
Comparison of Small RNA Profiles of Glycine max and Glycine soja at Early Developmental Stages

 , and
, and 
Abstract
:1. Introduction
2. Results
2.1. Construction of Small RNA Libraries from Glycine max and Glycine soja
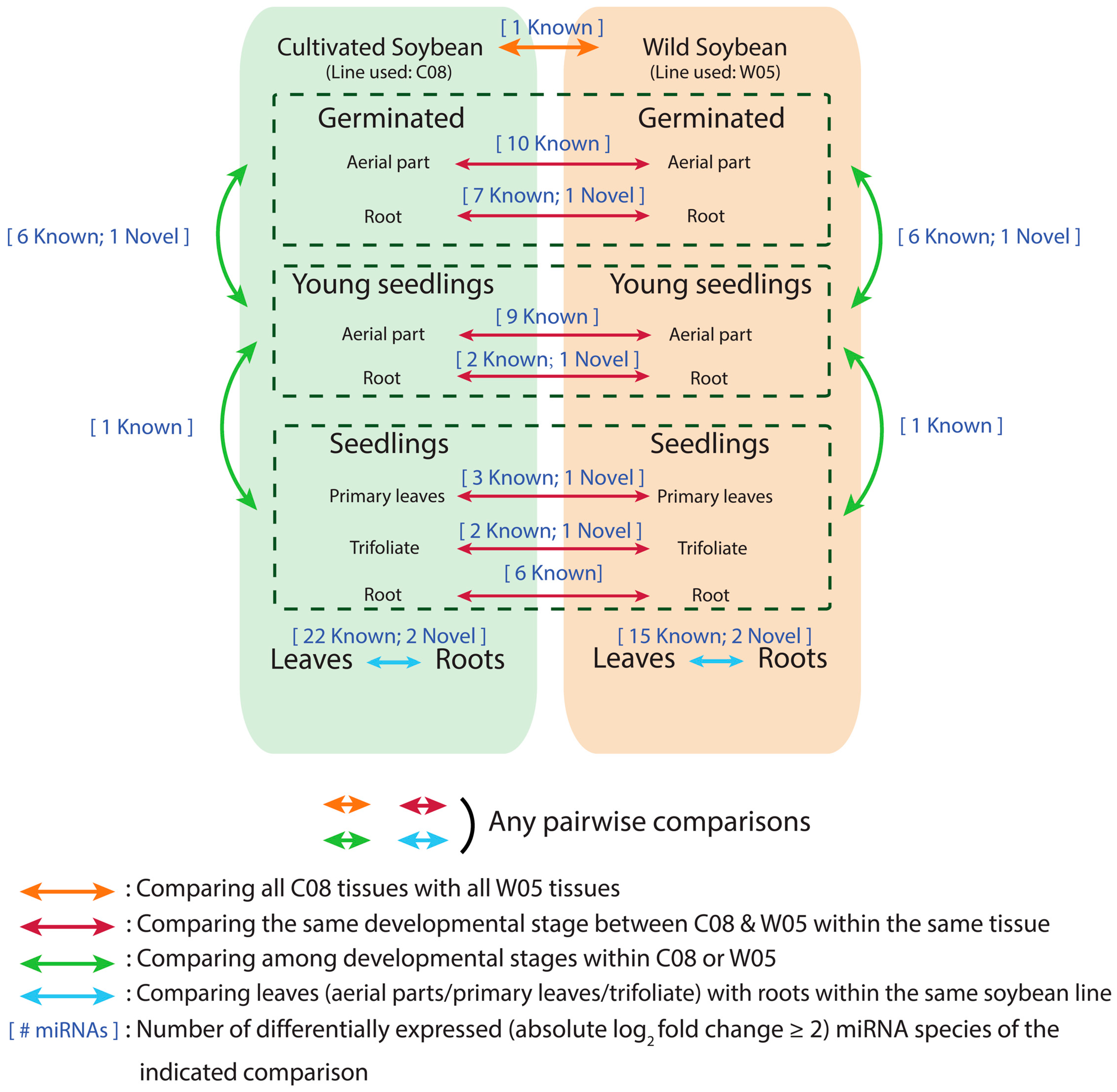
2.2. Identification of Novel miRNAs and Differential miRNA Expression in Glycine max and Glycine soja
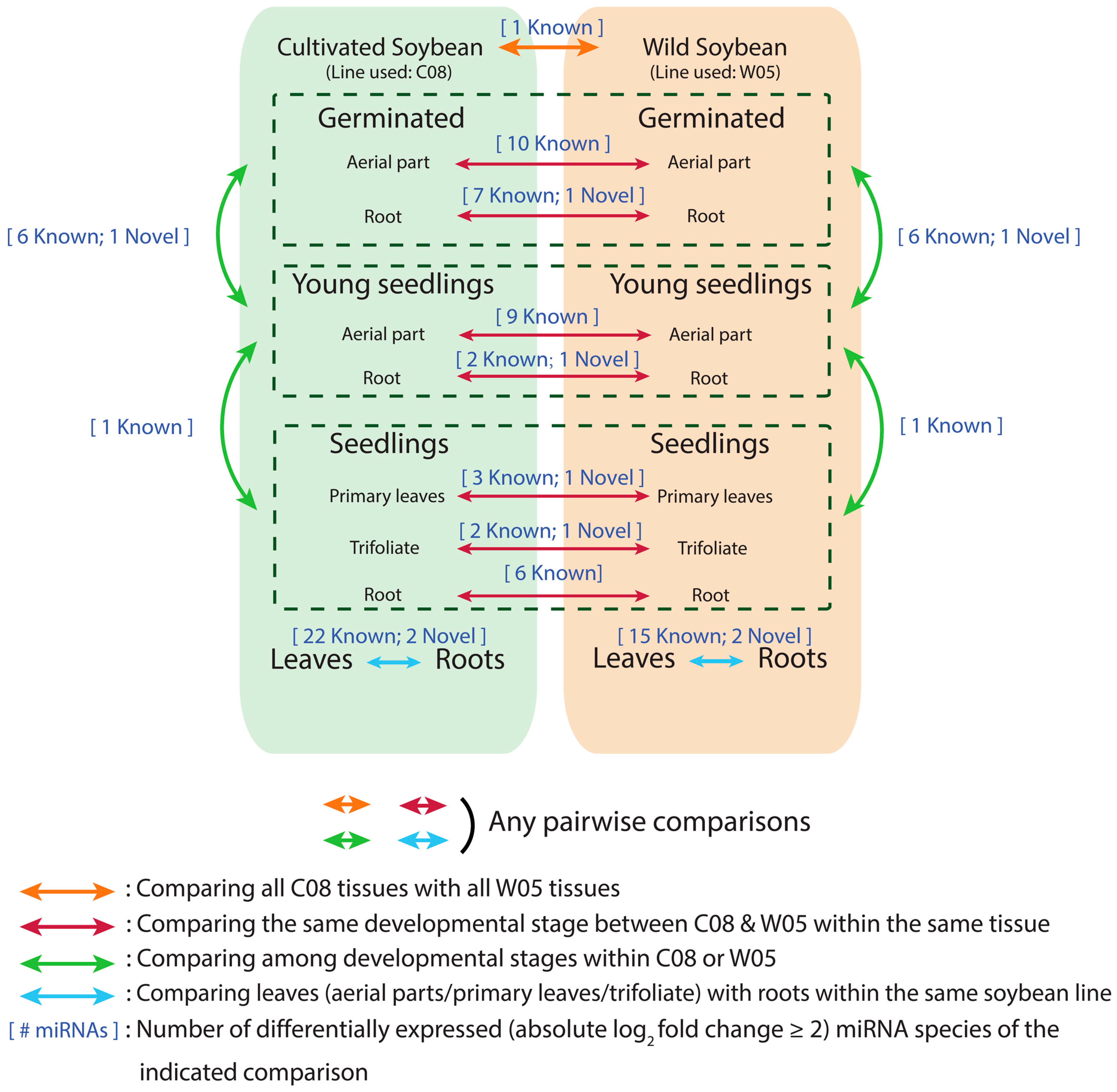
2.3. Tissue Types and Developmental Stages Are Significant Determinants of miRNA Profiles for Both Wild and Cultivated Soybeans
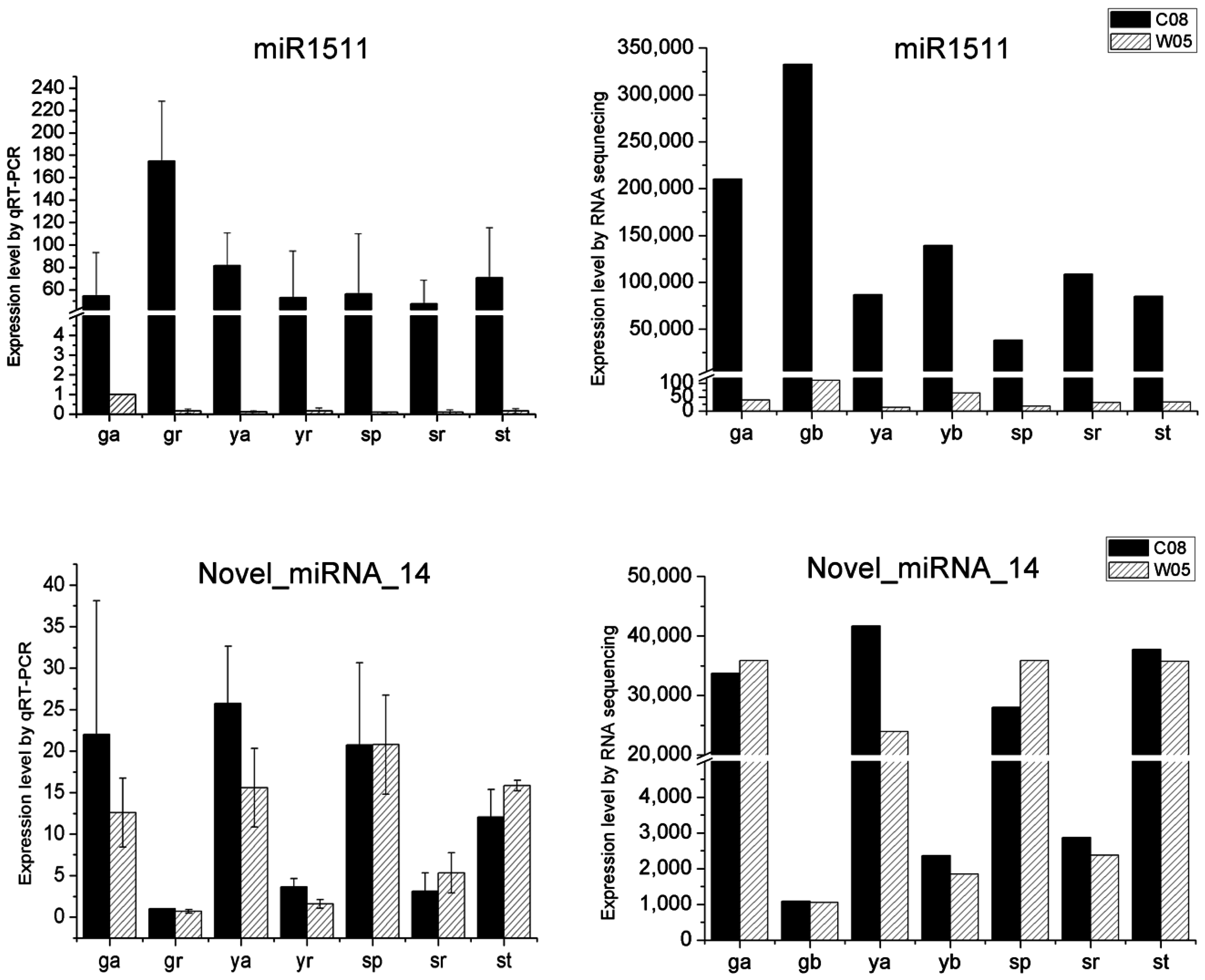
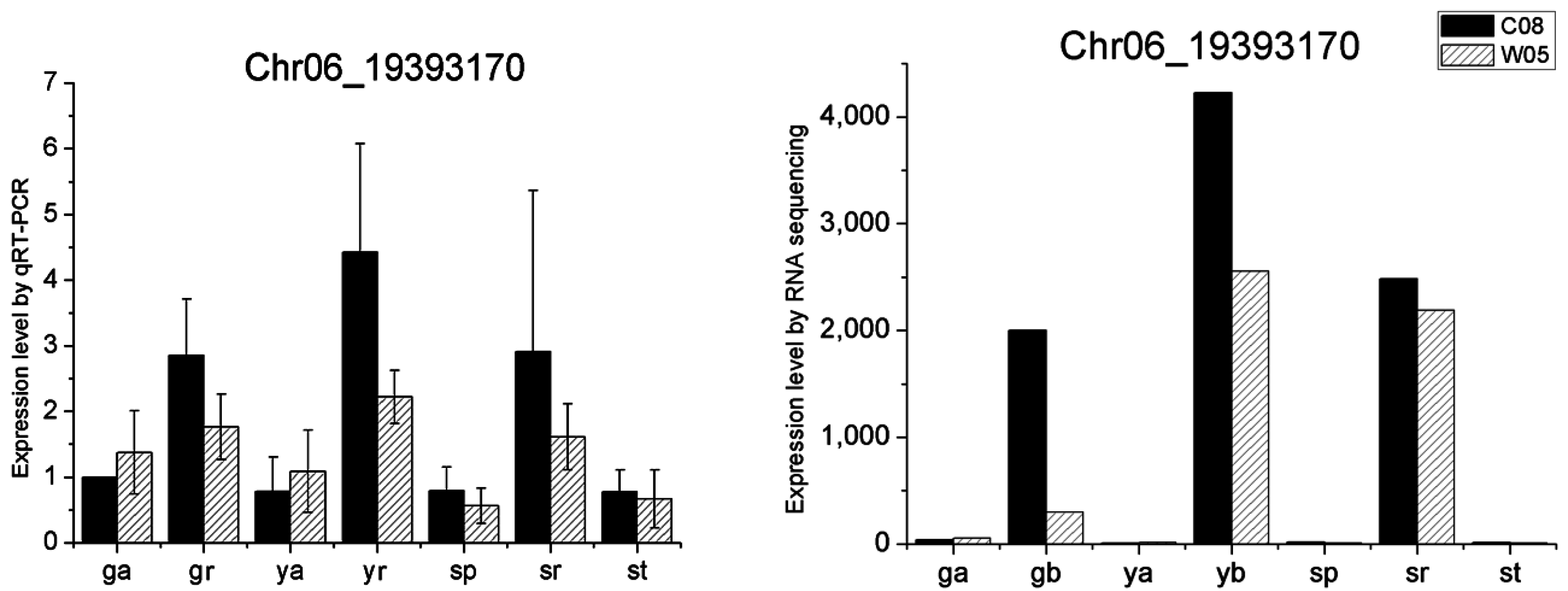
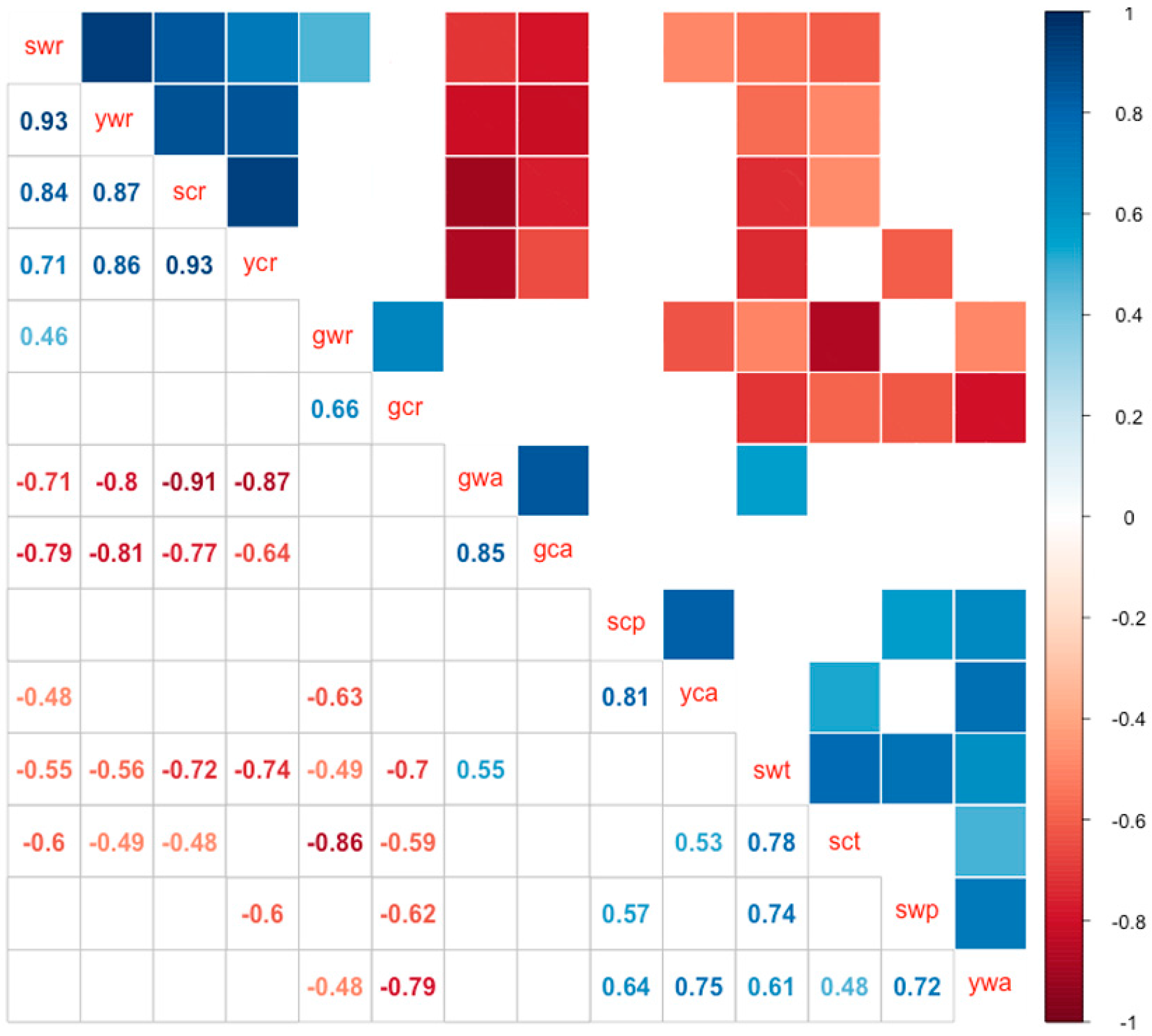
2.4. miRNA Profiles
2.5. Genome-Wide Identification of PHAS Loci and PhasiRNAs
2.6. Differential Expression of PhasiRNA in Different Tissues
3. Discussion
3.1. Differential Expressions of sRNAs between Cultivated and Wild Soybean and Their Biological Significance
3.2. PHAS Loci and phasiRNAs in Soybean
3.3. miRNA–phasiRNA Regulation in Soybean
4. Materials and Methods
4.1. Plant Materials
4.2. RNA Extraction and Sequencing of Small RNA
4.3. Reads Pre-Processing and Mapping for Downstream Analyses
4.4. Generation of rRNA, tRNA, Repeat Regions, snRNA/snoRNA Annotations
4.5. Soybean MicroRNA Discovery and Annotation
4.6. Normalization and Differential Gene Expression Calling of miRNAs
4.7. Target Prediction of miRNAs
4.8. Prediction of phasiRNA Regions
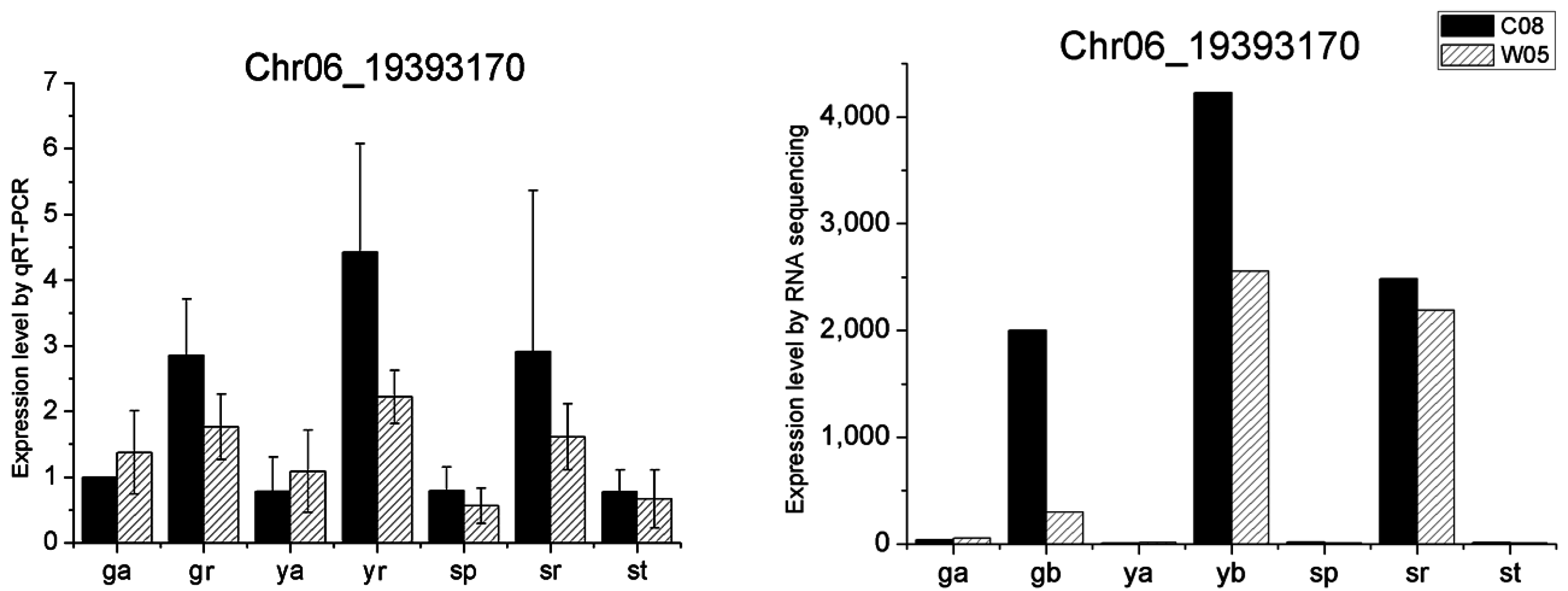
4.9. Quantitative RT-PCR
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Khraiwesh, B.; Zhu, J.K.; Zhu, J. Role of miRNAs and siRNAs in biotic and abiotic stress responses of plants. Biochim. Biophys. Acta 2012, 1819, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Xia, R.; Meyers, B.C.; Liu, Z.; Beers, E.P.; Ye, S.; Liu, Z. MicroRNA superfamilies descended from miR390 and their roles in secondary small interfering RNA biogenesis in eudicots. Plant Cell 2013, 25, 1555–1572. [Google Scholar] [CrossRef] [PubMed]
- Reinhart, B.J.; Weinstein, E.G.; Rhoades, M.W.; Bartel, B.; Bartel, D.P. MicroRNAs in plants. Genes Dev. 2002, 16, 1616–1626. [Google Scholar] [CrossRef] [PubMed]
- Khraiwesh, B.; Arif, M.A.; Seumel, G.I.; Ossowski, S.; Weigel, D.; Reski, R.; Frank, W. Transcriptional control of gene expression by microRNAs. Cell 2010, 140, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Zhai, J.; Jeong, D.H.; de Paoli, E.; Park, S.; Rosen, B.D.; Li, Y.; Gonzalez, A.J.; Yan, Z.; Kitto, S.L.; Grusak, M.A.; et al. MicroRNAs as master regulators of the plant NB-LRR defense gene family via the production of phased, trans-acting siRNAs. Genes Dev. 2011, 25, 2540–2553. [Google Scholar] [CrossRef] [PubMed]
- Fei, Q.; Xia, R.; Meyers, B.C. Phased, secondary, small interfering RNAs in posttranscriptional regulatory networks. Plant Cell 2013, 25, 2400–2415. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, M.; Peragine, A.; Park, M.Y.; Poethig, R.S. A pathway for the biogenesis of trans-acting siRNAs in Arabidopsis. Genes Dev. 2005, 19, 2164–2175. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.; Xie, Z.; Gustafson, A.M.; Carrington, J.C. MicroRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 2005, 121, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Felippes, F.F.; Weigel, D. Triggering the formation of tasiRNAs in Arabidopsis thaliana: The role of microRNA miR173. EMBO Rep. 2009, 10, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Gutmann, B.; Zhong, X.; Ye, Y.; Fisher, M.F.; Bai, F.; Castleden, I.; Song, Y.; Song, B.; Huang, J.; et al. Redefining the structural motifs that determine RNA binding and RNA editing by pentatricopeptide repeat proteins in land plants. Plant J. 2016. