
Differential Impacts of Alternative Splicing Networks on Apoptosis

Abstract
:1. Introduction
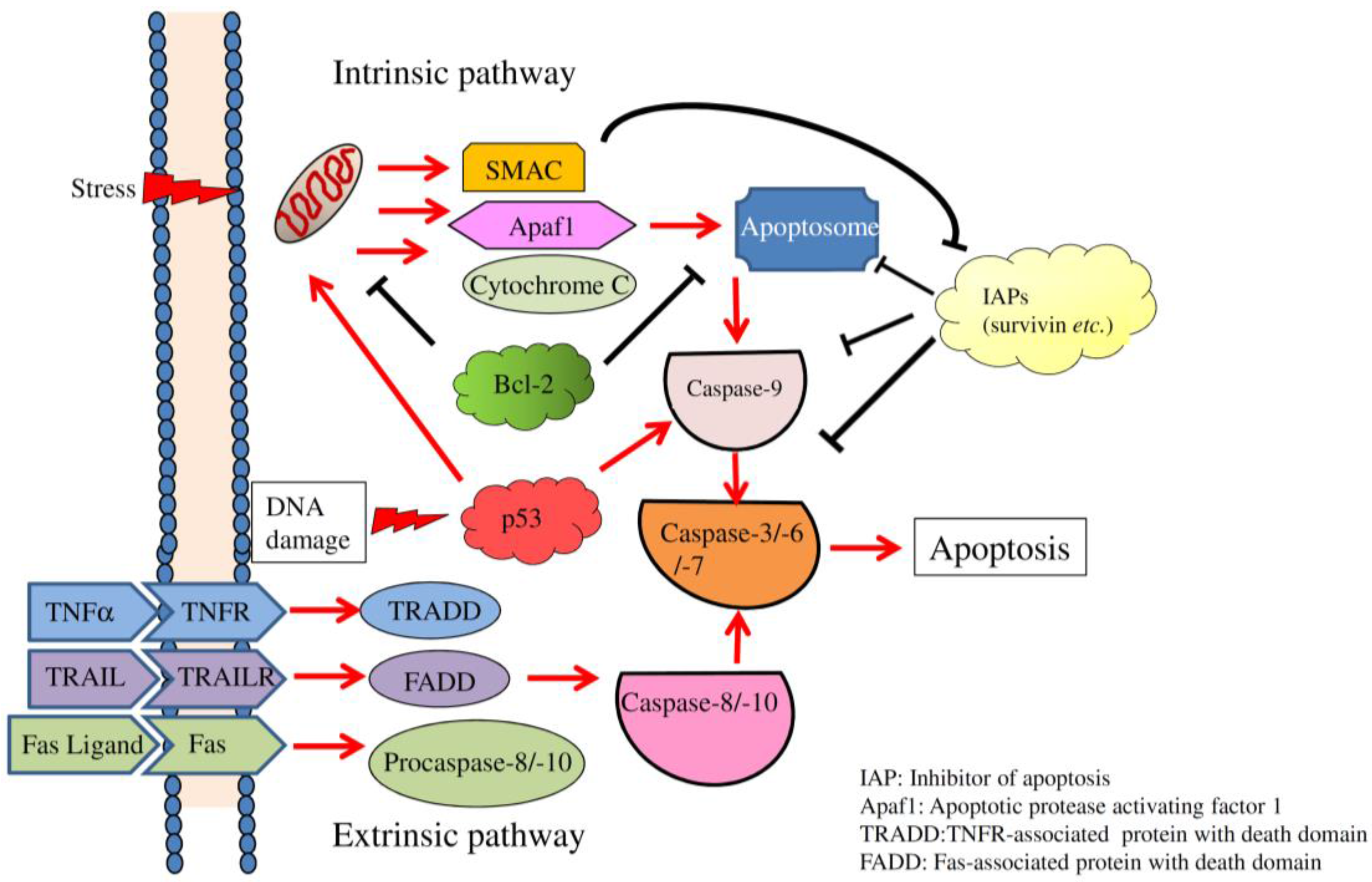
2. Overview of Apoptosis
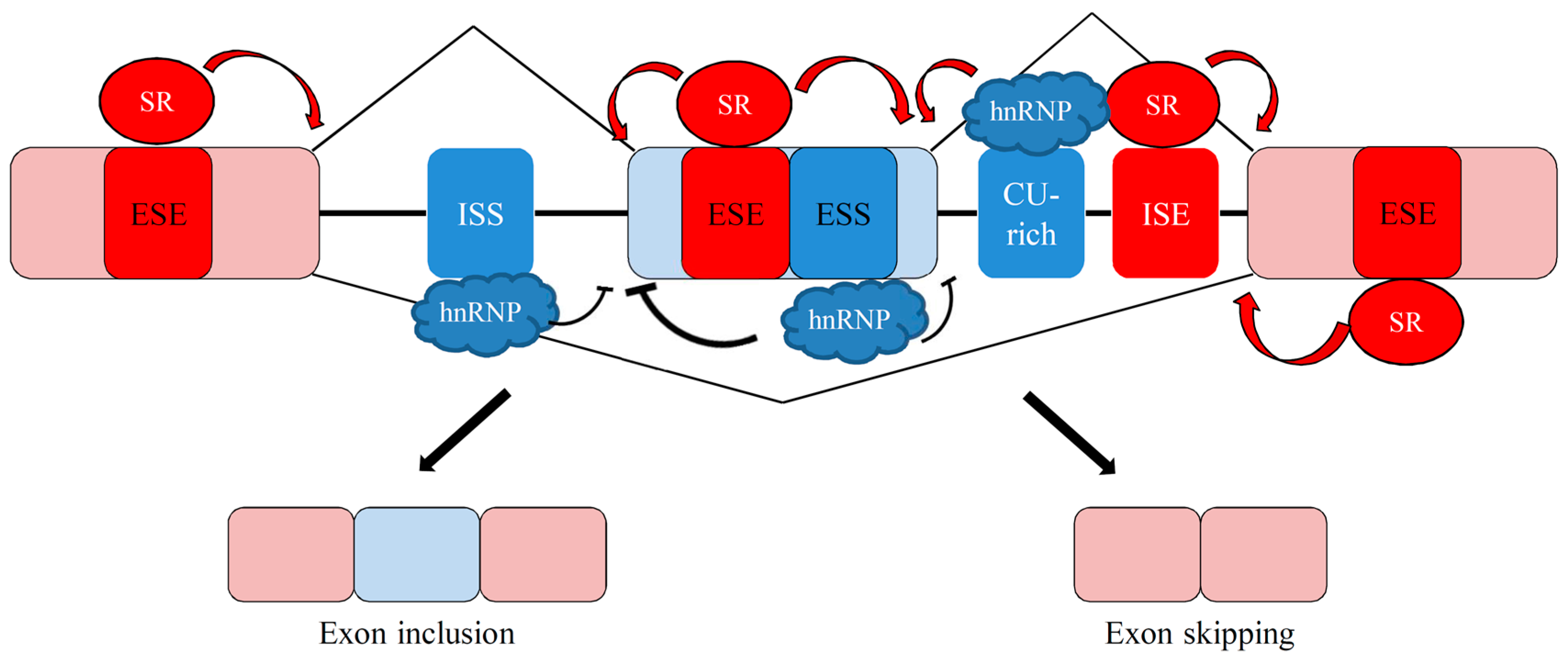
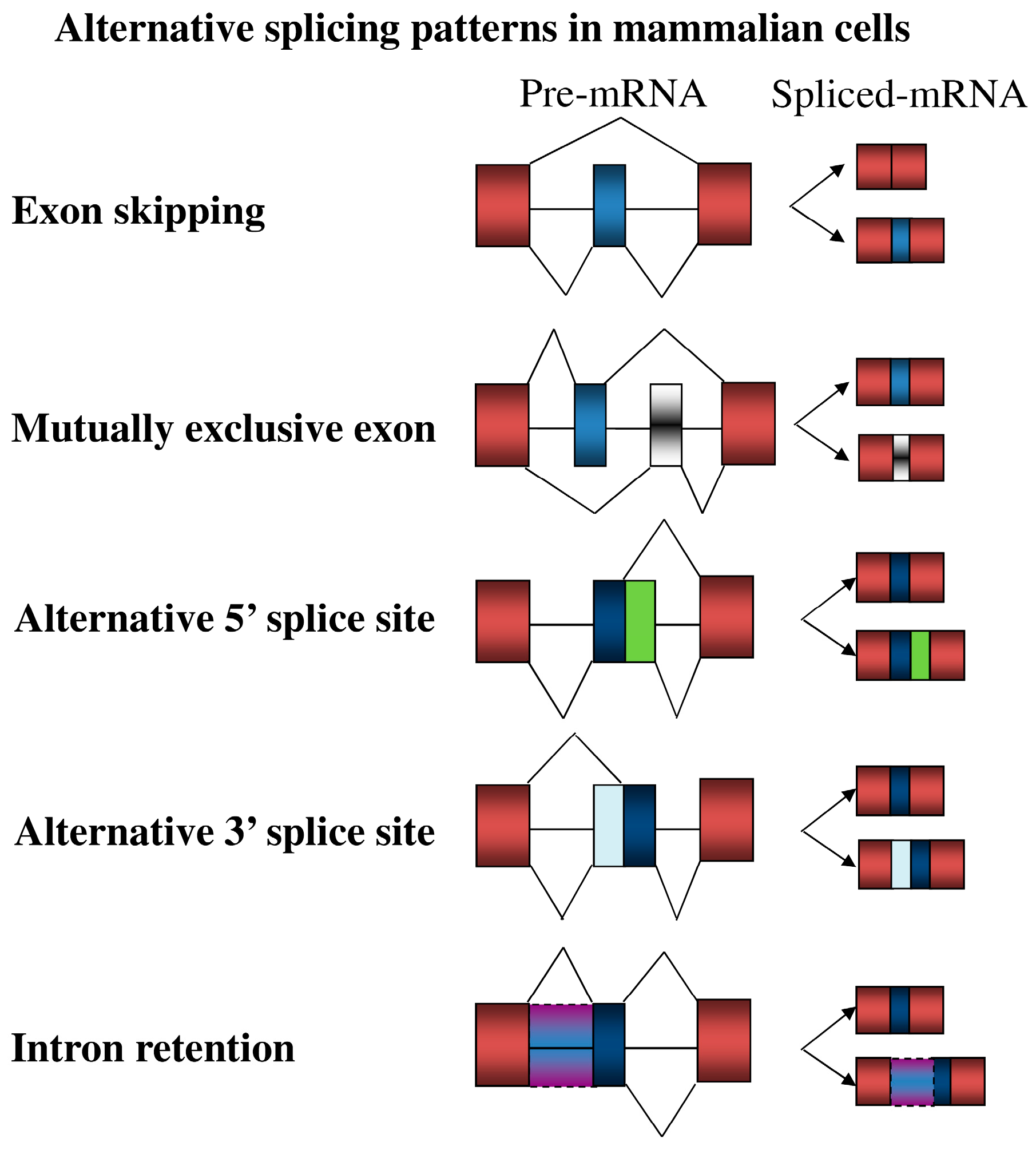
3. Overview of Alternative Splicing
4. Impacts of Alternative Splicing Events on Apoptosis
4.1. Apoptosis-Related Alternative Splicing Events
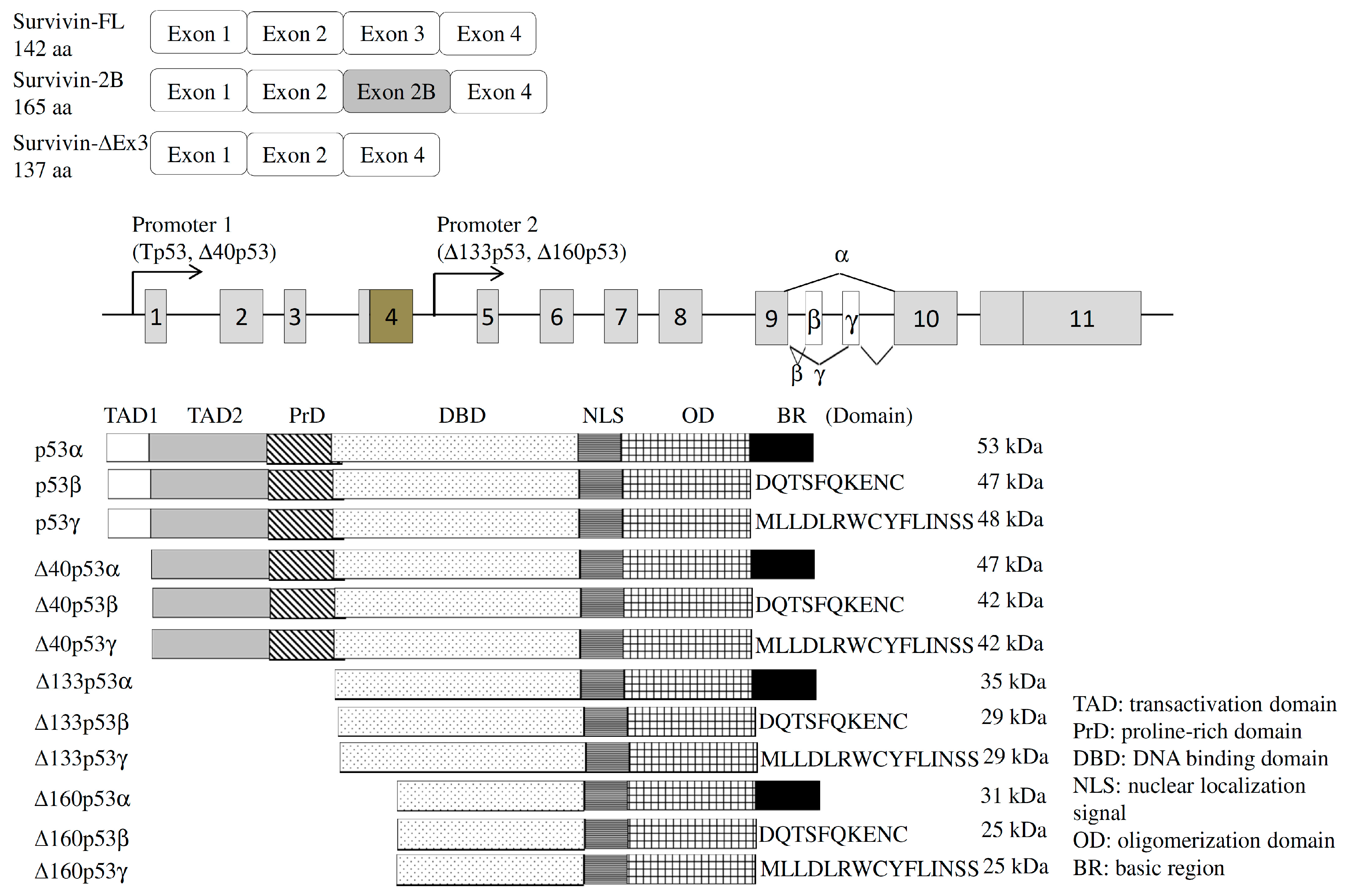
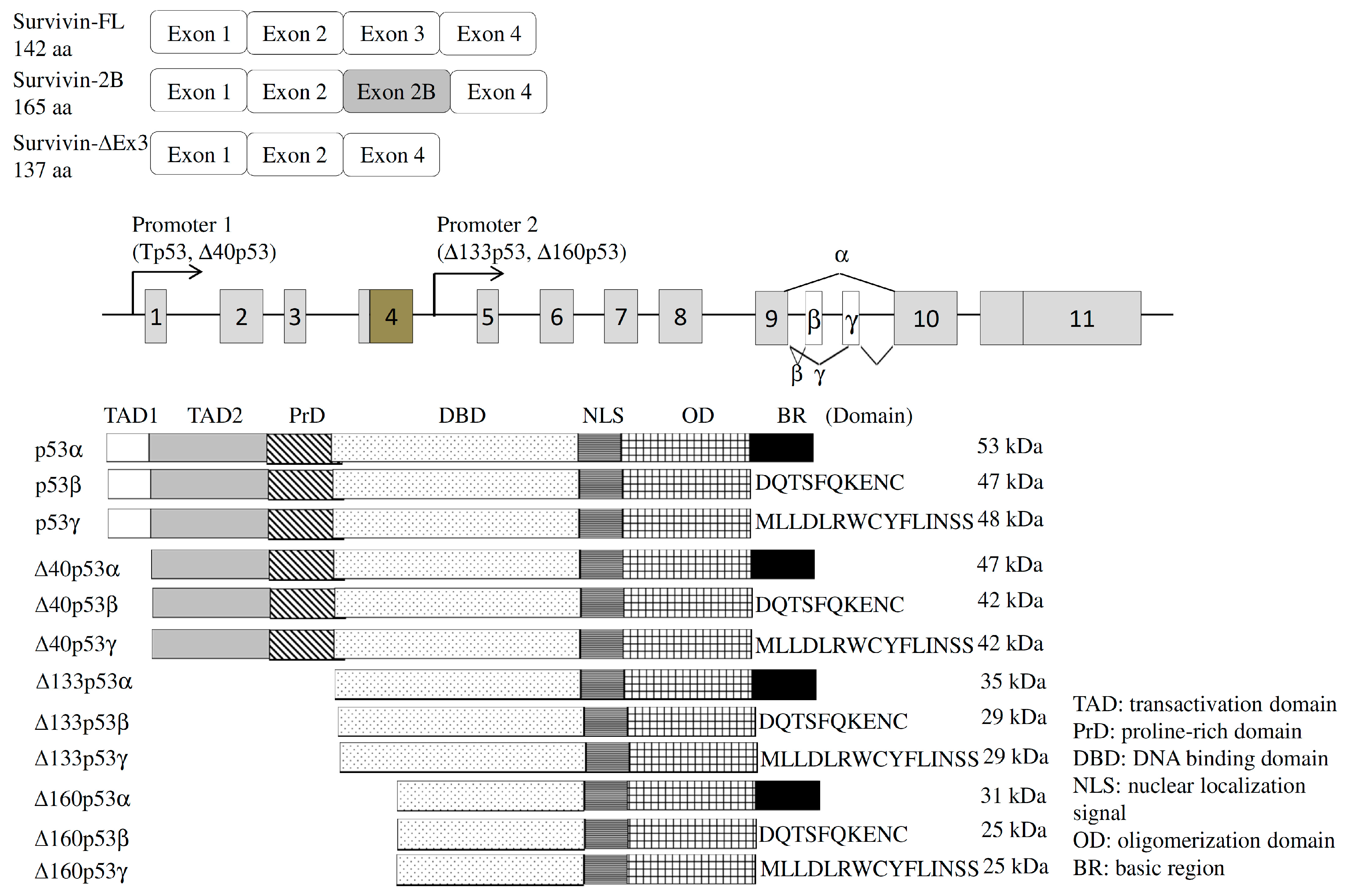
4.1.1. Survivin
4.1.2. Estrogen Receptor (ER)
4.1.3. Transient Receptor Potential Melastatin (TRPM)
4.1.4. Interleukin (IL)-15
4.2. Alternative Splicing of Apoptotic Factors
4.2.1. Tumor Protein p53 (TP53)
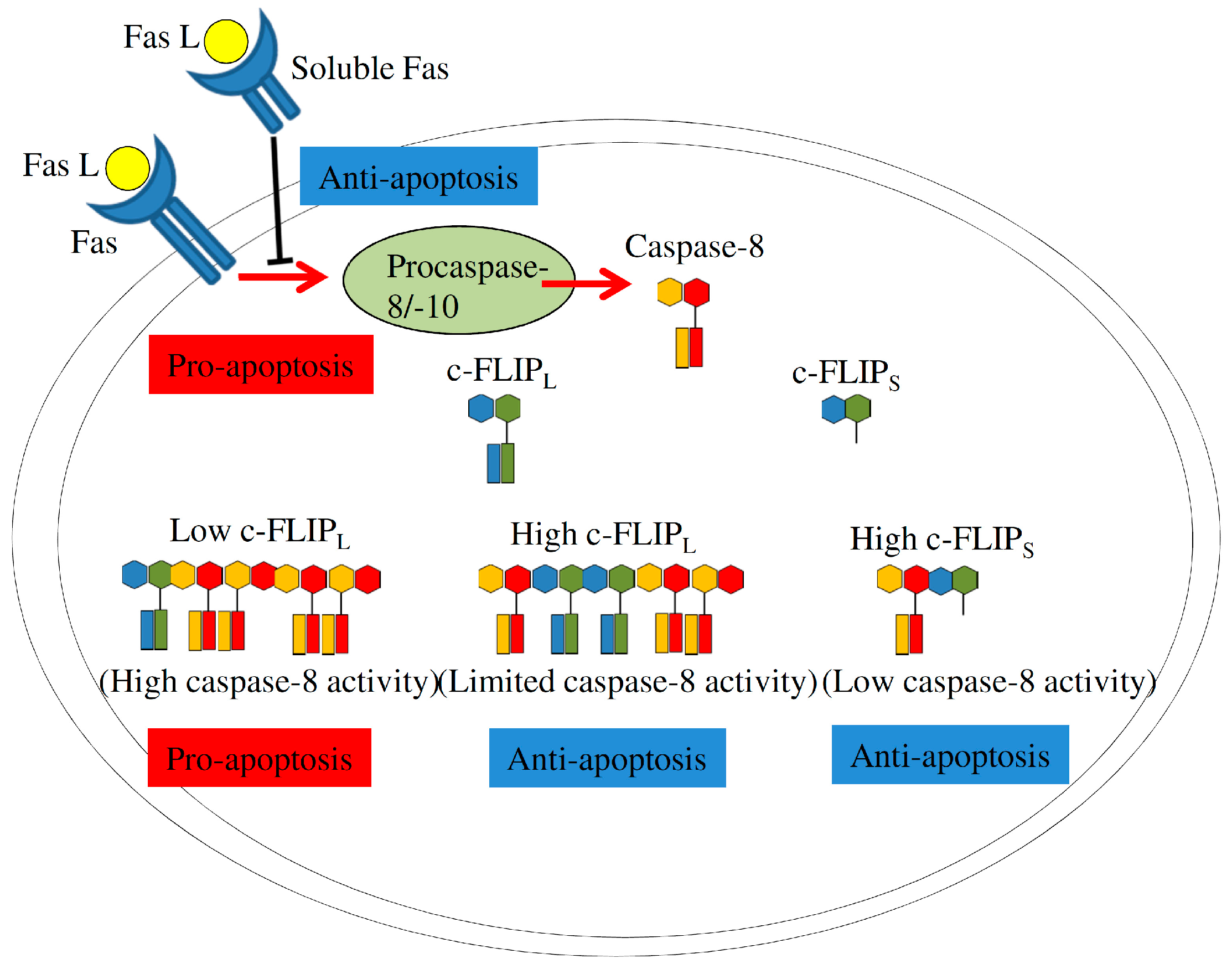
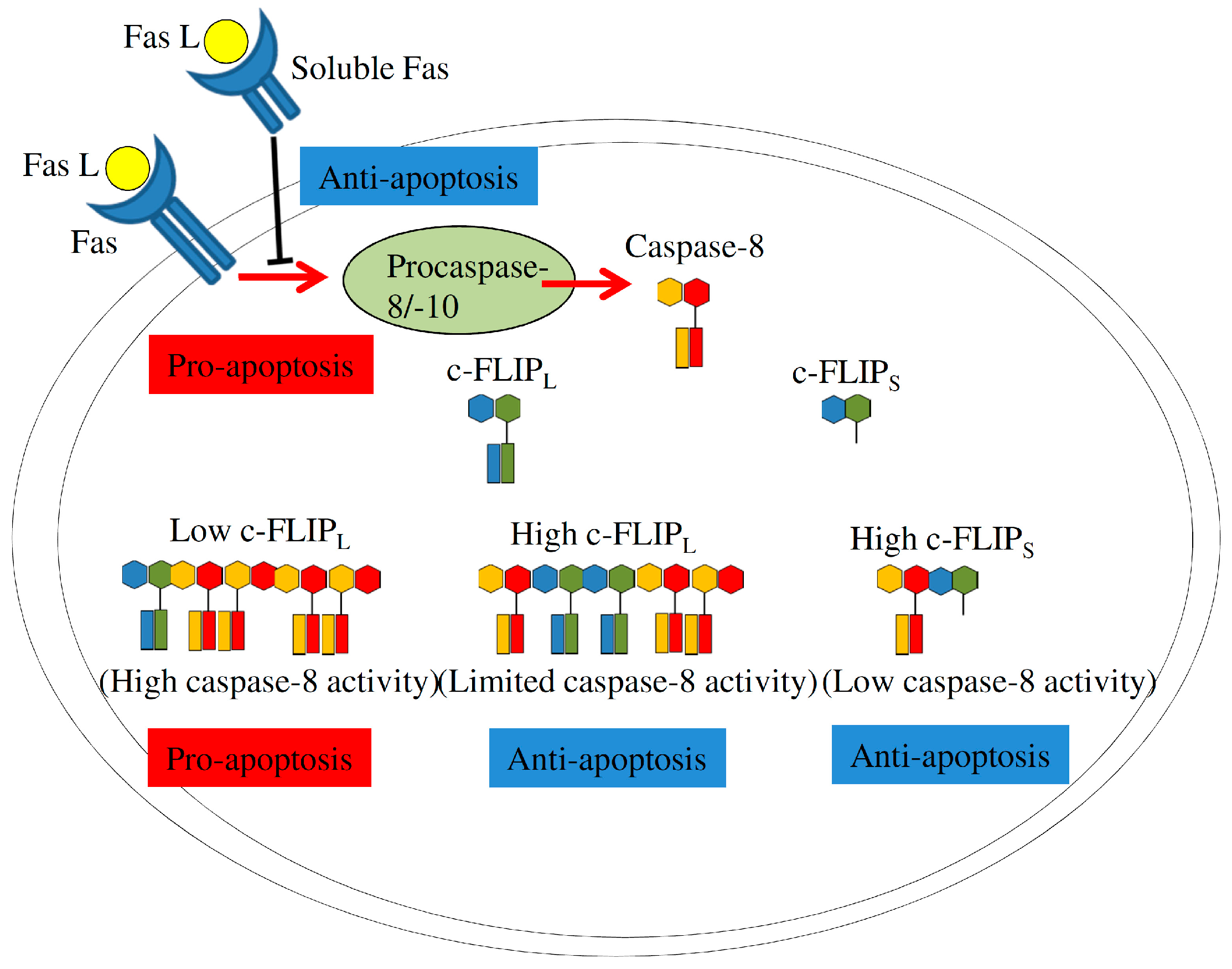
4.2.2. Fas Signaling
4.2.3. Bcl-2 Family
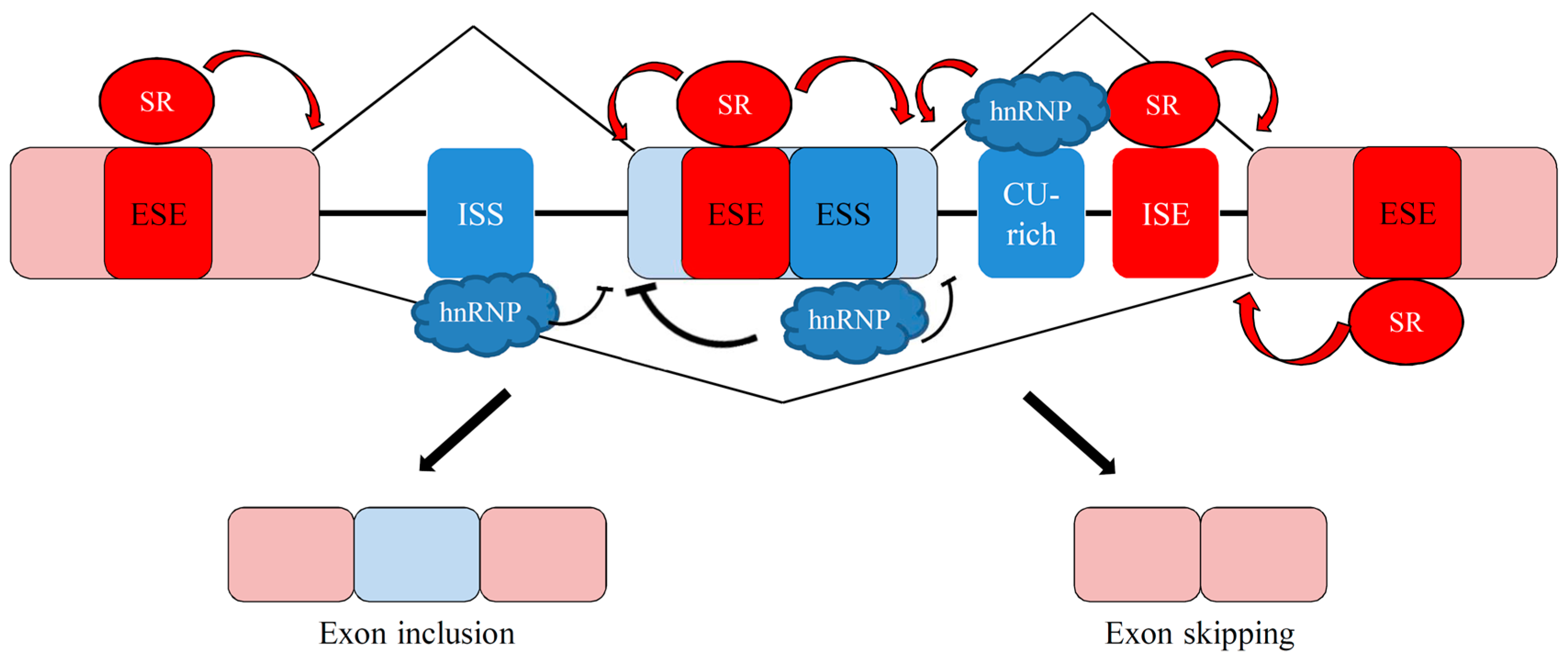
4.3. Splicing Factors Involved in Apoptosis-Related Splicing Events
4.3.1. Serine/Arginine (SR)-Rich Splicing Factors
4.3.2. Heterogeneous Nuclear Ribonucleoprotein (hnRNP) Family
4.3.3. RNA-Binding Motif Proteins (RBMPs)
5. Conclusions and Perspectives
Acknowledgments
Conflicts of Interest
References
- Yao, Y.; Gao, Z.; Liang, W.; Kong, L.; Jiao, Y.; Li, S.; Tao, Z.; Yan, Y.; Yang, J. Osthole promotes neuronal differentiation and inhibits apoptosis via Wnt/β-catenin signaling in an Alzheimer’s disease model. Toxicol. Appl. Pharmacol. 2015, 289, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Filatova, E.N.; Anisenkova, E.V.; Presnyakova, N.B.; Utkin, O.V. DR3 regulation of apoptosis of naive T-lymphocytes in children with acute infectious mononucleosis. Acta Microbiol. Immunol. Hung. 2016, 63, 339–357. [Google Scholar] [CrossRef] [PubMed]
- Kiraz, Y.; Adan, A.; Kartal Yandim, M.; Baran, Y. Major apoptotic mechanisms and genes involved in apoptosis. Tumour Biol. 2016, 37, 8471–8486. [Google Scholar] [CrossRef] [PubMed]
- Flusberg, D.A.; Sorger, P.K. Surviving apoptosis: Life-death signaling in single cells. Trends Cell Biol. 2015, 25, 446–458. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.Y.; Rainprecht, D.; Eichmair, E.; Messner, B.; Oehler, R. Serum-dependent processing of late apoptotic cells and their immunogenicity. Apoptosis 2015, 20, 1444–1456. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.J.; Kim, M.H.; Jeon, J.; Kim, O.Y.; Choi, Y.; Seo, J.; Hong, S.W.; Lee, W.H.; Jeon, S.G.; Gho, Y.S.; et al. Active immunization with extracellular vesicles derived from Staphylococcus aureus effectively protects against Staphylococcal Lung infections, mainly via Th1 cell-mediated immunity. PLoS ONE 2015, 10, e0136021. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.Y.; Dransfield, I.; Rossi, A.G.; Vermeren, S. Non-canonical PI3K-Cdc42-Pak-Mek-Erk signaling promotes immune-complex-induced apoptosis in human neutrophils. Cell Rep. 2016, 17, 374–386. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.P.; Foley, J.F.; Rubino, M.; Boyle, M.C.; Tandon, A.; Shah, R.; Archer, T.K. Brg1 enables rapid growth of the early embryo by suppressing genes that regulate apoptosis and cell growth arrest. Mol. Cell. Biol. 2016, 36, 1990–2010. [Google Scholar] [CrossRef] [PubMed]
- Moon, Y.J.; Yun, C.Y.; Choi, H.; Ka, S.O.; Kim, J.R.; Park, B.H.; Cho, E.S. Smad4 controls bone homeostasis through regulation of osteoblast/osteocyte viability. Exp. Mol. Med. 2016, 48, e256. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.F.; Xiang, L.; Zhou, Q.; Carralot, J.P.; Prunotto, M.; Niederfellner, G.; Pastan, I. Actinomycin D enhances killing of cancer cells by immunotoxin RG7787 through activation of the extrinsic pathway of apoptosis. Proc. Natl. Acad. Sci. USA 2016, 113, 10666–10671. [Google Scholar] [CrossRef] [PubMed]
