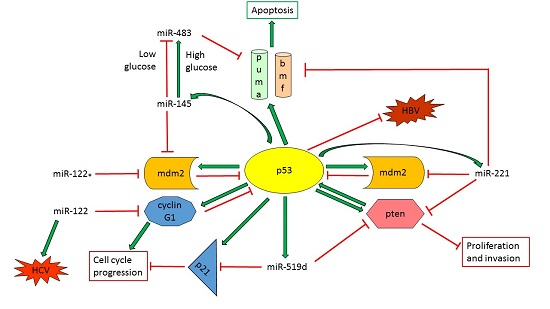
- Lupini, L.; Pepe, F.; Ferracin, M.; Braconi, C.; Callegari, E.; Pagotto, S.; Spizzo, R.; Zagatti, B.; Lanuti, P.; Fornari, F.; et al. Over-Expression of the miR-483-3p overcomes the miR-145/TP53 pro-apoptotic loop in hepatocellular carcinoma. Oncotarget 2016, 7, 31361–31371. [Google Scholar] [CrossRef] [PubMed]
- Veronese, A.; Lupini, L.; Consiglio, J.; Visone, R.; Ferracin, M.; Fornari, F.; Zanesi, N.; Alder, H.; D’Elia, G.; Gramantieri, L.; et al. Oncogenic role of miR-483-3p at the IGF2/483 locus. Cancer Res. 2010, 70, 3140–3149. [Google Scholar] [CrossRef] [PubMed]
- Ciafre, S.A.; Galardi, S.; Mangiola, A.; Ferracin, M.; Liu, C.G.; Sabatino, G.; Negrini, M.; Maira, G.; Croce, C.M.; Farace, M.G. Extensive modulation of a set of microRNAs in primary glioblastoma. Biochem. Biophys. Res. Commun. 2005, 334, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Jazdzewski, K.; Li, W.; Liyanarachchi, S.; Nagy, R.; Volinia, S.; Calin, G.A.; Liu, C.G.; Franssila, K.; Suster, S.; et al. The role of microRNA genes in papillary thyroid carcinoma. Proc. Natl. Acad. Sci. USA 2005, 102, 19075–19080. [Google Scholar] [CrossRef] [PubMed]
- Pallante, P.; Visone, R.; Ferracin, M.; Ferraro, A.; Berlingieri, M.T.; Troncone, G.; Chiappetta, G.; Liu, C.G.; Santoro, M.; Negrini, M.; et al. MicroRNA deregulation in human thyroid papillary carcinomas. Endocr. Relat. Cancer 2006, 13, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Fornari, F.; Gramantieri, L.; Ferracin, M.; Veronese, A.; Sabbioni, S.; Calin, G.A.; Grazi, G.L.; Giovannini, C.; Croce, C.M.; Bolondi, L.; et al. miR-221 controls CDKN1c/p57 and CDKN1b/p27 expression in human hepatocellular carcinoma. Oncogene 2008, 27, 5651–5661. [Google Scholar] [CrossRef] [PubMed]
- Galardi, S.; Mercatelli, N.; Giorda, E.; Massalini, S.; Frajese, G.V.; Ciafre, S.A.; Farace, M.G. miR-221 and miR-222 expression affects the proliferation potential of human prostate carcinoma cell lines by targeting p27kip1. J. Biol. Chem. 2007, 282, 23716–23724. [Google Scholar] [CrossRef] [PubMed]