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, T.A.; Howell, M.D.; Cuperus, J.T.; Li, D.; Hansen, J.E.; Alexander, A.L.; Chapman, E.J.; Fahlgren, N.; Allen, E.; Carrington, J.C. Specificity of ARGONAUTE7-miR390 interaction and dual functionality in TAS3 trans-acting siRNA formation. Cell 2008, 133, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.J.; Mittal, A.; Jia, F.; Rock, C.D. An autoregulatory feedback loop involving PAP1 and TAS4 in response to sugars in Arabidopsis. Plant Mol. Biol. 2012, 80, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Peragine, A.; Yoshikawa, M.; Wu, G.; Albrecht, H.L.; Poethig, R.S. SGS3 and SGS2/SDE1/RDR6 are required for juvenile development and the production of trans-acting siRNAs in Arabidopsis. Genes Dev. 2004, 18, 2368–2379. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, F.; Vaucheret, H.; Rajagopalan, R.; Lepers, C.; Gasciolli, V.; Mallory, A.C.; Hilbert, J.L.; Bartel, D.P.; Crete, P. Endogenous trans-acting siRNAs regulate the accumulation of Arabidopsis mRNAs. Mol. Cell 2004, 16, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Adenot, X.; Elmayan, T.; Lauressergues, D.; Boutet, S.; Bouche, N.; Gasciolli, V.; Vaucheret, H. DRB4-dependent TAS3 trans-acting siRNAs control leaf morphology through AGO7. Curr. Biol. 2006, 16, 927–932. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, T.A.; Yoo, S.J.; Fahlgren, N.; Gilbert, S.D.; Howell, M.D.; Sullivan, C.M.; Alexander, A.; Nguyen, G.; Allen, E.; Ahn, J.H.; et al. AGO1-mir173 complex initiates phased siRNA formation in plants. Proc. Natl. Acad. Sci. USA 2008, 105, 20055–20062. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.; Howell, M.D. miRNAs in the biogenesis of trans-acting siRNAs in higher plants. Semin. Cell Dev. Biol. 2010, 21, 798–804. [Google Scholar] [CrossRef] [PubMed]
- Howell, M.D.; Fahlgren, N.; Chapman, E.J.; Cumbie, J.S.; Sullivan, C.M.; Givan, S.A.; Kasschau, K.D.; Carrington, J.C. Genome-wide analysis of the RNA-DEPENDENT RNA POLYMERASE6/DICER-LIKE4 pathway in Arabidopsis reveals dependency on miRNA- and tasiRNA-directed targeting. Plant Cell 2007, 19, 926–942. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, R.; Vaucheret, H.; Trejo, J.; Bartel, D.P. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev. 2006, 20, 3407–3425. [Google Scholar] [CrossRef] [PubMed]
- Parent, J.S.; Bouteiller, N.; Elmayan, T.; Vaucheret, H. Respective contributions of Arabidopsis DCL2 and DCL4 to RNA silencing. Plant J. 2015, 81, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Lam, H.M.; Xu, X.; Liu, X.; Chen, W.; Yang, G.; Wong, F.L.; Li, M.W.; He, W.; Qin, N.; Wang, B.; et al. Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat. Genet. 2010, 42, 1053–1059. [Google Scholar] [CrossRef] [PubMed]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Lee, S.; Van, K.; Kim, T.H.; Jeong, S.C.; Choi, I.Y.; Kim, D.S.; Lee, Y.S.; Park, D.; Ma, J.; et al. Whole-genome sequencing and intensive analysis of the undomesticated soybean (Glycine soja Sieb. And Zucc.) genome. Proc. Natl. Acad. Sci. USA 2010, 107, 22032–22037. [Google Scholar] [CrossRef] [PubMed]