- Jo, A.R.; Jeong, H.S.; Kim, M.K.; Yun, H.Y.; Baek, K.J.; Kwon, N.S.; Kim, D.S. Geranylgeranylacetone induces apoptosis via the intrinsic pathway in human melanoma cells. Biomed. Pharmacother. 2016, 82, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Montagnani Marelli, M.; Marzagalli, M.; Moretti, R.M.; Beretta, G.; Casati, L.; Comitato, R.; Gravina, G.L.; Festuccia, C.; Limonta, P. Vitamin E δ-tocotrienol triggers endoplasmic reticulum stress-mediated apoptosis in human melanoma cells. Sci. Rep. 2016, 6, 30502. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Hong, S.H.; Park, C.; Kim, G.Y.; Leem, S.H.; Choi, S.H.; Keum, Y.S.; Hyun, J.W.; Kwon, T.K.; Hong, S.H.; et al. Hwang-Heuk-San induces apoptosis in HCT116 human colorectal cancer cells through the ROS-mediated activation of caspases and the inactivation of the PI3K/Akt signaling pathway. Oncol. Rep. 2016, 36, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Yanagi, T.; Shi, R.; Aza-Blanc, P.; Reed, J.C.; Matsuzawa, S. PCTAIRE1-knockdown sensitizes cancer cells to TNF family cytokines. PLoS ONE 2015, 10, e0119404. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Meng, D.; Wei, T.; Zhang, S.; Hu, Y.; Wang, M. Apoptosis and pro-inflammatory cytokine response of mast cells induced by influenza A viruses. PLoS ONE 2014, 9, e100109. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.W.; Huang, L.H.; Yan, S.; Jin, J.D.; Ren, S.Y. Cordycepin induces apoptosis in human liver cancer HepG2 cells through extrinsic and intrinsic signaling pathways. Oncol. Lett. 2016, 12, 995–1000. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, T.W.; Graveley, B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature 2010, 463, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Quesnel-Vallières, M.; Irimia, M.; Cordes, S.P.; Blencowe, B.J. Essential roles for the splicing regulator nSR100/SRRM4 during nervous system development. Genes Dev. 2015, 29, 746–759. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Matera, A.G.; Wang, Z. A day in the life of the spliceosome. Nat. Rev. Mol. Cell Biol. 2014, 15, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Arrigoni, A.; Ranzani, V.; Rossetti, G.; Panzeri, I.; Abrignani, S.; Bonnal, R.J.; Pagani, M. Analysis RNA-seq and Noncoding RNA. Methods Mol. Biol. 2016, 1480, 125–135. [Google Scholar] [PubMed]
- Li, J.; Yang, Z.; Li, Y.; Xia, J.; Li, D.; Li, H.; Ren, M.; Liao, Y.; Yu, S.; Chen, Y.; et al. Cell apoptosis, autophagy and necroptosis in osteosarcoma treatment. Oncotarget 2016, 7, 44763–44778. [Google Scholar] [PubMed]
- Kallenberger, S.M.; Beaudouin, J.; Claus, J.; Fischer, C.; Sorger, P.K.; Legewie, S.; Eils, R. Intra- and interdimeric caspase-8 self-cleavage controls strength and timing of CD95-induced apoptosis. Sci. Signal. 2014, 7, ra23. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, E.; Tang, M.; Yang, X.; Wang, Y.; Quan, Z.; Wu, X.; Luo, C. The SMAD2/3 pathway is involved in hepaCAM-induced apoptosis by inhibiting the nuclear translocation of SMAD2/3 in bladder cancer cells. Tumour Biol. 2016, 37, 10731–10743. [Google Scholar] [CrossRef] [PubMed]
