
Estrogenic Endocrine Disrupting Chemicals Influencing NRF1 Regulated Gene Networks in the Development of Complex Human Brain Diseases


Abstract
:1. Introduction
2. Epidemiologic and Experimental Evidence of Brain Health Deficits with Exposure to Estrogenic Endocrine Disruptors (EEDs)
2.1. Estrogens and Brain Health
2.2. Oral Contraceptives and Brain Health
2.3. Hormonal Replacement Therapy and Brain Health
2.4. Bisphenol A and Brain Health
2.5. Phthalates and Brain Health
2.6. Polychlorinated Biphenyls and Brain Health
2.7. Cadmium and Brain Health
2.8. Arsenic and Brain Health
2.9. Manganese and Brain Health
3. Mechanisms of Actions of Estrogenic Endocrine Disruptors (EEDs) on Brain Health Deficits
3.1. NRF1, Mitochondrial Dysfunction, and Neurodegenerative Diseases
3.2. NRF1-Mediated Regulation of Neurogenesis and Synaptogenesis
3.3. Understanding Sex Bias and NRF1 Regulated Genes-EEDs Interactions in AD
4. Conclusions
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- UNEP/WHO. State of the Science of Endocrine Disrupting Chemicals—2012; Bergman, A., Heindel, J.J., Jobling, S., Kidd, K.A., Zoeller, R.T., Eds.; WHO Press: Geneva, Switzerland, 2013; pp. 1–272. [Google Scholar]
- Roy, D.; Palangat, M.; Chen, C.W.; Thomas, R.T.; Colerangle, J.C.; Atkinson, A.; Yan, Z.J. Biochemical and molecular changes at the cellular levels in response to exposure of environmental estrogen-like chemicals. J. Toxicol. Environ. Health 1997, 49, 101–129. [Google Scholar]
- Diamanti-Kandarakis, E.; Bourguignon, J.-P.; Giudice, L.C.; Hauser, R.; Prins, G.S.; Soto, A.M.; Zoeller, R.T.; Gore, A.C. Endocrine-disrupting chemicals: An Endocrine Society scientific statement. Endocr. Rev. 2009, 30, 293–342. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.; Morgan, M.; Yoo, C.; Deoraj, A.; Roy, S.; Yadav, V.K.; Garoub, M.; Assaggaf, H.; Doke, M. Integrated bioinformatics, environmental epidemiologic and genomic approaches to identify environmental and molecular links between endometriosis and breast cancer. Int. J. Mol. Sci. 2015, 16, 25285–25322. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.M. Estradiol and the developing brain. Physiol. Rev. 2008, 88, 91–124. [Google Scholar] [CrossRef] [PubMed]
- Kajta, M.; Wojtowicz, A.K. Impact of endocrine-disrupting chemicals on neural development and the onset of neurological disorders. Pharmacol. Rep. 2013, 65, 1632–1639. [Google Scholar] [CrossRef]
- Masuo, Y.; Ishido, M. Neurotoxicity of endocrine disruptors: Possible involvement in brain development and neurodegeneration. J. Toxicol. Environ. Health B Crit. Rev. 2011, 14, 346–369. [Google Scholar] [CrossRef] [PubMed]
- Okoh, V.; Deoraj, A.; Roy, D. Estrogen-induced ROS mediated redox signaling contributes in the development of breast cancer. Biochem. Biophys. Acta 2011, 1815, 115–133. [Google Scholar] [PubMed]
- Satoh, J.I.; Kawana, N.; Yamamoto, Y. Pathway analysis of ChIP-Seq-Based NRF1 target genes suggests a logical hypothesis of their Involvement in the pathogenesis of neurodegenerative diseases. Gene Regul. Syst. Biol. 2013, 2013, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Pennacchio, L.A.; Loots, G.G.; Nobrega, M.A.; Ovcharenko, I. Predicting tissue-specific enhancers in the human genome. Genome Res. 2007, 17, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Burns, K.A.; Korach, K.S. Estrogen receptors and human disease: An update. Arch. Toxicol. 2012, 86, 1491–1504. [Google Scholar] [CrossRef] [PubMed]
- Gillies, G.E.; McArthur, S. Estrogen actions in the brain and the basis for differential action in men and women: A case for sex-specific medicines. Pharmacol. Rev. 2010, 62, 155–198. [Google Scholar] [CrossRef] [PubMed]
- Arevalo, M.-A.; Azcoitia, I.; Garcia-Segura, L.M. The neuroprotective actions of oestradiol and oestrogen receptors. Neuroscience 2015, 16, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, M.; Haque, A.; Banik, N.L.; Nagarkatti, P.; Nagarkatti, M.; Ray, S.K. Estrogen receptor agonists for attenuation of neuroinflammation and neurodegeneration. Brain Res. Bull. 2014, 109, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Green, P.S.; Simpkins, J.W. Neuroprotective effects of estrogens: Potential mechanisms of action. Int. J. Dev. Neurosci. 2000, 18, 347–358. [Google Scholar] [CrossRef]
- Hristian, C.; Ruber, J.G.; Alter, W.; Schugguel, T.; Chneeberger, S.; Ohannes, J.; Uber, C.H. Production and actions of estrogens. N. Engl. J. Med. 2002, 346, 340–352. [Google Scholar]
- Pozzi, S.; Benedusi, V.; Maggi, A.; Vegeto, E. Estrogen action in neuroprotection and brain inflammation. Ann. N. Y. Acad. Sci. 2006, 1089, 302–323. [Google Scholar] [CrossRef] [PubMed]
- Wise, P.M.; Dubal, D.B.; Wilson, M.E.; Rau, S.W.; Liu, Y. Estrogens: Trophic and protective factors in the adult brain. Front. Neuroendocrinol. 2001, 22, 33–66. [Google Scholar] [CrossRef] [PubMed]
- Hojo, Y.; Murakami, G.; Mukai, H.; Higo, S.; Hatanaka, Y.; Ogiue-Ikeda, M.; Ishii, H.; Kimoto, T.; Kawato, S. Estrogen synthesis in the brain-role in synaptic plasticity and memory. Stud. Surf. Sci. Catal. 2008, 77, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Ogiue-Ikeda, M.; Tanabe, N.; Mukai, H.; Hojo, Y.; Murakami, G.; Tsurugizawa, T.; Takata, N.; Kimoto, T.; Kawato, S. Rapid modulation of synaptic plasticity by estrogens as well as endocrine disrupters in hippocampal neurons. Brain Res. Rev. 2008, 57, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Mukai, H.; Kimoto, T.; Hojo, Y.; Kawato, S.; Murakami, G.; Higo, S.; Hatanaka, Y.; Ogiue-Ikeda, M. Modulation of synaptic plasticity by brain estrogen in the hippocampus. Biochim. Biophys. Acta—Gen. Subj. 2010, 1800, 1030–1044. [Google Scholar] [CrossRef] [PubMed]
- Parducz, A.; Hajszan, T.; MacLusky, N.J.; Hoyk, Z.; Csakvari, E.; Kurunczi, A.; Prange-Kiel, J.; Leranth, C. Synaptic remodeling induced by gonadal hormones: Neuronal plasticity as a mediator of neuroendocrine and behavioral responses to steroids. Neuroscience 2006, 138, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Leranth, C.; Petnehazy, O.; MacLusky, N.J. Gonadal hormones affect spine synaptic density in the CA1 hippocampal subfield of male rats. J. Neurosci. 2003, 23, 1588–1592. [Google Scholar] [PubMed]
- MacLusky, N.J.; Hajszan, T.; Prange-Kiel, J.; Leranth, C. Androgen modulation of hippocampal synaptic plasticity. Neuroscience 2006, 138, 957–965. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.; Beyer, C. Neuroprotection by estrogen in the brain: The mitochondrial compartment as presumed therapeutic target. J. Neurochem. 2009, 110, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Grimm, A.; Schmitt, K.; Lang, U.E.; Mensah-Nyagan, A.G.; Eckert, A. Improvement of neuronal bioenergetics by neurosteroids: Implications for age-related neurodegenerative disorders. Biochim. Biophys. Acta—Mol. Basis Dis. 2014, 1842, 2427–2438. [Google Scholar] [CrossRef] [PubMed]
- Simpkins, J.W.; Yi, K.D.; Yang, S.; Dykens, J.A. Mitochondrial mechanisms of estrogen neuroprotection. Biochim. Biophys. Acta 2011, 1800, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Barrett-Connor, E.; Goodman-Gruen, D. Cognitive function and endogenous sex hormones in older women. J. Am. Geriatr. Soc. 1999, 47, 1289–1293. [Google Scholar] [CrossRef] [PubMed]
- Cereda, E.; Barichella, M.; Cassani, E.; Caccialanza, R.; Pezzoli, G. Reproductive factors and clinical features of Parkinson’s disease. Parkinsonism Relat. Disord. 2013, 19, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Heys, M.; Jiang, C.; Cheng, K.K.; Zhang, W.; Yeung, S.L.A.; Lam, T.H.; Leung, G.M.; Schooling, C.M. Life long endogenous estrogen exposure and later adulthood cognitive function in a population of naturally postmenopausal women from Southern China: The Guangzhou biobank cohort study. Psychoneuroendocrinology 2011, 36, 864–873. [Google Scholar] [CrossRef] [PubMed]
- Low, L.F. Reproductive period and cognitive function in a representative sample of naturally postmenopausal women aged 60–64 years. Climacteric 2005, 8, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, M.E.; Lipton, R.B.; Santoro, N.; McConnell, D.S.; Derby, C.A.; Katz, M.J.; Baigi, K.; Saunders-Pullman, R. Endogenous estradiol is associated with verbal memory in nondemented older men. Brain Cogn. 2011, 76, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Manly, J.J.; Merchant, C.A.; Jacobs, D.M.; Small, S.A.; Bell, K.; Ferin, M.; Mayeux, R. Endogenous estrogen levels and Alzheimer’s disease among postmenopausal women. Neurology 2000, 54, 833–837. [Google Scholar] [CrossRef] [PubMed]
- De Jong, S.; Huisman, M.; Sutedja, N.; van Der Kooi, A.; de Visser, M.; Schelhaas, J.; van Der Schouw, Y.; Veldink, J.; van Den Berg, L. Endogenous female reproductive hormones and the risk of amyotrophic lateral sclerosis. J. Neurol. 2013, 260, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Geerlings, M.I.; Ruitenberg, A.; Witteman, J.C.; van Swieten, J.C.; Hofman, A.; van Duijn, C.M.; Breteler, M.M.; Launer, L.J. Reproductive period and risk of dementia in postmenopausal women. JAMA 2001, 285, 1475–1481. [Google Scholar] [CrossRef] [PubMed]
- Schupf, N.; Winsten, S.; Patel, B.; Pang, D.; Ferin, M.; Zigman, W.B.; Silverman, W.; Mayeux, R. Bioavailable estradiol and age at onset of Alzheimer’s disease in postmenopausal women with Down syndrome. Neurosci. Lett. 2006, 406, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Fox, M.; Berzuini, C.; Knapp, L.A. Cumulative estrogen exposure, number of menstrual cycles, and Alzheimer’s risk in a cohort of British women. Psychoneuroendocrinology 2013, 38, 2973–2982. [Google Scholar] [CrossRef] [PubMed]
- Griksiene, R.; Ruksenas, O. Effects of hormonal contraceptives on mental rotation and verbal fluency. Psychoneuroendocrinology 2011, 36, 1239–1248. [Google Scholar] [CrossRef] [PubMed]
- Pletzer, B.A.; Kerschbaum, H.H. 50 years of hormonal contraception—Time to find out, what it does to our brain. Front. Neurosci. 2014, 8, 256. [Google Scholar] [CrossRef] [PubMed]
- Beltz, A.M.; Hampson, E.; Berenbaum, S.A. Oral contraceptives and cognition: A role for ethinyl estradiol. Horm. Behav. 2015, 74, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Egan, K.R.; Gleason, C.E. Longer duration of hormonal contraceptive use predicts better cognitive outcomes later in life. J. Women’s Health 2012, 21, 1259–1266. [Google Scholar] [CrossRef] [PubMed]
- Warren, A.M.; Gurvich, C.; Worsley, R.; Kulkarni, J. A systematic review of the impact of oral contraceptives on cognition. Contraception 2014, 90, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Maki, P.M.; Henderson, V.W. Hormone therapy, dementia, and cognition: The Women’s Health Initiative ten years on. Climacteric 2012, 15, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Cao, G.L.; Marshall, C.; Hu, G. Hypothesis: Multiple factors are associated with the lack of any beneficial effects of oestrogen-replacement therapy in the late postmenopausal stage. Clin. Exp. Pharmacol. Physiol. 2010, 37, 873–876. [Google Scholar] [CrossRef] [PubMed]
- Shumaker, S.A.; Legault, C.; Rapp, S.R.; Thal, L.; Wallace, R.B.; Ockene, J.K.; Hendrix, S.L.; Jackson, R.D.; Kotchen, J.M.; Wassertheil-Smoller, S.; et al. Estrogen plus progestin and the incidence of dementia and mild cognitive impairment in postmenopausal women. JAMA 2003, 289, 2651–2662. [Google Scholar] [CrossRef] [PubMed]
- Shumaker, S.A.; Legault, C.; Kuller, L.; Rapp, S.R.; Thal, L.; Lane, D.S.; Fillit, H.; Stefanick, M.L.; Hendrix, S.L.; Lewis, C.E.; et al. Conjugated equine estrogens and incidence of probable dementia and mild cognitive impairment in postmenopausal women: Women’s Health Initiative Memory Study. JAMA 2004, 291, 2947–2958. [Google Scholar] [CrossRef] [PubMed]
- Resnick, S.M.; Maki, P.M.; Rapp, S.R.; Espeland, M.; Brunner, R.; Coker, L.H.; Granek, I.; Hogan, P.; Ockene, J.K.; Shumaker, S. Effects of combination estrogen plus progestin hormone treatment on cognition and affect. J. Clin. Endocrinol. Metab. 2006, 91, 1802–1810. [Google Scholar] [CrossRef] [PubMed]
- Resnick, S.M.; Espeland, M.A.; An, Y.; Maki, P.M.; Coker, L.H.; Jackson, R.; Stefanick, M.L.; Wallace, R.; Rapp, S.R. Effects of conjugated equine estrogens on cognition and affect in postmenopausal women with prior hysterectomy. J. Clin. Endocrinol. Metab. 2009, 94, 4152–4161. [Google Scholar] [CrossRef] [PubMed]
- Yaffe, K.; Vittinghoff, E.; Ensrud, K.E.; Johnson, K.C.; Diem, S.; Hanes, V.; Grady, D. Effects of ultra-low-dose transdermal estradiol on cognition and health-related quality of life. Arch. Neurol. 2006, 63, 945–950. [Google Scholar] [CrossRef] [PubMed]
- Viscoli, C.M.; Brass, L.M.; Kernan, W.N.; Sarrel, P.M.; Suissa, S.; Horwitz, R.I. Estrogen therapy and risk of cognitive decline: Results from the Women’s Estrogen for Stroke Trial (WEST). Am. J. Obstet. Gynecol. 2005, 192, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Almeida, O.P.; Lautenschlager, N.T.; Vasikaran, S.; Leedman, P.; Gelavis, A.; Flicker, L. A 20-week randomized controlled trial of estradiol replacement therapy for women aged 70 years and older: Effect on mood, cognition and quality of life. Neurobiology 2006, 27, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Pefanco, M.A.; Kenny, A.M.; Kaplan, R.F.; Kuchel, G.; Walsh, S.; Kleppinger, A.; Prestwood, K. The effect of 3-year treatment with 0.25 mg/day of micronized 17 β-estradiol on cognitive function in older postmenopausal women. J. Am. Geriatr. Soc. 2007, 55, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Phillips, S.M.; Sherwin, B.B. Effects of estrogen on memory function in surgically menopausal women. Psychoneuroendocrinology 1992, 17, 485–495. [Google Scholar] [CrossRef]
- Sherwin, B.B. Estrogen and/or androgen replacement therapy and cognitive functioning in surgically menopausal women. Psychoneuroendocrinology 1988, 13, 345–357. [Google Scholar] [CrossRef]
- Shao, H.; Breitner, J.C.S.; Whitmer, R.A.; Wang, J.; Hayden, K.; Wengreen, H.; Corcoran, C.; Tschanz, J.; Norton, M.; Munger, R.; et al. Hormone therapy and Alzheimer disease dementia: New findings from the Cache County Study. Neurology 2012, 79, 1846–1852. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Jackson, J.W.; Grodstein, F.; Blacker, D.; Weuve, J. Postmenopausal hormone therapy is not associated with risk of all-cause dementia and Alzheimer’s disease. Epidemiol. Rev. 2014, 36, 83–103. [Google Scholar] [CrossRef] [PubMed]
- Lundin, J.I.; Ton, T.G.; LaCroix, A.Z.; Longstreth, W.; Franklin, G.M.; Swanson, P.D.; Smith-Weller, T.; Racette, B.A.; Checkoway, H. Formulations of hormone therapy and risk of Parkinson’s disease. Mov. Disord. 2014, 29, 1631–1636. [Google Scholar] [CrossRef] [PubMed]
- Blitshteyn, S.; Crook, J.E.; Jaeckle, K.A. Is there an association between meningioma and hormone replacement therapy? J. Clin. Oncol. 2008, 26, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Henderson, V.W.; Ala, T.; Sainani, K.L.; Bernstein, A.L.; Stephenson, B.S.; Rosen, A.C.; Farlow, M.R. Raloxifene for women with Alzheimer disease: A randomized controlled pilot trial. Neurology 2015, 85, 1937–1944. [Google Scholar] [CrossRef] [PubMed]
- Agency for Toxic Substances and Disease Registry (ATSDR). ATSDR Toxicological Profile for Phenol; Agency for Toxic Substances and Disease Registry (ATSDR): Atlanta, GA, USA, 2008; pp. 1–269.
- Calafat, A.M.; Ye, X.; Wong, L.Y.; Reidy, J.A.; Needham, L.L. Exposure of the U.S. population to Bisphenol A and 4-tertiary-octylphenol: 2003–2004. Environ. Health Perspect. 2008, 116, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Rubin, B.S. Bisphenol A: An endocrine disruptor with widespread exposure and multiple effects. J. Steroid Biochem. Mol. Biol. 2011, 127, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Le, H.H.; Carlson, E.M.; Chua, J.P.; Belcher, S.M. Bisphenol A is released from polycarbonate drinking bottles and mimics the neurotoxic actions of estrogen in developing cerebellar neurons. Toxicol. Lett. 2008, 176, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Nakashima, M.N.; Takahashi, M.; Kuroda, N.; Nakashima, K. Determination of bisphenol A in rat brain by microdialysis and column switching high-performance liquid chromatography with fluorescence detection. Biomed. Chromatogr. 2002, 16, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.S.; Sapienza, P.P.; Ross, I.A.; Johnson, W.; Luu, H.M.D.; Hutter, J.C. Distribution of bisphenol A in the neuroendocrine organs of female rats. Toxicol. Ind. Health 2004, 20, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Huttenlocher, P.R.; Dabholkar, A.S. Regional differences in synaptogenesis in human cerebral cortex. J. Comp. Neurol. 1997, 387, 167–178. [Google Scholar] [CrossRef]
- Eriksson, P.S.; Perfilieva, E.; Björk-Eriksson, T.; Alborn, A.M.; Nordborg, C.; Peterson, D.A.; Gage, F.H. Neurogenesis in the adult human hippocampus. Nat. Med. 1998, 4, 1313–1317. [Google Scholar] [CrossRef] [PubMed]
- Bowman, R.E.; Luine, V.; Diaz Weinstein, S.; Khandaker, H.; DeWolf, S.; Frankfurt, M. Bisphenol-A exposure during adolescence leads to enduring alterations in cognition and dendritic spine density in adult male and female rats. Horm. Behav. 2015, 69, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Eilam-Stock, T.; Serrano, P.; Frankfurt, M.; Luine, V. Bisphenol-A impairs memory and reduces dendritic spine density in adult male rats. Behav. Neurosci. 2012, 126, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, T.; Frankfurt, M.; Luine, V. Estrogen-induced memory enhancements are blocked by acute bisphenol A in adult female rats: Role of dendritic spines. Endocrinology 2012, 153, 3357–3367. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Niu, R.; Zhu, Y.; Han, H.; Luo, G.; Zhou, B.; Wang, J. Changes in memory and synaptic plasticity induced in male rats after maternal exposure to bisphenol A. Toxicology 2014, 322, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Yang, Y.; Xu, X.; Hu, Y. Effects of uterine and lactational exposure to di-(2-ethylhexyl) phthalate on spatial memory and NMDA receptor of hippocampus in mice. Horm. Behav. 2015, 71, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Liu, X.; Zhang, Q.; Zhang, G.; Lu, Y.; Ruan, Q.; Dong, F.; Yang, Y. Sex-specific effects of bisphenol-A on memory and synaptic structural modification in hippocampus of adult mice. Horm. Behav. 2013, 63, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Elsworth, J.D.; Jentsch, J.D.; Groman, S.M.; Roth, R.H.; Redmond, E.D.; Leranth, C. Low circulating levels of bisphenol-A induce cognitive deficits and loss of asymmetric spine synapses in dorsolateral prefrontal cortex and hippocampus of adult male monkeys. J. Comp. Neurol. 2015, 523, 1248–1257. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.J.; Park, H.R.; Kim, T.H.; Yang, W.J.; Lee, J.J.; Choi, S.Y.; Oh, S.B.; Lee, E.; Park, J.H.; Kim, H.P.; et al. High dose bisphenol A impairs hippocampal neurogenesis in female mice across generations. Toxicology 2012, 296, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.E.; Park, H.R.; Gong, E.J.; Choi, S.Y.; Kim, H.S.; Lee, J. Exposure to bisphenol A appears to impair hippocampal neurogenesis and spatial learning and memory. Food Chem. Toxicol. 2011, 49, 3383–3389. [Google Scholar] [CrossRef] [PubMed]
- Kinch, C.D.; Ibhazehiebo, K.; Jeong, J.-H.; Habibi, H.R.; Kurrasch, D.M. Low-dose exposure to bisphenol A and replacement bisphenol S induces precocious hypothalamic neurogenesis in embryonic zebrafish. Proc. Natl. Acad. Sci. USA 2015, 112, 1475–1480. [Google Scholar] [CrossRef] [PubMed]
- Gursoy, E.; Cardounel, A.; Kalimi, M. The environmental estrogenic compound bisphenol A exerts estrogenic effects on mouse hippocampal (HT-22) cells: Neuroprotection against glutamate and amyloid β protein toxicity. Neurochem. Int. 2001, 38, 181–186. [Google Scholar] [CrossRef]
- Stump, D.G.; Beck, M.J.; Radovsky, A.; Garman, R.H.; Freshwater, L.L.; Sheets, L.P.; Marty, M.S.; Waechter, J.M.; Dimond, S.S.; van Miller, J.P.; et al. Developmental neurotoxicity study of dietary bisphenol A in Sprague-Dawley rats. Toxicol. Sci. 2010, 115, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Ishido, M.; Masuo, Y.; Terasaki, M.; Morita, M. Rat hyperactivity by bisphenol A, but not by its derivatives, 3-hydroxybisphenol A or bisphenol A 3,4-quinone. Toxicol. Lett. 2011, 206, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Viberg, H.; Fredriksson, A.; Buratovic, S.; Eriksson, P. Dose-dependent behavioral disturbances after a single neonatal Bisphenol A dose. Toxicology 2011, 290, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Kimura, E.; Matsuyoshi, C.; Miyazaki, W.; Benner, S.; Hosokawa, M.; Yokoyama, K.; Kakeyama, M.; Tohyama, C. Prenatal exposure to bisphenol A impacts neuronal morphology in the hippocampal CA1 region in developing and aged mice. Arch. Toxicol. 2016, 90, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Yin, N.; Yao, X.; Qin, Z.; Wang, Y.L.; Faiola, F. Assessment of Bisphenol A (BPA) neurotoxicity in vitro with mouse embryonic stem cells. J. Environ. Sci. 2015, 36, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Mersha, M.D.; Patel, B.M.; Patel, D.; Richardson, B.N.; Dhillon, H.S. Effects of BPA and BPS exposure limited to early embryogenesis persist to impair non-associative learning in adults. Behav. Brain Funct. 2015, 11, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Son, T.G.; Park, H.R.; Kim, S.J.; Kim, H.S.; Kim, H.S.; Kim, T.S.; Jung, K.K.; Han, S.Y.; Lee, J. Potencies of bisphenol A on the neuronal differentiation and hippocampal neurogenesis. J. Toxicol. Environ. Health A 2009, 72, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- U.S. Environmental Protection Agency. Bisphenol A; CASRN 80-05-7; U.S. Environmental Protection Agency: Washington, DC, USA, 1988; pp. 1–7.