- Li, W.Y.; Shao, G.; Lam, H.M. Ectopic expression of GmPAP3 alleviates oxidative damage caused by salinity and osmotic stresses. New Phytol. 2008, 178, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.X.; Liu, Y.F.; Hu, X.Y.; Zhang, W.K.; Ma, B.; Chen, S.Y.; Zhang, J.S. Identification of miRNAs and their target genes in developing soybean seeds by deep sequencing. BMC Plant Biol. 2011, 11, 5. [Google Scholar] [CrossRef] [PubMed]
- Arikit, S.; Xia, R.; Kakrana, A.; Huang, K.; Zhai, J.; Yan, Z.; Valdes-Lopez, O.; Prince, S.; Musket, T.A.; Nguyen, H.T.; et al. An atlas of soybean small RNAs identifies phased siRNAs from hundreds of coding genes. Plant Cell 2014, 26, 4584–4601. [Google Scholar] [CrossRef] [PubMed]
- Joshi, T.; Yan, Z.; Libault, M.; Jeong, D.H.; Park, S.; Green, P.J.; Sherrier, D.J.; Farmer, A.; May, G.; Meyers, B.C.; et al. Prediction of novel miRNAs and associated target genes in Glycine max. BMC Bioinform. 2010, 11 (Suppl. 1), S14. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, S.; Oldroyd, G.E. GRAS-domain transcription factors that regulate plant development. Plant Signal. Behav. 2009, 4, 698–700. [Google Scholar] [CrossRef] [PubMed]
- Nuruzzaman, M.; Sharoni, A.M.; Kikuchi, S. Roles of NAC transcription factors in the regulation of biotic and abiotic stress responses in plants. Front. Microbiol. 2013, 4, 248. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.M.; Li, Y.H.; Wu, S.H. Bioinformatic prediction and experimental validation of a microRNA-directed tandem trans-acting siRNA cascade in Arabidopsis. Proc. Natl. Acad. Sci. USA 2007, 104, 3318–3323. [Google Scholar] [CrossRef] [PubMed]
- Barkan, A.; Small, I. Pentatricopeptide repeat proteins in plants. Annu. Rev. Plant Biol. 2014, 65, 415–442. [Google Scholar] [CrossRef] [PubMed]
- Htwe, N.M.; Luo, Z.Q.; Jin, L.G.; Nadon, B.; Wang, K.J.; Qiu, L.J. Functional marker development of miR1511-InDel and allelic diversity within the genus Glycine. BMC Genom. 2015, 16, 467. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Shi, L.; Wang, Y.; Chen, L.; Cai, Z.; Jin, J.; Li, X. Identification and dynamic regulation of microRNAs involved in salt stress responses in functional soybean nodules by high-throughput sequencing. Int. J. Mol. Sci. 2013, 14, 2717–2738. [Google Scholar] [CrossRef] [PubMed]
- Lelandais-Briere, C.; Naya, L.; Sallet, E.; Calenge, F.; Frugier, F.; Hartmann, C.; Gouzy, J.; Crespi, M. Genome-wide Medicago truncatula small RNA analysis revealed novel microRNAs and isoforms differentially regulated in roots and nodules. Plant Cell 2009, 21, 2780–2796. [Google Scholar] [CrossRef] [PubMed]
- Pelaez, P.; Trejo, M.S.; Iniguez, L.P.; Estrada-Navarrete, G.; Covarrubias, A.A.; Reyes, J.L.; Sanchez, F. Identification and characterization of microRNAs in Phaseolus vulgaris by high-throughput sequencing. BMC Genom. 2012, 13, 83. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Park, M.Y.; Conway, S.R.; Wang, J.W.; Weigel, D.; Poethig, R.S. The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell 2009, 138, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Teotia, S.; Tang, G. To bloom or not to bloom: Role of microRNAs in plant flowering. Mol. Plant 2015, 8, 359–377. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Poethig, R.S. Temporal regulation of shoot development in Arabidopsis thaliana by miR156 and its target SPL3. Development 2006, 133, 3539–3547. [Google Scholar] [CrossRef] [PubMed]