- Le Sage, C.; Nagel, R.; Egan, D.A.; Schrier, M.; Mesman, E.; Mangiola, A.; Anile, C.; Maira, G.; Mercatelli, N.; Ciafre, S.A.; et al. Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO J. 2007, 26, 3699–3708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Kang, C.; You, Y.; Pu, P.; Yang, W.; Zhao, P.; Wang, G.; Zhang, A.; Jia, Z.; Han, L.; et al. Co-suppression of miR-221/222 cluster suppresses human glioma cell growth by targeting p27Kip1 in vitro and in vivo. Int. J. Oncol. 2009, 34, 1653–1660. [Google Scholar] [PubMed]
- Gramantieri, L.; Fornari, F.; Ferracin, M.; Veronese, A.; Sabbioni, S.; Calin, G.A.; Grazi, G.L.; Croce, C.M.; Bolondi, L.; Negrini, M. MicroRNA-221 targets bmf in hepatocellular carcinoma and correlates with tumor multifocality. Clin. Cancer Res. 2009, 15, 5073–5081. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Z.; Zhang, J.X.; Zhang, A.L.; Shi, Z.D.; Han, L.; Jia, Z.F.; Yang, W.D.; Wang, G.X.; Jiang, T.; You, Y.P.; et al. miR-221 and miR-222 target puma to induce cell survival in glioblastoma. Mol. Cancer 2010, 9, 229. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yao, L.; Li, G.; Ma, D.; Sun, C.; Gao, S.; Zhang, P.; Gao, F. miR-221 promotes epithelial-mesenchymal transition through targeting pten and forms a positive feedback loop with β-catenin/C-jun signaling pathway in extra-hepatic cholangiocarcinoma. PLoS ONE 2015, 10, e0141168. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cao, J.; Zhao, X. miR-221 facilitates the TGFβ1-induced epithelial-mesenchymal transition in human bladder cancer cells by targeting stmn1. BMC Urol. 2015, 15, 36. [Google Scholar] [CrossRef] [PubMed]
- Su, A.; He, S.; Tian, B.; Hu, W.; Zhang, Z. MicroRNA-221 mediates the effects of PDGF-BB on migration, proliferation, and the epithelial-mesenchymal transition in pancreatic cancer cells. PLoS ONE 2013, 8, e71309. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Z.; Han, L.; Zhang, A.L.; Fu, Y.C.; Yue, X.; Wang, G.X.; Jia, Z.F.; Pu, P.Y.; Zhang, Q.Y.; Kang, C.S. MicroRNA-221 and microRNA-222 regulate gastric carcinoma cell proliferation and radioresistance by targeting pten. BMC Cancer 2010, 10, 367. [Google Scholar]