- Baluchamy, S.; Ravichandran, P.; Periyakaruppan, A.; Ramesh, V.; Hall, J.C.; Zhang, Y.; Jejelowo, O.; Gridley, D.S.; Wu, H.; Ramesh, G.T. Induction of cell death through alteration of oxidants and antioxidants in lung epithelial cells exposed to high energy protons. J. Biol. Chem. 2010, 285, 24769–24774. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Ye, F.; Zhu, W.; Hu, D.; Xiao, C.; Nan, J.; Su, S.; Wang, Y.; Liu, M.; Gao, K.; et al. Cardiac ankyrin repeat protein attenuates cardiomyocyte apoptosis by upregulation of Bcl-2 expression. Biochim. Biophys. Acta 2016, 4889, 30254. [Google Scholar] [CrossRef] [PubMed]
- Akgul, C.; Moulding, D.A.; Edwards, S.W. Alternative splicing of Bcl-2-related genes: Functional consequences and potential therapeutic applications. Cell. Mol. Life Sci. 2004, 61, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Würstle, M.L.; Laussmann, M.A.; Rehm, M. The central role of initiator caspase-9 in apoptosis signal transduction and the regulation of its activation and activity on the apoptosome. Exp. Cell Res. 2012, 318, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.R.; Park, B.S.; Kim, S.Y.; Gu, A.; Kim da, H.; Lee, J.S.; Kim, I.S. Cytokine secreted by S100A9 via TLR4 in monocytes delays neutrophil apoptosis by inhibition of caspase 9/3 pathway. Cytokine 2016, 86, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Gai, W.T.; Yu, D.P.; Wang, X.S.; Wang, P.T. Anti-cancer effect of ursolic acid activates apoptosis through ROCK/PTEN mediated mitochondrial translocation of cofilin-1 in prostate cancer. Oncol. Lett. 2016, 12, 2880–2885. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhou, Q.; Tan, S. Targeting miRNAs associated with surface expression of death receptors to modulate TRAIL resistance in breast cancer. Cancer Lett. 2016. [Google Scholar] [CrossRef] [PubMed]
- Ling, G.; Zhang, T.; Zhang, P.; Sun, J.; He, Z. Synergistic and complete reversal of the multidrug resistance of mitoxantrone hydrochloride by three-in-one multifunctional lipid-sodium glycocholate nanocarriers based on simultaneous BCRP and Bcl-2 inhibition. Int. J. Nanomed. 2016, 11, 4077–4091. [Google Scholar]
- Yang, A.; Wilson, N.S.; Ashkenazi, A. Proapoptotic DR4 and DR5 signaling in cancer cells: Toward clinical translation. Curr. Opin. Cell Biol. 2010, 22, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Mohammadpour, H.; Pourfathollah, A.A.; Nikougoftar Zarif, M.; Shahbazfar, A.A. Irradiation enhances susceptibility of tumor cells to the antitumor effects of TNF-α activated adipose derived mesenchymal stem cells in breast cancer model. Sci. Rep. 2016, 6, 28433. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hawkins, O.E.; Vilgelm, A.E.; Pawlikowski, J.S.; Ecsedy, J.A.; Sosman, J.A.; Kelley, M.C.; Richmond, A. Combining an aurora kinase inhibitor and a death receptor ligand/agonist antibody triggers apoptosis in melanoma cells and prevents tumor growth in preclinical mouse models. Clin. Cancer Res. 2015, 21, 5338–5348. [Google Scholar] [CrossRef] [PubMed]
- Dickens, L.S.; Boyd, R.S.; Jukes-Jones, R.; Hughes, M.A.; Robinson, G.L.; Fairall, L.; Schwabe, J.W.; Cain, K.; Macfarlane, M. A death effector domain chain DISC model reveals a crucial role for caspase-8 chain assembly in mediating apoptotic cell death. Mol. Cell 2012, 47, 291–305. [Google Scholar] [CrossRef] [PubMed]
- Krieg, A.; Krieg, T.; Wenzel, M.; Schmitt, M.; Ramp, U.; Fang, B.; Gabbert, H.E.; Gerharz, C.D.; Mahotka, C. TRAIL-β and TRAIL-γ: Two novel splice variants of the human TNF-related apoptosis-inducing ligand (TRAIL) without apoptotic potential. Br. J. Cancer 2003, 88, 918–927. [Google Scholar] [CrossRef] [PubMed]