- Diaz Weinstein, S.; Villafane, J.J.; Juliano, N.; Bowman, R.E. Adolescent exposure to Bisphenol-A increases anxiety and sucrose preference but impairs spatial memory in rats independent of sex. Brain Res. 2013, 1529, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.A.; Javurek, A.B.; Painter, M.S.; Ellersieck, M.R.; Welsh, T.H.; Camacho, L.; Lewis, S.M.; Vanlandingham, M.M.; Ferguson, S.A.; Rosenfeld, C.S. Effects of developmental exposure to bisphenol A on spatial navigational learning and memory in rats: A CLARITY-BPA study. Horm. Behav. 2016, 80, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.M.; Yolton, K.; Dietrich, K.N.; Hornung, R.; Ye, X.; Calafat, A.M.; Lanphear, B.P. Prenatal bisphenol A exposure and early childhood behavior. Environ. Health Perspect. 2009, 117, 1945–1952. [Google Scholar] [CrossRef] [PubMed]
- Rebuli, M.E.; Camacho, L.; Adonay, M.E.; Reif, D.M.; Aylor, D.L.; Patisaul, H.B. Impact of low-dose oral exposure to bisphenol A (BPA) on Juvenile and adult rat exploratory and anxiety behavior: A CLARITY-BPA Consortium Study. Toxicol. Sci. 2015, 148, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Wang, S.; Li, Z.; Wei, R.; Zhang, L.; Liu, H.; Wang, C.; Niu, R.; Wang, J. Maternal bisphenol a diet induces anxiety-like behavior in female juvenile with neuroimmune activation. Toxicol. Sci. 2014, 140, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Carr, R.; Bertasi, F.; Betancourt, A.; Bowers, S.; Gandy, B.S.; Ryan, P.; Willard, S. Effect of neonatal rat bisphenol a exposure on performance in the Morris water maze. J. Toxicol. Environ. Health A 2003, 66, 2077–2088. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, K.; Narita, M.; Narita, M.; Akama, H.; Suzuki, T. Memory impairment associated with a dysfunction of the hippocampal cholinergic system induced by prenatal and neonatal exposures to bisphenol-A. Neurosci. Lett. 2007, 418, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Kawai, K.; Nozaki, T.; Nishikata, H.; Aou, S.; Takii, M.; Kubo, C. Aggressive behavior and serum testosterone concentration during the maturation process of male mice: The effects of fetal exposure to bisphenol A. Environ. Health Perspect. 2003, 111, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Nagao, T.; Saito, Y.; Usumi, K.; Kuwagata, M.; Imai, K. Reproductive function in rats exposed neonatally to bisphenol A and estradiol benzoate. Reprod. Toxicol. 1999, 13, 303–311. [Google Scholar] [CrossRef]
- Narita, M.; Miyagawa, K.; Mizuo, K.; Yoshida, T.; Suzuki, T. Prenatal and neonatal exposure to low-dose of bisphenol-A enhance the morphine-induced hyperlocomotion and rewarding effect. Neurosci. Lett. 2006, 402, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Miyagawa, K.; Mizuo, K.; Yoshida, T.; Suzuki, T. Changes in central dopaminergic systems and morphine reward by prenatal and neonatal exposure to bisphenol-A in mice: Evidence for the importance of exposure period. Addict. Biol. 2007, 12, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Harley, K.G.; Gunier, R.B.; Kogut, K.; Johnson, C.; Bradman, A.; Calafat, A.M.; Eskenazi, B. Prenatal and early childhood bisphenol A concentrations and behavior in school-aged children. Environ. Res. 2013, 126, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Sarrouilhe, D.; Dejean, C. Autism spectrum disorders and bisphenol A: Is serotonin the lacking link in the chain? Encephale 2016. [Google Scholar] [CrossRef]
- Kondolot, M.; Ozmert, E.N.; Ascı, A.; Erkekoglu, P.; Oztop, D.B.; Gumus, H.; Kocer-Gumusel, B.; Yurdakok, K. Plasma phthalate and bisphenol a levels and oxidant-antioxidant status in autistic children. Environ. Toxicol. Pharmacol. 2016, 43, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Repossi, G.; Dain, A.; Tarres, M.C.; Das, U.N.; Eynard, A.R. Beneficial action of resveratrol: How and why? Nutrition 2015, 32, 1–5. [Google Scholar]
- Arbuckle, T.E.; Davis, K.; Boylan, K.; Fisher, M.; Fu, J. Bisphenol A, phthalates and lead and learning and behavioral problems in Canadian children 6–11 years of age: CHMS 2007–2009. Neurotoxicology 2016, 54, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Yolton, K.; Xu, Y.; Strauss, D.; Altaye, M.; Calafat, A.M.; Khoury, J. Prenatal exposure to bisphenol A and phthalates and infant neurobehavior. Neurotoxicol. Teratol. 2011, 33, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Roen, E.L.; Wang, Y.; Calafat, A.M.; Wang, S.; Margolis, A.; Herbstman, J.; Hoepner, L.A.; Rauh, V.; Perera, F.P. Bisphenol A exposure and behavioral problems among inner city children at 7–9 years of age. Environ. Res. 2015, 142, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.M.; Kalkbrenner, A.E.; Calafat, A.M.; Yolton, K.; Ye, X.; Dietrich, K.N.; Lanphear, B.P. Impact of early-life bisphenol A exposure on behavior and executive function in children. Pediatrics 2011, 128, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Miodovnik, A.; Engel, S.M.; Zhu, C.; Ye, X.; Soorya, L.V.; Silva, M.J.; Calafat, A.M.; Wolff, M.S. Endocrine disruptors and childhood social impairment. Neurotoxicology 2011, 32, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Bondy, M.; Lee Ligon, B. Epidemiology and etiology of intracranial meningiomas: A review. J. Neurooncol. 1996, 29, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Duan, B.; Hu, X.; Zhao, H.; Qin, J.; Luo, J. The relationship between urinary bisphenol A levels and meningioma in Chinese adults. Int. J. Clin. Oncol. 2013, 18, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.Y.; Gorski, J. High concentrations of bisphenol A induce cell growth and prolactin secretion in an estrogen-responsive pituitary tumor cell line. Toxicol. Appl. Pharmacol. 2000, 162, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.M.; Raisuddin, S.; Rhee, J.S.; Ki, J.S.; Kim, I.C.; Lee, J.S. Modulatory effect of environmental endocrine disruptors on N-ras oncogene expression in the hermaphroditic fish, Kryptolebias marmoratus. Comp. Biochem. Physiol.—C Toxicol. Pharmacol. 2008, 147, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Mikhail, Y.; Cheryl, S. Alkylphenol xenoestrogens with varying carbon chain lengths differentially and potently activate signaling and functional responses in GH3/B6/F10 somatomam. Environ. Health Perspect. 2009, 117, 723. [Google Scholar]
- Ghisari, M.; Bonefeld-Jorgensen, E.C. Impact of environmental chemicals on the thyroid hormone function in pituitary rat GH3 cells. Mol. Cell. Endocrinol. 2005, 244, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Agency for Toxic Substances and Disease Registry (ATSDR). Toxicological Profile for Di-N-Octylphthalate; Agency for Toxic Substances and Disease Registry (ATSDR): Atlanta, GA, USA, 1997.
- Agency for Toxic Substances and Disease Registry (ATSDR). Toxicological Profile: Di-(2-Ethylhexyl) Phthalate (DEHP); Agency for Toxic Substances and Disease Registry (ATSDR): Atlanta, GA, USA, 2002.
- Agency for Toxic Substances and Disease Registry (ATSDR). Toxicological profile for di-N-butyl phthalate. Agency Toxic Subst. Dis. Regist. 2001, 18, 187–191. [Google Scholar]
- Agency for Toxic Substances and Disease Registry (ATSDR). Toxicological Profile for Diethyl Phthalate; Agency for Toxic Substances and Disease Registry (ATSDR): Atlanta, GA, USA, 1995; pp. 1–158.
- Heudorf, U.; Mersch-Sundermann, V.; Angerer, J. Phthalates: Toxicology and exposure. Int. J. Hyg. Environ. Health 2007, 210, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Liu, S.; Gao, P.; Cao, P.; Xu, H. Effect of diisobutyl phthalate on learning and memory behavior and apoptosis of hippocampus cells in mice. Wei Sheng Yan Jiu 2013, 42, 57–60. (In Chinese) [Google Scholar] [PubMed]
- Zeliger, H.I. Exposure to lipophilic chemicals as a cause of neurological impairments, neurodevelopmental disorders and neurodegenerative diseases. Interdiscip. Toxicol. 2013, 6, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Holahan, M.R. Reduced hippocampal dendritic spine density and BDNF expression following acute postnatal exposure to di-(2-ethylhexyl) phthalate in male long evans rats. PLoS ONE 2014, 9, e109522. [Google Scholar] [CrossRef] [PubMed]
- Holahan, M.R.; Smith, C.A. Phthalates and neurotoxic effects on hippocampal network plasticity. Neurotoxicology 2015, 48, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Jiang, L.; Chen, L.; Chen, H.S.; Li, X. Neurotoxicity of dibutyl phthalate in brain development following perinatal exposure: A study in rats. Environ. Toxicol. Pharmacol. 2013, 36, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Betz, A.J.; Jayatilaka, S.; Joshi, J.; Ramanan, S.; Debartolo, D.; Pylypiw, H.; Franke, E. Chronic exposure to benzyl butyl phthalate (BBP) alters social interaction and fear conditioning in male adult rats: Alterations in amygdalar MeCP2, ERK1/2 and ER?? Neuroendocrinol. Lett. 2013, 34, 347–358. [Google Scholar] [PubMed]
- Lin, H.; Yuan, K.; Li, L.; Liu, S.; Li, S.; Hu, G.; Lian, Q.Q.; Ge, R.S. In utero exposure to diethylhexyl phthalate affects rat brain development: A behavioral and genomic approach. Int. J. Environ. Res. Public Health 2015, 12, 13696–13710. [Google Scholar] [CrossRef] [PubMed]
- Shiue, I. Arsenic, heavy metals, phthalates, pesticides, hydrocarbons and polyfluorinated compounds but not parabens or phenols are associated with adult remembering condition: US NHANES, 2011–2012. Environ. Sci. Pollut. Res. Int. 2015, 22, 6381–6386. [Google Scholar] [CrossRef] [PubMed]
- Factor-Litvak, P.; Insel, B.; Calafat, A.M.; Liu, X.; Perera, F.; Rauh, V.A.; Whyatt, R.M. Persistent associations between maternal prenatal exposure to phthalates on child IQ at age 7 years. PLoS ONE 2014, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Masuo, Y.; Morita, M.; Oka, S.; Ishido, M. Motor hyperactivity caused by a deficit in dopaminergic neurons and the effects of endocrine disruptors: A study inspired by the physiological roles of PACAP in the brain. Regul. Pept. 2004, 123, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Larsson, M.; Weiss, B.; Janson, S.; Sundell, J.; Bornehag, C.G. Associations between indoor environmental factors and parental-reported autistic spectrum disorders in children 6–8 years of age. Neurotoxicology 2009, 30, 822–831. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Ban, J.-B.; Zhang, N.; Zu, Y.-K.; Sun, W.-X. Perinatal exposure to di-(2-ethylhexyl)-phthalate leads to cognitive dysfunction and phospho-tau level increase in aged rats. Environ. Toxicol. 2014, 29, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Agency for Toxic Substances and Disease Registry (ATSDR). Toxicological Profile for Polychlorinated Biphenyls (PCBs); Agency for Toxic Substances and Disease Registry (ATSDR): Atlanta, GA, USA, 2000; pp. 1–948.
- Reilly, M.P.; Weeks, C.D.; Topper, V.Y.; Thompson, L.M.; Crews, D.; Gore, A.C. The effects of prenatal PCBs on adult female paced mating reproductive behaviors in rats. Horm. Behav. 2007, 51, 364–372. [Google Scholar]
- Lee, D.W.; Notter, S.A.; Thiruchelvam, M.; Dever, D.P.; Fitzpatrick, R.; Kostyniak, P.J.; Cory-slechta, D.A.; Opanashuk, L.A. Subchronic polychlorinated biphenyl (aroclor 1254) exposure produces oxidative damage and neuronal death of ventral midbrain dopaminergic systems. Toxicol. Sci. 2012, 125, 496–508. [Google Scholar] [CrossRef] [PubMed]
- Zahara, A.R.D.; Michel, N.L.; Flahr, L.M.; Ejack, L.E.; Morrissey, C.A. Latent cognitive effects from low-level polychlorinated biphenyl exposure in juvenile European starlings (Sturnus vulgaris). Environ. Toxicol. Chem. 2015, 34, 2513–2520. [Google Scholar] [CrossRef] [PubMed]
- Elnar, A.A.; Allouche, A.; Desor, F.; Yen, F.T.; Soulimani, R.; Oster, T. Lactational exposure of mice to low levels of non-dioxin-like polychlorinated biphenyls increases susceptibility to neuronal stress at a mature age. Neurotoxicology 2015, 53, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Hilgier, W.; Łazarewicz, J.W.; Strużynska, L.; Frontczak-Baniewicz, M.; Albrecht, J. Repeated exposure of adult rats to Aroclor 1254 induces neuronal injury and impairs the neurochemical manifestations of the NMDA receptor-mediated intracellular signaling in the hippocampus. Neurotoxicology 2012, 33, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Formisano, L.; Guida, N.; Laudati, G.; Mascolo, L.; Di Renzo, G.; Canzoniero, L.M.T. MS-275 inhibits aroclor 1254-induced SH-SY5Y neuronal cell toxicity by preventing the formation of the HDAC3/REST complex on the synapsin-1 promoter. J. Pharmacol. Exp. Ther. 2015, 352, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.; Lim, J.; Kim, S.S.; Jeong, H.M.; Jung, K.K.; Kang, I.H.; Lee, K.Y.; Choi, H.J. Aroclor1254 interferes with estrogen receptor-mediated neuroprotection against β-amyloid toxicity in cholinergic SN56 cells. Neurochem. Int. 2011, 59, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Szczepkowska, A.; Lagaraine, C.; Robert, V.; Młynarczuk, J.; Dufourny, L.; Thiéry, J.C.; Skipor, J. PCB153 (2,2′,4,4′,5,5′-hexachlorobiphenyl) differentially affects the VEGF/VEGFR system depending on photoperiod in the ovine choroid plexus. Ecotoxicol. Environ. Saf. 2016, 124, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Weisskopf, M.; Knekt, P.; O’Reilly, E.; Lyytinen, J.; Reunanen, A.; Laden, F.; Altshul, L.; Ascherio, A. Polychlorinated biphenyls in prospectively collected serum and parkinson’s disease risk. Mov. Disord. 2012, 27, 1659–1665. [Google Scholar] [CrossRef] [PubMed]
- Steenland, K.; Hein, M.J.; Cassinelli, R.T.; Prince, M.M.; Nilsen, N.B.; Whelan, E.A.; Waters, M.A.; Ruder, A.M.; Schnorr, T.M. Polychlorinated biphenyls and neurodegenerative disease mortality in an occupational cohort. Epidemiology 2006, 17, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Hatcher-Martin, J.M.; Gearing, M.; Steenland, K.; Levey, A.I.; Miller, G.W.; Pennell, K.D. Association between polychlorinated biphenyls and Parkinson’s disease neuropathology. Neurotoxicology 2012, 33, 1298–1304. [Google Scholar] [CrossRef] [PubMed]
- Eubig, P.A.; Aguiar, A.; Schantz, S.L. Lead and PCBs as risk factors for attention defcit/hyperactivity disorder. Environ. Health Perspect. 2010, 118, 1654–1667. [Google Scholar] [CrossRef] [PubMed]
- Orenstein, S.T.C.; Thurston, S.W.; Bellinger, D.C.; Schwartz, J.D.; Amarasiriwardena, C.J.; Altshul, L.M.; Korrick, S.A. Prenatal organochlorine and methylmercury exposure and memory and learning in school-age children in communities near the new bedford harbor superfund site, Massachusetts. Environ. Health Perspect. 2014, 122, 1253–1259. [Google Scholar] [CrossRef] [PubMed]
- Boucher, O.; Muckle, G.; Jacobson, J.L.; Carter, R.C.; Kaplan-Estrin, M.; Ayotte, P.; Dewailly, É.; Jacobson, S.W. Domain-specific effects of prenatal exposure to PCBs, mercury, and lead on infant cognition: Results from the environmental contaminants and child development study in nunavik. Environ. Health Perspect. 2014, 122, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, E.F.; Shrestha, S.; Gomez, M.I.; McCaffrey, R.J.; Zimmerman, E.A.; Kannan, K.; Hwang, S. An polybrominated diphenyl ethers (PBDEs), polychlorinated biphenyls (PCBs) and neuropsychological status among older adults in New York. Neurotoxicology 2012, 33, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, E.F.; Belanger, E.E.; Gomez, M.I.; Cayo, M.; McCaffrey, R.J.; Seegal, R.F.; Jansing, R.L.; Hwang, S.A.; Hicka, H.E. Polychlorinated biphenyl exposure and neuropsychological status among older residents of upper Hudson River communities. Environ. Health Perspect. 2008, 116, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Schantz, S.L.; Gasior, D.M.; Polverejan, E.; McCaffrey, R.J.; Sweeney, A.M.; Humphrey, H.E.B.; Gardiner, J.C. Impairments of memory and learning in older adults exposed to polychlorinated biphenyls via consumption of Great Lakes Fish. Environ. Health Perspect. 2001, 109, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Peper, M.; Klett, M.; Morgenstern, R. Neuropsychological effects of chronic low-dose exposure to polychlorinated biphenyls (PCBs): A cross-sectional study. Environ. Health 2005, 4, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ronchetti, S.A.; Novack, G.V.; Bianchi, M.S.; Crocco, M.C.; Duvilanski, B.H.; Cabilla, J.P. In vivo xenoestrogenic actions of cadmium and arsenic in anterior pituitary and uterus. Reproduction 2016, 152, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Hu, P.; Wang, H.-L.; Wang, M.; Chen, J.-T.; Tang, J.-L.; Ruan, D.-Y. Effects of Cd2+ on AMPA receptor-mediated synaptic transmission in rat hippocampal CA1 area. Toxicol. Lett. 2008, 176, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, L.; Luo, Y.; Huang, S. MAPK and mTOR pathways are involved in cadmium-induced neuronal apoptosis. J. Neurochem. 2008, 105, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.H.; Ge, G.; Gao, K.; Pang, Y.; Chai, R.C.; Jia, X.H.; Kong, J.G.; Yu, A.C.-H. Calcium signaling involvement in cadmium-induced astrocyte cytotoxicity and cell death through activation of MAPK and PI3K/Akt signaling pathways. Neurochem. Res. 2015, 40, 1929–1944. [Google Scholar] [CrossRef] [PubMed]
- Chow, E.S.H.; Hui, M.N.Y.; Lin, C.C.; Cheng, S.H. Cadmium inhibits neurogenesis in zebrafish embryonic brain development. Aquat. Toxicol. 2008, 87, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Jiang, C.; Xu, H.; Sun, Y.; Hu, F.; Bian, J.; Liu, X.; Gu, J.; Liu, Z. Cadmium-induced apoptosis in primary rat cerebral cortical neurons culture is mediated by a calcium signaling pathway. PLoS ONE 2013, 8, e64330. [Google Scholar] [CrossRef] [PubMed]
- Hossain, S.; Liu, H.-N.; Nguyen, M.; Shore, G.; Almazan, G. Cadmium exposure induces mitochondria-dependent apoptosis in oligodendrocytes. Neurotoxicology 2009, 30, 544–554. [Google Scholar] [CrossRef] [PubMed]
- Notarachille, G.; Arnesano, F.; Calò, V.; Meleleo, D. Heavy metals toxicity: Effect of cadmium ions on amyloid β protein 1–42. Possible implications for Alzheimer’s disease. Biometals 2014, 27, 371–388. [Google Scholar] [CrossRef] [PubMed]
- Ciesielski, T.; Bellinger, D.C.; Schwartz, J.; Hauser, R.; Wright, R.O. Associations between cadmium exposure and neurocognitive test scores in a cross-sectional study of US adults. Environ. Health 2013, 12, 214–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, S.; Jin, Y.; Unverzagt, F.W.; Ma, F.; Hall, K.S.; Murrell, J.R.; Cheng, Y.; Shen, J.; Ying, B.; Ji, R.; et al. Trace element levels and cognitive function in rural elderly Chinese. J. Gerontol. Ser. A-Biol. Sci. Med. Sci. 2008, 63, 635–641. [Google Scholar] [CrossRef]
- Roos, P.M.; Vesterberg, O.; Syversen, T.; Flaten, T.P.; Nordberg, M. Metal concentrations in cerebrospinal fluid and blood plasma from patients with amyotrophic lateral sclerosis. Biol. Trace Elem. Res. 2013, 151, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, F.; Kagawa, Y.; Kawabata, T.; Kaneko, Y.; Chimedregzen, U.; Purvee, B.; Otgon, J. A high accumulation of hair minerals in Mongolian people: 2nd report; influence of manganese, iron, lead, cadmium and aluminum to oxidative stress, Parkinsonism and arthritis. Curr. Aging Sci. 2011, 4, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Jumpponen, M.; Rönkkömäki, H.; Pasanen, P.; Laitinen, J. Occupational exposure to solid chemical agents in biomass-fired power plants and associated health effects. Chemosphere 2014, 104, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Agency for Toxic Substances and Disease Registry (ATSDR). Toxicological Profile for Arsenic. Health Effects; Agency for Toxic Substances and Disease Registry (ATSDR): Atlanta, GA, USA, 2007.
- Agency for Toxic Substances and Disease Registry (ATSDR). Toxicological Profile for Arsenic; Agency for Toxic Substances and Disease Registry (ATSDR): Atlanta, GA, USA, 2013.