- Gandikota, M.; Birkenbihl, R.P.; Hohmann, S.; Cardon, G.H.; Saedler, H.; Huijser, P. The miRNA156/157 recognition element in the 3’ UTR of the Arabidopsis SBP box gene SPL3 prevents early flowering by translational inhibition in seedlings. Plant J. 2007, 49, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.H.; Ju, Y.; Seo, P.J.; Lee, J.H.; Park, C.M. The SOC1-SPL module integrates photoperiod and gibberellic acid signals to control flowering time in Arabidopsis. Plant J. 2012, 69, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Lauter, N.; Kampani, A.; Carlson, S.; Goebel, M.; Moose, S.P. MicroRNA172 down-regulates glossy15 to promote vegetative phase change in maize. Proc. Natl. Acad. Sci. USA 2005, 102, 9412–9417. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Zhang, X.; Williams, S.J.; Ve, T.; Bernoux, M.; Sohn, K.H.; Jones, J.D.; Dodds, P.N.; Kobe, B. Crystallization and preliminary X-ray diffraction analyses of the tir domains of three TIR-NB-LRR proteins that are involved in disease resistance in Arabidopsis thaliana. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2013, 69, 1275–1280. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Pignatta, D.; Bendix, C.; Brunkard, J.O.; Cohn, M.M.; Tung, J.; Sun, H.; Kumar, P.; Baker, B. MicroRNA regulation of plant innate immune receptors. Proc. Natl. Acad. Sci. USA 2012, 109, 1790–1795. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, M.; Iki, T.; Tsutsui, Y.; Miyashita, K.; Poethig, R.S.; Habu, Y.; Ishikawa, M. 3′ fragment of miR173-programmed RISC-cleaved RNA is protected from degradation in a complex with RISC and SGS3. Proc. Natl. Acad. Sci. USA 2013, 110, 4117–4122. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.K.; Hagedorn, P.H.; de Torres-Zabala, M.; Grant, M.R.; Rung, J.H.; Collinge, D.B.; Lyngkjaer, M.F. Transcriptional regulation by an NAC (NAM-ATAF1,2-CUC2) transcription factor attenuates ABA signalling for efficient basal defence towards Blumeria graminis f. sp. hordei in Arabidopsis. Plant J. 2008, 56, 867–880. [Google Scholar] [PubMed]
- Qi, X.; Li, M.W.; Xie, M.; Liu, X.; Ni, M.; Shao, G.; Song, C.; Kay-Yuen Yim, A.; Tao, Y.; Wong, F.L.; et al. Identification of a novel salt tolerance gene in wild soybean by whole-genome sequencing. Nat. Commun. 2014, 5, 4340. [Google Scholar] [CrossRef] [PubMed]
- Vidal, E.A.; Araus, V.; Lu, C.; Parry, G.; Green, P.J.; Coruzzi, G.M.; Gutierrez, R.A. Nitrate-responsive miR393/AFB3 regulatory module controls root system architecture in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2010, 107, 4477–4482. [Google Scholar] [CrossRef] [PubMed]
- Andika, I.B.; Maruyama, K.; Sun, L.; Kondo, H.; Tamada, T.; Suzuki, N. Differential contributions of plant dicer-like proteins to antiviral defences against potato virus × in leaves and roots. Plant J. 2015, 81, 781–793. [Google Scholar] [CrossRef] [PubMed]
- Andika, I.B.; Kondo, H.; Tamada, T. Evidence that RNA silencing-mediated resistance to beet necrotic yellow vein virus is less effective in roots than in leaves. Mol. Plant Microbe Interact. 2005, 18, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Jagadeeswaran, G.; Zheng, Y.; Li, Y.F.; Shukla, L.I.; Matts, J.; Hoyt, P.; Macmil, S.L.; Wiley, G.B.; Roe, B.A.; Zhang, W.; et al. Cloning and characterization of small RNAs from Medicago truncatula reveals four novel legume-specific microRNA families. New Phytol. 