
p53 as a Regulator of Lipid Metabolism in Cancer

Abstract
:1. Introduction
2. Regulation of Lipid Metabolism by Wild-Type p53
2.1. Glucose-6-Phosphate Dehydrogenase (G6PD)
2.2. Sterol Regulatory Element-Binding Protein-1 (SREBP-1)
2.3. Sirtuin 1 (SIRT1)
2.4. Aromatase
2.5. Acyl-CoA Dehydrogenase Family Member 11 (Acad11)
2.6. Lipin 1
2.7. Malonyl-CoA Decarboxylase (MCD)
2.8. Dehydrogenase/Reductase 3 (DHRS3)
2.9. Caveolin 1
3. Roles of Mutant p53 in the Lipid Metabolism
4. Summary and Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Keijer, J.; van Dartel, D.A. Reprogrammed metabolism of cancer cells as a potential therapeutic target. Curr. Pharm. Des. 2014, 20, 2580–2594. [Google Scholar] [CrossRef] [PubMed]
- Hirschey, M.D.; DeBerardinis, R.J.; Diehl, A.M.; Drew, J.E.; Frezza, C.; Green, M.F.; Jones, L.W.; Ko, Y.H.; Le, A.; Lea, M.A.; et al. Dysregulated metabolism contributes to oncogenesis. Semin. Cancer Biol. 2015, 35 (Suppl.), S129–S150. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.R.; Schulze, A. Lipid metabolism in cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef] [PubMed]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef] [PubMed]
- Zahra Bathaie, S.; Ashrafi, M.; Azizian, M.; Tamanoi, F. Mevalonate pathway and human cancers. Curr. Mol. Pharm. 2016, 9. [Google Scholar] [CrossRef]
- Ribas, V.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Mitochondria, cholesterol and cancer cell metabolism. Clin. Transl. Med. 2016, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Bell, E.H.; Mischel, P.; Chakravarti, A. Targeting SREBP-1-driven lipid metabolism to treat cancer. Curr. Pharm. Des. 2014, 20, 2619–2626. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [PubMed]
- Rivlin, N.; Brosh, R.; Oren, M.; Rotter, V. Mutations in the p53 tumor suppressor gene: Important milestones at the various steps of tumorigenesis. Genes Cancer 2011, 2, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Berkers, C.R.; Maddocks, O.D.; Cheung, E.C.; Mor, I.; Vousden, K.H. Metabolic regulation by p53 family members. Cell Metab. 2013, 18, 617–633. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, I.; Ezra, O.; Rivlin, N.; Molchadsky, A.; Madar, S.; Goldfinger, N.; Rotter, V. p53, a novel regulator of lipid metabolism pathways. J. Hepatol. 2012, 56, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Tumor suppressor p53 and its mutants in cancer metabolism. Cancer Lett. 2015, 356, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 2011, 13, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, J.; Liang, Y.; Wu, R.; Zhao, Y.; Hong, X.; Lin, M.; Yu, H.; Liu, L.; Levine, A.J.; et al. Tumour-associated mutant p53 drives the Warburg effect. Nat. Commun. 2013, 4, 2935. [Google Scholar] [CrossRef] [PubMed]