- Luo, J.; Qiu, Z.; Zhang, L.; Shu, W. Arsenite exposure altered the expression of NMDA receptor and postsynaptic signaling proteins in rat hippocampus. Toxicol. Lett. 2012, 211, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Qiu, Z.; Shu, W.; Zhang, Y.; Zhang, L.; Chen, J. Effects of arsenic exposure from drinking water on spatial memory, ultra-structures and NMDAR gene expression of hippocampus in rats. Toxicol. Lett. 2009, 184, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Qu, L.; Gao, Y.; Sun, H.; Wang, H.; Liu, X.; Sun, D. Role of PTEN-Akt-CREB signaling pathway in nervous system impairment of rats with chronic arsenite exposure. Biol. Trace Elem. Res. 2016, 170, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Shavali, S.; Sens, D.A. Synergistic neurotoxic effects of arsenic and dopamine in human dopaminergic neuroblastoma SH-SY5Y cells. Toxicol. Sci. 2007, 102, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Zarazúa, S.; Bürger, S.; Delgado, J.M.; Jiménez-Capdeville, M.E.; Schliebs, R. Arsenic affects expression and processing of amyloid precursor protein (APP) in primary neuronal cells overexpressing the Swedish mutation of human APP. Int. J. Dev. Neurosci. 2011, 29, 389–396. [Google Scholar] [CrossRef] [PubMed]
- O’Bryant, S.E.; Edwards, M.; Menon, C.; Gong, G.; Barber, R. Long-term low-level arsenic exposure is associated with poorer neuropsychological functioning: A project frontier study. Int. J. Environ. Res. Public Health 2011, 8, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.; Hall, J.; Gong, G.; O’Bryant, S.E. Arsenic exposure, AS3MT polymorphism, and neuropsychological functioning among rural dwelling adults and elders: A cross-sectional study. Environ. Health 2014, 13, 225. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-H.; Lee, D.-W.; Park, K.S.; Joung, H. Serum trace metal levels in Alzheimer’s disease and normal control groups. Am. J. Alzheimers Dis. Other Dement. 2014, 29, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.; Johnson, L.; Mauer, C.; Barber, R.; Hall, J.; O’Bryant, S. Regional specific groundwater arsenic levels and neuropsychological functioning: A cross-sectional study. Int. J. Environ. Health Res. 2014, 24, 546–557. [Google Scholar] [CrossRef] [PubMed]
- Dearth, R.K.; Hiney, J.K.; Srivastava, V.K.; Hamilton, A.M.; Dees, W.L. Prepubertal exposure to elevated manganese results in estradiol regulated mammary gland ductal differentiation and hyperplasia in female rats. Exp. Biol. Med. 2014, 239, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Guilarte, T.R.; McGlothan, J.L.; Degaonkar, M.; Chen, M.-K.; Barker, P.B.; Syversen, T.; Schneider, J.S. Evidence for cortical dysfunction and widespread manganese accumulation in the nonhuman primate brain following chronic manganese exposure: A 1H-MRS and MRI study. Toxicol. Sci. 2006, 94, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Guilarte, T.R.; Chen, M.K. Manganese inhibits NMDA receptor channel function: Implications to psychiatric and cognitive effects. Neurotoxicology 2007, 28, 1147–1152. [Google Scholar] [CrossRef] [PubMed]
- Neal, A.P.; Guilarte, T.R. Mechanisms of lead and manganese neurotoxicity. Toxicol. Res. 2013, 2, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Guilarte, T.R.; Burton, N.C.; McGlothan, J.L.; Verina, T.; Zhou, Y.; Alexander, M.; Pham, L.; Griswold, M.; Wong, D.F.; Syversen, T.; et al. Impairment of nigrostriatal dopamine neurotransmission by manganese is mediated by pre-synaptic mechanism(s): Implications to manganese-induced parkinsonism. J. Neurochem. 2008, 107, 1236–1247. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.S.; Decamp, E.; Koser, A.J.; Fritz, S.; Gonczi, H.; Syversen, T.; Guilarte, T.R. Effects of chronic manganese exposure on cognitive and motor functioning in non-human primates. Brain Res. 2006, 1118, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Bowler, R.M.; Gocheva, V.; Harris, M.; Ngo, L.; Abdelouahab, N.; Wilkinson, J.; Doty, R.L.; Park, R.; Roels, H.A. Prospective study on neurotoxic effects in manganese-exposed bridge construction welders. Neurotoxicology 2011, 32, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.G.; Criswell, S.R.; Racette, B.A.; Simpson, C.D.; Sheppard, L.; Checkoway, H.; Sheppard, L. Neurological outcomes associated with low-level manganese exposure in an inception cohort of asymptomatic welding trainees. Scand. J. Work. Environ. Health 2015, 41, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Wasserman, G.A.; Liu, X.; Parvez, F.; Factor-Litvak, P.; Ahsan, H.; Levy, D.; Kline, J.; van Geen, A.; Mey, J.; Slavkovich, V.; et al. Arsenic and manganese exposure and children’s intellectual function. Neurotoxicology 2011, 32, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Bowler, R.M.; Abdelouahab, N.; Harris, M.; Gocheva, V.; Roels, H.A. Motor function in adults of an Ohio community with environmental manganese exposure. Neurotoxicology 2011, 32, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.F.; Menezes-Filho, J.A.; de Matos, V.P.; Bessa, J.R.; Coelho-Santos, J.; Viana, G.F.S.; Argollo, N.; Abreu, N. Elevated airborne manganese and low executive function in school-aged children in Brazil. Neurotoxicology 2014, 45, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Oulhote, Y.; Mergler, D.; Barbeau, B.; Bellinger, D.C.; Bouffard, T.; Brodeur, M.-È.; Saint-Amour, D.; Legrand, M.; Sauvé, S.; Bouchard, M.F. Neurobehavioral function in school-age children exposed to manganese in drinking water. Environ. Health Perspect. 2014, 122, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Guilarte, T.R. Manganese and Parkinson’s disease: A critical review and new findings. Environ. Health Perspect. 2010, 118, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Hozumi, I.; Hasegawa, T.; Honda, A.; Ozawa, K.; Hayashi, Y.; Hashimoto, K.; Yamada, M.; Koumura, A.; Sakurai, T.; Kimura, A.; et al. Patterns of levels of biological metals in CSF differ among neurodegenerative diseases. J. Neurol. Sci. 2011, 303, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Koc, E.R.; Ilhan, A.; Ayturk, Z.; Acar, B.; Gurler, M.; Altuntas, A.; Karapirli, M.; Bodur, A.S. A comparison of hair and serum trace elements in patients with Alzheimer disease and healthy participants. Turk. J. Med. Sci. 2015, 45, 1034–1039. [Google Scholar] [CrossRef] [PubMed]