2009, 184, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Liu, X.; Sun, Y.; Yiu, S.M.; Lim, B.L. Global small RNA analysis in fast-growing Arabidopsis thaliana with elevated concentrations of ATP and sugars. BMC Genom. 2014, 15, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin, E.; Jouannet, V.; Herz, A.; Lokerse, A.S.; Weijers, D.; Vaucheret, H.; Nussaume, L.; Crespi, M.D.; Maizel, A. miR390, Arabidopsis TAS3 tasiRNAs, and their AUXIN RESPONSE FACTOR targets define an autoregulatory network quantitatively regulating lateral root growth. Plant Cell 2010, 22, 1104–1117. [Google Scholar] [CrossRef] [PubMed]
- Aushubel, F.; Brent, R.; Kingston, R.; Moore, D.; Seidman, J.; Smith, J.; Struhl, K. Current Protocols in Molecular Biology; John Wiley & Sons, Inc.: New York, NY, USA, 1995. [Google Scholar]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Wong, T.; Zhu, J.; Liu, C.-M.; Zhu, X.; Wu, E.; Lee, L.-K.; Lin, H.; Zhu, W.; Cheung, D.W. SOAP3-dp: Fast, accurate and sensitive GPU-based short read aligner. PLoS ONE 2013, 8, e65632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagesen, K.; Hallin, P.; Rodland, E.A.; Staerfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.; Hubley, R.; Green, P. Repeatmasker Open-4.0. Available online: http://www.Repeatmasker.Org (accessed on 30 November 2016).
- Nawrocki, E.P.; Burge, S.W.; Bateman, A.; Daub, J.; Eberhardt, R.Y.; Eddy, S.R.; Floden, E.W.; Gardner, P.P.; Jones, T.A.; Tate, J.; et al. Rfam 12.0: Updates to the RNA families database. Nucleic Acids Res. 2015, 43, D130–D137. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Sun, Y. miR-prefer: An accurate, fast and easy-to-use plant miRNA prediction tool using small RNA-seq data. Bioinformatics 2014, 30, 2837–2839. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. Degseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server. Nucleic Acids Res. 2011, 39, W155–W159. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-M.; Chen, L.-T.; Patel, K.; Li, Y.-H.; Baulcombe, D.C.; Wu, S.-H. 22-Nucleotide RNAs trigger secondary siRNA biogenesis in plants. Proc. Natl. Acad. Sci. USA 2010, 107, 15269–15274. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chr | PHAS Start | PHAS End | Accession | Gene Name | Homologous Gene | Annotation |
|---|---|---|---|---|---|---|
| Chr02 | 45,229,200 | 45,229,892 | C08 | NA | NA | NA |
| Chr03 | 35,270,999 | 35,271,544 | C08 | Glyma.03G136600 | AT2G41750 | DTW domain-containing protein |
| Chr04 | 47,362,862 | 47,363,575 | C08 | NA | NA | NA |
| Chr04 | 49,035,971 | 49,037,012 | C08,W05 | Glyma.04G219600 | AT5G36930 | Disease resistance protein (TIR-NB-LRR class) family |
| Chr05 | 656,827 | 657,414 | C08 | Glyma.05G006900 | AT3G14470 | NB-ARC domain-containing disease resistance protein |
| Chr05 | 38,268,854 | 38,269,817 | C08,W05 | Glyma.05G198500 | AT5G23570 | SGS3, XS zinc finger domain-containing protein |
| Chr06 | 11,932,996 | 11,934,011 | C08,W05 | Glyma.06G146200 | AT5G36930 | Disease resistance protein (TIR-NB-LRR class) family |
| Chr06 | 19,392,730 | 19,393,674 | C08,W05 | Glyma.06G205100 | AT5G17680 | Disease resistance protein (TIR-NB-LRR class), putative |
| Chr06 | 39,170,442 | 39,171,407 | W05 | Glyma.06G239000 | AT5G17680 | Disease resistance protein (TIR-NB-LRR class), putative |
| Chr06 | 43,287,288 | 43,287,977 | C08,W05 | Glyma.06G254300 | AT3G14470 | NB-ARC domain-containing disease resistance protein |