- Garofalo, M.; Quintavalle, C.; Di Leva, G.; Zanca, C.; Romano, G.; Taccioli, C.; Liu, C.G.; Croce, C.M.; Condorelli, G. MicroRNA signatures of trail resistance in human non-small cell lung cancer. Oncogene 2008, 27, 3845–3855. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Roy, S.; Nuovo, G.; Ramaswamy, B.; Miller, T.; Shapiro, C.; Jacob, S.T.; Majumder, S. Anti-MicroRNA-222 (anti-miR-222) and -181b suppress growth of tamoxifen-resistant xenografts in mouse by targeting timp3 protein and modulating mitogenic signal. J. Biol. Chem. 2011, 286, 42292–42302. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.E.; Ghoshal, K.; Ramaswamy, B.; Roy, S.; Datta, J.; Shapiro, C.L.; Jacob, S.; Majumder, S. MicroRNA-221/222 confers tamoxifen resistance in breast cancer by targeting p27kip1. J. Biol. Chem. 2008, 283, 29897–29903. [Google Scholar] [CrossRef] [PubMed]
- Fornari, F.; Milazzo, M.; Galassi, M.; Callegari, E.; Veronese, A.; Miyaaki, H.; Sabbioni, S.; Mantovani, V.; Marasco, E.; Chieco, P.; et al. P53/mdm2 feedback loop sustains miR-221 expression and dictates the response to anticancer treatments in hepatocellular carcinoma. Mol. Cancer Res. 2014, 12, 203–216. [Google Scholar] [CrossRef] [PubMed]
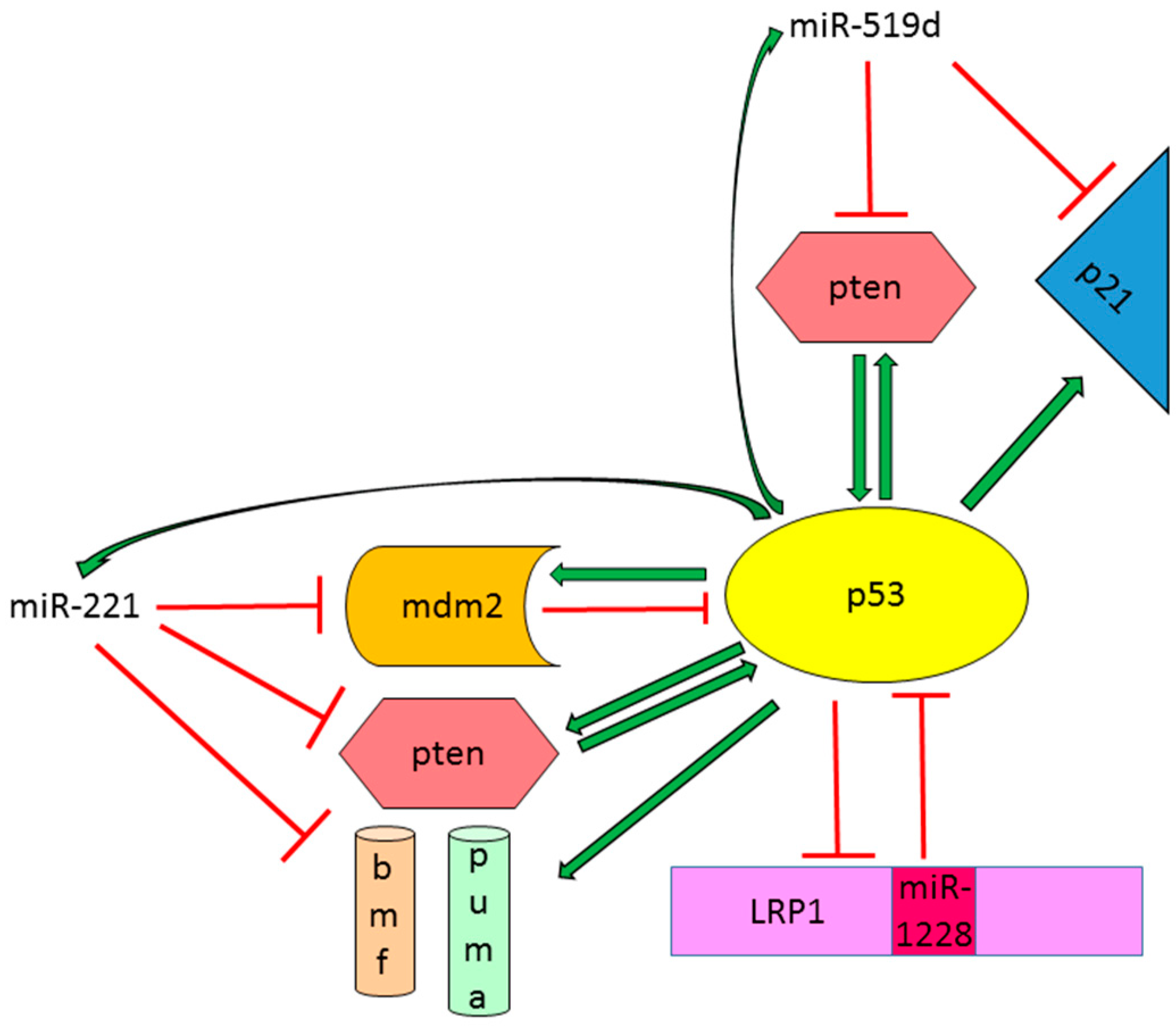
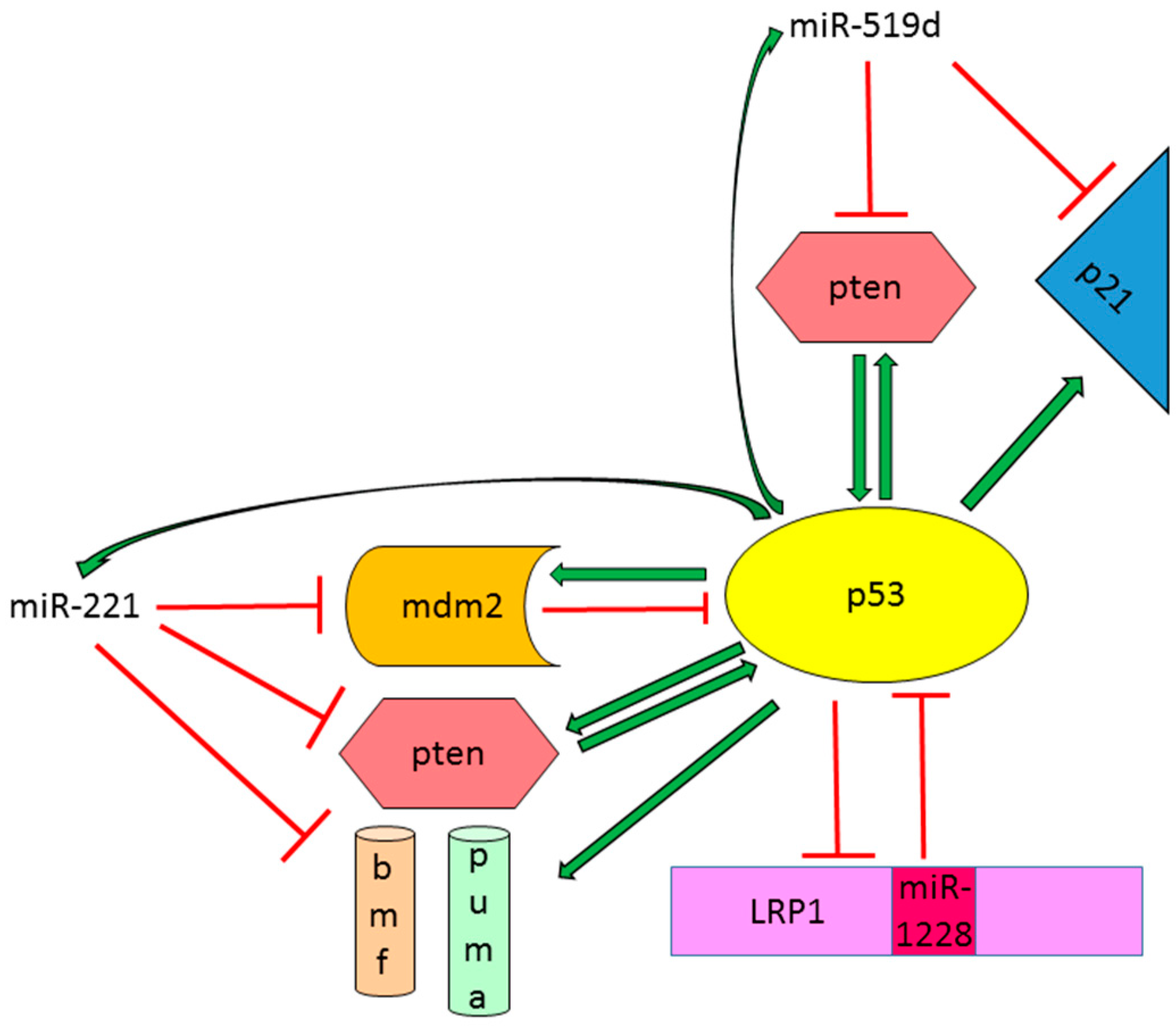
- Fornari, F.; Milazzo, M.; Chieco, P.; Negrini, M.; Marasco, E.; Capranico, G.; Mantovani, V.; Marinello, J.; Sabbioni, S.; Callegari, E.; et al. In hepatocellular carcinoma miR-519d is up-regulated by p53 and DNA hypomethylation and targets CDKN1a/p21, PTEN, Akt3 and TIMP2. J. Pathol. 2012, 227, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dai, J.; Deng, H.; Wan, H.; Liu, M.; Wang, J.; Li, S.; Li, X.; Tang, H. miR-1228 promotes the proliferation and metastasis of hepatoma cells through a p53 forward feedback loop. Br. J. Cancer 2015, 112, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Fornari, F.; Ferracin, M.; Trere, D.; Milazzo, M.; Marinelli, S.; Galassi, M.; Venerandi, L.; Pollutri, D.; Patrizi, C.; Borghi, A.; et al. Circulating microRNAs, miR-939, miR-595, miR-519d and miR-494, identify cirrhotic patients with hcc. PLoS ONE 2015, 10, e0141448. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Sevignani, C.; Dumitru, C.D.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M.; et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar] [CrossRef] [PubMed]