- Miyake, Y.; Tanaka, K.; Fukushima, W.; Sasaki, S.; Kiyohara, C.; Tsuboi, Y.; Yamada, T.; Oeda, T.; Miki, T.; Kawamura, N.; et al. Dietary intake of metals and risk of Parkinson’s disease: A case-control study in Japan. J. Neurol. Sci. 2011, 306, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Kihira, T.; Sakurai, I.; Yoshida, S.; Wakayama, I.; Takamiya, K.; Okumura, R.; Iinuma, Y.; Iwai, K.; Kajimoto, Y.; Hiwatani, Y.; et al. Neutron activation analysis of scalp hair from ALS patients and residents in the Kii Peninsula, Japan. Biol. Trace Elem. Res. 2015, 164, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Arain, M.S.; Afridi, H.I.; Kazi, T.G.; Talpur, F.N.; Arain, M.B.; Kazi, A.; Arain, S.A.; Ali, J. Correlation of aluminum and manganese concentration in scalp hair samples of patients having neurological disorders. Environ. Monit. Assess. 2015, 187, 4172. [Google Scholar] [CrossRef] [PubMed]
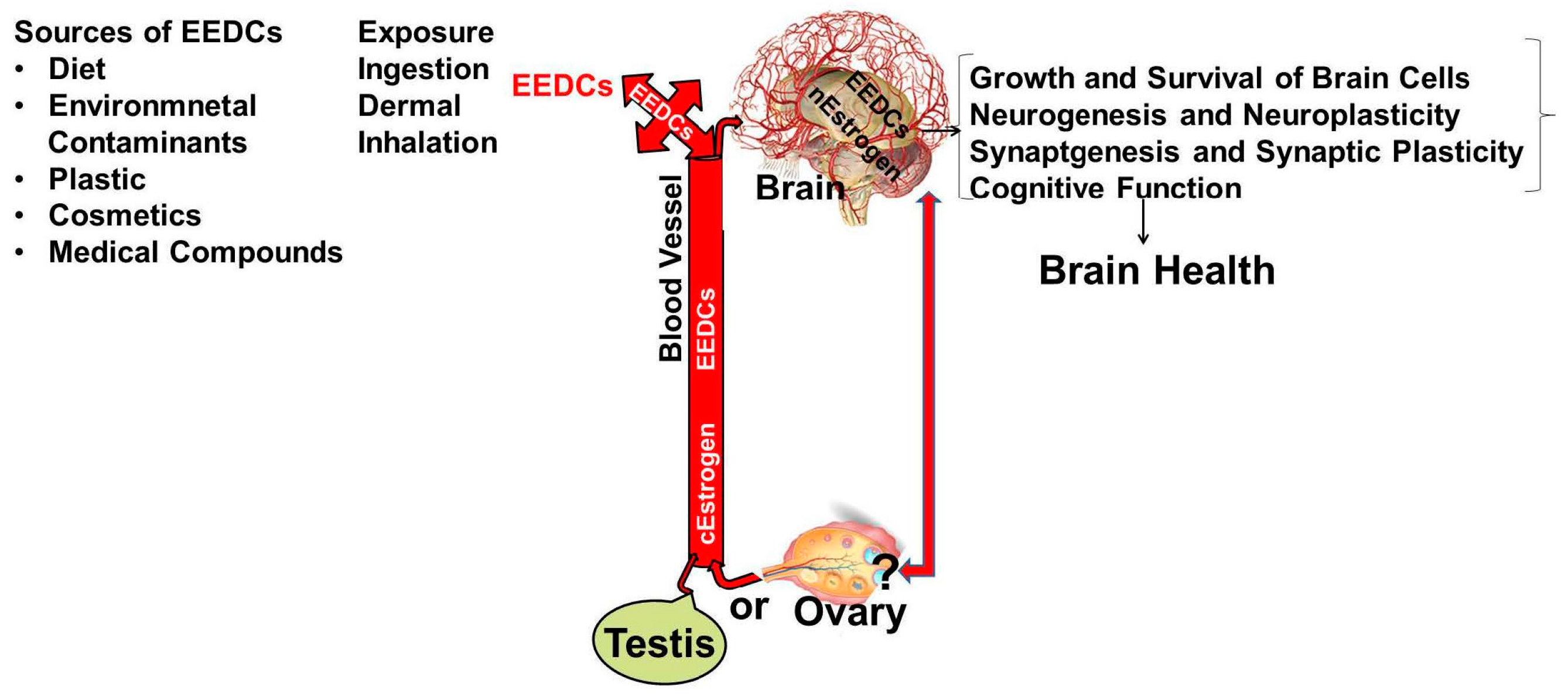
- Hess, R.A.; Carnes, K. The role of estrogen in testis and the male reproductive tract: A review and species comparison. Anim. Reprod. 2004, 1, 5–30. [Google Scholar]
- Kenealy, B.P.; Kapoor, A.; Guerriero, K.A.; Keen, K.L.; Garcia, J.P.; Kurian, J.R.; Ziegler, T.E.; Terasawa, E. Neuroestradiol in the hypothalamus contributes to the regulation of gonadotropin releasing hormone release. J. Neurosci. 2013, 33, 19051–19059. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, A.L.; Nelson, L.H.; Saldanha, C.J. Centrally synthesized estradiol is a potent anti-inflammatory in the injured zebra finch brain. Endocrinology 2016, 157, 2041–2051. [Google Scholar] [CrossRef] [PubMed]
- Brann, D.W.; Dhandapani, K.; Wakade, C.; Mahesh, V.B.; Khan, M.M. Neurotrophic and neuroprotective actions of estrogen: Basic mechanisms and clinical implications. Steroids 2007, 72, 381–405. [Google Scholar] [CrossRef] [PubMed]
- Lanting, C.I.; Huisman, M.; Muskiet, F.A.J.; van der Paauw, C.G.; Essed, C.E.; Boersma, E.R. Polychlorinated biphenyls in adipose tissue, liver, and brain from nine stillborns of varying gestational Ages. Pediatr. Res. 1998, 44, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Darbre, P.D. Metalloestrogens: An emerging class of inorganic xenoestrogens with potential to add to the oestrogenic burden of the human breast. J. Appl. Toxicol. 2006, 26, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.R. Nanotoxicology and metalloestrogens: Possible involvement in breast cancer. Toxics 2015, 3, 390–413. [Google Scholar] [CrossRef]
- Harvey, P.W.; Everett, D.J. Regulation of endocrine-disrupting chemicals: Critical overview and deficiencies in toxicology and risk assessment for human health. Best Pract. Res. Clin. Endocrinol. 2006, 20, 145–165. [Google Scholar] [CrossRef] [PubMed]
- Jeng, H.A. Exposure to endocrine disrupting chemicals and male reproductive health. Front. Public Health 2014, 2, 18033–18037. [Google Scholar] [CrossRef] [PubMed]
- Klenke, U.; Constantin, S.; Wray, S. BPA directly decreases GnRH neuronal activity via non-canonical pathway. Endocrinology 2016, 157, 1980–1990. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Suk, K.; Kim, I.K.; Jang, I.S.; Park, J.W.; Johnson, V.J.; Kwon, T.K.; Choi, B.J.; Kim, S.H. Signaling pathways of bisphenol A-induced apoptosis in hippocampal neuronal cells: Role of calcium-induced reactive oxygen species, mitogen-activated protein kinases, and nuclear factor-κB. J. Neurosci. Res. 2008, 86, 2932–2942. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, Y.K.; Shin, T.Y.; Kim, S.H. Neurotoxic effects of bisphenol AF on calcium-induced ROS and MAPKs. Neurotox. Res. 2013, 23, 249–259. [Google Scholar] [CrossRef] [PubMed]
- El-Missiry, M.A.; Othman, A.I.; Al-Abdan, M.A.; El-Sayed, A.A. Melatonin ameliorates oxidative stress, modulates death receptor pathway proteins, and protects the rat cerebrum against bisphenol-A-induced apoptosis. J. Neurol. Sci. 2014, 347, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, Z.; Han, H.; Luo, G.; Zhou, B.; Wang, S.; Wang, J. Impairment of object recognition memory by maternal bisphenol A exposure is associated with inhibition of Akt and ERK/CREB/BDNF pathway in the male offspring hippocampus. Toxicology 2016, 341–343, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Tiwari, S.K.; Seth, B.; Yadav, A.; Singh, A.; Mudawal, A.; Chauhan, L.K.S.; Gupta, S.K.; Choubey, V.; Tripathi, A.; et al. Activation of autophagic flux against xenoestrogen bisphenol-A-induced hippocampal neurodegeneration via AMP kinase (AMPK)/mammalian target of rapamycin (mTOR) pathways. J. Biol. Chem. 2015, 290, 21163–21184. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, Y.; Fang, F.; Chen, D.; Gao, Y.; Liu, J.; Gao, R.; Wang, J.; Xiao, H. Bisphenol A disrupts glucose transport and neurophysiological role of IR/IRS/AKT/GSK3β axis in the brain of male mice. Environ. Toxicol. Pharmacol. 2016, 43, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Scinicariello, F.; Feroe, A.G.; Attanasio, R. Urinary phthalates and leukocyte telomere length: An analysis of NHANES 1999–2002. EBioMedicine 2015, 6, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Miodovnik, A.; Edwards, A.; Bellinger, D.C.; Hauser, R. Developmental neurotoxicity of ortho-phthalate diesters: Review of human and experimental evidence. Neurotoxicology 2014, 41, 112–122. [Google Scholar] [CrossRef] [PubMed]
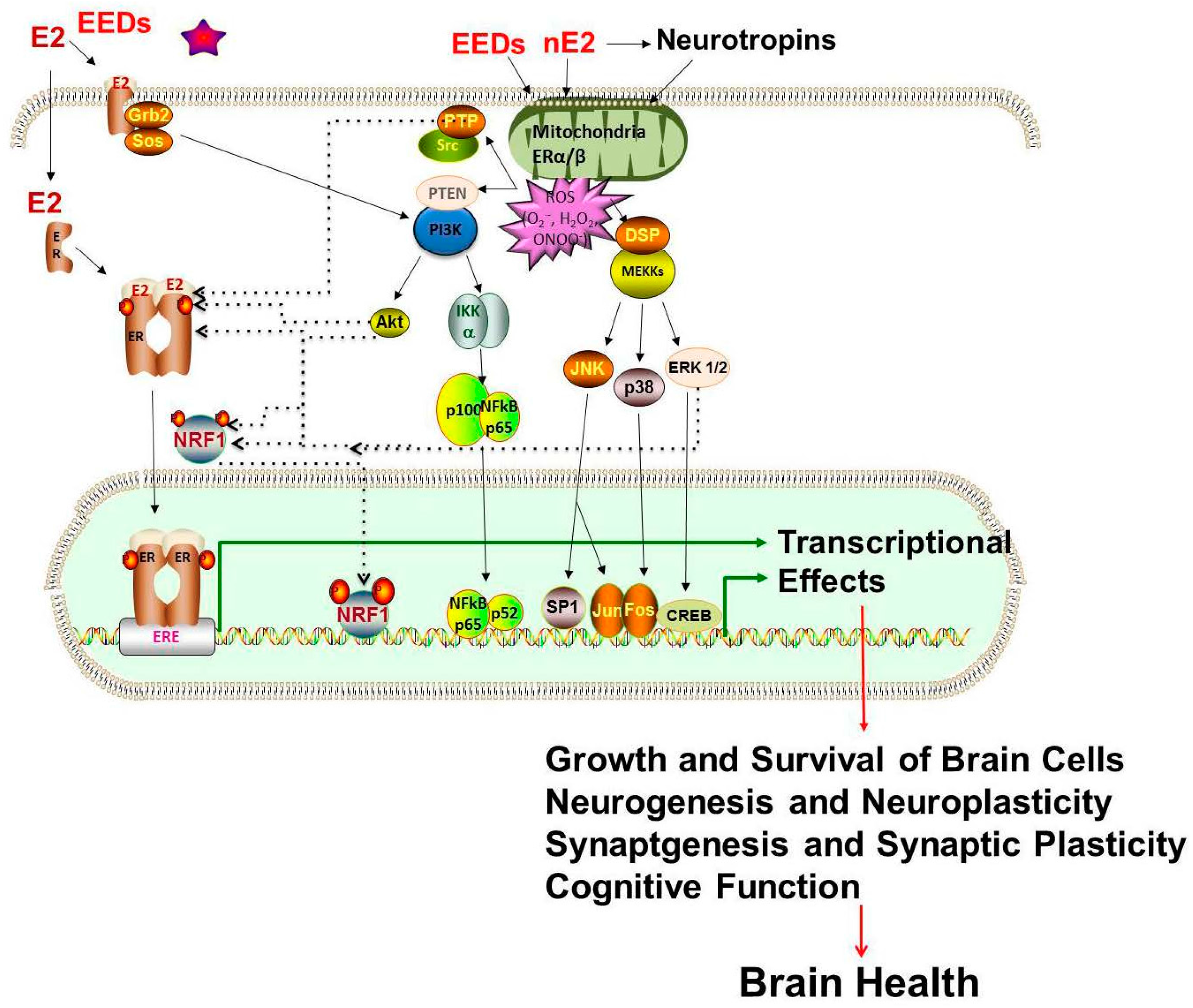
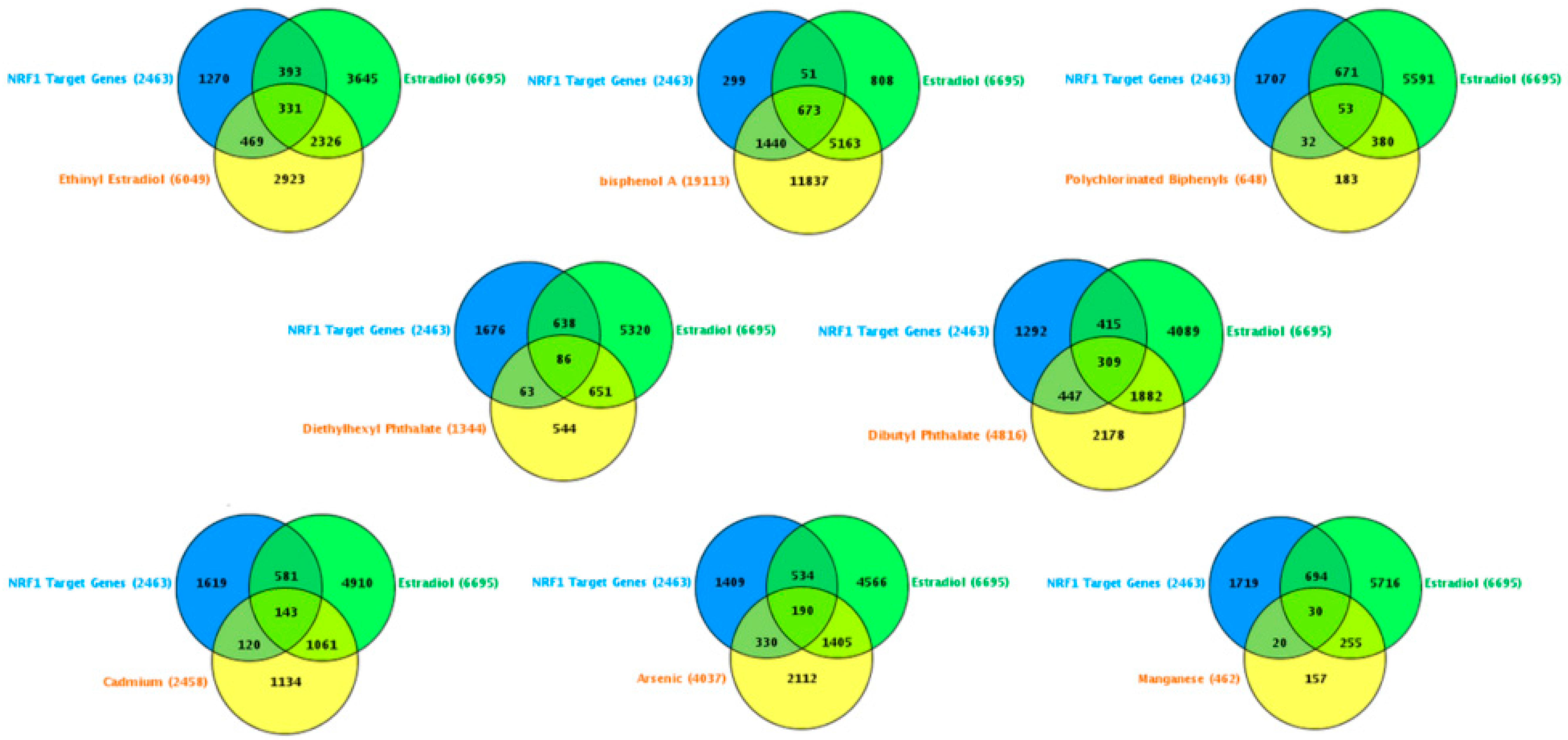
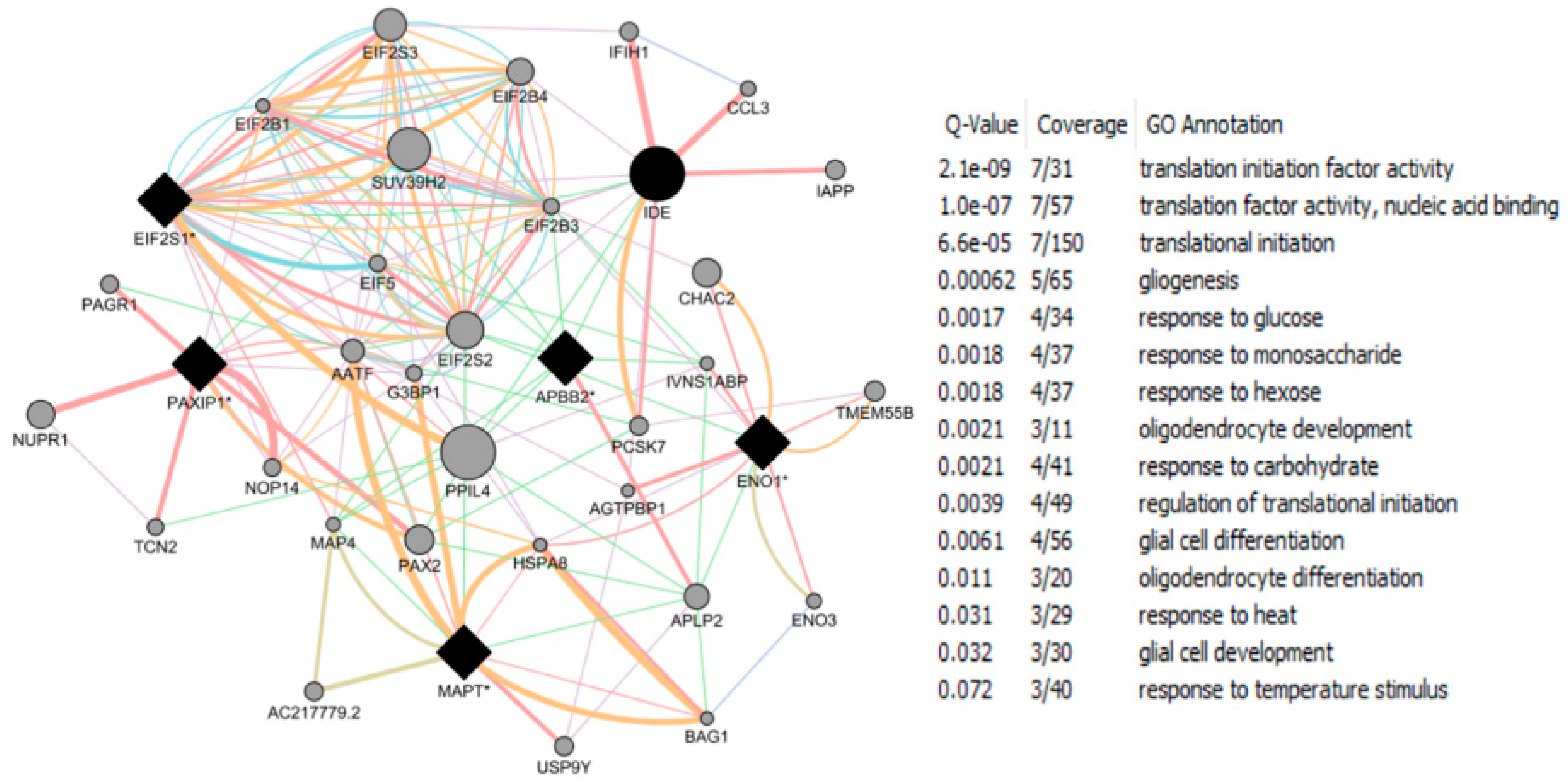
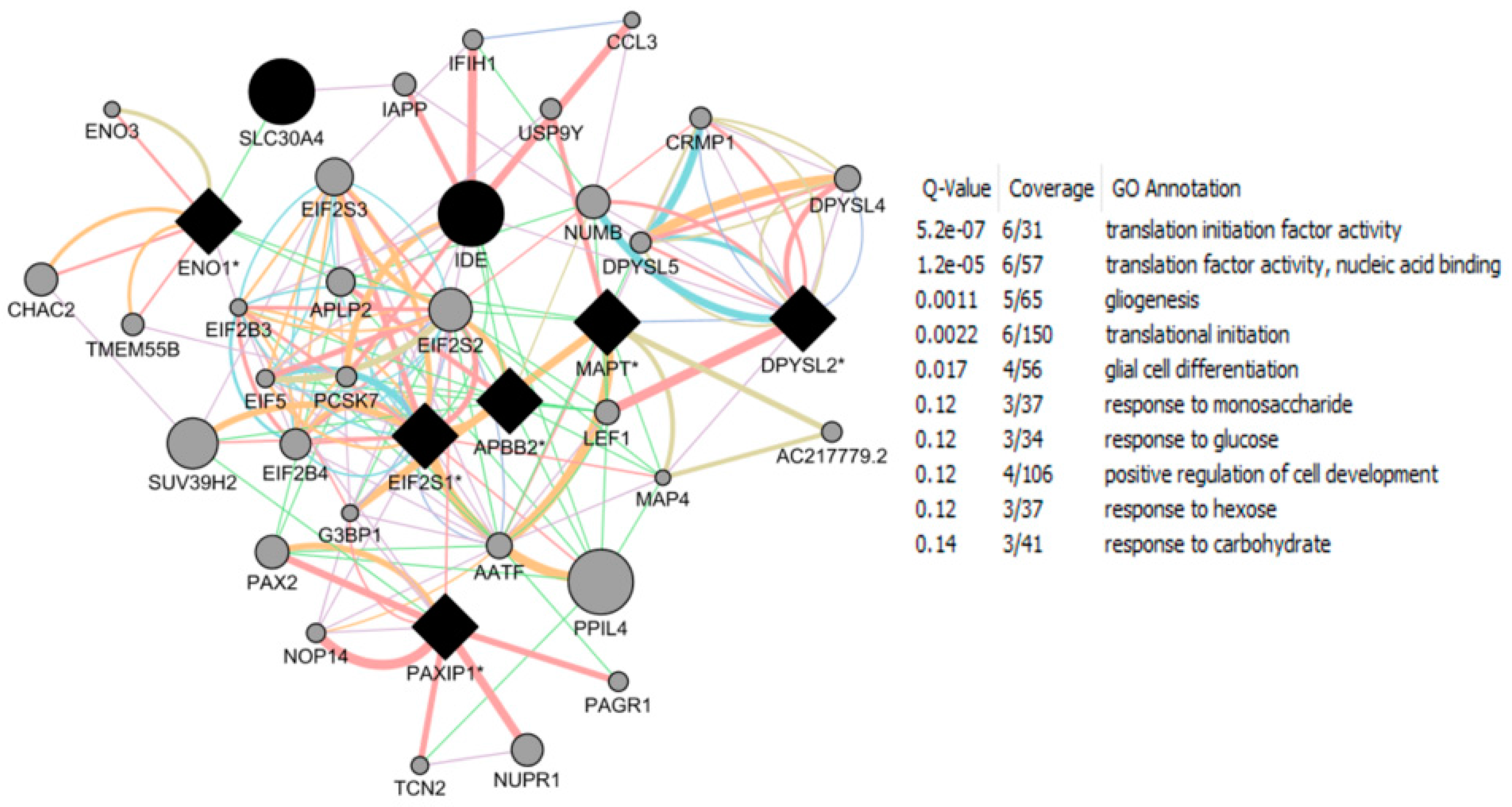
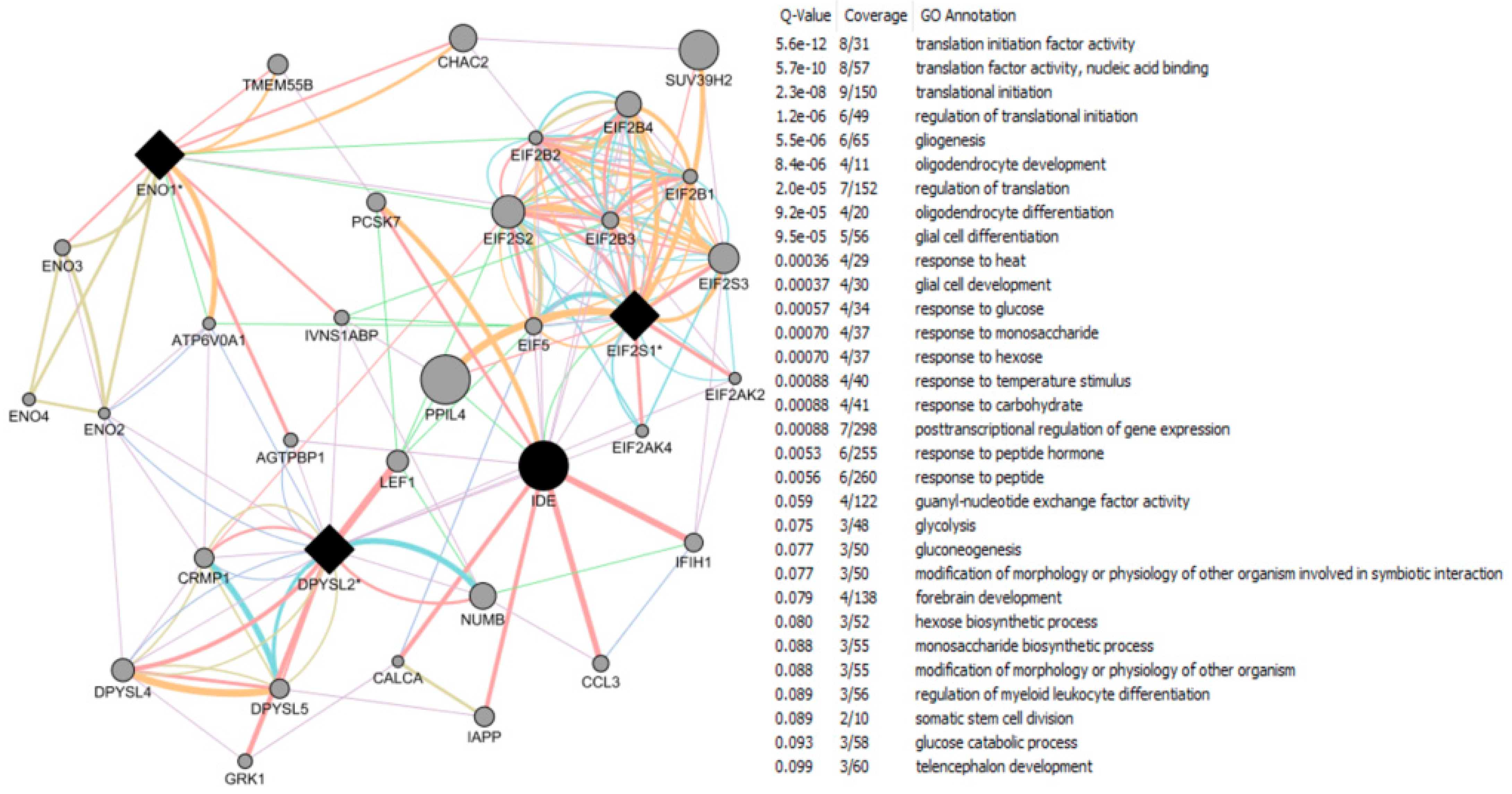
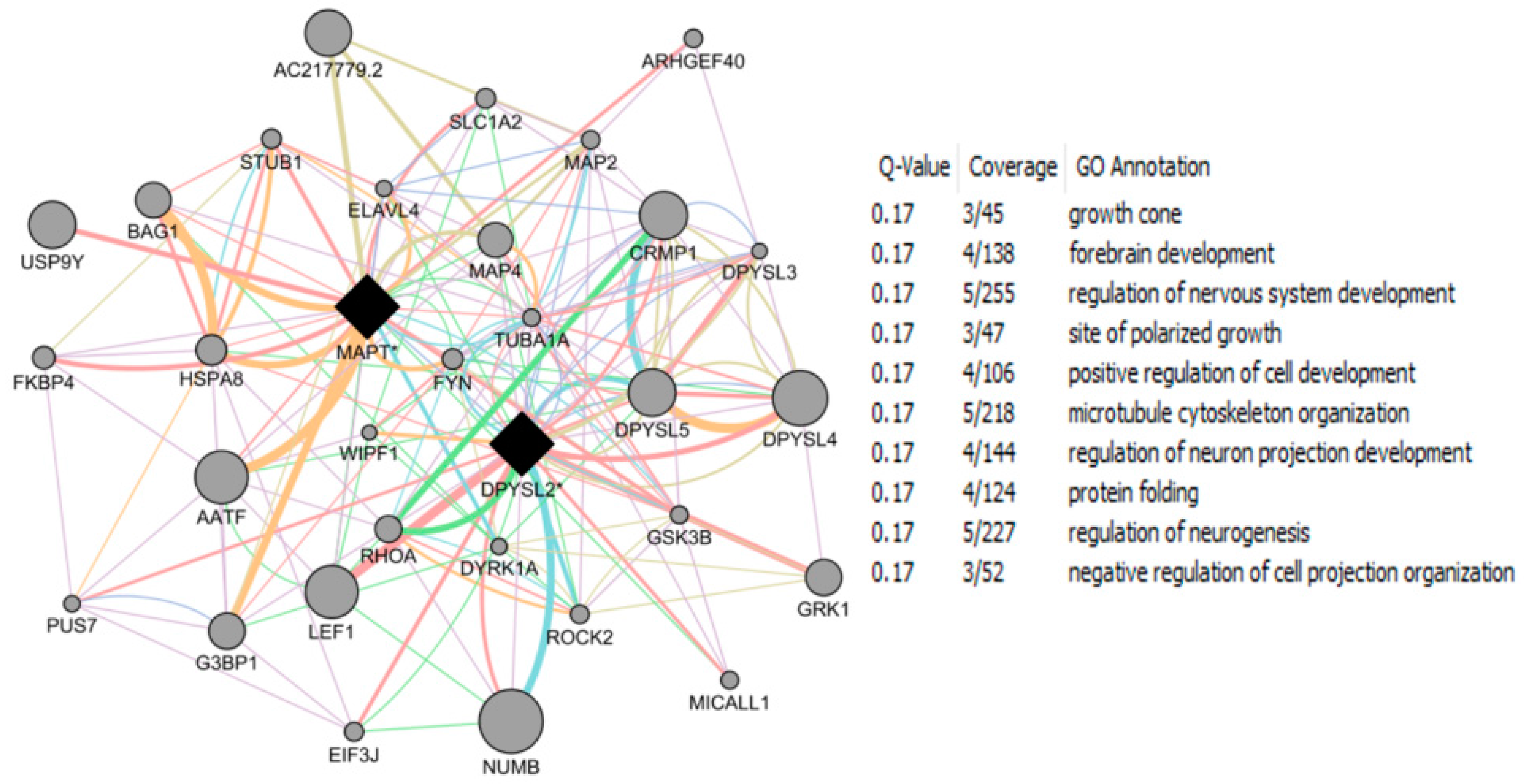
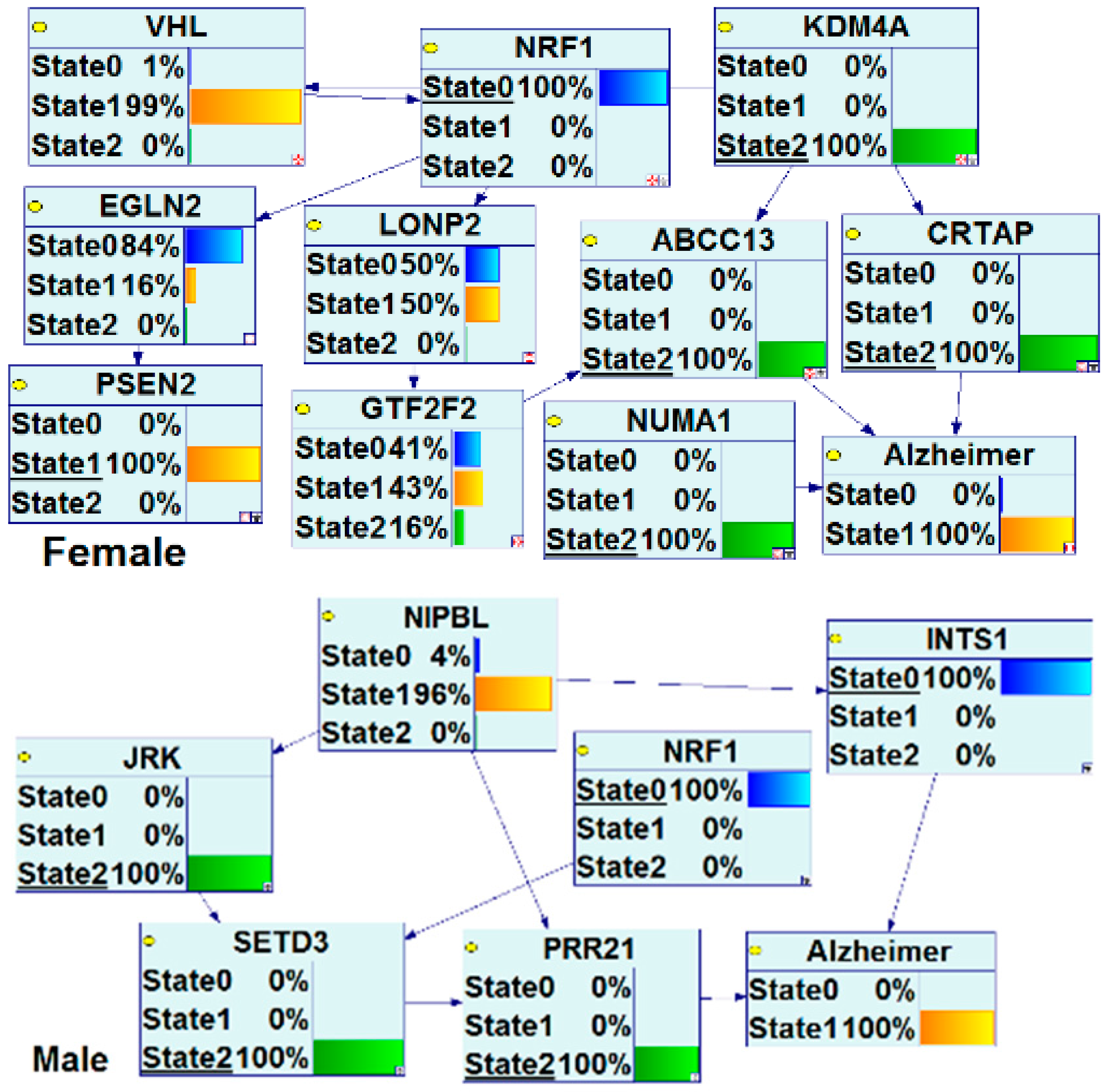
- Roy, D.; Tamuli, R. NRF1 (Nuclear Respiratory Factor 1). Atlas Genet Cytogenet Oncol Haematol. 2008. Available online: http://AtlasGeneticsOncology.org/Genes/NRF1ID44233ch7q32.html (accessed on 12 September 2016).