| Chr06 | 47,410,720 | 47,411,412 | C08,W05 | Glyma.06G285500 | AT5G17680 | Disease resistance protein (TIR-NB-LRR class), putative |
| Chr07 | 4,028,350 | 4,029,084 | C08,W05 | Glyma.07G048000 | AT4G35580 | NTL9 NAC transcription factor-like 9 |
| Chr08 | 41,923,481 | 41,923,921 | C08,W05 | Glyma.08G301200 | AT5G36930 | Disease resistance protein (TIR-NB-LRR class) family |
| Chr08 | 41,923,963 | 41,924,634 | C08 | Glyma.08G301200 | AT5G36930 | Disease resistance protein (TIR-NB-LRR class) family |
| Chr09 | 2,041,836 | 2,042,507 | C08,W05 | Glyma.09G025300 | AT3G03300 | ATDCL2,DCL2 dicer-like 2 |
| Chr09 | 2,737,112 | 2,737,899 | C08,W05 | Glyma.09G032400 | NA | NA |
| Chr09 | 47,565,020 | 47,565,481 | W05 | Glyma.09G256600 | AT3G22470 | Pentatricopeptide repeat (PPR) superfamily protein |
| Chr11 | 30,572,116 | 30,573,147 | C08,W05 | Glyma.11G212800 | AT3G14460 | LRR and NB-ARC domains-containing disease resistance protein |
| Chr12 | 31,231,341 | 31,232,533 | C08,W05 | Glyma.12G163000 | AT1G31540 | Disease resistance protein (TIR-NB-LRR class) family |
| Chr12 | 39,530,806 | 39,531,225 | C08 | Glyma.12G236500 | AT4G27220 | NB-ARC domain-containing disease resistance protein |
| Chr13 | 19,268,532 | 19,269,329 | C08 | NA | NA | NA |
| Chr13 | 29,861,566 | 29,862,427 | C08 | Glyma.13G184800 | AT3G14460 | LRR and NB-ARC domains-containing disease resistance protein |
| Chr13 | 29,875,305 | 29,876,627 | C08,W05 | Glyma.13G184900 | AT3G14470 | NB-ARC domain-containing disease resistance protein |
| Chr13 | 30,174,985 | 30,175,782 | C08,W05 | Glyma.13G187900 | AT3G14470 | NB-ARC domain-containing disease resistance protein |
| Chr13 | 30,207,736 | 30,208,554 | C08,W05 | Glyma.13G188300 | AT3G14470 | NB-ARC domain-containing disease resistance protein |
| Chr13 | 30,389,026 | 30,390,362 | C08,W05 | Glyma.13G190300 | AT3G14470 | NB-ARC domain-containing disease resistance protein |
| Chr13 | 30,664,791 | 30,665,882 | C08 | Glyma.13G193300 | AT3G14470 | NB-ARC domain-containing disease resistance protein |
| Chr13 | 30,727,236 | 30,728,889 | C08,W05 | Glyma.13G194100 | AT3G14470 | NB-ARC domain-containing disease resistance protein |
| Chr13 | 30,915,467 | 30,916,705 | C08,W05 | Glyma.13G195600 | AT3G14460 | LRR and NB-ARC domains-containing disease resistance protein |
| Chr14 | 3,878,256 | 3,878,843 | C08 | NA | NA | NA |
| Chr15 | 43,699,964 | 43,700,551 | C08,W05 | Glyma.15G232400 | AT5G41540 | Disease resistance protein (TIR-NB-LRR class) family |
| Chr15 | 43,746,080 | 43,746,499 | C08 | Glyma.15G232800 | AT3G14460 | LRR and NB-ARC domains-containing disease resistance protein |
| Chr15 | 44,546,466 | 44,547,735 | C08,W05 | NA | NA | NA |
| Chr16 | 1,452,507 | 1,453,157 | C08,W05 | Glyma.16G016600 | AT4G35580 | NTL9 NAC transcription factor-like 9 |
| Chr16 | 1,453,270 | 1,453,905 | C08 | Glyma.16G016600 | AT4G35580 | NTL9 NAC transcription factor-like 9 |
| Chr16 | 1,461,742 | 1,462,416 | C08,W05 | Glyma.16G016700 | AT4G35580 | NTL9 NAC transcription factor-like 9 |
| Chr16 | 1,462,506 | 1,463,177 | C08 | Glyma.16G016700 | AT4G35580 | NTL9 NAC transcription factor-like 9 |
| Chr16 | 2,941,895 | 2,942,482 | C08,W05 | Glyma.16G031100 | AT2G46100 | Nuclear transport factor 2 (NTF2) family protein |
| Chr16 | 4,855,988 | 4,856,743 | C08,W05 | Glyma.16G050500 | AT1G12820 | AFB3 auxin signaling F-box 3 protein |
| Chr16 | 5,770,754 | 5,771,618 | C08,W05 | Glyma.16G058900 | NA | NA |