- Picarda, G.; Surget, S.; Guiho, R.; Téletchéa, S.; Berreur, M.; Tirode Pellat-Deceunynck, C.; Heymann, D.; Trichet, V.; Rédini, F. A functional, new short isoform of death receptor 4 in Ewing’s sarcoma cell lines may be involved in TRAIL sensitivity/resistance mechanisms. Mol. Cancer Res. 2012, 10, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Rautureau, G.J.; Day, C.L.; Hinds, M.G. Intrinsically disordered proteins in Bcl-2 regulated apoptosis. Int. J. Mol. Sci. 2010, 11, 1808–1824. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Li, X.; Cheng, Y.; Wu, W.; Xie, Z.; Xi, Q.; Han, J.; Wu, G.; Fang, J.; Feng, Y. BCLAF1 and its splicing regulator SRSF10 regulate the tumorigenic potential of colon cancer cells. Nat. Commun. 2014, 5, 4581. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Moore, M.J. The spliceosome: Disorder and dynamics defined. Curr. Opin. Struct. Biol. 2014, 24, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Papasaikas, P.; Tejedor, J.R.; Vigevani, L.; Valcárcel, J. Functional splicing network reveals extensive regulatory potential of the core spliceosomal machinery. Mol. Cell 2015, 57, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Cyphert, T.J.; Suchanek, A.L.; Griffith, B.N.; Salati, L.M. Starvation actively inhibits splicing of glucose-6-phosphate dehydrogenase mRNA via a bifunctional ESE/ESS element bound by hnRNP K. Biochim. Biophys. Acta 2013, 1829, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Erkelenz, S.; Mueller, W.F.; Evans, M.S.; Busch, A.; Schöneweis, K.; Hertel, K.J.; Schaal, H. Position-dependent splicing activation and repression by SR and hnRNP proteins rely on common mechanisms. RNA 2013, 19, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.D.; Ares, M., Jr. Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.C.; Tarn, W.Y. RBM4 down-regulates PTB and antagonizes its activity in muscle cell–specific alternative splicing. J. Cell Biol. 2011, 193, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Martini, E.; Schneider, E.; Neufert, C.; Neurath, M.F.; Becker, C. Survivin is a guardian of the intestinal stem cell niche and its expression is regulated by TGF-β. Cell Cycle 2016, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Lee, J.Y.; Jung, C.L.; Bae, I.H.; Suh, K.H.; Ahn, Y.G.; Jin, D.H.; Kim, T.W.; Suh, Y.A.; Jang, S.J. A novel antagonist to the inhibitors of apoptosis (IAPs) potentiates cell death in EGFR-overexpressing non-small-cell lung cancer cells. Cell Death Dis. 2014, 5, e1477. [Google Scholar] [CrossRef] [PubMed]
- Turkkila, M.; Andersson, K.M.; Amu, S.; Brisslert, M.; Erlandsson, M.C.; Silfverswärd, S.; Bokarewa, M.I. Suppressed diversity of survivin splicing in active rheumatoid arthritis. Arthritis Res. Ther. 2015, 17, 175. [Google Scholar] [CrossRef] [PubMed]
- Faversani, A.; Vaira, V.; Moro, G.P.; Tosi, D.; Lopergolo, A.; Schultz, D.C.; Rivadeneira, D.; Altieri, D.C.; Bosari, S. Survivin family proteins as novel molecular determinants of doxorubicin resistance in organotypic human breast tumors. Breast Cancer Res. 2014, 16, R55. [Google Scholar] [CrossRef] [PubMed]
- Tazo, Y.; Hara, A.; Onda, T.; Saegusa, M. Bifunctional roles of survivin-ΔEx3 and survivin-2B for susceptibility to apoptosis in endometrial carcinomas. J. Cancer Res. Clin. Oncol. 2014, 140, 2027–2037. [Google Scholar] [CrossRef] [PubMed]