- Okoh, V.O.; Garba, N.A.; Penney, R.B.; Das, J.; Deoraj, A.; Singh, K.P.; Sarkar, S.; Felty, Q.; Yoo, C.; Jackson, R.M.; et al. Redox signalling to nuclear regulatory proteins by reactive oxygen species contributes to oestrogen-induced growth of breast cancer cells. Br. J. Cancer 2015, 112, 1687–1702. [Google Scholar] [CrossRef] [PubMed]
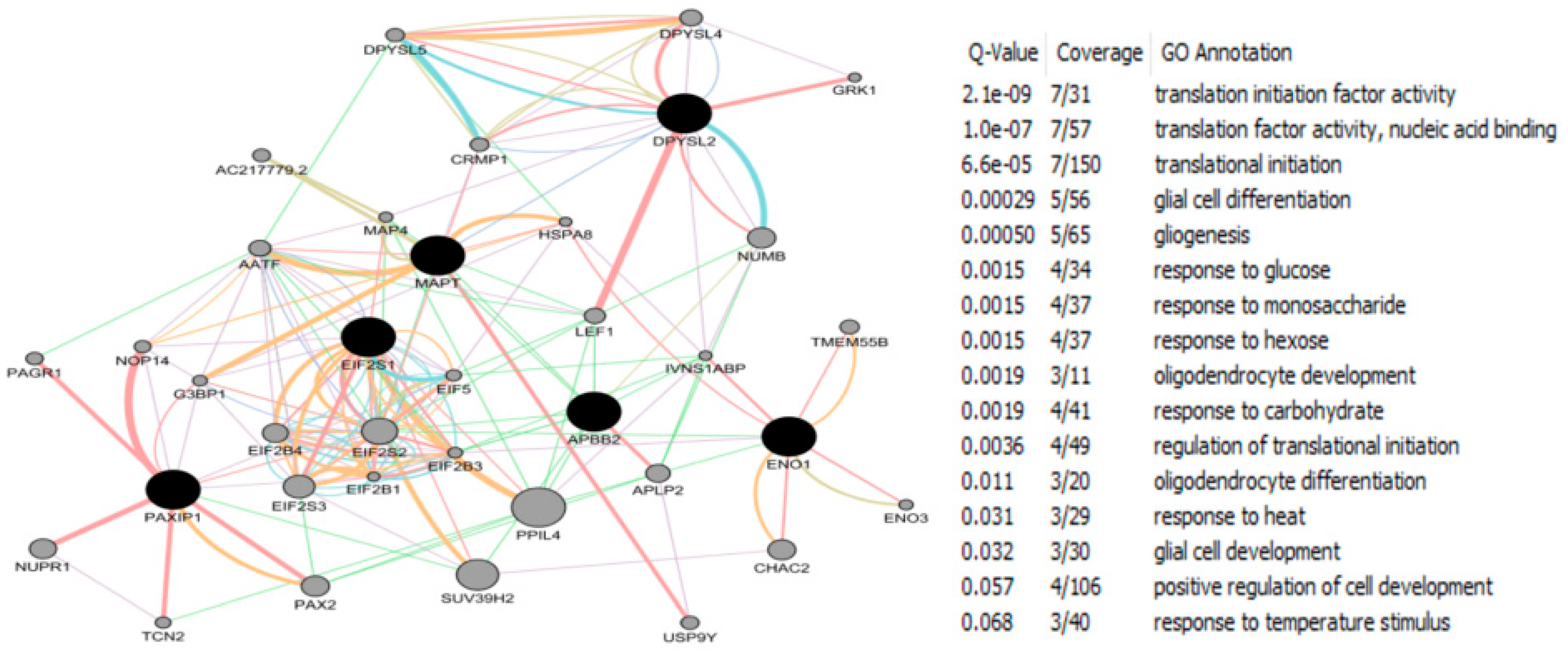
- Chandrasekaran, S.; Bonchev, D. Network topology analysis of post-mortem brain microarrays identifies more Alzheimer’s related genes and MicroRNAs and points to novel routes for fighting with the disease. PLoS ONE 2016, 11, e0144052. [Google Scholar] [CrossRef] [PubMed]
- Sheng, B.; Wang, X.; Su, B.; Lee, H.; Casadesus, G.; Perry, G.; Zhu, X. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J. Neurochem. 2012, 120, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Johri, A.; Beal, F. Mitochondrial dysfunction in neurodegenerative diseases. J. Pharmacol. Exp. Ther. 2012, 342, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Corona, J.C.; Duchen, M.R. Impaired mitochondrial homeostasis and neurodegeneration: Towards new therapeutic targets? J. Bioenerg. Biomembr. 2014, 47, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Velarde, M.C. Mitochondrial and sex steroid hormone crosstalk during aging. Longev. Healthspan 2014, 3, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Scarpulla, R. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 2008, 88, 611–638. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.S.; Johar, K.; Wong-Riley, M.T.T. Bigenomic transcriptional regulation of all thirteen cytochrome c oxidase subunit genes by specificity protein 1. Open Biol. 2013, 3, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Thau, N.; Knippenberg, S.; Körner, S.; Rath, K.J.; Dengler, R.; Petri, S. Decreased mRNA expression of PGC-1α and PGC-1α-regulated factors in the SOD1G93A ALS mouse model and in human sporadic ALS. J. Neuropathol. Exp. Neurol. 2012, 71, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, R.K.; Adhihetty, P.; Shukla, S.; Hennessy, T.; Calingasan, N.; Yang, L.; Starkov, A.; Kiaei, M.; Cannella, M.; Sassone, J.; et al. Impaired PGC-1α function in muscle in Huntington’s disease. Hum. Mol. Genet. 2009, 18, 3048–3065. [Google Scholar] [CrossRef] [PubMed]
- Weydt, P.; Pineda, V.V.; Torrence, A.E.; Libby, R.T.; Satterfield, T.F.; Lazarowski, E.; Gilbert, M.L.; Morton, G.J.; Bammler, T.K.; Strand, A.D.; et al. Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1α in Huntington’s disease neurodegeneration. Cell Metab. 2006, 4, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Gong, B.; Chen, F.; Pan, Y.; Arrieta-Cruz, I.; Yoshida, Y.; Haroutunian, V.; Pasinetti, G.M. SCFFbx2-E3-ligase-mediated degradation of BACE1 attenuates Alzheimer’s disease amyloidosis and improves synaptic function. Aging Cell 2010, 9, 1018–1031. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Haroutunian, V.; Katsel, P.; Cardozo, C.P.; Ho, L.; Buxbaum, J.D.; Pasinetti, G.M. PGC-1α expression decreases in the Alzheimer disease brain as a function of dementia. Arch. Neurol. 2009, 66, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Hagl, S.; Grewal, R.; Ciobanu, I.; Helal, A.; Khayyal, M.T.; Muller, W.E.; Eckert, G.P. Rice bran extract compensates mitochondrial dysfunction in a cellular model of early Alzheimer’s disease. J. Alzheimers Dis. 2015, 43, 927–938. [Google Scholar] [PubMed]
- Austin, S.; St-Pierre, J. PGC1α and mitochondrial metabolism–emerging concepts and relevance in ageing and neurodegenerative disorders. J. Cell Sci. 2012, 12, 4963–4971. [Google Scholar] [CrossRef] [PubMed]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; et al. Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. 2001, 21, 3017–3023. [Google Scholar] [PubMed]
- Nagy, Z.; Esiri, M.M.; LeGris, M.; Matthews, P.M. Mitochondrial enzyme expression in the hippocampus in relation to Alzheimer-type pathology. Acta Neuropathol. 1999, 97, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. Brain aging, Alzheimer’s disease, and mitochondria. Biochim. Biophys. Acta 2011, 1812, 1630–1639. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. Mitochondria and cell bioenergetics: Increasingly recognized components and a possible etiologic cause of Alzheimer’s disease. Antioxid. Redox Signal. 2012, 16, 1434–1455. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. β-Apptists and tauists, it is time for sermon from the book of biogenesis. J. Neurochem. 2012, 120, 347–349. [Google Scholar] [CrossRef] [PubMed]
- Brannval, K.; Korhonen, L.; Lindholm, D. Estrogen-receptor-dependent regulation of neural stem cell proliferation and differentiation. Mol. Cell. Neurosci. 2002, 21, 512–520. [Google Scholar] [CrossRef]
- Okada, M.; Murase, K.; Makino, A.; Nakajima, M.; Kaku, T.; Furukawa, S.; Furukawa, Y. Effects of estrogens on proliferation and differentiation of neural stem/progenitor cells. Biomed. Res. 2008, 29, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Makino, A.; Nakajima, M.; Okuyama, S.; Furukawa, S.; Furukawa, Y. Estrogen stimulates proliferation and differentiation of neural stem/progenitor cells through different signal transduction pathways. Int. J. Mol. Sci. 2010, 11, 4114–4123. [Google Scholar] [CrossRef] [PubMed]
- Ishido, M.; Suzuki, J. Classification of phthalates based on an in vitro neurosphere assay using rat mesencephalic neural stem cells. J. Toxicol. Sci. 2014, 39, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Tofighi, R.; Wan Ibrahim, W.N.; Rebellato, P.; Andersson, P.L.; Uhlén, P.; Ceccatelli, S. Non-dioxin-like polychlorinated biphenyls interfere with neuronal differentiation of embryonic neural stem cells. Toxicol. Sci. 2011, 124, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, V.N.; Hei, T.K. Induction of apoptotic death and retardation of neuronal differentiation of human neural stem cells by sodium arsenite treatment. Exp. Cell Res. 2013, 319, 875–887. [Google Scholar] [CrossRef] [PubMed]
- Tamm, C.; Sabri, F.; Ceccatelli, S. Mitochondrial-mediated apoptosis in neural stem cells exposed to manganese. Toxicol. Sci. 2008, 101, 310–320. [Google Scholar] [CrossRef] [PubMed]
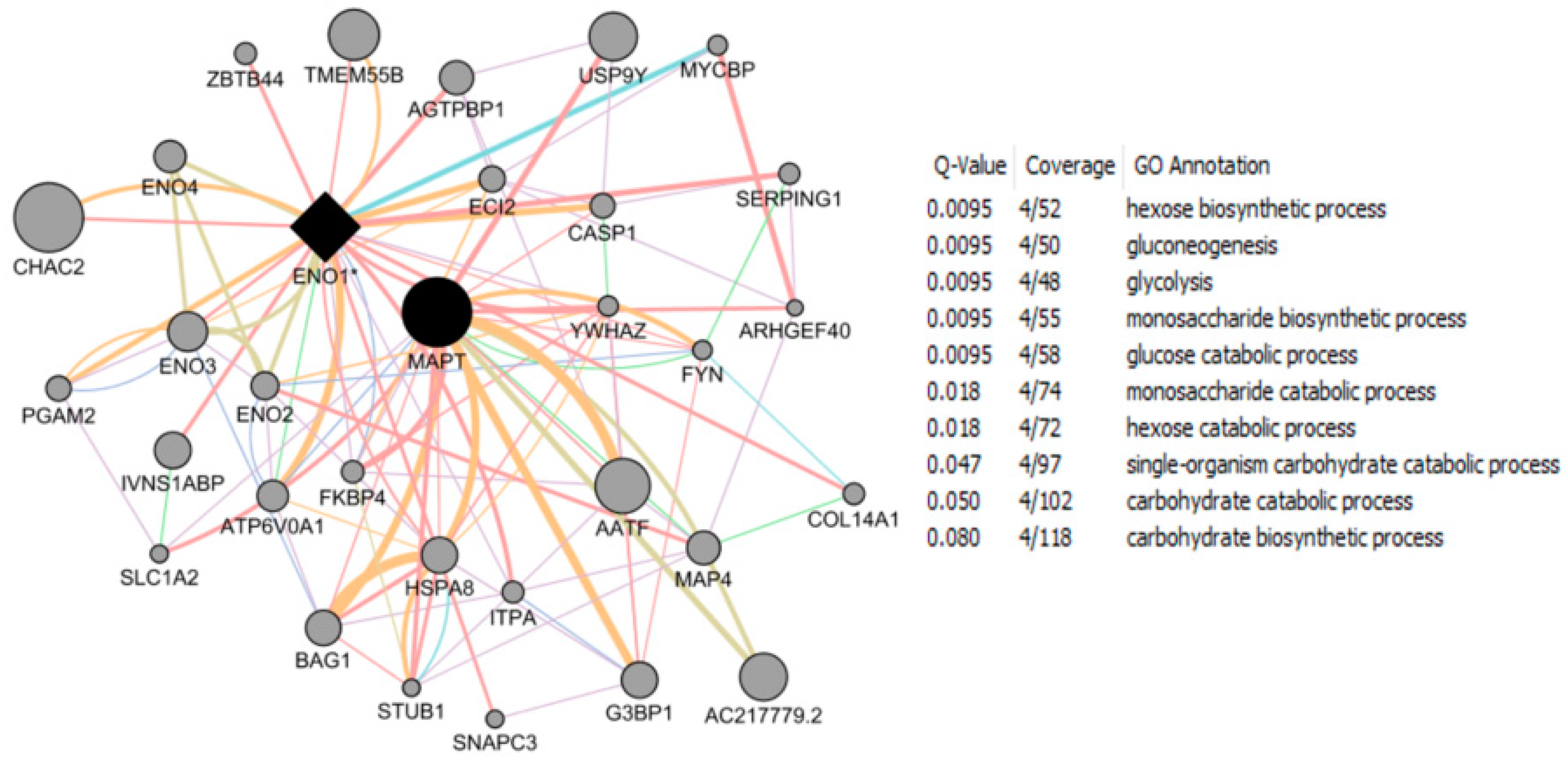
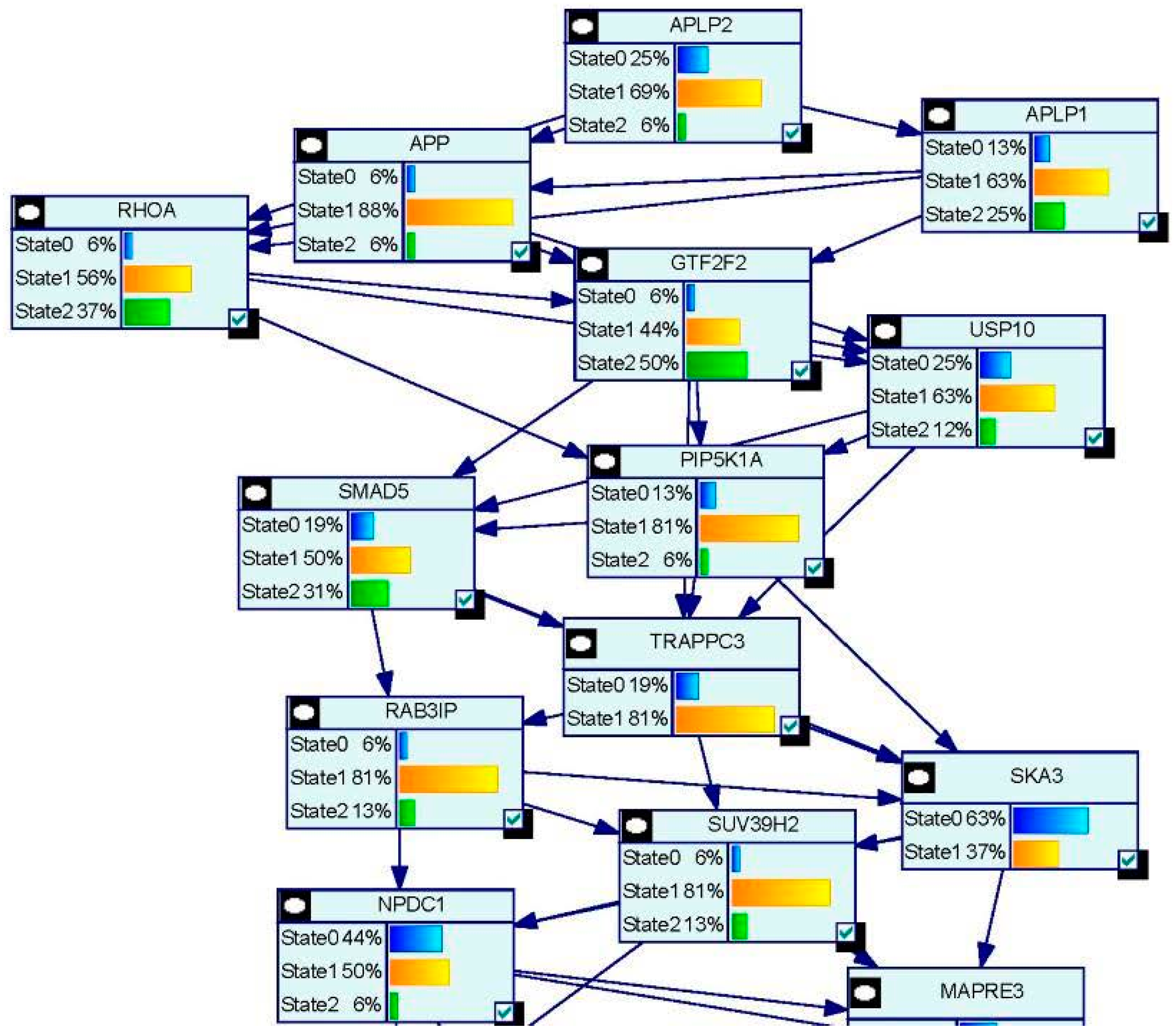
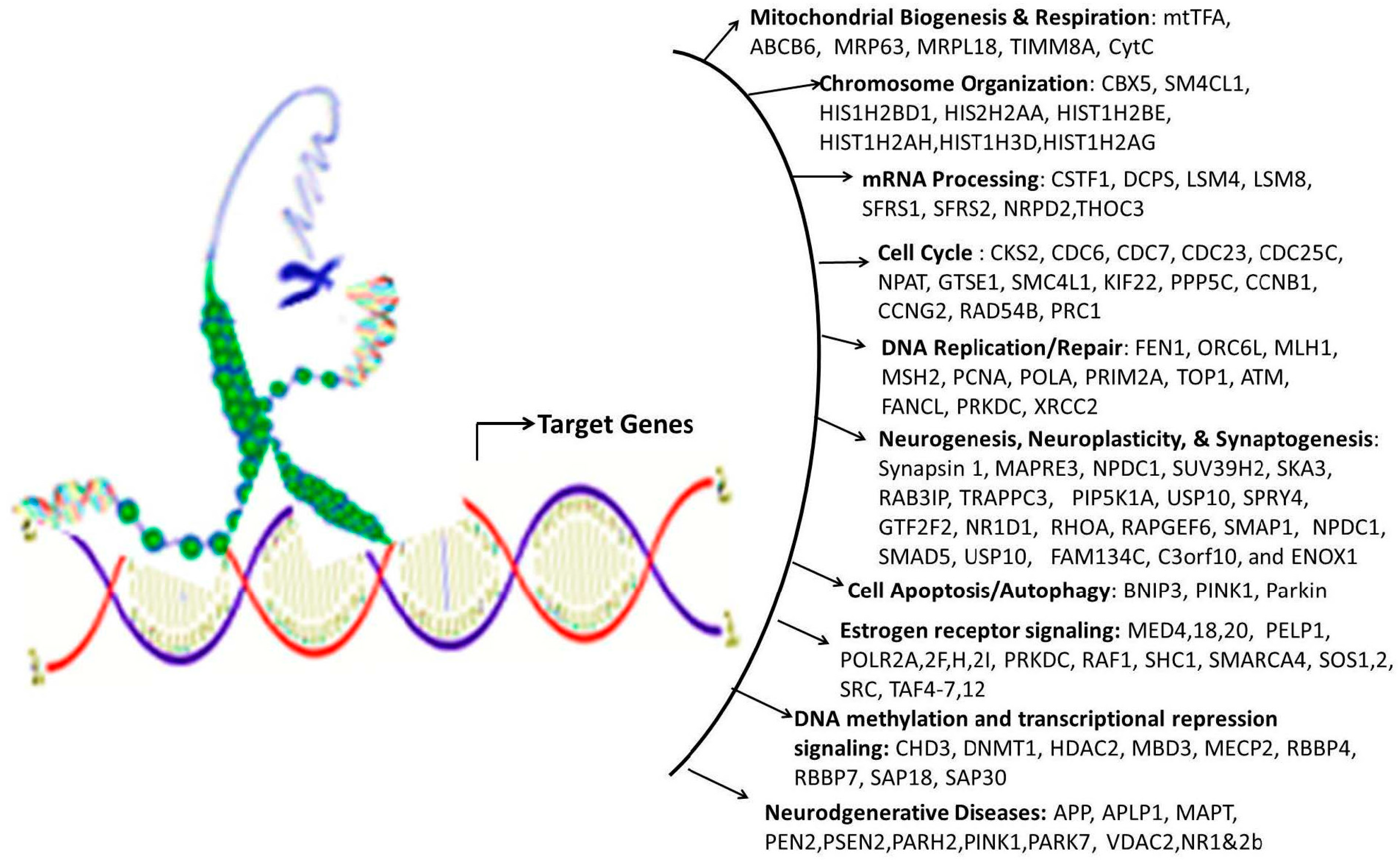
- Wang, J.L.; Chang, W.T.; Tong, C.W.; Kohno, K.; Huang, A.M. Human synapsin I mediates the function of nuclear respiratory factor 1 in neurite outgrowth in neuroblastoma IMR-32 cells. J. Neurosci. Res. 2009, 87, 2255–2263. [Google Scholar] [CrossRef] [PubMed]
- Tong, C.W.; Wang, J.L.; Jiang, M.S.; Hsu, C.H.; Chang, W.T.; Huang, A.M. Novel genes that mediate nuclear respiratory factor 1-regualted neurite outgrowth in neuroblastoma IMR-32 cells. Gene 2013, 515, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.L.; Tong, C.W.; Chang, W.T.; Huang, A.M. Novel genes FAM134C, C3orf10 and ENOX1 are regulated by NRF-1 and differentially regulate neurite outgrowth in neuroblastoma cells and hippocampal neurons. Gene 2013, 529, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Danysz, W.; Parsons, C.G. Alzheimer’s disease, β-amyloid, glutamate, NMDA receptors and memantine—Searching for the connections. Br. J. Pharmacol. 2012, 167, 324–352. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.S.; Liang, H.L.; Wong-Riley, M.T.T. Nuclear respiratory factor 1 co-regulates AMPA glutamate receptor subunit 2 and cytochrome c oxidase: Tight coupling of glutamatergic transmission and energy metabolism in neurons. J. Neurochem. 2009, 108, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Priya, A.; Johar, K.; Wong-Riley, M.T.T. Nuclear respiratory factor 2 regulates the expression of the same NMDA receptor subunit genes as NRF-1: Both factors act by a concurrent and parallel mechanism to couple energy metabolism and synaptic transmission. Biochim. Biophys. Acta—Mol. Cell Res. 2013, 1833, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Andreoli, V.; De Marco, E.V.; Trecroci, F.; Cittadella, R.; Di Palma, G.; Gambardella, A. Potential involvement of GRIN2B encoding the NMDA receptor subunit NR2B in the spectrum of Alzheimer’s disease. J. Neural Transm. 2014, 121, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Domcke, S.; Bardet, A.F.; Adrian Ginno, P.; Hartl, D.; Burger, L.; Schübeler, D. Competition between DNA methylation and transcription factors determines binding of NRF1. Nature 2015, 528, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Lavigne, M.D.; Vatsellas, G.; Polyzos, A.; Mantouvalou, E.; Sianidis, G.; Maraziotis, I.; Agelopoulos, M.; Thanos, D. Composite macroH2A/NRF-1 nucleosomes suppress noise and generate robustness in gene expression. Cell Rep. 2015, 11, 1090–1101. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, R.; Peter, C.J.; Jiang, Y.; Roussos, P.; Vogel-Ciernia, A.; Shen, E.Y.; Mitchell, A.C.; Mao, W.; Whittle, C.; Dincer, A.; et al. Conserved higher-order chromatin regulates NMDA receptor gene expression and cognition. Neuron 2014, 84, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.S.; Wong-Riley, M.T.T. The kinesin superfamily protein KIF17 is regulated by the same transcription factor (NRF-1) as its cargo NR2B in neurons. Biochim. Biophys. Acta—Mol. Cell Res. 2011, 1813, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Yeo, M.; Berglund, K.; Hanna, M.; Guo, J.U.; Kittur, J.; Torres, M.D.; Abramowitz, J.; Busciglio, J.; Gao, Y.; Birnbaumer, L.; et al. Bisphenol A delays the perinatal chloride shift in cortical neurons by epigenetic effects on the Kcc2 promoter. Proc. Natl. Acad. Sci. USA 2013, 110, 4315–4320. [Google Scholar] [CrossRef] [PubMed]
- Stansfield, K.H.; Bichell, T.J.; Bowman, A.B.; Guilarte, T.R. BDNF and Huntingtin protein modifications by manganese: Implications for striatal medium spiny neuron pathology in manganese neurotoxicity. J. Neurochem. 2014, 131, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Verina, T.; Schneider, J.S.; Guilarte, T.R. Manganese exposure induces α-synuclein aggregation in the frontal cortex of non-human primates. Toxicol. Lett. 2013, 217, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Guilarte, T.R.; Burton, N.C.; Verina, T.; Prabhu, V.V.; Becker, K.G.; Syversen, T.; Schneider, J.S. Increased APLP1 expression and neurodegeneration in the frontal cortex of manganese-exposed non-human primates. J. Neurochem. 2008, 105, 1948–1959. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Hou, Y.; Mattson, M.P. Mitochondria and neuroplasticity. ASN Neuro 2010, 2, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Arambula, S.E.; Belcher, S.M.; Planchart, A.; Turner, S.D.; Patisaul, H.B. Impact of low dose oral exposure to bisphenol a (BPA) on the neonatal rat hypothalamic and hippocampal transcriptome: A CLARITY-BPA consortium study. Endocrinology 2016, 157, 3856–3872. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Casanova, R.; Resnick, S.M.; Manson, J.E.; Baker, L.D.; Padual, C.B.; Kuller, L.H.; Bryan, R.N.; Espeland, M.A.; Davatzikos, C. Effects of hormone therapy on brain volumes changes of postmenopausal women revealed by optimally-discriminative voxel-based morphometry. PLoS ONE 2016, 11, e0150834. [Google Scholar] [CrossRef] [PubMed]
- Henderson, V.W.; John, J.A.; Hodis, H.N.; McCleary, C.A.; Stanczyk, F.Z.; Shoupe, D.; Kono, N.; Dustin, L.; Allayee, H.; Mack, W.J. Cognitive effects of estradiol after menopause: A randomized trial of the timing hypothesis. Neurology 2016, 87, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Rosario, E.R.; Chang, L.; Head, E.H.; Stanczyk, F.Z.; Pike, C.J. Brain levels of sex steroid hormones in men and women during normal aging and in Alzheimer’s disease. Neurobiol. Aging 2011, 32, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Foy, M.R.; Baudry, M.; Diaz Brinton, R.; Thompson, R.F. Estrogen and hippocampal plasticity in rodent models. J. Alzheimers. Dis. 2008, 15, 589–603. [Google Scholar] [PubMed]
- Leuba, G.; Vernay, A.; Kraftsik, R.; Tardif, E.; Riederer, B.M.; Savioz, A. Pathological reorganization of NMDA receptors subunits and postsynaptic protein PSD-95 distribution in Alzheimer’s disease. Curr. Alzheimer Res. 2014, 11, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Stein, J.L.; Hua, X.; Morra, J.H.; Lee, S.; Hibar, D.P.; Ho, A.J.; Leow, A.D.; Toga, A.W.; Sul, J.H.; Kang, H.M.; et al. Genome-wide analysis reveals novel genes influencing temporal lobe structure with relevance to neurodegeneration in Alzheimer’s disease. Neuroimage 2010, 51, 542–554. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Jia, J. Association between NR2B subunit gene (GRIN2B) promoter polymorphisms and sporadic Alzheimer’s disease in the North Chinese population. Neurosci. Lett. 2009, 450, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Snyder, E.M.; Nong, Y.; Almeida, C.G.; Paul, S.; Moran, T.; Choi, E.Y.; Nairn, A.C.; Salter, M.W.; Lombroso, P.J.; Gouras, G.K.; et al. Regulation of NMDA receptor trafficking by amyloid-β. Nat. Neurosci. 2005, 8, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.C.; Keeney, P.M.; Algarzae, N.K.; Ladd, A.C.; Thomas, R.R.; Bennett, J.P., Jr. Mitochondrial DNA copy numbers in pyramidal neurons are decreased and mitochondrial biogenesis transcriptome signaling is disrupted in Alzheimer’s Disease hippocampi. J. Alzheimer’s Dis. 2014, 40, 319–330. [Google Scholar]
- Renbaum, P.; Beeri, R.; Gabai, E.; Amiel, M.; Gal, M.; Ehrengruber, M.U.; Levy-Lahad, E. Egr-1 upregulates the Alzheimer’s disease presenilin-2 gene in neuronal cells. Gene 2003, 318, 113–124. [Google Scholar] [CrossRef]
- Hendrickx, A.; Pierrot, N.; Tasiaux, B.; Schakman, O.; Brion, J.P.; Kienlen-Campard, P.; De Smet, C.; Octave, J.N. Epigenetic induction of EGR-1 expression by the amyloid precursor protein during exposure to novelty. PLoS ONE 2013, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Atwood, C.S.; Bowen, R.L. A Unified hypothesis of early- and late-onset Alzheimer’s disease pathogenesis. J. Alzheimer’s Dis. 2015, 47, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; An, S.S.A.; Kim, S. Mutations in presenilin 2 and its implications in Alzheimer’s disease and other dementia-associated disorders. Clin. Interv. Aging 2015, 10, 1163–1172. [Google Scholar] [PubMed]
- Watanabe, H.; Iqbal, M.; Zheng, J.; Wines-Samuelson, M.; Shen, J. Partial loss of presenilin impairs age-dependent neuronal survival in the cerebral cortex. J. Neurosci. 2014, 34, 15912–15922. [Google Scholar] [CrossRef] [PubMed]
- Duplan, E.; Sevalle, J.; Viotti, J.; Goiran, T.; Bauer, C.; Renbaum, P.; Levy-Lahad, E.; Gautier, C.A.; Corti, O.; Leroudier, N.; et al. Parkin differently regulates presenilin-1 and presenilin-2 functions by direct control of their promoter transcription. J. Mol. Cell Biol. 2013, 5, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Ko, H.S.; Kang, H.; Lee, Y.; Lee, Y.I.L.; Pletinkova, O.; Troconso, J.C.; Dawson, V.L.; Dawson, T.M. PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in parkinson’s disease. Cell 2011, 144, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Charniak, E. Bayesian networks without tears. AI Mag. 1991, 12, 50–63. [Google Scholar]
- Hokama, M.; Oka, S.; Leon, J.; Ninomiya, T.; Honda, H.; Sasaki, K.; Iwaki, T.; Ohara, T.; Sasaki, T.; LaFerla, F.M.; et al. Altered expression of diabetes-related genes in Alzheimer’s disease brains: The Hisayama study. Cereb. Cortex 2014, 24, 2476–2488. [Google Scholar] [CrossRef] [PubMed]
- Silander, T.; Myllymaki, P. A simple approach for finding the globally optimal Bayesian network structure. In Proceedings of the Twenty-Second Conference on Uncertainty in Artificial Intelligence, Cambridge, MA, USA, 13–16 July 2006.