| Chr16 | 10,648,349 | 10,648,999 | C08,W05 | Glyma.16G087100 | AT5G36930 | Disease resistance protein (TIR-NB-LRR class) family |
| Chr16 | 30,784,351 | 30,785,128 | C08,W05 | Glyma.16G147100 | NA | NA |
| Chr16 | 32,109,362 | 32,110,117 | C08,W05 | Glyma.16G161900 | AT1G12775 | Pentatricopeptide repeat (PPR) superfamily protein |
| Chr16 | 32,167,989 | 32,168,613 | C08 | Glyma.16G162800 | AT1G62670 | RPF2 RNA processing factor 2 |
| Chr16 | 32,182,422 | 32,182,883 | C08 | Glyma.16G163100 | AT1G12700 | ATP binding;nucleic acid binding;helicases |
| Chr16 | 32,453,347 | 32,453,892 | W05 | Glyma.16G165400 | AT1G12300 | Tetratricopeptide repeat (TPR)-like superfamily protein |
| Chr16 | 33,378,642 | 33,379,535 | W05 | Glyma.16G173200 | NA | NA |
| Chr16 | 35,681,818 | 35,682,321 | W05 | Glyma.16G194800 | AT1G63130 | Tetratricopeptide repeat (TPR)-like superfamily protein |
| Chr16 | 35,687,005 | 35,687,634 | C08,W05 | Glyma.16G195000 | AT1G12775 | Pentatricopeptide repeat (PPR) superfamily protein |
| Chr16 | 35,692,915 | 35,693,523 | C08,W05 | Glyma.16G195100 | AT1G12700 | ATP binding;nucleic acid binding;helicases |
| Chr16 | 35,883,305 | 35,883,850 | W05 | Glyma.16G197600 | AT1G62910 | Pentatricopeptide repeat (PPR) superfamily protein |
| Chr16 | 36,088,255 | 36,088,695 | C08,W05 | Glyma.16G199700 | AT1G62930 | Tetratricopeptide repeat (TPR)-like superfamily protein |
| Chr16 | 36,970,009 | 36,970,575 | C08 | Glyma.16G210600 | AT5G36930 | Disease resistance protein (TIR-NB-LRR class) family |
| Chr17 | 40,464,414 | 40,465,001 | C08,W05 | Glyma.17G249500 | NA | NA |
| Chr19 | 34,719,617 | 34,720,288 | C08,W05 | Glyma.19G100200 | AT1G12820 | AFB3 auxin signaling F-box 3 protein |
| Chr19 | 40,040,875 | 40,041,548 | C08,W05 | NA | NA | NA |
| Chr20 | 31,854,969 | 31,855,514 | W05 | NA | NA | NA |
| Gene Name | Annotation | Number of phasiRNAs | Fold Change of Expression Level (W05/C08) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Ga | Gr | Ya | Yr | Sp | Sr | St | |||
| Glyma.04G219600 | Disease resistance protein (TIR-NB-LRR class) | 4 | 1.1 | 0.4 | 0.6 | 1.1 | 0.7 | 1.2 | 0.5 |
| Glyma.06G146200 | Disease resistance protein (TIR-NB-LRR class) | 4 | 1.4 | 0.4 | 0.9 | 1.1 | 0.3 | 0.6 | 0.3 |
| Glyma.06G205100 | Disease resistance protein (TIR-NB-LRR class) | 2 | 1.1 | 0.3 | 1.4 | 0.8 | 1.0 | 1.5 | 0.4 |
| Glyma.06G285500 | Disease resistance protein (TIR-NB-LRR class) | 1 | 1.8 | 0.5 | 1.6 | 1.5 | 1.5 | 1.2 | 1.1 |
| Glyma.08G301200 | Disease resistance protein (TIR-NB-LRR class) | 1 | 0.9 | 0.5 | 0.5 | 0.7 | 0.6 | 1.0 | 0.4 |
| Glyma.12G163000 | Disease resistance protein (TIR-NB-LRR class) | 2 | 0.6 | 0.5 | 0.9 | 1.1 | 1.3 | 1.1 | 0.6 |
| Glyma.13G187900 | NB-ARC domain-containing disease resistance protein | 4 | 3.6 | 2.2 | 2.5 | 2.2 | 1.5 | 2.3 | 3.5 |
| Glyma.11G212800 | LRR and NB-ARC domains-containing disease resistance protein | 1 | 0.6 | 0.5 | 0.6 | 1.0 | 0.8 | 1.5 | 0.5 |
| Glyma.07G048000 | NTL9 NAC transcription factor-like 9 | 4 | 2.0 | 1.9 | 1.8 | 2.5 | 1.6 | 3.2 | 1.7 |
| Glyma.16G016600 | NTL9 NAC transcription factor-like 9 | 1 | 1.1 | 0.3 | 0.4 | 1.0 | 0.3 | 1.1 | 0.3 |
| Glyma.09G025300 | ATDCL2, DCL2 | 2 | 1.0 | 0.3 | 0.9 | 1.2 | 0.6 | 1.4 | 1.0 |
| Glyma.16G050500 | AFB3 auxin signaling F-box 3 | 1 | 2.7 | 0.9 | 1.0 | 0.8 | 0.5 | 0.7 | 1.3 |
| Glyma.16G161900 | Pentatricopeptide repeat (PPR) superfamily protein | 1 | 1.0 | 0.5 | 0.8 | 1.0 | 0.6 | 1.2 | 0.7 |