- Waligórska-Stachura, J.; Andrusiewicz, M.; Sawicka-Gutaj, N.; Kubiczak, M.; Jankowska, A.; Liebert, W.; Czarnywojtek, A.; Waśko, R.; Blanco-Gangoo, A.R.; Ruchała, M. Evaluation of survivin splice variants in pituitary tumors. Pituitary 2015, 18, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.; Deoraj, A.; Felty, Q.; Roy, D. Environmental estrogen-like endocrine disrupting chemicals and breast cancer. Mol. Cell. Endocrinol. 2016, 7207, 30411–30417. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, H.J.; Bao, Q.C.; Wang, L.; Guo, T.K.; Chen, W.L.; Xu, L.L.; Zhou, H.S.; Bian, J.L.; Yang, Y.R.; et al. NRF2 promotes breast cancer cell proliferation and metastasis by increasing RhoA/ROCK pathway signal transduction. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Divekar, S.D.; Tiek, D.M.; Fernandez, A.; Riggins, R.B. Estrogen-related receptor β (ERRβ)—Renaissance receptor or receptor renaissance? Nucl. Recept. Signal. 2016, 14, e002. [Google Scholar] [PubMed]
- Huang, P.C.; Kuo, W.W.; Shen, C.Y.; Chen, Y.F.; Lin, Y.M.; Ho, T.J.; Padma, V.V.; Lo, J.F.; Huang, C.Y.; Huang, C.Y. Anthocyanin attenuates doxorubicin-induced cardiomyotoxicity via estrogen receptor-α/β and stabilizes HSF1 to inhibit the IGF-IIR apoptotic pathway. Int. J. Mol. Sci. 2016, 17, 1588. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Wang, L.; James, T.; Jung, Y.; Kim, I.; Tan, R.; Hoffmann, F.M.; Xu, W. Reciprocal regulation of ERα and ERβ stability and activity by Diptoindonesin G. Chem. Biol. 2015, 22, 1608–1621. [Google Scholar] [CrossRef] [PubMed]
- Diao, Y.; Azatyan, A.; Rahman, M.F.; Zhao, C.; Zhu, J.; Dahlman-Wright, K.; Zaphiropoulos, P.G. Blockade of the Hedgehog pathway downregulates estrogen receptor α signaling in breast cancer cells. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Piperigkou, Z.; Bouris, P.; Onisto, M.; Franchi, M.; Kletsas, D.; Theocharis, A.D.; Karamanos, N.K. Estrogen receptor β modulates breast cancer cells functional properties, signaling and expression of matrix molecules. Matrix Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Hirschfeld, M.; Ouyang, Y.Q.; Jaeger, M.; Erbes, T.; Orlowska-Volk, M.; Zur Hausen, A.; Stickeler, E. HNRNP G and HTRA2-Β1 regulate estrogen receptor α expression with potential impact on endometrial cancer. BMC Cancer 2015, 15, 86. [Google Scholar] [CrossRef] [PubMed]
- Backes, F.J.; Walker, C.J.; Goodfellow, P.J.; Hade, E.M.; Agarwal, G.; Mutch, D.; Cohn, D.E.; Suarez, A.A. Estrogen receptor-α as a predictive biomarker in endometrioid endometrial cancer. Gynecol. Oncol. 2016, 141, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Liu, Z.; Wu, J.; Liu, J.H.; Hyder, S.M.; Antoniou, E.; Lubahn, D.B. Identification and characterization of two novel splicing isoforms of human estrogen-related receptor β. J. Clin. Endocrinol. Metab. 2006, 91, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Wong, Y.C.; Wang, X.H.; Ling, M.T.; Ng, C.F.; Chen, S.; Chan, F.L. Orphan nuclear receptor estrogen-related receptor-β suppresses in vitro and in vivo growth of prostate cancer cells via p21(WAF1/CIP1) induction and as a potential therapeutic target in prostate cancer. Oncogene 2008, 27, 3313–3328. [Google Scholar] [CrossRef] [PubMed]
- Komiya, Y.; Runnels, L.W. TRPM channels and magnesium in early embryonic development. Int. J. Dev. Biol. 2015, 59, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Frühwald, J.; Camacho Londoño, J.; Dembla, S.; Mannebach, S.; Lis, A.; Drews, A.; Wissenbach, U.; Oberwinkler, J.; Philipp, S.E. Alternative splicing of a protein domain indispensable for function of transient receptor potential melastatin 3 (TRPM3) ion channels. J. Biol. Chem. 2012, 287, 36663–36672. [Google Scholar] [CrossRef] [PubMed]