- Kollareddy, M.; Dimitrova, E.; Vallabhaneni, K.C.; Chan, A.; Le, T.; Chauhan, K.M.; Carrero, Z.I.; Ramakrishnan, G.; Watabe, K.; Haupt, Y.; et al. Regulation of nucleotide metabolism by mutant p53 contributes to its gain-of-function activities. Nat. Commun. 2015, 6, 7389. [Google Scholar] [CrossRef] [PubMed]
- Yahagi, N.; Shimano, H.; Matsuzaka, T.; Najima, Y.; Sekiya, M.; Nakagawa, Y.; Ide, T.; Tomita, S.; Okazaki, H.; Tamura, Y.; et al. p53 activation in adipocytes of obese mice. J. Biol. Chem. 2003, 278, 25395–25400. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, S.; Fergusson, M.M.; Finkel, T. Nutrient availability regulates sirt1 through a forkhead-dependent pathway. Science 2004, 306, 2105–2108. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhao, X.; Gao, X.; Mei, Y.; Wu, M. A new role of p53 in regulating lipid metabolism. J. Mol. Cell Biol. 2013, 5, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; LaGory, E.L.; Kenzelmann Broz, D.; Bieging, K.T.; Brady, C.A.; Link, N.; Abrams, J.M.; Giaccia, A.J.; Attardi, L.D. Analysis of p53 transactivation domain mutants reveals ACAD11 as a metabolic target important for p53 pro-survival function. Cell Rep. 2015, 10, 1096–1109. [Google Scholar] [CrossRef] [PubMed]
- Assaily, W.; Rubinger, D.A.; Wheaton, K.; Lin, Y.; Ma, W.; Xuan, W.; Brown-Endres, L.; Tsuchihara, K.; Mak, T.W.; Benchimol, S. ROS-mediated p53 induction of lpin1 regulates fatty acid oxidation in response to nutritional stress. Mol. Cell 2011, 44, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; He, Y.; Jin, A.; Tikunov, A.P.; Zhou, L.; Tollini, L.A.; Leslie, P.; Kim, T.H.; Li, L.O.; Coleman, R.A.; et al. Ribosomal protein-Mdm2-p53 pathway coordinates nutrient stress with lipid metabolism by regulating mcd and promoting fatty acid oxidation. Proc. Natl. Acad. Sci. USA 2014, 111, E2414–E2422. [Google Scholar] [CrossRef] [PubMed]
- Deisenroth, C.; Itahana, Y.; Tollini, L.; Jin, A.; Zhang, Y. p53-inducible DHRS3 is an endoplasmic reticulum protein associated with lipid droplet accumulation. J. Biol. Chem. 2011, 286, 28343–28356. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, R.D.; Rother, K.; Muller, G.A.; Engeland, K. The retinal dehydrogenase/reductase retSDR1/DHRS3 gene is activated by p53 and p63 but not by mutants derived from tumors or EEC/ADULT malformation syndromes. Cell Cycle 2010, 9, 2177–2188. [Google Scholar] [CrossRef] [PubMed]