- Vrba, L.; Jensen, T.J.; Garbe, J.C.; Heimark, R.L.; Cress, A.E.; Dickinson, S.; Stampfer, M.R.; Futscher, B.W. Role for DNA methylation in the regulation of miR-200c and miR-141 expression in normal and cancer cells. PLoS ONE 2010, 5, e8697. [Google Scholar] [CrossRef] [PubMed]
- Elmen, J.; Lindow, M.; Schutz, S.; Lawrence, M.; Petri, A.; Obad, S.; Lindholm, M.; Hedtjarn, M.; Hansen, H.F.; Berger, U.; et al. Lna-Mediated microRNA silencing in non-human primates. Nature 2008, 452, 896–899. [Google Scholar] [CrossRef] [PubMed]
- Esau, C.; Davis, S.; Murray, S.F.; Yu, X.X.; Pandey, S.K.; Pear, M.; Watts, L.; Booten, S.L.; Graham, M.; McKay, R.; et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006, 3, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Huang, J.; Ma, L.; Shan, J.; Shen, J.; Yang, Z.; Liu, L.; Luo, Y.; Yao, C.; Qian, C. MicroRNA-122 confers sorafenib resistance to hepatocellular carcinoma cells by targeting IGF-1r to regulate RAS/RAF/ERK signaling pathways. Cancer Lett. 2016, 371, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Fabbri, M.; Calin, G.A. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat. Rev. Drug Discov. 2013, 12, 847–865. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Li, C.H.; Chan, S.L.; Xu, F.; Feng, L.; Wang, Y.; Jiang, J.D.; Sung, J.J.; Cheng, C.H.; Chen, Y. A small-molecule modulator of the tumor-suppressor miR34a inhibits the growth of hepatocellular carcinoma. Cancer Res. 2014, 74, 6236–6247. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Callegari, E.; Elamin, B.K.; Giannone, F.; Milazzo, M.; Altavilla, G.; Fornari, F.; Giacomelli, L.; D’Abundo, L.; Ferracin, M.; Bassi, C.; et al. Liver tumorigenicity promoted by microRNA-221 in a mouse transgenic model. Hepatology 2012, 56, 1025–1033. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA Name | miRNA/p53 Relationship | miRNA Functions | Target Genes |
|---|---|---|---|
| miR-34a | P53-effector miRNA | Tumor suppressor miRNA decreasing tumor invasion and metastasis OncomiRNA in β-catenin mutated patients increasing tumor progression and development | MET HNF4A, CCND1 |