- Bidaux, G.; Beck, B.; Zholos, A.; Gordienko, D.; Lemonnier, L.; Flourakis, M.; Roudbaraki, M.; Borowiec, A.S.; Fernández, J.; Delcourt, P.; et al. Regulation of activity of transient receptor potential melastatin 8 (TRPM8) channel by its short isoforms. J. Biol. Chem. 2012, 287, 2948–2962. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Wang, Z.; Yang, Z.; Tao, L.; Liu, Q.; Yi, L.U.; Wang, X. Overexpression of short TRPM8 variant α promotes cell migration and invasion, and decreases starvation-induced apoptosis in prostate cancer LNCaP cells. Oncol. Lett. 2015, 10, 1378–1384. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.J.; Huang, Y.C.; Cheng, P.J.; Lee, P.T.; Hsiao, H.S.; Kuo, M.L. Interleukin-15 enhances the expansion and function of natural killer T cells from adult peripheral and umbilical cord blood. Cytokine 2015, 76, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Washizu, J.; Nakamura, N.; Enomoto, A.; Yoshikai, Y. Translational efficiency is up-regulated by alternative exon in murine IL-15 mRNA. J. Immunol. 1998, 160, 936–942. [Google Scholar] [PubMed]
- Zhao, L.; Hu, B.; Zhang, Y.; Song, Y.; Lin, D.; Liu, Y.; Mei, Y.; Sandikin, D.; Sun, W.; Zhuang, M.; et al. An activation-induced IL-15 isoform is a natural antagonist for IL-15 function. Sci. Rep. 2016, 6, 25822. [Google Scholar] [CrossRef] [PubMed]
- Laptenko, O.; Tong, D.R.; Manfredi, J.; Prives, C. The tail that wags the dog: How the disordered C-terminal domain controls the transcriptional activities of the p53 tumor-suppressor protein. Trends Biochem. Sci. 2016, 41, 1022–1034. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Weiss, W.A. Alternative splicing in cancer: Implications for biology and therapy. Oncogene 2015, 34, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; An, S.S. Role of p53 isoforms and aggregations in cancer. Medicine 2016, 95, e3993. [Google Scholar] [CrossRef] [PubMed]
- Marcel, V.; Fernandes, K.; Terrier, O.; Lane, D.P.; Bourdon, J.C. Modulation of p53β and p53γ expression by regulating the alternative splicing of TP53 gene modifies cellular response. Cell Death Differ. 2014, 21, 1377–1387. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Mondal, A.M.; Horikawa, I.; Nguyen, G.H.; Kumamoto, K.; Sohn, J.J.; Bowman, E.D.; Mathe, E.A.; Schetter, A.J.; Pine, S.R. p53 Isoforms Δ133p53 and p53β are endogenous regulators of replicative cellular senescence. Nat. Cell Biol. 2009, 11, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Silden, E.; Hjelle, S.M.; Wergeland, L.; Sulen, A.; Andresen, V.; Bourdon, J.C.; Micklem, D.R.; McCormack, E.; Gjertsen, B.T. Expression of TP53 isoforms p53β or p53γ enhances chemosensitivity in TP53null cell lines. PLoS ONE 2013, 8, e56276. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Liu, F.; Yan, J.; Zhang, Y.; Yan, J.; Shang, H.; Dou, Q.; Zhao, Q.; Song, Y. Trans-splicing repair of mutant p53 suppresses the growth of hepatocellular carcinoma cells in vitro and in vivo. Sci. Rep. 2015, 5, 8705. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Xu, C.; Zhao, H.; Xia, P.; Song, R.; Gu, J.; Liu, X.; Bian, J.; Yuan, Y.; Liu, Z. Osteoprotegerin induces apoptosis of osteoclasts and osteoclast precursor cells via the fas/fas ligand pathway. PLoS ONE 2015, 10, e0142519. [Google Scholar] [CrossRef] [PubMed]
- Paronetto, M.P.; Bernardis, I.; Volpe, E.; Bechara, E.; Sebestyén, E.; Eyras, E.; Valcárcel, J. Regulation of FAS exon definition and apoptosis by the Ewing sarcoma protein. Cell Rep. 2014, 7, 1211–1226. [Google Scholar] [CrossRef] [PubMed]