- Bist, A.; Fielding, C.J.; Fielding, P.E. p53 regulates caveolin gene transcription, cell cholesterol, and growth by a novel mechanism. Biochemistry 2000, 39, 1966–1972. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, P.P.; Sonati, F.; Rivi, R.; Mason, P.; Grosveld, F.; Luzzatto, L. Targeted disruption of the housekeeping gene encoding glucose 6-phosphate dehydrogenase (G6PD): G6PD is dispensable for pentose synthesis but essential for defense against oxidative stress. EMBO J. 1995, 14, 5209–5215. [Google Scholar] [PubMed]
- Stanton, R.C. Glucose-6-phosphate dehydrogenase, NADPH, and cell survival. IUBMB Life 2012, 64, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, X.; Zhang, X.; Fan, R.; Gu, H.; Shi, Y.; Liu, H. Glucose-6-phosphate dehydrogenase expression is correlated with poor clinical prognosis in esophageal squamous cell carcinoma. Eur. J. Surg. Oncol. 2015, 41, 1293–1299. [Google Scholar] [CrossRef] [PubMed]
- Eberle, D.; Hegarty, B.; Bossard, P.; Ferre, P.; Foufelle, F. SREBP transcription factors: Master regulators of lipid homeostasis. Biochimie 2004, 86, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D.; Bashmakov, Y.; Shimomura, I.; Shimano, H. Regulation of sterol regulatory element binding proteins in livers of fasted and refed mice. Proc. Natl. Acad. Sci. USA 1998, 95, 5987–5992. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Prins, R.M.; Dang, J.; Kuga, D.; Iwanami, A.; Soto, H.; Lin, K.Y.; Huang, T.T.; Akhavan, D.; Hock, M.B.; et al. EGFR signaling through an Akt-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci. Signal. 2009, 2. [Google Scholar] [CrossRef] [PubMed]
- Ettinger, S.L.; Sobel, R.; Whitmore, T.G.; Akbari, M.; Bradley, D.R.; Gleave, M.E.; Nelson, C.C. Dysregulation of sterol response element-binding proteins and downstream effectors in prostate cancer during progression to androgen independence. Cancer Res. 2004, 64, 2212–2221. [Google Scholar] [CrossRef] [PubMed]
- Schug, T.T.; Li, X. Sirtuin 1 in lipid metabolism and obesity. Ann. Med. 2011, 43, 198–211. [Google Scholar] [CrossRef] [PubMed]
- Purushotham, A.; Schug, T.T.; Xu, Q.; Surapureddi, S.; Guo, X.; Li, X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009, 9, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, S.; Blander, G.; Tse, J.G.; Krieger, M.; Guarente, L. SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol. Cell 2007, 28, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Ponugoti, B.; Kim, D.H.; Xiao, Z.; Smith, Z.; Miao, J.; Zang, M.; Wu, S.Y.; Chiang, C.M.; Veenstra, T.D.; Kemper, J.K. SIRT1 deacetylates and inhibits SREBP-1C activity in regulation of hepatic lipid metabolism. J. Biol. Chem. 2010, 285, 33959–33970. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.K.; Yang, F.; Jiang, K.; Ji, J.Y.; Watts, J.L.; Purushotham, A.; Boss, O.; Hirsch, M.L.; Ribich, S.; Smith, J.J.; et al. Conserved role of SIRT1 orthologs in fasting-dependent inhibition of the lipid/cholesterol regulator SREBP. Genes Dev. 2010, 24, 1403–1417. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.E.; Thorburn, A.W.; Britt, K.L.; Hewitt, K.N.; Wreford, N.G.; Proietto, J.; Oz, O.K.; Leury, B.J.; Robertson, K.M.; Yao, S.; et al. Aromatase-deficient (ArKO) mice have a phenotype of increased adiposity. Proc. Natl. Acad. Sci. USA 2000, 97, 12735–12740. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Pei, Z.; Mohsen, A.W.; Watkins, P.; Murdoch, G.; van Veldhoven, P.P.; Ensenauer, R.; Vockley, J. Identification and characterization of new long chain acyl-CoA dehydrogenases. Mol. Genet. Metab. 2011, 102, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Peterfy, M.; Phan, J.; Xu, P.; Reue, K. Lipodystrophy in the FLD mouse results from mutation of a new gene encoding a nuclear protein, lipin. Nat. Genet. 2001, 27, 121–124. [Google Scholar] [PubMed]
- Finck, B.N.; Gropler, M.C.; Chen, Z.; Leone, T.C.; Croce, M.A.; Harris, T.E.; Lawrence, J.C., Jr.; Kelly, D.P. Lipin 1 is an inducible amplifier of the hepatic PGC-1α/PPARα regulatory pathway. Cell Metab. 2006, 4, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Laurent, G.; German, N.J.; Saha, A.K.; de Boer, V.C.; Davies, M.; Koves, T.R.; Dephoure, N.; Fischer, F.; Boanca, G.; Vaitheesvaran, B.; et al. SIRT4 coordinates the balance between lipid synthesis and catabolism by repressing malonyl CoA decarboxylase. Mol. Cell 2013, 50, 686–698. [Google Scholar] [CrossRef] [PubMed]
- Lindstrom, M.S.; Jin, A.; Deisenroth, C.; White Wolf, G.; Zhang, Y. Cancer-associated mutations in the Mdm2 zinc finger domain disrupt ribosomal protein interaction and attenuate Mdm2-induced p53 degradation. Mol. Cell. Biol. 2007, 27, 1056–1068. [Google Scholar] [CrossRef] [PubMed]
- Zolfaghari, R.; Chen, Q.; Ross, A.C. DHRS3, a retinal reductase, is differentially regulated by retinoic acid and lipopolysaccharide-induced inflammation in THP-1 cells and rat liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G578–G588. [Google Scholar] [CrossRef] [PubMed]
- Beilstein, F.; Carriere, V.; Leturque, A.; Demignot, S. Characteristics and functions of lipid droplets and associated proteins in enterocytes. Exp. Cell Res. 2016, 340, 172–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Rudick, M.; Anderson, R.G. Multiple functions of caveolin-1. J. Biol. Chem. 2002, 277, 41295–41298. [Google Scholar] [CrossRef] [PubMed]