- Cheng, A.; Wan, R.; Yang, J.L.; Kamimura, N.; Son, T.G.; Ouyang, X.; Luo, Y.; Okun, E. Involvement of PGC-1α in the formation and maintenance of neuronal dendritic spines. Nat. Commun. 2012, 3, 1250. [Google Scholar] [CrossRef] [PubMed]
- Kunkle, B.W.; Yoo, C.; Roy, D. Reverse engineering of modified genes by bayesian network analysis defines molecular determinants critical to the development of glioblastoma. PLoS ONE 2013, 8, e64140. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Reference | EDC | Year | Epidemiological Study Type | Effect on Brain | Brain Disease |
|---|---|---|---|---|---|
| Manly et al., 2000 | Endogenous estrogen | 2000 | Case-control | Lower estradiol levels in women associated with greater risk of AD. | AD |
| Geerlings et al., 2001 | Endogenous estrogen | 2001 | Prospective cohort | Longer exposure to endogenous estrogen associated with AD and dementia. | Dementia and AD |
| Schupf et al., 2006 | Endogenous estrogen | 2006 | Prospective-cohort | Women with low bioavailable estrogen were more likely to develop AD. | AD |
| De Jong et al., 2012 | Endogenous estrogen | 2012 | Case-Control | Longer reproductive time-span and exposure to endogenous estrogen decreases ALS risk. | ALS |
| Fox et al., 2013 | Endogenous estrogen | 2013 | Retrospective cohort study | Longer duration of endogenous estrogen exposure may have a protective effect against AD risk. | AD |
| Cereda et al., 2013 | Endogenous estrogen | 2013 | Cross-sectional | Age of PD onset was positively associated with duration of exposure to endogenous estrogens. | PD |
| Shumaker et al., 2003 | Hormonal replacement therapy | 2003 | Clinical trial | Estrogen progestin HRT increased risk for dementia and did not prevent cognitive impairment. | Dementia |
| Shumaker et al., 2004 | Hormonal replacement therapy | 2004 | Clinical trial | Estrogen only HRT increased risk of dementia and cognitive impairment. | Dementia |
| Shao et al., 2012 | Hormonal replacement therapy | 2012 | Cohort study | Increased Alzheimer’s disease risk amongst women who used HRT more than five years after menopause, but observed a decreased risk of AD if used within five years of menopause. | AD |
| Lundin et al., 2014 | Hormonal replacement therapy | 2014 | Cohort study | Increased in PD risk observed depending on type of hormonal therapy. | PD |
| Weisskopf et al., 2012 | Polychlorinated biphenyls | 2012 | Nested case-control | PCB exposure not associated with PD development. | PD |
| Steenland et al., 2006 | Polychlorinated biphenyls | 2006 | Retrospective mortality study | In PCB exposed plant workers, higher death rates from PD were observed in women. | PD |
| Hatcher-Martin et al., 2012 | Polychlorinated biphenyls | 2012 | Case-control | In post-mortem brain tissue from PD, AD, and controls, PCB levels were higher in PD brain tissue. | PD |
| Roos et al., 2013 | Cadmium | 2013 | Cross-sectional | Elevated heavy metals, including cadmium, were higher in ALS patients. | ALS |
| Komatsu et al., 2011 | Cadmium | 2011 | Case-control | Elevated cadmium hair levels were associated with Parkinson-like symptoms. | PD |
| Park et al., 2014 | Arsenic | 2014 | Cross-sectional | No difference in serum arsenic levels in AD patients and controls. | AD |
| Hozumi et al., 2011 | Manganese | 2011 | Cross-sectional | Higher levels of manganese found among PD patients. | PD |
| Koc et al., 2015 | Manganese | 2015 | Cross-sectional | Higher manganese levels found in hair samples of AD patients compared to controls. | AD |
| Miyake et al., 2011 | Manganese | 2011 | Case-control | In PD patients, no association found with increased manganese intake. | PD |
| Roos et al., 2013 | Manganese | 2013 | Case-control | Elevated manganese levels observed in ALS patients. | ALS |
| Kumudini et al., 2014 | Manganese | 2014 | Case-control | No association between blood manganese levels in PD patients’ vs. controls. | PD |
| Garzillo et al., 2014 | Manganese | 2014 | Case-control | No association between blood manganese levels in ALS patients’ vs. controls. | ALS |
| Kihira et al., 2015 | Manganese | 2015 | Case-control | Elevated manganese levels in hair observed in ALS patients’ vs. controls. | ALS |
| Arain et al., 2015 | Manganese | 2015 | Case-control | Higher levels of manganese and aluminum in those suffering from neurodegenerative disease. | Neurodegenerative disease. |
| Baker et al., 2015 | Manganese | 2015 | Prospective cohort | Low level manganese exposure causes sub-clinical brain changes before symptoms occur. | Neurodegenerative disease |
| Study Reference | EDC | Year | Epidemiological Study Type | Study Population | Measurement of Exposure | Outcome | Results | Confounders | Comments | Brain Health Indicator |
|---|---|---|---|---|---|---|---|---|---|---|
| Barret-Connor and Goodman-Gruen, 1999 | Endogenous estrogen | 1999 | Cross-sectional | 393 females (ages 55 to 89). | Serum estradiol and estrone levels. Bioavailable and total. | Partial correlation (p-value) and Linear Regression (β, p-value). | No associations between neuropsychological tests and endogenous estrogen exposure. | Smoking status, alcohol use, body mass index, mood, age, education. | Does not account for past exogenous estrogen use, only present use. | Memory performance |
| Low et al., 2005 | Endogenous estrogen | 2005 | Cross-sectional | 760 women (ages 60–64). | Reproductive time period as a surrogate of endogenous estrogen exposure. | Linear regression (β, p-value). | No significant associations found between performance on cognitive and memory tests and endogenous estrogen exposure. | Age, education, verbal intelligence, health and mood variables, lifestyle variables, reproductive variables. | Study accounts for exogenous estrogen use. | Memory performance |
| Heys et al., 2011 | Endogenous estrogen | 2011 | Cross-sectional | 11094 women (age > 50 years). | Proxies of endogenous estrogen exposure. | Multivariate Linear regression (p-value, 95% CI). | Longer reproductive period associated with higher cognitive delayed recall scores (p-value = 0.001; 95% CI, 0.008–0.02) and mini-mental state exam sores (p-value < 0.001; 95% CI, 0.04–0.07. | Age, education, childhood and adulthood socio-economic position and physical activity. | Accounts for exogenous estrogen use. | Memory performance |
| Zimmerman et al., 2011 | Endogenous estrogen | 2011 | Cross-sectional | 181 men (mean age = 81 years). | Estradiol and testosterone levels. | Linear Regression (β, p-value). | Men with higher levels of total estradiol performed better on verbal memory assessments (β = 0.17, p-value < 0.02), compared to men with lower levels of total estradiol. | Age, education, body mass index, and cardiovascular comorbidities. | Accounts for exogenous estrogen use. | Memory performance |
| Oral Contraceptives and Memory Performance | ||||||||||
| Beltz et al., 2015 | Ethinyl estradiol | 2015 | Cross-sectional | 136 men, 93 normally cycling women, 148 OC users. 18–30 years of age. | History of OC use. | Hierarchal Regression. | Ethinyl estradiol was found to affect memory, but only amongst homogenous groups of OC users. | Age and vocabulary. | Study does not account for endogenous estrogen exposure. | Memory Performance |
| Egan and Gleason, 2012 | Ethinyl estradiol | 2012 | Cross-sectional | 261 cognitively normal women, 40–65 years of age. | History of OC use. | MANCOVA (u, CI, p-value). | OC users performed better on Visuo-spatial ability (u = 0.75, CI 0.23–1.28, p = 0.005) and speed and flexibility (u = 0.52, CI 0.16–1.04, p = 0.007). | Age, years of education, socioeconomic status. | Study compares OC users to non-users. | Memory Performance |
| Griksiene and Ruksenas, 2011 | Ethinyl estradiol | 2011 | Cross-sectional | 43 females, 23 OC users, 20 non-users, ages 19 to 24 years of age. | Salivary 17- β estradiol and progesterone levels. | ANOVA. | OC use negatively affects verbal and spatial abilities. | Not stated. | Does not differentiate between the type of OC. | Memory Performance |
| Hormone Replacement Therapy and Memory Performance | ||||||||||
| Resnick et al., 2006 | Hormonal replacement therapy | 2006 | Randomized, double-blind, placebo–controlled clinical trial | 1416 postmenopausal women, 65 years of age and older. | Estrogen + Progestin (CEE + MPA) or Placebo. | Change is cognitive function and affect. Generalized Linear Models (p-value). | CEE + MPA had a negative effect on verbal memory (p < 0.01) and positive effect on figural memory (p = 0.012) compared to placebo. No significant influence on positive affect, negative affect, or depressive symptoms. | Age, time at enrollment, education, race, BMI, smoking status, alcoholic drinks/week, history of cardiovascular disease, hypertension, diabetes, prior HT use. | Beneficial and detrimental effects observed. | Memory Performance |
| Resnick et al., 2009 | Hormonal replacement therapy | 2009 | Randomized, double-blind, placebo-controlled clinical trial | 866 postmenopausal women with prior hysterectomy, 65 years of age and older, free of probable dementia. | 0.625 mg of CEE daily or placebo. | Annual rates of change in specific cognitive functions and affects. Linear Mixed Models (p-value). | Compared with placebo, CEE treatment was associated with lower spatial rotational ability (p < 0.01) which diminished over time. CEE did not affect other cognitive functions and affects. | Age, time at enrollment, education, race, BMI, smoking status, alcoholic drinks/week, history of cardiovascular disease, hypertension, diabetes, prior HT use. | No improvements in memory over time. | Memory performance |
| Almeida et al., 2006 | Hormonal replacement therapy. | 2006 | Randomized, double-blind, placebo-controlled trial | 115 Postmenopausal women, 65 years of age and older. | 2 mg estradiol or placebo. | Changes in cognitive function and affect. | Higher dosages of estradiol containing HRT treatment did not improve cognitive function. | Age, marital status, education, previous HRT use, daily living, age at menarche, age at menopause, cognitive factors and complaints, plasma estradiol and homocysteine levels. | No significant improvements in memory. | Memory performance. |
| Pefanco et al., 2007 | Hormonal replacement therapy | 2007 | Randomized, placebo-controlled trial | 57 postmenopausal women, 65 years of age and older. | 0.20 mg of micronized 17-β estradiol or placebo. | Neuropsychological measures of memory, language, mood, and executive function Repeated Measures Analysis (p-value). | No differences were found between ET and placebo on any of the neurocognitive measures or depression instruments, nor were there any differences when the groups were stratified according to age. | Age, education, income, daily living, estradiol levels, estrone levels, hysterectomy. | No significant improvements in memory. | Memory performance. |
| Viscoli et al., 2005 | Hormone replacement therapy. | 2005 | Randomized, double-blind trial. | 644 postmenopausal women. | 1 mg of 17-β estradiol or placebo. | Results of MMSE and domain measures. Generalized Linear Models (p-values). | Estrogen therapy did not have a significant effect on cognitive measures over time. | Age, race, education level, chronic diseases, prior estrogen therapy use, hysterectomy, depression, stroke. | No significant improvement in memory. | Memory performance. |
| Yaffe et al., 2006 | Hormone replacement therapy. | 2006 | Randomized, placebo-controlled, double-blind trial | 417 postmenopausal women, 60 to 80 years of age. | Weekly transdermal patch of 0.014 mg/day estradiol or placebo. | Results of global cognitive function, verbal and visuospatial memory, language, executive function, and semantic memory. Linear mixed models (p-value). | Estrogen therapy did not have a significant effect on cognitive measures over time. | Age, education level, race, BMI, smoking status, depression status, presence of hot flashes, current estradiol level. | No significant improvement in memory. | Memory performance. |
| Bisphenol A, Maternal Exposure, and Child Behavior | ||||||||||
| Harley et al., 2013 | Bisphenol-A | 2013 | Prospective Cohort Study | 592 mothers and their children, followed prenatally up to 9 years of age. | Urinary BPA concentrations (µg/L). Child and maternal exposure. | Child behavior assessment results. Multivariable linear regression (β, 95% CI). | Prenatal urinary BPA concentrations were associated with increased internalizing problems in boys at age 7 (β = 1.8, 95% CI: 0.3–3.3). Childhood urinary exposure was associated with increased externalizing behavior in girls at age 7 (β = 1.2, 95% CI: 0.3–2.1 and β = 1.0, 95% CI: 0.1–2.0) and increased inattention and hyperactivity behaviors in both boys and girls at age 7. | Maternal age, race/ethnicity, education level, marital status, country of birth, years of US residency, number siblings, family income, maternal depression, pesticide metabolites. | Behavioral results from both teacher and mother reported. | Behavior |
| Braun et al., 2009 | Bisphenol-A | 2009 | Prospective Cohort Study | 249 mothers and their children followed prenatally up to 2 years of age. | Urinary BPA concentrations (µg/L). Child and maternal exposure. | Child behavior assessment. Multivariable linear regression (β, 95% CI). | Prenatal BPA concentrations associated with externalizing scores among females (β = 6.0, 95% CI 0.1–12.0). BPA concentrations at 16 weeks associated with externalizing behavior among all children (β = 2.9, 95% CI 0.2–5.7). | Maternal age, race, education, marital status, SES, maternal depression. | Female appear to be more affected. | Behavior |
| Yolton et al., 2011 | Bisphenol-A | 2011 | Prospective Cohort Study | 350 mothers and their children up to 5 weeks. | Urinary BPA concentrations (ng/mL). | Neurobehavioral outcomes. Multivariable linear regression (p-value). | No significant associations between gestational exposure to BPA and infant neurobehavior. Higher BPA exposure associated with greater hypotonia at 26 weeks (p-value = 0.09). | Maternal race, household income, marital status, maternal depression, maternal BMI, maternal cotinine levels, infant weight, NICU stay after birth. | Phthalates also measured. | Behavior |
| Roen et al., 2015 | Bisphenol-A | 2015 | Prospective Cohort Study | 250 mothers and their children followed prenatally up to 7–9 years of age. | Urinary BPA concentrations (µg/L). | Neurobehavioral outcomes. Poisson Regression (β, p-value). | Among boys, high prenatal BPA concentrations was associated with increase internalizing (β = 0.41, p < 0.0001) and externalizing scores (β = 0.40, p < 0.0001). High postnatal BPA concentrations was associated with increased internalizing (β = 0.30, p = 0.0002) and externalizing scores (β = 0.33, p < 0.0001). | Prenatal and postnatal BPA concentration, child age, ethnicity, gestational age, maternal intelligence, maternal education, demoralization, tobacco smoke exposure, and phthalate exposure. | Possible sex specific and timing of exposure mechanism. | Behavior |