- Proussakova, O.V.; Rabaya, N.A.; Moshnikova, A.B.; Telegina, E.S.; Turanov, A.; Nanazashvili, M.G.; Beletsky, I.P. Oligomerization of soluble Fas antigen induces its cytotoxicity. J. Biol. Chem. 2003, 278, 36236–36241. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, J.M.; Majós, N.; Bonnal, S.; Martínez, C.; Castelo, R.; Guigó, R.; Bilbao, D.; Valcárcel, J. Regulation of Fas alternative splicing by antagonistic effects of TIA-1 and PTB on exon definition. Mol. Cell 2005, 19, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, J.M. Hu antigen R (HuR) functions as an alternative pre-mRNA splicing regulator of Fas apoptosis-promoting receptor on exon definition. J. Biol. Chem. 2008, 283, 19077–19084. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, J.M. Heterogeneous ribonucleoprotein C displays a repressor activity mediated by T-cell intracellular antigen-1-related/like protein to modulate Fas exon 6 splicing through a mechanism involving Hu antigen R. Nucleic Acids Res. 2010, 38, 8001–8014. [Google Scholar] [CrossRef] [PubMed]
- Julien, P.; Miñana, B.; Baeza-Centurion, P.; Valcárcel, J.; Lehner, B. The complete local genotype-phenotype landscape for the alternative splicing of a human exon. Nat. Commun. 2016, 7, 11558. [Google Scholar] [CrossRef] [PubMed]
- Tejedor, J.R.; Papasaikas, P.; Valcárcel, J. Genome-wide identification of Fas/CD95 alternative splicing regulators reveals links with iron homeostasis. Mol. Cell 2015, 57, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Villamizar, O.; Chambers, C.B.; Riberdy, J.M.; Persons, D.A.; Wilber, A. Long noncoding RNA Saf and splicing factor 45 increase soluble Fas and resistance to apoptosis. Oncotarget 2016, 7, 13810–13826. [Google Scholar] [PubMed]
- Hughes, M.A.; Powley, I.R.; Jukes-Jones, R.; Horn, S.; Feoktistova, M.; Fairall, L.; Schwabe, J.W.; Leverkus, M.; Cain, K.; MacFarlane, M. Co-operative and hierarchical binding of c-FLIP and caspase-8: A unified model defines how c-FLIP isoforms differentially control cell fate. Mol. Cell 2016, 61, 834–849. [Google Scholar] [CrossRef] [PubMed]
- Ram, D.R.; Ilyukha, V.; Volkova, T.; Buzdin, A.; Tai, A.; Smirnova, I.; Poltorak, A. Balance between short and long isoforms of cFLIP regulates Fas-mediated apoptosis in vivo. Proc. Natl. Acad. Sci. USA 2016, 113, 1606–1611. [Google Scholar] [CrossRef] [PubMed]
- Hatok, J.; Racay, P. Bcl-2 family proteins: Master regulators of cell survival. Biomol. Concepts 2016, 7, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Risberg, K.; Redalen, K.R.; Sønstevold, L.; Bjørnetrø, T.; Sølvernes, J.; Ree, A.H. Pro-survival responses to the dual inhibition of anti-apoptotic Bcl-2 family proteins and mTOR-mediated signaling in hypoxic colorectal carcinoma cells. BMC Cancer 2016, 16, 531. [Google Scholar] [CrossRef] [PubMed]
- Shkreta, L.; Toutant, J.; Durand, M.; Manley, J.L.; Chabot, B. SRSF10 connects DNA damage to the alternative splicing of transcripts encoding apoptosis, cell-cycle control, and DNA repair factors. Cell Rep. 2016, 17, 1990–2003. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Mao, C.; Ming, X. Modulation of Bcl-x alternative splicing induces apoptosis of human hepatic stellate cells. BioMed Res. Int. 2016, 2016, 7478650. [Google Scholar] [CrossRef] [PubMed]