- Murata, M.; Peranen, J.; Schreiner, R.; Wieland, F.; Kurzchalia, T.V.; Simons, K. VIP21/caveolin is a cholesterol-binding protein. Proc. Natl. Acad. Sci. USA 1995, 92, 10339–10343. [Google Scholar] [CrossRef] [PubMed]
- Fielding, C.J.; Bist, A.; Fielding, P.E. Intracellular cholesterol transport in synchronized human skin fibroblasts. Biochemistry 1999, 38, 2506–2513. [Google Scholar] [CrossRef] [PubMed]
- Freeman, M.R.; Yang, W.; di Vizio, D. Caveolin-1 and prostate cancer progression. Adv. Exp. Med. Biol. 2012, 729, 95–110. [Google Scholar] [PubMed]
- Anwar, S.L.; Wahyono, A.; Aryandono, T.; Haryono, S.J. Caveolin-1 in breast cancer: Single molecule regulation of multiple key signaling pathways. Asian Pac. J. Cancer Prev. 2015, 16, 6803–6812. [Google Scholar] [CrossRef] [PubMed]
- Nwosu, Z.C.; Ebert, M.P.; Dooley, S.; Meyer, C. Caveolin-1 in the regulation of cell metabolism: A cancer perspective. Mol. Cancer 2016, 15, 71. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Reumers, J.; Couceiro, J.R.; de Smet, F.; Gallardo, R.; Rudyak, S.; Cornelis, A.; Rozenski, J.; Zwolinska, A.; Marine, J.C.; et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 2011, 7, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Do, P.M.; Varanasi, L.; Fan, S.; Li, C.; Kubacka, I.; Newman, V.; Chauhan, K.; Daniels, S.R.; Boccetta, M.; Garrett, M.R.; et al. Mutant p53 cooperates with ETS2 to promote etoposide resistance. Genes Dev. 2012, 26, 830–845. [Google Scholar] [CrossRef] [PubMed]
- Walerych, D.; Lisek, K.; Sommaggio, R.; Piazza, S.; Ciani, Y.; Dalla, E.; Rajkowska, K.; Gaweda-Walerych, K.; Ingallina, E.; Tonelli, C.; et al. Proteasome machinery is instrumental in a common gain-of-function program of the p53 missense mutants in cancer. Nat. Chem. Biol. 2016, 18, 897–909. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerod, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Wang, J.; Zhao, M.; Xie, T.X.; Tanaka, N.; Sano, D.; Patel, A.A.; Ward, A.M.; Sandulache, V.C.; Jasser, S.A.; et al. Gain-of-function mutant p53 promotes cell growth and cancer cell metabolism via inhibition of AMPK activation. Mol. Cell 2014, 54, 960–974. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.H.; Gammon, S.R.; Knippers, J.D.; Paulsen, S.R.; Rubink, D.S.; Winder, W.W. Phosphorylation-activity relationships of AMPK and acetyl-CoA carboxylase in muscle. J. Appl. Physiol. 2002, 92, 2475–2482. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, S.; Mihaylova, M.M.; Zheng, B.; Hou, X.; Jiang, B.; Park, O.; Luo, Z.; Lefai, E.; Shyy, J.Y.; et al. AMPK phosphorylates and inhibits srebp activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011, 13, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Parrales, A.; Ranjan, A.; Iyer, S.V.; Padhye, S.; Weir, S.J.; Roy, A.; Iwakuma, T. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat. Cell Biol. 2016, 18, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.V.; Parrales, A.; Begani, P.; Narkar, A.; Adhikari, A.S.; Martinez, L.A.; Iwakuma, T. Allele-specific silencing of mutant p53 attenuates dominant-negative and gain-of-function activities. Oncotarget 2016, 7, 5401–5415. [Google Scholar] [PubMed]
- Zhu, H.B.; Yang, K.; Xie, Y.Q.; Lin, Y.W.; Mao, Q.Q.; Xie, L.P. Silencing of mutant p53 by siRNA induces cell cycle arrest and apoptosis in human bladder cancer cells. World J. Surg. Oncol. 2013, 11, 22. [Google Scholar] [CrossRef] [PubMed]
- Bossi, G.; Lapi, E.; Strano, S.; Rinaldo, C.; Blandino, G.; Sacchi, A. Mutant p53 gain of function: Reduction of tumor malignancy of human cancer cell lines through abrogation of mutant p53 expression. Oncogene 2006, 25, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.Y.; Vidnovic, N.; Ellisen, L.W.; Leong, C.O. Mutant p53 mediates survival of breast cancer cells. Br. J. Cancer 2009, 101, 1606–1612. [Google Scholar] [CrossRef] [PubMed]
- Sanfilippo, K.M.; Keller, J.; Gage, B.F.; Luo, S.; Wang, T.F.; Moskowitz, G.; Gumbel, J.; Blue, B.; O’Brian, K.; Carson, K.R. Statins are associated with reduced mortality in multiple myeloma. J. Clin. Oncol. 2016, 19, JCO683482. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.; Hill, M.M.; Roberts, M.J.; Gardiner, R.A.; Brown, A.J. Statins: Protectors or pretenders in prostate cancer? Trends Endocrinol. Metab. 2014, 25, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Baandrup, L.; Dehlendorff, C.; Friis, S.; Olsen, J.H.; Kjaer, S.K. Statin use and risk for ovarian cancer: A Danish nationwide case-control study. Br. J. Cancer 2015, 112, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.L.; Hsieh, M.C.; Chow, J.M.; Liu, S.H.; Chang, C.L.; Wu, S.Y. Statins improve outcomes of nonsurgical curative treatments in hepatocellular carcinoma patients. Medicine 2016, 95, e4639. [Google Scholar] [CrossRef] [PubMed]
- Goldvaser, H.; Rizel, S.; Hendler, D.; Neiman, V.; Shepshelovich, D.; Shochat, T.; Sulkes, A.; Brenner, B.; Yerushalmi, R. The association between treatment for metabolic disorders and breast cancer characteristics. Int. J. Endocrinol. 2016, 2016, 4658469. [Google Scholar] [CrossRef] [PubMed]