| Glyma.16G058900 | NA | 3 | 1.0 | 0.4 | 0.9 | 1.0 | 0.6 | 1.1 | 0.5 |
| Glyma.16G147100 | NA | 3 | 0.9 | 1.4 | 0.6 | 1.0 | 0.4 | 1.1 | 0.6 |
| Chr15_44546856_44547257 | NA | 4 | 0.6 | 0.2 | 0.6 | 0.8 | 0.4 | 0.9 | 0.6 |
| Glyma.09G032400 | NA | 5 | 1.1 | 0.5 | 0.7 | 0.8 | 0.7 | 1.0 | 0.5 |
| Gene Name | Annotation | Number of phasiRNA | Leaf/Root (Germinated Seedlings) | Leaf/Root (Young Seedlings) | Leaf/Root (Seedlings) | Leaf/Root (Three Stages) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| C08 | W05 | C08 | W05 | C08 | W05 | C08 | W05 | |||
| Glyma.04G219600 | Disease resistance protein (TIR-NB-LRR class) | 4 | 2.2 | 5.4 | 2.6 | 1.6 | 5.3 | 3.9 | 2.9 | 3.0 |
| Glyma.06G146200 | Disease resistance protein (TIR-NB-LRR class) | 4 | 0.8 | 1.8 | 1.2 | 1.0 | 2.9 | 1.2 | 1.4 | 1.3 |
| Glyma.06G205100 | Disease resistance protein (TIR-NB-LRR class) | 2 | 0.04 | 0.13 | 0.01 | 0.01 | 0.02 | 0.02 | 0.03 | 0.04 |
| Glyma.06G285500 | Disease resistance protein (TIR-NB-LRR class) | 1 | 7.1 | 27.9 | 6.1 | 6.8 | 10.6 | 13.1 | 7.3 | 12.0 |
| Glyma.08G301200 | Disease resistance protein (TIR-NB-LRR class) | 1 | 0.1 | 0.3 | 0.6 | 0.4 | 1.5 | 1.1 | 0.5 | 0.5 |
| Glyma.12G163000 | Disease resistance protein (TIR-NB-LRR class) | 2 | 19.9 | 34.2 | 7.0 | 6.9 | 12.6 | 15.6 | 11.1 | 11.2 |
| Glyma.13G187900 | NB-ARC domain-containing disease resistance protein | 4 | 1.1 | 1.4 | 1.4 | 1.6 | 4.8 | 2.7 | 2.2 | 1.9 |
| Glyma.11G212800 | LRR and NB-ARC domains-containing disease resistance protein | 1 | 1.2 | 1.3 | 0.7 | 0.4 | 1.7 | 1.0 | 1.2 | 0.8 |
| Glyma.07G048000 | NTL9 NAC transcription factor-like 9 | 4 | 1.3 | 1.2 | 0.4 | 0.3 | 1.0 | 0.6 | 0.8 | 0.6 |
| Glyma.16G016600 | NTL9 NAC transcription factor-like 9 | 1 | 1.0 | 3.2 | 1.2 | 0.6 | 2.5 | 0.7 | 1.5 | 1.2 |
| Glyma.09G025300 | ATDCL2, DCL2 | 2 | 0.4 | 1.4 | 0.6 | 0.5 | 1.4 | 0.6 | 0.7 | 0.7 |
| Glyma.16G050500 | AFB3 auxin signaling F-box 3 | 1 | 0.1 | 0.3 | 0.3 | 0.4 | 0.7 | 0.5 | 0.5 | 0.4 |
| Glyma.16G161900 | Pentatricopeptide repeat (PPR) superfamily protein | 1 | 3.4 | 7.5 | 2.7 | 2.3 | 6.8 | 3.1 | 3.9 | 3.9 |
| Glyma.16G058900 | NA | 3 | 0.8 | 1.6 | 1.1 | 0.9 | 2.3 | 1.4 | 1.1 | 1.3 |
| Glyma.16G147100 | NA | 3 | 0.4 | 0.9 | 1.0 | 0.5 | 1.6 | 0.8 | 0.8 | 0.7 |
| Chr15_44546856_44547257 | NA | 4 | 1.3 | 3.8 | 3.0 | 2.2 | 5.8 | 2.6 | 2.4 | 2.9 |
| Glyma.09G032400 | NA | 5 | 1.3 | 2.0 | 3.1 | 2.7 | 4.4 | 2.8 | 2.5 | 2.5 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Y.; Mui, Z.; Liu, X.; Yim, A.K.-Y.; Qin, H.; Wong, F.-L.; Chan, T.-F.; Yiu, S.-M.; Lam, H.-M.; Lim, B.L. Comparison of Small RNA Profiles of Glycine max and Glycine soja at Early Developmental Stages. Int. J. Mol. Sci. 2016, 17, 2043. https://doi.org/10.3390/ijms17122043
Sun Y, Mui Z, Liu X, Yim AK-Y, Qin H, Wong F-L, Chan T-F, Yiu S-M, Lam H-M, Lim BL. Comparison of Small RNA Profiles of Glycine max and Glycine soja at Early Developmental Stages. International Journal of Molecular Sciences. 2016; 17(12):2043. https://doi.org/10.3390/ijms17122043
Chicago/Turabian StyleSun, Yuzhe, Zeta Mui, Xuan Liu, Aldrin Kay-Yuen Yim, Hao Qin, Fuk-Ling Wong, Ting-Fung Chan, Siu-Ming Yiu, Hon-Ming Lam, and Boon Leong Lim. 2016. "Comparison of Small RNA Profiles of Glycine max and Glycine soja at Early Developmental Stages" International Journal of Molecular Sciences 17, no. 12: 2043. https://doi.org/10.3390/ijms17122043