| Braun et al., 2011 | Bisphenol-A | 2011 | Prospective Cohort Study | 244 mothers and their children followed from gestation up to 3 years of age. | Urinary BPA concentrations (µg/L). | Neurobehavioral outcomes. Multivariate linear regression (β, 95% CI). | BPA detected in >97% of gestational and childhood samples. Each 10-fold increase in gestational BPA concentrations was associated with more anxious and depressed behavior. The effects were more pronounced in boys. | Mother’s race, education, marital status, household income, maternal depressive behavior, phthalate exposure, tobacco smoke. | Childhood exposure was not significantly associated with behavioral modifications. | Behavior |
| Miodovnik et al., 2011 | Bisphenol-A | 2011 | Prospective Cohort Study | 404 mothers and their children followed from gestation up to 9 years of age. | Urinary BPA concentrations (µg/L). | Neurobehavioral outcomes. General linear models (β, 95% CI). | No associations found with BPA and outcomes. | Maternal age, maternal IQ, marital status, maternal education, child race, sex, child IQ, age at exam, urinary creatinine. | Significant associations found with phthalate exposure. | Behavior |
| Phthalates, Behavioral Disorders, and IQ | ||||||||||
| Yolton et al., 2011 | Phthalates | 2011 | Prospective Cohort Study | 350 mothers and their children up to 5 weeks. | Urinary phthalate concentrations (ng/mL). | Neurobehavioral outcomes. Multivariable linear regression (p-value). | Prenatal exposure to dibutyl phthalate was associated with improved behavioral organization in 5 week old infants characterized by decreased arousal (p = 0.04), increased self-regulation (p = 0.052), and decreased handling (p = 0.02). Prenatal exposure to diethylhexyl phthalate at 26 weeks was associated with nonoptimal reflexes (p = 0.02) in male infants. | Maternal race, household income, marital status, maternal depression, maternal BMI, maternal cotinine levels, infant weight, NICU stay after birth. | Results are inconsistent between phthalate types. | Behavior |
| Miodovnik et al., 2011 | Phthalates | 2011 | Prospective Cohort Study | 404 mothers and their children followed from gestation up to 9 years of age. | Urinary phthalate concentrations (µg/L). | Neurobehavioral outcomes. General linear models (β, 95% CI). | Increased phthalate exposure associated with greater social deficits (β = 1.52, 95% CI: 0.25–2.8). | Maternal age, maternal IQ, marital status, maternal education, child race, sex, child IQ, age at exam, urinary creatinine. | Significant associations found with phthalate exposure. | Behavior |
| Factor-Litvak et al., 2014 | Phthalates | 2014 | Prospective Cohort Study | 328 inner-city mothers and their children followed up to 7 years of age. | Urinary phthalate concentrations (ng/mL). | IQ test results. Linear regression models (β, 95% CI). | Prenatal metabolite concentrations of DnBP and DiBP inversely associated with IQ (β = −2.69, 95% CI −4.33, −1.05). Inverse associations seen between IQ, cognitive function, and maternal prenatal metabolite concentrations. | Race, maternal education, marital status, income, parity, gestational age, birth weight, child sex, breastfeeding history, tobacco smoke exposure, prenatal alcohol, hardships during pregnancy, maternal depression. | Phthalate-specific effects were observed. | Child IQ and cognitive function. |
| Reference | EDC | Study Type | Effects on Brain Health |
|---|---|---|---|
| Bowman et al., 2015 | Bisphenol-A | Adolescent male and female adolescent rats | In male rats: Decrease in non-spatial memory and object recognition. |
| In both sexes: Decreased spine density on apical and basal dendrites on pyramidal cells in CA1 of the hippocampus. | |||
| Eilam-Stock et al., 2012 | Bisphenol-A | Adult male rats | Significantly impaired visual and spatial memory. |
| Decreased spine density on pyramidal cells in the CA1 and mPFC. | |||
| Decrease expression of PSD-95, increased expression of pCREB. | |||
| Elsworth et al., 2015 | Bisphenol-A | Adult male vervet monkeys | Decreased working memory and accuracy. |
| Decreased excitatory synaptic outputs on dendritic spines of pyramidal neurons in pfc and hippocampus. | |||
| Inagaki et al., 2012 | Bisphenol-A | Ovariectomized female rats | Exposure to BPA alters E-induced enhancements of spatial and nonspatial memory. |
| In the hippocampus, BPA blocked E2 induced increases in basal spine density. | |||
| Wang et al., 2014 | Bisphenol-A | Postnatal male rats born from BPA exposed female rats | Maternal exposure affected locomotor activity, exploratory habits, emotional behavior. |
| Increase reference and working memory errors. | |||
| Decreased mRNA and protein expression of synaptophysin, PSD-95, spinophilin, GluR1, and NMDAR1 in hippocampus. | |||
| Adverse effects on synaptic structure. | |||
| Xu et al., 2013 | Bisphenol-A | Male and female adult mice | Sex-specific effects of BPA exposure on spatial and passive avoidance memory. |
| Reduced synaptic density and adversely affected structure of synaptic interface. | |||
| In hippocampus, down-regulation of snynapsin I, PSD-95, NDMA receptor subunit NR1 and AMPA receptor subunit GluR1 in male mice. | |||
| Jang et al., 2012 | Bisphenol-A | Postnatal female rats born from BPA expose female rats | Decrease in newly generated cells in the hippocampus. |
| Negatively affected memory attention. | |||
| Lower levels of phospho-ERK, BDNF, phosphor-CREB in hippocampus. | |||
| Increased DNA methylation of Crtc1. | |||
| Kim et al., 2011 | Bisphenol-A | Young adult mice | High dose BPA exposure decreased the number of newly generated cells in the hippocampus, while low dose exposure had the opposite effect. |
| High dose BPA exposure was shown to impair learning and memory performance significantly. | |||
| Diaz Weinstein et al., 2013 | Bisphenol-A | Adolescent male and female rats | Decreased spatial memory. |
| Adverse locomotor activity. | |||
| Increased anxiety. | |||
| Elsworth et al., 2015 | Bisphenol-A | Pregnant female rhesus monkeys | Abnormal fetal brain development. |
| Fetuses of female monkeys showed a decrease in midbrain dopamine neurons and a reduction in spine synapses in the CA1 region of the hippocampus. | |||
| Johnson et al., 2015 | Bisphenol-A | Pregnant female rats and offspring | In the offspring: Developmental exposure disrupted navigational learning and navigational memory. |
| Kumar and Kumar Thakur 2014 | Bisphenol-A | Pregnant female mice and male offspring | In the offspring: Impaired spatial memory. |
| Increased dendritic spine density in the cerebral cortex and hippocampus. | |||
| Upregulation of synaptic proteins Nrxn1 and Nlgn3. | |||
| Matsuda et al., 2013 | Bisphenol-A | Pregnant female mice and offspring | In the offspring: Enhanced fear memory. |
| Increased serotonin metabolites. | |||
| Increased expression of Tph2, Slc6a4, and Maoa. | |||
| Xu et al., 2014 | Bisphenol-A | Pregnant female rats and offspring | In the offspring: Interference with estrogen receptor signaling in the developing hippocampus. |
| Zhang et al., 2014 | Bisphenol-A | Adult male mice | Enhanced the acquisition and retention of fear memory by the increased levels of histone acetylation, NMDA receptor, and phosphorylation of the ERK1/2 in the hippocampus of male mice. |
| Stump et al., 2010 | Bisphenol-A | Pregnant female rats and offspring | No adverse effects reported. |
| Ishido et al., 2011 | Bisphenol-A | Postnatal male mice | Apparent hyperactivity observed. |
| Viberg et al., 2011 | Bisphenol-A | Postnatal mice | Alterations in spontaneous behavior and cognitive function observed. |
| Kimura et al., 2015 | Bisphenol-A | Prenatal mice | In utero exposure to BPA reduced spine densities in the hippocampal CA1 region of the brain in prenatal mice. |
| Kim et al., 2009 | Bisphenol-A | Neonatal and postnatal mice | Stimulation of neuronal differentiation and possible disruption of neonatal brain development observed. |
| Dai et al., 2015 | Phthalates | Young mice | Negatively affected locomotion activity and memory. |
| Betz et al., 2013 | Phthalates | Young mice | Negatively affects learning and social behavior. |
| Li et al., 2013 | Phthalates | Young mice | Induced apoptotic cell death, synaptic loss, and synaptic dysfunction. |
| Elnar et al., 2015 | Phthalates | Young mice | Early exposure to PCBs induced neuronal susceptibility to amyloid stress. Lower expression of synaptic proteins. |
| Reilly et al., 2015 | PCBs | Young mice | Negatively affects social behavior. |
| Zahara et al., 2015 | PCBs | Juvenile Avians | Reduction in learning ability. |
| Hilgier et al., 2012 | PCBs | Adult Rats | Exposure resulted in neuronal injury and loss. |
| Lee et al., 2012 | PCBs | Adult mice | Exposure resulted in hyperactivity and dopaminergic neuron degeneration. |
| 17 β-Estradiol (E2) Interacting NRF1 Target Genes | ||
| KEGG Pathway | Number of Genes | Annotated Genes |
| Disease | 86 | ADCY9|AKT2|ALDOA|AP1S1|CCNT2|CDC42|CDK1|CDKN2B|CHMP2A|CHMP2B|CHMP5|CREB1|CSK|CSNK1A1|CTBP1|CYB5A|ENO1|EPS15L1|ERCC2|EXT2|FASN|FZD4|GLB1|GTF2E2|HDAC2|HDAC3|HDAC4|HMMR|HRAS|HSP90AA1|IRS1|MAP2K7|MKNK1|MMADHC|MTHFD1|NAMPT|NPM1|NUP107|NUP50|OS9|P4HB|PAPSS1|PFKFB4|PIP5K1B|PLCG1|POLR2A|POM121|PPIA|PRKAR2A|PSENEN|PYGL|RAC1|RAE1|RAF1|RAN|RBX1|RHOA|RPL10|RPL13A|RPL14|RPL36|RPS13|RPS16|RPS21|RPS29|RPS9|SHC1|SLC25A10|SLC37A4|SMARCA4|SNW1|SOS1|SRC|STUB1|STX1A|TAF12|TAF5|TCEB2|TGIF1|TNKS2|TPR|UBA52|WNT5A|XRCC5|XRCC6|YWHAZ |
| Metabolism | 86 | ACADM|ADCY9|ADI1|AGPAT5|AKR7A2|ALAS1|ALDOA|AMACR|ATP5O|AUH|BCAT1|BCAT2|BSG|CBR1|COX5B|COX6C|CTPS1|CYB5A|CYCS|DTYMK|ELOVL4|ENO1|ERCC2|ETFB|ETFDH|EXT2|FASN|FDPS|GCLC|GLB1|GNG5|GPD1L|GSR|GSS|GSTM3|HDAC3|HMMR|HSD17B4|HSP90AA1|INPP5A|INPP5K|IP6K2|LDHA|LPCAT3|LSS|MGST3|MMADHC|MPC2|MTHFD1|NAMPT|NDUFA3|NDUFB4|NUBP1|NUP107|NUP50|OAZ2|ODC1|P4HB|PAICS|PAPSS1|PDHB|PFKFB4|PIP5K1B|PLA2G12A|PLCG1|PLD1|POM121|PPAT|PRKAR2A|PSAP|PYGL|RAE1|RAN|SDHA|SGMS1|SIN3B|SLC16A1|SLC25A10|SLC37A4|SLC44A2|STX1A|SUCLA2|TPMT|TPR|UBA52|VAPB |
| Gene Expression | 77 | CCNT2|CDKN2B|CNOT1|DHX9|DNMT1|EEF1A1|EEF1D|EEF1G|EIF2S1|EIF2S2|EIF3K|EIF4E|EIF4G1|ERCC2|ESRRA|EXOSC2|EZH2|FARSB|GEMIN4|GTF2E2|HARS|HDAC2|HNRNPA1|HNRNPH1|HNRNPU|HSPA8|LSM2|MARS2|MED20|MED4|NR1D1|PCBP2|POLR1A|POLR2A|POLRMT|PPA1|PTBP1|RAN|RBBP4|RBBP7|RNPS1|RPL10|RPL13A|RPL14|RPL36|RPS13|RPS16|RPS21|RPS29|RPS9|RUNX2|SAP18|SARS|SEC61G|SET|SIN3B|SNW1|SRRM1|SRSF1|TAF12|TAF5|TARS|TCEB2|TGIF1|THRA|TRAM1|U2AF1|UBA52|UPF1|UPF2|VARS2|XPO5|YWHAZ|ZNF610|ZNF658|ZNF711|ZNF750 |
| Signal Transduction | 72 | ABI1|ADCY9|AKT2|ARHGDIA|BRK1|CASP2|CCNT2|CDC42|CDK1|CDKN2B|CREB1|CRK|CRKL|CSK|CSNK1A1|CTBP1|DAAM1|DGKH|E2F1|EIF4E|EIF4G1|EPS15L1|FSTL3|FZD4|GNG5|GPR37|HDAC2|HDAC3|HDAC4|HRAS|HSP90AA1|IRS1|JAK2|LFNG|MAPK7|MEF2A|MKNK1|OS9|P4HB|PFN1|PIP5K1B|PLCG1|PRDM4|PRKAR2A|PSAP|PSENEN|PTCH1|PTK2|RAC1|RAF1|RBX1|RELA|RHOA|RIPK2|ROCK1|RPS6KA5|RPS6KB1|SHC1|SMARCA4|SNW1|SOCS1|SOS1|SRC|STARD13|STUB1|TGIF1|THBS2|TNKS2|TRIO|UBA52|WNT5A|YWHAZ |
| Immune System | 67 | ABI1|ADCY9|AKT2|ANAPC1|ANAPC11|AP1S1|ARPC2|ARPC5|BRK1|CASP2|CDC34|CDC42|CDK1|CREB1|CRK|CRKL|CSK|CTSD|DHX9|DYNLL1|EIF4E|EIF4G1|FADD|FZR1|HRAS|HSP90AA1|IP6K2|IRS1|JAK2|KIF18A|KIF4A|MAP2K7|MAPK7|MEF2A|NUP107|NUP50|PCBP2|PELI1|PLCG1|PLD1|POM121|PRKAR2A|PRKDC|PTK2|PVR|RAC1|RAE1|RAF1|RBX1|RELA|RIPK2|RNF19B|RPS6KA5|SEC61G|SHC1|SOCS1|SOS1|SRC|STUB1|TCEB2|TPR|TUBB4B|UBA52|UBE2D4|XRCC5|XRCC6|YWHAZ |
| Cell Cycle | 62 | ANAPC1|ANAPC11|APITD1|ATM|AURKA|AURKB|BUB1|CCNB1|CCNE1|CDC7|CDK1|CDKN2B|CDKN2D|CDT1|CENPN|CKS1B|CLASP2|DKC1|DSN1|DYNLL1|E2F1|ERCC6L|FBXO5|FZR1|GOLGA2|HSP90AA1|KIF18A|LIN37|MAD2L1|MCM2|MCM4|MCM6|MLH3|NEK2|NPM1|NUP107|NUP50|OIP5|ORC6|PCNA|PLK4|POLA1|POM121|PPP1R12A|RAB1B|RAD50|RAE1|RBBP4|RBBP7|RBBP8|RBL2|RSF1|SDCCAG8|SET|SMC4|SPC24|SPDL1|TPR|TUBB|TUBB4B|UBA52|ZWINT |
| Metabolic pathways | 62 | ACADM|ADI1|ALAS1|ALDOA|AMACR|ATP5O|AUH|BCAT1|BCAT2|CBR1|COX5B|COX6A2|COX6C|CTPS1|DAD1|DGKH|DNMT1|DTYMK|ENO1|EXT2|FASN|FBL|FDPS|FUT8|GAA|GCLC|GFPT1|GLB1|GSS|HSD17B4|INPP5A|INPP5K|LAP3|LDHA|LSS|MECR|MTHFD1|NDUFA3|NDUFB4|ODC1|PAICS|PAPSS1|PDHB|PGAM5|PGAP1|PIGA|PIP5K1B|PLA2G12A|PLCG1|PLD1|POLA1|POLE4|POLG2|POLR1A|POLR2A|PPAT|SDHA|SGMS1|SHMT2|SUCLA2|SUFU|TGDS |
| Metabolism of proteins | 48 | B3GNTL1|CCT3|CCT6A|CCT8|CTSD|DAD1|DDIT3|DNAJB9|DNAJC3|DOHH|EEF1A1|EEF1D|EEF1G|EIF2S1|EIF2S2|EIF3K|EIF4E|EIF4G1|EIF5A|FBXW5|FUT8|GFPT1|GLB1|GRPEL2|HSPA9|HSPD1|IGFBP3|MANEA|PFDN1|PGAP1|PIGA|RPL10|RPL13A|RPL14|RPL36|RPS13|RPS16|RPS21|RPS29|RPS9|SEC61G|SHC1|STX1A|THBS2|TRAM1|TSPYL2|TUBB4B|UBA52 |
| Developmental Biology | 42 | ABLIM2|AKT2|ARPC2|ARPC5|CDC42|CDK1|CLASP2|CREB1|CRMP1|CTNNA2|DPYSL2|HDAC3|HRAS|HSP90AA1|HSPA8|ITGA1|KIF4A|MED11|MED18|MED20|MED4|MEF2A|MYH10|MYH14|NRTN|PFN1|PLCG1|PSENEN|PTK2|RAC1|RAF1|RHOA|ROCK1|RPS6KA5|RPS6KA6|SCN3B|SDCBP|SIAH2|SOS1|SPTAN1|SRC|TRIO|TUBB4B |
| Mitotic M-M/G1 phases | 35 | ANAPC1|ANAPC11|APITD1|AURKB|BUB1|CCNB1|CDC7|CDK1|CDT1|CENPN|CLASP2|DSN1|ERCC6L|FBXO5|GOLGA2|KIF18A|MAD2L1|MCM2|MCM4|MCM6|NUP107|NUP50|ORC6|POLA1|POM121|RAB1B|RAE1|SET|SMC4|SPC24|SPDL1|TPR|TUBB4B|UBA52|ZWINT |
| Ethinyl Estradiol Interacting Common E2- and NRF1-Target Genes | ||
| KEGG Pathway | Number of Genes | Annotated Genes |
| Metabolism | 49 | ACADM|ADCY9|AGPAT5|ALAS1|ALDOA|ATP5O|BCAT1|BSG|CBR1|COX5B|COX6C|CTPS1|CYB5A|CYCS|ENO1|ERCC2|ETFDH|EXT2|FASN|FDPS|GCLC|GLB1|GSR|GSS|GSTM3|HDAC3|HSD17B4|HSP90AA1|INPP5A|LDHA|LSS|MGST3|MTHFD1|NAMPT|NUP50|ODC1|P4HB|PAPSS1|PDHB|PPAT|PSAP|PYGL|RAE1|RAN|SGMS1|SIN3B|SLC16A1|SLC25A10|SLC37A4 |
| Disease | 48 | ADCY9|AKT2|ALDOA|AP1S1|CDC42|CDK1|CHMP2A|CREB1|CSK|CSNK1A1|CYB5A|ENO1|ERCC2|EXT2|FASN|GLB1|GTF2E2|HDAC2|HDAC3|HRAS|HSP90AA1|MAP2K7|MTHFD1|NAMPT|NPM1|NUP50|OS9|P4HB|PAPSS1|PSENEN|PYGL|RAE1|RAN|RBX1|RHOA|RPL36|RPS16|RPS9|SLC25A10|SLC37A4|SMARCA4|STUB1|TAF12|TAF5|TCEB2|WNT5A|XRCC5|YWHAZ |
| Gene Expression | 43 | DNMT1|EEF1A1|EEF1G|EIF2S1|EIF2S2|EIF3K|EIF4E|ERCC2|EZH2|FARSB|GTF2E2|HARS|HDAC2|HNRNPA1|HNRNPH1|HNRNPU|HSPA8|MED4|NR1D1|PCBP2|POLR1A|POLRMT|RAN|RBBP4|RBBP7|RNPS1|RPL36|RPS16|RPS9|SAP18|SARS|SEC61G|SET|SIN3B|SRSF1|TAF12|TAF5|TARS|TCEB2|THRA|UPF1|VARS2|YWHAZ |
| Immune System | 36 | ABI1|ADCY9|AKT2|ANAPC1|ANAPC11|AP1S1|ARPC2|ARPC5|CASP2|CDC42|CDK1|CREB1|CRK|CSK|CTSD|DYNLL1|EIF4E|FADD|FZR1|HRAS|HSP90AA1|MAP2K7|MAPK7|NUP50|PCBP2|PTK2|PVR|RAE1|RBX1|SEC61G|SOCS1|STUB1|TCEB2|TUBB4B|XRCC5|YWHAZ |
| Cell Cycle | 35 | ANAPC1|ANAPC11|AURKB|BUB1|CCNB1|CCNE1|CDC7|CDK1|CDT1|CKS1B|DYNLL1|E2F1|FZR1|GOLGA2|HSP90AA1|LIN37|MAD2L1|MCM2|MCM4|MCM6|NPM1|NUP50|ORC6|PCNA|PLK4|PPP1R12A|RAE1|RBBP4|RBBP7|RBBP8|RSF1|SET|SPC24|SPDL1|TUBB4B |
| Metabolic pathways | 33 | ACADM|ALAS1|ALDOA|ATP5O|BCAT1|CBR1|COX5B|COX6C|CTPS1|DNMT1|ENO1|EXT2|FASN|FDPS|GCLC|GLB1|GSS|HSD17B4|INPP5A|LDHA|LSS|MECR|MTHFD1|ODC1|PAPSS1|PDHB|PGAM5|PIGA|POLE4|POLG2|POLR1A|PPAT|SGMS1 |
| Signal Transduction | 33 | ABI1|ADCY9|AKT2|CASP2|CDC42|CDK1|CREB1|CRK|CSK|CSNK1A1|E2F1|EIF4E|HDAC2|HDAC3|HRAS|HSP90AA1|MAPK7|OS9|P4HB|PFN1|PRDM4|PSAP|PSENEN|PTCH1|PTK2|RBX1|RHOA|SMARCA4|SOCS1|STUB1|TRIO|WNT5A|YWHAZ |
| Metabolism of proteins | 28 | B3GNTL1|CCT3|CCT6A|CCT8|CTSD|DDIT3|DNAJC3|EEF1A1|EEF1G|EIF2S1|EIF2S2|EIF3K|EIF4E|EIF5A|FBXW5|GLB1|GRPEL2|HSPA9|HSPD1|IGFBP3|PFDN1|PIGA|RPL36|RPS16|RPS9|SEC61G|TSPYL2|TUBB4B |