- Nishi, K.; Suzuki, K.; Sawamoto, J.; Tokizawa, Y.; Iwase, Y.; Yumita, N.; Ikeda, T. Inhibition of fatty acid synthesis induces apoptosis of human pancreatic cancer cells. Anticancer Res. 2016, 36, 4655–4660. [Google Scholar] [CrossRef] [PubMed]
- Camarda, R.; Zhou, A.Y.; Kohnz, R.A.; Balakrishnan, S.; Mahieu, C.; Anderton, B.; Eyob, H.; Kajimura, S.; Tward, A.; Krings, G.; et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat. Med. 2016, 22, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.H.; Sengupta, K.; Li, C.; Kim, H.S.; Cao, L.; Xiao, C.; Kim, S.; Xu, X.; Zheng, Y.; Chilton, B.; et al. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell 2008, 14, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Firestein, R.; Blander, G.; Michan, S.; Oberdoerffer, P.; Ogino, S.; Campbell, J.; Bhimavarapu, A.; Luikenhuis, S.; de Cabo, R.; Fuchs, C.; et al. The SIRT1 deacetylase suppresses intestinal tumorigenesis and colon cancer growth. PLoS ONE 2008, 3, e2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McPherson, S.J.; Wang, H.; Jones, M.E.; Pedersen, J.; Iismaa, T.P.; Wreford, N.; Simpson, E.R.; Risbridger, G.P. Elevated androgens and prolactin in aromatase-deficient mice cause enlargement, but not malignancy, of the prostate gland. Endocrinology 2001, 142, 2458–2467. [Google Scholar] [CrossRef] [PubMed]
- Fowler, K.A.; Gill, K.; Kirma, N.; Dillehay, D.L.; Tekmal, R.R. Overexpression of aromatase leads to development of testicular Leydig cell tumors: An in vivo model for hormone-mediated testicularcancer. Am. J. Pathol. 2000, 156, 347–353. [Google Scholar] [CrossRef]
- Diaz-Cruz, E.S.; Sugimoto, Y.; Gallicano, G.I.; Brueggemeier, R.W.; Furth, P.A. Comparison of increased aromatase versus ERα in the generation of mammary hyperplasia and cancer. Cancer Res. 2011, 71, 5477–5487. [Google Scholar] [CrossRef] [PubMed]
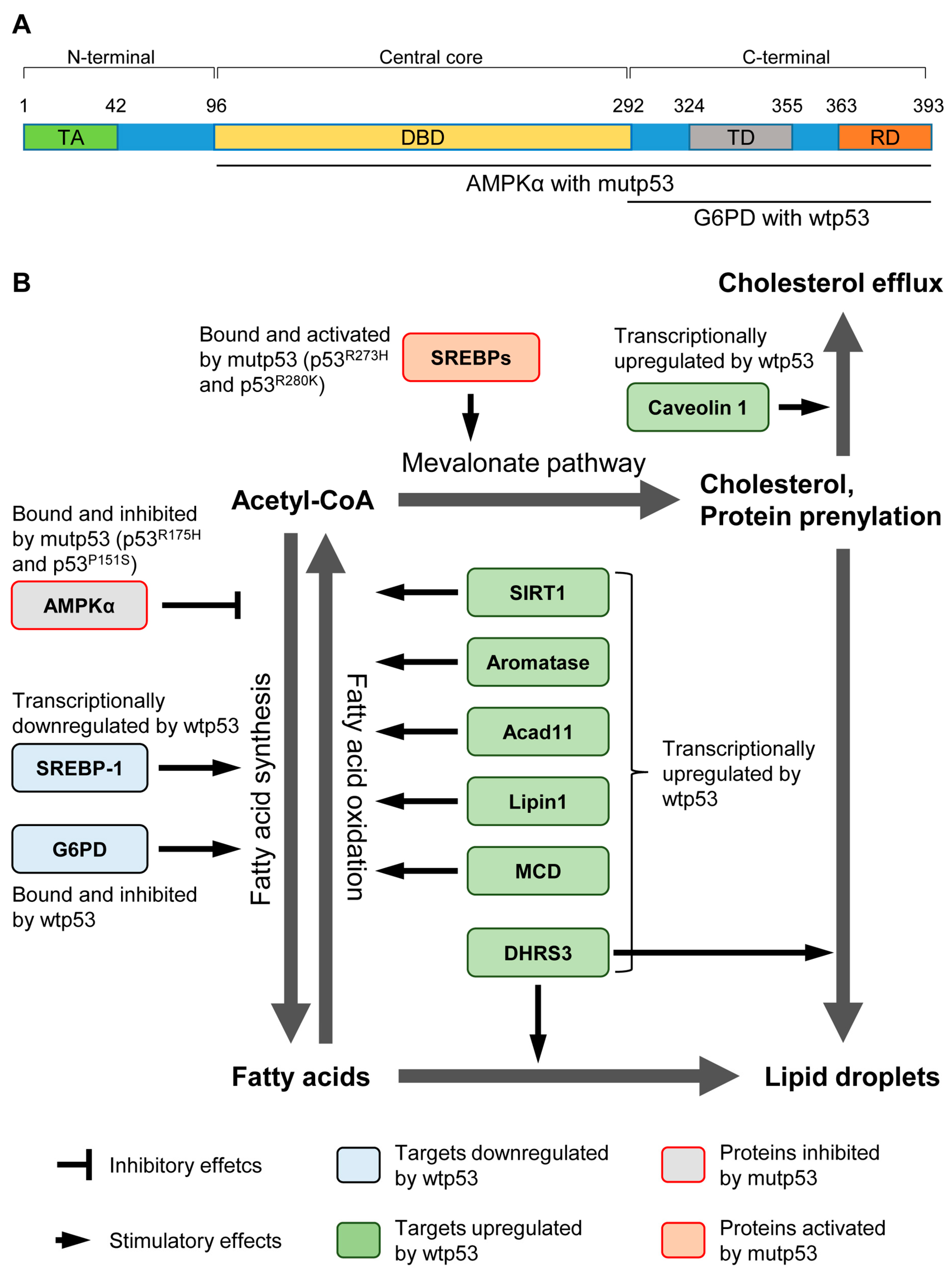

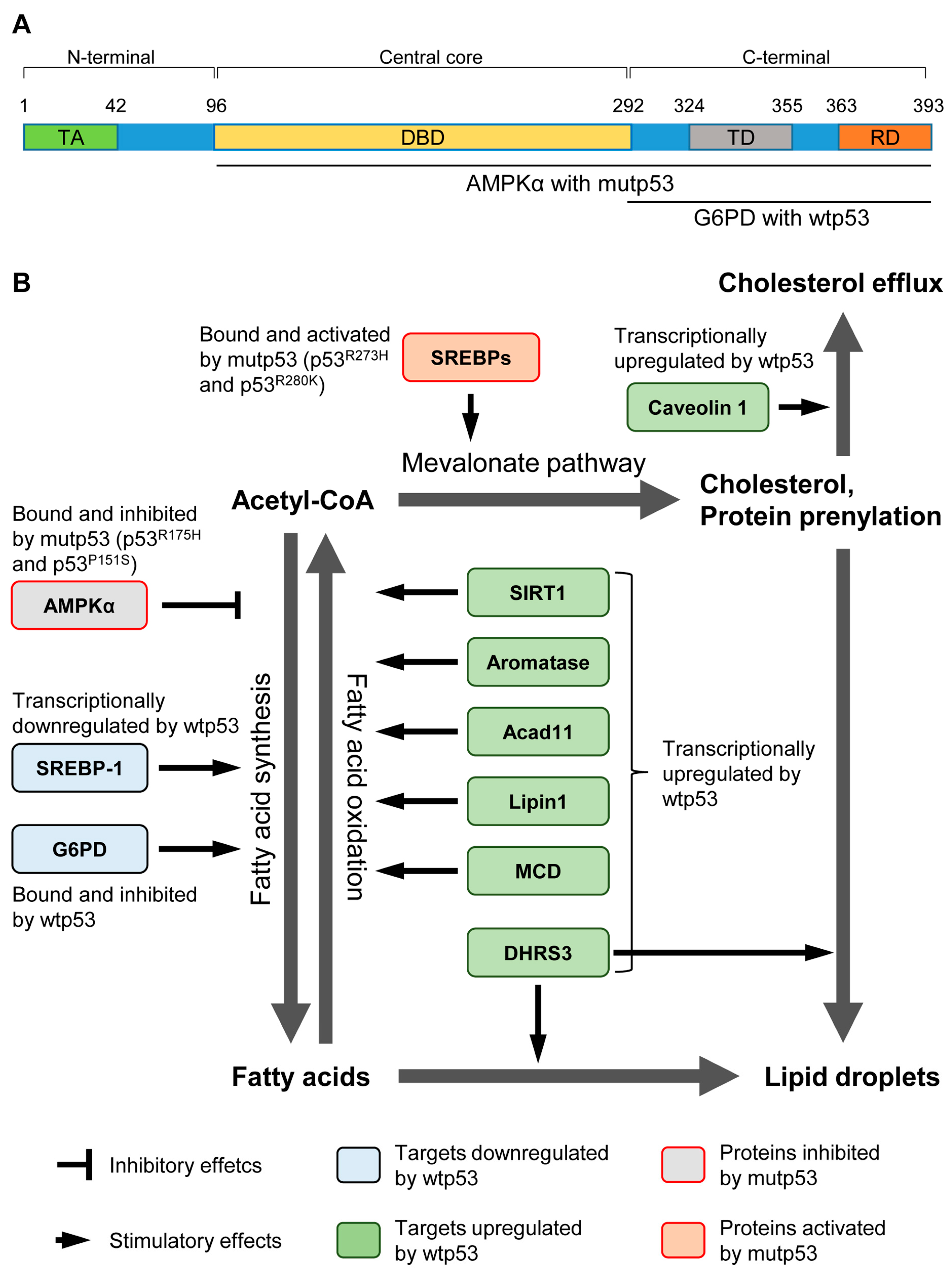{kind=link}
| Targets | Effects of Wtp53 | Biological Consequence | Reference |
|---|---|---|---|
| G6PD | Inhibit the activity by physical binding. | Loss of p53 activates G6PD and the pentose pathway, leading to lipid accumulation in the liver. | [16] |
| SREBP-1 | Transcriptionally repress the expression. | Disruption of p53 in ob/ob mice restores the expression of lipogenic enzymes regulated by SREBP-1. | [20] |
| SIRT1 | A complex of p53 and Foxo3a transactivates SIRT1. | In p53−/− mice, nutrient starvation fails to increase SIRT1. It remains unclear whether increased lipid accumulation in p53−/− mice is due to attenuated SIRT1 levels. | [21] |