| miR-125a-5p miR-125b | P53-effector miRNAs | Tumor suppressor miRNAs inducing cell cycle arrest and reducing tumor growth | SIRT7 |
| miR-200b/a miR-194-1/215 miR-194-2/192 | P53-effector miRNAs | Tumor suppressor miRNAs inducing epithelial-to-mesenchymal transition | ZEB1, ZEB2 |
| miR-26a | P53-activator miRNA | Tumor suppressor miRNAs reducing cell viability | ERα, CCND2, CCNE2 |
| miR-18a | P53-effector miRNA | Tumor suppressor miRNA reducing the risk of HCC development in female HBV patients | ERα |
| miR-23a | P53-effector miRNA | Tumor suppressor miRNA inducing apoptotic cell death | XIAP |
| miR-122 | P53-regulator miRNA | Tumor suppressor miRNA decreasing cell cycle progression and invasion and promoting apoptosis. Inhibition of HBV transcription. Induction of HCV replication. | CCNG1 |
| miR-122* | P53-regulator miRNA | Tumor suppressor miRNA increasing apoptosis and decreasing tumor growth | MDM2 |
| miR-145-5p | P53-regulator and effector miRNA | Tumor suppressor miRNA increasing apoptosis and decreasing tumor growth | MDM2 |
| miR-221 | P53-regulator and effector miRNA | Oncogenic miRNA increasing cell proliferation and tumor growth. Regulation of apoptotic cell death is dependent on TP53 status | CDKN2B, CDKN2C, PTEN, TIMP3, MDM2 |
| miR-519d | P53-regulator and effector miRNA | Oncogenic miRNA increasing cell proliferation and invasion and reducing apoptotic cell death | PTEN, AKT3, CDN2A |
| miR-1228 | P53-regulator and effector miRNA | Oncogenic miRNA increasing cell proliferation, invasion and metastasis | TP53 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pollutri, D.; Gramantieri, L.; Bolondi, L.; Fornari, F. TP53/MicroRNA Interplay in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2016, 17, 2029. https://doi.org/10.3390/ijms17122029
Pollutri D, Gramantieri L, Bolondi L, Fornari F. TP53/MicroRNA Interplay in Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2016; 17(12):2029. https://doi.org/10.3390/ijms17122029
Chicago/Turabian StylePollutri, Daniela, Laura Gramantieri, Luigi Bolondi, and Francesca Fornari. 2016. "TP53/MicroRNA Interplay in Hepatocellular Carcinoma" International Journal of Molecular Sciences 17, no. 12: 2029. https://doi.org/10.3390/ijms17122029