| Developmental Biology | 25 | AKT2|ARPC2|ARPC5|CDC42|CDK1|CREB1|CTNNA2|HDAC3|HRAS|HSP90AA1|HSPA8|ITGA1|MED11|MED4|MYH10|NRTN|PFN1|PSENEN|PTK2|RHOA|RPS6KA6|SDCBP|SIAH2|TRIO|TUBB4B |
| Bisphenol A Interacting Common E2- and NRF1-Target Genes | ||
| KEGG Pathway | Number of Genes | Annotated Genes |
| Metabolism | 85 | ACADM|ADCY9|ADI1|AGPAT5|AKR7A2|ALAS1|ALDOA|AMACR|ATP5O|BCAT1|BCAT2|BSG|CBR1|COX5B|COX6C|CTPS1|CYB5A|CYCS|DTYMK|ELOVL4|ENO1|ERCC2|ETFB|ETFDH|EXT2|FASN|FDPS|GCLC|GLB1|GNG5|GPD1L|GSR|GSS|GSTM3|HDAC3|HMMR|HSD17B4|HSP90AA1|INPP5A|INPP5K|IP6K2|LDHA|LPCAT3|LSS|MGST3|MMADHC|MPC2|MTHFD1|NAMPT|NDUFA3|NDUFB4|NUBP1|NUP107|NUP50|OAZ2|ODC1|P4HB|PAICS|PAPSS1|PDHB|PFKFB4|PIP5K1B|PLA2G12A|PLCG1|PLD1|POM121|PPAT|PRKAR2A|PSAP|PYGL|RAE1|RAN|SDHA|SGMS1|SIN3B|SLC16A1|SLC25A10|SLC37A4|SLC44A2|STX1A|SUCLA2|TPMT|TPR|UBA52|VAPB |
| Disease | 84 | ADCY9|AKT2|ALDOA|AP1S1|CCNT2|CDC42|CDK1|CDKN2B|CHMP2A|CHMP2B|CHMP5|CREB1|CSK|CSNK1A1|CTBP1|CYB5A|ENO1|ERCC2|EXT2|FASN|FZD4|GLB1|GTF2E2|HDAC2|HDAC3|HDAC4|HMMR|HRAS|HSP90AA1|IRS1|MAP2K7|MKNK1|MMADHC|MTHFD1|NAMPT|NPM1|NUP107|NUP50|OS9|P4HB|PAPSS1|PFKFB4|PIP5K1B|PLCG1|POLR2A|POM121|PPIA|PRKAR2A|PSENEN|PYGL|RAC1|RAE1|RAF1|RAN|RBX1|RHOA|RPL13A|RPL14|RPL36|RPS13|RPS16|RPS21|RPS29|RPS9|SHC1|SLC25A10|SLC37A4|SMARCA4|SNW1|SOS1|SRC|STUB1|STX1A|TAF12|TAF5|TCEB2|TGIF1|TNKS2|TPR|UBA52|WNT5A|XRCC5|XRCC6|YWHAZ |
| Gene Expression | 71 | CCNT2|CDKN2B|CNOT1|DHX9|DNMT1|EEF1A1|EEF1D|EEF1G|EIF2S1|EIF2S2|EIF3K|EIF4E|EIF4G1|ERCC2|ESRRA|EXOSC2|EZH2|FARSB|GEMIN4|GTF2E2|HARS|HDAC2|HNRNPA1|HNRNPH1|HNRNPU|HSPA8|LSM2|MARS2|MED20|MED4|NR1D1|PCBP2|POLR1A|POLR2A|PPA1|PTBP1|RAN|RBBP4|RBBP7|RNPS1|RPL13A|RPL14|RPL36|RPS13|RPS16|RPS21|RPS29|RPS9|RUNX2|SAP18|SARS|SEC61G|SET|SIN3B|SNW1|SRRM1|SRSF1|TAF12|TAF5|TARS|TCEB2|TGIF1|THRA|TRAM1|U2AF1|UBA52|UPF1|UPF2|XPO5|YWHAZ|ZNF711 |
| Signal Transduction | 68 | ABI1|ADCY9|AKT2|ARHGDIA|BRK1|CASP2|CCNT2|CDC42|CDK1|CDKN2B|CREB1|CRK|CRKL|CSK|CSNK1A1|CTBP1|DAAM1|DGKH|E2F1|EIF4E|EIF4G1|FSTL3|FZD4|GNG5|GPR37|HDAC2|HDAC3|HDAC4|HRAS|HSP90AA1|IRS1|JAK2|LFNG|MAPK7|MKNK1|OS9|P4HB|PFN1|PIP5K1B|PLCG1|PRKAR2A|PSAP|PSENEN|PTCH1|PTK2|RAC1|RAF1|RBX1|RELA|RHOA|RIPK2|ROCK1|RPS6KA5|RPS6KB1|SHC1|SMARCA4|SNW1|SOCS1|SOS1|SRC|STARD13|STUB1|TGIF1|THBS2|TNKS2|UBA52|WNT5A|YWHAZ |
| Immune System | 66 | ABI1|ADCY9|AKT2|ANAPC1|ANAPC11|AP1S1|ARPC2|ARPC5|BRK1|CASP2|CDC34|CDC42|CDK1|CREB1|CRK|CRKL|CSK|CTSD|DHX9|DYNLL1|EIF4E|EIF4G1|FADD|FZR1|HRAS|HSP90AA1|IP6K2|IRS1|JAK2|KIF18A|KIF4A|MAP2K7|MAPK7|NUP107|NUP50|PCBP2|PELI1|PLCG1|PLD1|POM121|PRKAR2A|PRKDC|PTK2|PVR|RAC1|RAE1|RAF1|RBX1|RELA|RIPK2|RNF19B|RPS6KA5|SEC61G|SHC1|SOCS1|SOS1|SRC|STUB1|TCEB2|TPR|TUBB4B|UBA52|UBE2D4|XRCC5|XRCC6|YWHAZ |
| Cell Cycle | 61 | ANAPC1|ANAPC11|ATM|AURKA|AURKB|BUB1|CCNB1|CCNE1|CDC7|CDK1|CDKN2B|CDKN2D|CDT1|CENPN|CKS1B|CLASP2|DKC1|DSN1|DYNLL1|E2F1|ERCC6L|FBXO5|FZR1|GOLGA2|HSP90AA1|KIF18A|LIN37|MAD2L1|MCM2|MCM4|MCM6|MLH3|NEK2|NPM1|NUP107|NUP50|OIP5|ORC6|PCNA|PLK4|POLA1|POM121|PPP1R12A|RAB1B|RAD50|RAE1|RBBP4|RBBP7|RBBP8|RBL2|RSF1|SDCCAG8|SET|SMC4|SPC24|SPDL1|TPR|TUBB|TUBB4B|UBA52|ZWINT |
| Metabolic pathways | 58 | ACADM|ADI1|ALAS1|ALDOA|AMACR|ATP5O|BCAT1|BCAT2|CBR1|COX5B|COX6C|CTPS1|DAD1|DGKH|DNMT1|DTYMK|ENO1|EXT2|FASN|FBL|FDPS|FUT8|GAA|GCLC|GFPT1|GLB1|GSS|HSD17B4|INPP5A|INPP5K|LDHA|LSS|MECR|MTHFD1|NDUFA3|NDUFB4|ODC1|PAICS|PAPSS1|PDHB|PGAM5|PGAP1|PIGA|PIP5K1B|PLA2G12A|PLCG1|PLD1|POLA1|POLG2|POLR1A|POLR2A|PPAT|SDHA|SGMS1|SHMT2|SUCLA2|SUFU|TGDS |
| Metabolism of proteins | 47 | B3GNTL1|CCT3|CCT6A|CCT8|CTSD|DAD1|DDIT3|DNAJB9|DNAJC3|DOHH|EEF1A1|EEF1D|EEF1G|EIF2S1|EIF2S2|EIF3K|EIF4E|EIF4G1|EIF5A|FBXW5|FUT8|GFPT1|GLB1|GRPEL2|HSPA9|HSPD1|IGFBP3|MANEA|PFDN1|PGAP1|PIGA|RPL13A|RPL14|RPL36|RPS13|RPS16|RPS21|RPS29|RPS9|SEC61G|SHC1|STX1A|THBS2|TRAM1|TSPYL2|TUBB4B|UBA52 |
| Developmental Biology | 41 | ABLIM2|AKT2|ARPC2|ARPC5|CDC42|CDK1|CLASP2|CREB1|CRMP1|CTNNA2|DPYSL2|HDAC3|HRAS|HSP90AA1|HSPA8|ITGA1|KIF4A|MED11|MED18|MED20|MED4|MYH10|MYH14|NRTN|PFN1|PLCG1|PSENEN|PTK2|RAC1|RAF1|RHOA|ROCK1|RPS6KA5|RPS6KA6|SCN3B|SDCBP|SIAH2|SOS1|SPTAN1|SRC|TUBB4B |
| Pathways in cancer | 34 | AKT2|ARAF|CCNE1|CDC42|CDKN2B|CKS1B|CKS2|CRK|CRKL|CTBP1|CTNNA2|CYCS|E2F1|FADD|FZD4|HDAC2|HRAS|HSP90AA1|MSH2|PLCG1|PLD1|PTCH1|PTK2|RAC1|RAF1|RASSF1|RBX1|RELA|RHOA|SOS1|SUFU|TCEB2|TPR|WNT5A |
| Dibutyl Phthalate Interacting Common E2- and NRF1-Target Genes | ||
| KEGG Pathway | Number of Genes | Annotated Genes |
| Metabolism | 56 | ACADM|ADCY9|ADI1|AKR7A2|ALAS1|ALDOA|AMACR|ATP5O|BCAT1|BCAT2|BSG|CBR1|CYB5A|CYCS|DTYMK|ENO1|ETFB|ETFDH|EXT2|FASN|FDPS|GCLC|GLB1|GNG5|GSR|GSS|HDAC3|HMMR|HSD17B4|HSP90AA1|INPP5A|IP6K2|LDHA|LPCAT3|LSS|MGST3|MTHFD1|NAMPT|NDUFA3|NDUFB4|NUBP1|NUP50|OAZ2|ODC1|PAPSS1|PLA2G12A|PLD1|POM121|PRKAR2A|PYGL|RAE1|RAN|SDHA|SGMS1|SLC25A10|UBA52 |
| Disease | 45 | ADCY9|ALDOA|CCNT2|CDK1|CREB1|CSNK1A1|CTBP1|CYB5A|ENO1|EXT2|FASN|GLB1|HDAC3|HMMR|HSP90AA1|MTHFD1|NAMPT|NPM1|NUP50|PAPSS1|POLR2A|POM121|PRKAR2A|PYGL|RAE1|RAF1|RAN|RHOA|RPL13A|RPL14|RPL36|RPS13|RPS21|RPS29|SLC25A10|SMARCA4|SNW1|SRC|STUB1|TCEB2|TGIF1|TNKS2|UBA52|WNT5A|XRCC5 |
| Gene Expression | 39 | CCNT2|DNMT1|EEF1A1|EEF1D|EIF2S1|EIF2S2|EIF4E|EIF4G1|ESRRA|EXOSC2|EZH2|GEMIN4|HNRNPA1|HNRNPU|HSPA8|LSM2|MED20|POLR1A|POLR2A|PTBP1|RAN|RBBP4|RNPS1|RPL13A|RPL14|RPL36|RPS13|RPS21|RPS29|SET|SNW1|SRSF1|TARS|TCEB2|TGIF1|THRA|U2AF1|UBA52|XPO5 |
| Metabolic pathways | 38 | ACADM|ADI1|ALAS1|ALDOA|AMACR|ATP5O|BCAT1|BCAT2|CBR1|COX6A2|DNMT1|DTYMK|ENO1|EXT2|FASN|FBL|FDPS|GCLC|GFPT1|GLB1|GSS|HSD17B4|INPP5A|LDHA|LSS|MTHFD1|NDUFA3|NDUFB4|ODC1|PAPSS1|PGAM5|PLA2G12A|PLD1|POLE4|POLR1A|POLR2A|SDHA|SGMS1 |
| Cell Cycle | 34 | ANAPC1|ANAPC11|APITD1|ATM|AURKB|CCNB1|CCNE1|CDC7|CDK1|CDKN2D|CDT1|CENPN|CKS1B|CLASP2|FBXO5|FZR1|HSP90AA1|MCM2|MCM4|MCM6|NPM1|NUP50|OIP5|ORC6|PCNA|POM121|PPP1R12A|RAE1|RBBP4|SET|SPC24|SPDL1|UBA52|ZWINT |
| Signal Transduction | 32 | ADCY9|ARHGDIA|CCNT2|CDK1|CREB1|CSNK1A1|CTBP1|DAAM1|EIF4E|EIF4G1|GNG5|HDAC3|HSP90AA1|JAK2|LFNG|MEF2A|PRDM4|PRKAR2A|PTCH1|RAF1|RHOA|SMARCA4|SNW1|SOCS1|SRC|STUB1|TGIF1|THBS2|TNKS2|TRIO|UBA52|WNT5A |
| Immune System | 31 | ADCY9|ANAPC1|ANAPC11|ARPC2|ARPC5|CDK1|CREB1|CTSD|EIF4E|EIF4G1|FADD|FZR1|HSP90AA1|IP6K2|JAK2|MEF2A|NUP50|PLD1|POM121|PRKAR2A|PRKDC|PVR|RAE1|RAF1|RNF19B|SOCS1|SRC|STUB1|TCEB2|UBA52|XRCC5 |
| Metabolism of proteins | 25 | CCT3|CTSD|DDIT3|DNAJB9|DNAJC3|EEF1A1|EEF1D|EIF2S1|EIF2S2|EIF4E|EIF4G1|FBXW5|GFPT1|GLB1|HSPA9|HSPD1|PFDN1|RPL13A|RPL14|RPL36|RPS13|RPS21|RPS29|THBS2|UBA52 |
| Mitotic M-M/G1 phases | 23 | ANAPC1|ANAPC11|APITD1|AURKB|CCNB1|CDC7|CDK1|CDT1|CENPN|CLASP2|FBXO5|MCM2|MCM4|MCM6|NUP50|ORC6|POM121|RAE1|SET|SPC24|SPDL1|UBA52|ZWINT |
| Developmental Biology | 21 | ARPC2|ARPC5|CDK1|CLASP2|CREB1|CTNNA2|DPYSL2|HDAC3|HSP90AA1|HSPA8|ITGA1|MED20|MEF2A|MYH10|NRTN|RAF1|RHOA|SCN3B|SDCBP|SRC|TRIO |
| Diethylhexyl Phthalate Interacting Common E2- and NRF1-Target Genes | ||
| KEGG Pathway | Number of Genes | Annotated Genes |
| Metabolism | 21 | ACADM|ALAS1|ALDOA|BCAT2|BSG|CYCS|FASN|FDPS|GSR|GSTM3|HSD17B4|HSP90AA1|LDHA|LSS|MMADHC|NDUFA3|ODC1|PAPSS1|PLD1|PYGL|SLC37A4 |
| Metabolic pathways | 15 | ACADM|ALAS1|ALDOA|BCAT2|DNMT1|FASN|FDPS|HSD17B4|LDHA|LSS|NDUFA3|ODC1|PAPSS1|PLD1|POLE4 |
| Disease | 15 | AKT2|ALDOA|FASN|HDAC2|HDAC4|HSP90AA1|IRS1|MMADHC|NPM1|PAPSS1|PYGL|RBX1|RPL36|RPS13|SLC37A4 |
| Immune System | 12 | AKT2|ANAPC11|CDC34|DYNLL1|EIF4G1|HSP90AA1|IRS1|KIF18A|PLD1|RBX1|SEC61G|TUBB4B |
| Metabolism of proteins | 11 | CCT3|CCT6A|DNAJB9|EIF4G1|GRPEL2|HSPA9|HSPD1|RPL36|RPS13|SEC61G|TUBB4B |
| Cell Cycle | 9 | ANAPC11|CCNB1|CCNE1|DYNLL1|HSP90AA1|KIF18A|NPM1|TUBB4B|ZWINT |
| Cellular responses to stress | 8 | ANAPC11|CBX2|CCNE1|CYCS|GSR|HSP90AA1|HSPA8|RBX1 |
| Pathways in cancer | 8 | AKT2|CCNE1|CYCS|HDAC2|HSP90AA1|MSH2|PLD1|RBX1 |
| Developmental Biology | 8 | AKT2|DPYSL2|HSP90AA1|HSPA8|ITGA1|SCN3B|SIAH2|TUBB4B |
| Cell cycle | 5 | ANAPC11|CCNB1|CCNE1|HDAC2|RBX1 |
| Polychlorinated Biphenyls Interacting Common E2- and NRF1-Target Gene | ||
| KEGG Pathway | Number of Genes | Annotated Genes |
| Cell Cycle | 13 | AURKA|AURKB|CDK1|CDT1|CENPN|CKS1B|FBXO5|MCM2|MCM6|PCNA|PLK4|SMC4|ZWINT |
| Mitotic M-M/G1 phases | 9 | AURKB|CDK1|CDT1|CENPN|FBXO5|MCM2|MCM6|SMC4|ZWINT |
| DNA Replication | 4 | CDT1|MCM2|MCM6|PCNA |
| Cell cycle | 4 | CDK1|MCM2|MCM6|PCNA |
| DNA replication | 3 | MCM2|MCM6|PCNA |
| DNA replication and repair | 3 | CDK1|PCNA|UNG |
| Pancreatic cancer | 3 | ARAF|CDC42|RAF1 |
| Cadmium Interacting Common E2- and NRF1-Target Genes | ||
| KEGG Pathway | Number of Genes | Annotated Genes |
| Metabolism | 26 | ACADM|ALDOA|AUH|BCAT1|COX5B|COX6C|CYCS|DTYMK|ENO1|FASN|GCLC|GSR|GSS|GSTM3|HSD17B4|HSP90AA1|INPP5K|LDHA|MGST3|OAZ2|PAICS|RAN|SDHA|SGMS1|SLC16A1|SLC37A4 |
| Gene Expression | 25 | CCNT2|CDKN2B|DNMT1|EEF1A1|EEF1D|EEF1G|EXOSC2|HARS|HNRNPA1|HNRNPH1|HSPA8|LSM2|MARS2|MED20|RAN|RPL10|RPL13A|RPL14|RUNX2|SRRM1|TAF12|TCEB2|THRA|XPO5|YWHAZ |
| Disease | 24 | ALDOA|CCNT2|CDC42|CDK1|CDKN2B|CREB1|CTBP1|ENO1|FASN|HSP90AA1|IRS1|OS9|PPIA|RAC1|RAF1|RAN|RPL10|RPL13A|RPL14|SHC1|SLC37A4|TAF12|TCEB2|YWHAZ |
| Signal Transduction | 21 | ARHGDIA|CCNT2|CDC42|CDK1|CDKN2B|CREB1|CTBP1|E2F1|FSTL3|GPR37|HSP90AA1|IRS1|OS9|PTK2|RAC1|RAF1|RELA|ROCK1|RPS6KB1|SHC1|YWHAZ |
| Metabolic pathways | 20 | ACADM|ALDOA|AUH|BCAT1|COX5B|COX6C|DNMT1|DTYMK|ENO1|FASN|GAA|GCLC|GSS|HSD17B4|INPP5K|LDHA|PAICS|SDHA|SGMS1|SHMT2 |
| Immune System | 18 | CDC42|CDK1|CREB1|CTSD|HSP90AA1|IRS1|KIF4A|PELI1|PTK2|PVR|RAC1|RAF1|RELA|RNF19B|SHC1|TCEB2|UBE2D4|YWHAZ |
| Cell Cycle | 17 | AURKA|BUB1|CCNB1|CCNE1|CDK1|CDKN2B|CKS1B|E2F1|FBXO5|HSP90AA1|MAD2L1|MCM2|MCM4|MCM6|NEK2|OIP5|SMC4 |
| Pathways in cancer | 15 | CCNE1|CDC42|CDKN2B|CKS1B|CTBP1|CYCS|E2F1|HSP90AA1|MSH2|PTK2|RAC1|RAF1|RASSF1|RELA|TCEB2 |
| Developmental Biology | 15 | CDC42|CDK1|CREB1|HSP90AA1|HSPA8|KIF4A|MED20|MYH10|MYH14|NRTN|PTK2|RAC1|RAF1|ROCK1|SPTAN1 |
| Metabolism of proteins | 12 | CTSD|DDIT3|DNAJB9|EEF1A1|EEF1D|EEF1G|EIF5A|HSPD1|RPL10|RPL13A|RPL14|SHC1 |
| Arsenic Interacting Common E2- and NRF1-Target Genes | ||
| KEGG Pathway | Number of Genes | Annotated Genes |
| Metabolism | 33 | ADCY9|ALDOA|CYCS|ENO1|ERCC2|ETFB|ETFDH|FASN|GCLC|GSR|GSS|GSTM3|HSD17B4|HSP90AA1|INPP5A|IP6K2|LDHA|MMADHC|NUBP1|P4HB|PAICS|PLCG1|PLD1|POM121|PPAT|PRKAR2A|SDHA|SGMS1|SLC44A2|STX1A|SUCLA2|TPMT|UBA52 |
| Immune System | 30 | ADCY9|ARPC2|CASP2|CDC42|CDK1|CREB1|CTSD|FZR1|HRAS|HSP90AA1|IP6K2|JAK2|KIF4A|MAPK7|PCBP2|PELI1|PLCG1|PLD1|POM121|PRKAR2A|PRKDC|PTK2|PVR|RAF1|SOS1|SRC|TUBB4B|UBA52|UBE2D4|XRCC5 |
| Disease | 27 | ADCY9|ALDOA|CDC42|CDK1|CDKN2B|CREB1|CTBP1|ENO1|ERCC2|FASN|HDAC4|HRAS|HSP90AA1|MMADHC|NPM1|P4HB|PLCG1|POM121|PRKAR2A|RAF1|RPL36|SMARCA4|SOS1|SRC|STX1A|UBA52|XRCC5 |
| Signal Transduction | 24 | ADCY9|CASP2|CDC42|CDK1|CDKN2B|CREB1|CTBP1|E2F1|HDAC4|HRAS|HSP90AA1|JAK2|MAPK7|P4HB|PLCG1|PRKAR2A|PTCH1|PTK2|RAF1|ROCK1|SMARCA4|SOS1|SRC|UBA52 |
| Developmental Biology | 22 | ABLIM2|ARPC2|CDC42|CDK1|CREB1|HRAS|HSP90AA1|HSPA8|KIF4A|MED18|MYH10|MYH14|NRTN|PLCG1|PTK2|RAF1|ROCK1|SCN3B|SIAH2|SOS1|SRC|TUBB4B |
| Metabolic pathways | 20 | ALDOA|COX6A2|DNMT1|ENO1|FASN|FUT8|GCLC|GSS|HSD17B4|INPP5A|LAP3|LDHA|PAICS|PLCG1|PLD1|PPAT|SDHA|SGMS1|SUCLA2|SUFU |
| Cell Cycle | 19 | ATM|AURKA|CCNB1|CCNE1|CDK1|CDKN2B|CDKN2D|E2F1|FZR1|HSP90AA1|MAD2L1|NPM1|PCNA|POM121|SET|SPDL1|TUBB|TUBB4B|UBA52 |
| Gene Expression | 19 | CDKN2B|CNOT1|DNMT1|EEF1G|ERCC2|EZH2|HNRNPA1|HSPA8|LSM2|PCBP2|PTBP1|RNPS1|RPL36|RUNX2|SET|UBA52|UPF1|VARS2|ZNF610 |
| Pathways in cancer | 17 | CCNE1|CDC42|CDKN2B|CTBP1|CYCS|E2F1|HRAS|HSP90AA1|MSH2|PLCG1|PLD1|PTCH1|PTK2|RAF1|RASSF1|SOS1|SUFU |
| Cellular responses to stress | 16 | ATM|CCNE1|CDKN2B|CDKN2D|CYCS|E2F1|EHMT1|EZH2|FZR1|GSR|HSP90AA1|HSPA8|MAPK7|P4HB|PRDX5|UBA52 |
| Manganese Interacting Common E2- and NRF1-Target Genes | ||
| KEGG Pathway | Number of Genes | Annotated Genes |
| Cellular responses to stress | 6 | ATM|CYCS|GSR|HSPA8|P4HB|RELA |
| Developmental Biology | 5 | CREB1|HSPA8|SCN3B|SPTAN1|SRC |
| Tuberculosis | 4 | CREB1|CYCS|RELA|SRC |
| p53 signaling pathway | 3 | ATM|CYCS|PPM1D |
| Small cell lung cancer | 3 | CKS2|CYCS|RELA |
| Apoptosis | 3 | ATM|CYCS|RELA |
| Cell cycle | 3 | ATM|BUB1|MCM4 |
| Endocrine Disrupting Chemical (EDC) | Individual EDC Responsive Modified Genes Common to Both NRF1 and E2 Target. * Indicates E2 Responsive |
|---|---|
| Alzheimer’s Disease (AD) | |
| 17 β-estradiol | 6 genes: APBB2|DPYSL2|EIF2S1|ENO1|MAPT|PAXIP1 |
| Ethinyl Estradiol | 6 genes: APBB2 *|EIF2S1 *|ENO1 *|IDE|MAPT *|PAXIP1 * |
| Bisphenol A | 8 genes: APBB2 *|DPYSL2 *|EIF2S1 *|ENO1 *|IDE|MAPT *|PAXIP1 *|SLC30A4 |
| Dibutyl Phthalate | 4 genes: DPYSL2 *|EIF2S1 *|ENO1 *|IDE |
| Diethylhexyl Phthalate | 2 genes: DPYSL2 *|MAPT * |
| Cadmium | 2 genes: ENO1 *|SLC30A4 |
| Arsenic | 2 genes: ENO1 *|MAPT |
| Manganese | 1 gene: ENO1 * |
| Parkinson’s Disease (PD) | |
| 17 β-estradiol | 8 genes: HSPA9|MAPT|RPL14 |
| Ethinyl Estradiol | 3 genes: HSPA9 *|MAPT *|PINK1 |
| Bisphenol A | 8 genes: GAK|HSPA9 *|MAPT *|PARK2|PARK7|PINK1|RPL14 *|VPS35 |
| Dibutyl Phthalate | 4 genes: HSPA9 *|PARK2|PARK7|RPL14 * |
| Diethylhexyl Phthalate | 3 genes: HSPA9 *|MAPT *|PARK2 |
| Cadmium | 3 genes: PARK2|PINK1|RPL14 * |
| Arsenic | 4 genes: GAK|HSPA9 *|MAPT *|PARK2 |
| Manganese | 2 genes: PARK2|PARK7 |
| Huntington’s Disease (HD) | |
| 17 β-estradiol | 2 genes: AIFM1|P6K2 |
| Ethinyl Estradiol | 1 gene: AIFM1 * |
| Bisphenol A | 2 genes: AIFM1 *|IP6K2 * |
| Dibutyl Phthalate | 1 gene: IP6K2 * |
| Cadmium | 1 gene: AIFM1 * |
| Arsenic | 1 gene: IP6K2 * |
| Amyotrophic Lateral Sclerosis (ALS) | |
| 17 β-estradiol | 1 gene: GSR|CHMP2B |
| Ethinyl Estradiol | 1 gene: GSR * |
| Bisphenol A | 2 genes: GSR *|CHMP2B *|UNC13A |
| Dibutyl Phthalate | 1 gene: GSR * |
| Diethylhexyl Phthalate | 1 gene: GSR * |
| Cadmium | 1 gene: GSR * |
| Arsenic | 1 gene: GSR * |
| Manganese | 1 gene: GSR |
| Autism Spectrum Disorder (ASD) | |
| 17 β-estradiol | 3 genes: CIRBP|PCDH9|GTF2I |
| Ethinyl Estradiol | 2 genes: CIRBP *|GTF2I * |
| Bisphenol A | 3 genes: CIRBP *|GTF2I *|PCDH9 * |
| Polychlorinated Biphenyls | 1 gene: CIRBP * |
| Cadmium | 1 gene: CIRBP * |
| Arsenic | 1 gene: PCDH9 * |
| Brain Neoplasms | |
| 17 β-estradiol | 3 genes: PCNA|PTCH1|RELA |
| Ethinyl Estradiol | 2 genes: PCNA *|PTCH1 * |
| Bisphenol A | 4 genes: EML4|PCNA *|PTCH1 *|RELA * |
| Dibutyl Phthalate | 2 genes: PCNA *|PTCH1 * |
| Polychlorinated Biphenyls | 1 gene: PCNA |
| Cadmium | 1 gene: RELA * |
| Arsenic | 2 genes: PCNA *|PTCH1 * |
| Manganese | 1 gene: RELA * |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Preciados, M.; Yoo, C.; Roy, D. Estrogenic Endocrine Disrupting Chemicals Influencing NRF1 Regulated Gene Networks in the Development of Complex Human Brain Diseases. Int. J. Mol. Sci. 2016, 17, 2086. https://doi.org/10.3390/ijms17122086
Preciados M, Yoo C, Roy D. Estrogenic Endocrine Disrupting Chemicals Influencing NRF1 Regulated Gene Networks in the Development of Complex Human Brain Diseases. International Journal of Molecular Sciences. 2016; 17(12):2086. https://doi.org/10.3390/ijms17122086
Chicago/Turabian StylePreciados, Mark, Changwon Yoo, and Deodutta Roy. 2016. "Estrogenic Endocrine Disrupting Chemicals Influencing NRF1 Regulated Gene Networks in the Development of Complex Human Brain Diseases" International Journal of Molecular Sciences 17, no. 12: 2086. https://doi.org/10.3390/ijms17122086