| Aromatase | Transcriptionally increase the expression. | p53−/− mice have lower levels of aromatase, resulting in higher levels of testosterone and lipid accumulation, which is nullified by transgenic expression of aromatase. | [22] |
| Acad11 | Transcriptionally increase the expression. | Although Acad11 plays a key role in p53-mediated OXPHOS and cell survival upon glucose starvation, it is unclear whether increased Acad11 levels by p53 enhance fatty acid β-oxidation and how enhanced fatty acid β-oxidation contributes to cell survival. | [23] |
| Lipin1 | Transcriptionally increase the expression. | Glucose restriction in C2C12 cells phosphorylates p53, leading to upregulation of Lipin1 and fatty acid oxidation. | [24] |
| MCD | Transcriptionally increase the expression. | Mdm2C305F mice show attenuated MCD induction and increased fatty acid accumulation in the liver under ribosomal stress, due to lack of inhibitory effects of RPs on Mdm2 and reduction in the p53 activity. | [25] |
| DHRS3 | Transcriptionally increase the expression. | Activation of p53 upregulates DHRS3 which is associated with lipid droplets accumulation. | [26,27] |
| Caveolin 1 | Transcriptionally increase the expression. | Overexpression of p53 upregulates Caveolin 1, leading to redution in intracellular free choleserol and viable cell growth. | [28] |
| Targets | Effects of Mutp53 | Biological Consequence | References |
|---|---|---|---|
| SREBPs | Bind and activate the transcription activity. | In breast cancer cells expressing mutp53, increased activities of SREBPs enhance the mevalonate pathway and accelerate growth in the 3D culture. | [58] |
| AMPK | Bind and inhibit the kinase activity. | GOF p53 mutants bind to and inhibit AMPK activity. It remains unclear how significantly the mutp53’s inhibitory effect on AMPK contributes to fatty acid synthesis and tumor progression. | [59] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parrales, A.; Iwakuma, T. p53 as a Regulator of Lipid Metabolism in Cancer. Int. J. Mol. Sci. 2016, 17, 2074. https://doi.org/10.3390/ijms17122074
Parrales A, Iwakuma T. p53 as a Regulator of Lipid Metabolism in Cancer. International Journal of Molecular Sciences. 2016; 17(12):2074. https://doi.org/10.3390/ijms17122074
Chicago/Turabian StyleParrales, Alejandro, and Tomoo Iwakuma. 2016. "p53 as a Regulator of Lipid Metabolism in Cancer" International Journal of Molecular Sciences 17, no. 12: 2074. https://doi.org/10.3390/ijms17122074