- Pedrotti, S.; Busà, R.; Compagnucci, C.; Sette, C. The RNA recognition motif protein RBM11 is a novel tissue-specific splicing regulator. Nucleic Acids Res. 2012, 40, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Bielli, P.; Bordi, M.; di Biasio, V.; Sette, C. Regulation of BCL-X splicing reveals a role for the polypyrimidine tract binding protein (PTBP1/hnRNP I) in alternative 5′ splice site selection. Nucleic Acids Res. 2014, 42, 12070–12081. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, D.; Qian, H.; Tsai, Y.S.; Shao, S.; Liu, Q.; Dominguez, D.; Wang, Z. The splicing factor RBM4 controls apoptosis, proliferation, and migration to suppress tumor progression. Cancer Cell 2014, 26, 374–389. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, Q.; Han, L.; Tian, N.; Liang, Q.; Li, Y.; Zhao, X.; Du, C.; Tian, Y. Pro-apoptotic effects of splice-switching oligonucleotides targeting Bcl-x pre-mRNA in human glioma cell lines. Oncol. Rep. 2016, 35, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Reyna, D.E.; Gavathiotis, E. Self-regulation of BAX-induced cell death. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Bleicken, S.; Jeschke, G.; Stegmueller, C.; Salvador-Gallego, R.; García-Sáez, A.J.; Bordignon, E. Structural model of active Bax at the membrane. Mol. Cell 2014, 56, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Haferkamp, B.; Zhang, H.; Lin, Y.; Yeap, X.; Bunce, A.; Sharpe, J.; Xiang, J. BaxΔ2 is a novel Bax isoform unique to microsatellite unstable tumors. J. Biol. Chem. 2012, 287, 34722–34729. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lin, Y.; Mañas, A.; Zhao, Y.; Denning, M.F.; Ma, L.; Xiang, J. BaxΔ2 promotes apoptosis through caspase-8 activation in microsatellite-unstable colon cancer. Mol. Cancer Res. 2014, 12, 1225–1232. [Google Scholar] [CrossRef] [PubMed]
- Juan, W.C.; Roca, X.; Ong, S.T. Identification of cis-acting elements and splicing factors involved in the regulation of BIM Pre-mRNA splicing. PLoS ONE 2014, 9, e95210. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Chen, G.G.; Yun, J.P.; Chun, S.Y.; Zheng, Z.Z.; Ho, R.L.; Chak, E.C.; Xia, N.S.; Lai, P.B. Identification and characterization of BH3 domain protein Bim and its isoforms in human hepatocellular carcinomas. Apoptosis 2007, 12, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- Leu, S.; Lin, Y.M.; Wu, C.H.; Ouyang, P. Loss of Pnn expression results in mouse early embryonic lethality and cellular apoptosis through SRSF1-mediated alternative expression of Bcl-xS and ICAD. J. Cell Sci. 2012, 125, 3164–3172. [Google Scholar] [CrossRef] [PubMed]
- Augis, V.; Airiau, K.; Josselin, M.; Turcq, B.; Mahon, F.X.; Belloc, F. A single nucleotide polymorphism in cBIM is associated with a slower achievement of major molecular response in chronic myeloid leukaemia treated with imatinib. PLoS ONE 2013, 8, e78582. [Google Scholar] [CrossRef] [PubMed]
- Cáceres, J.F.; Kornblihtt, A.R. Alternative splicing: Multiple control mechanisms and involvement in human disease. Trends Genet. 2002, 18, 186–193. [Google Scholar] [CrossRef]
- Pandit, S.; Zhou, Y.; Shiue, L.; Coutinho-Mansfield, G.; Li, H.; Qiu, J.; Huang, J.; Yeo, G.W.; Ares, M., Jr.; Fu, X.D. Genome-wide analysis reveals SR protein cooperation and competition in regulated splicing. Mol. Cell 2013, 50, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Skrdlant, L.; Stark, J.M.; Lin, R.J. Myelodysplasia-associated mutations in serine/arginine-rich splicing factor SRSF2 lead to alternative splicing of CDC25C. BMC Mol. Biol. 2016, 17, 18. [Google Scholar] [CrossRef] [PubMed]
- Komeno, Y.; Huang, Y.J.; Qiu, J.; Lin, L.; Xu, Y.; Zhou, Y.; Chen, L.; Monterroza, D.D.; Li, H.; DeKelver, R.C.; et al. SRSF2 is essential for hematopoiesis, and its myelodysplastic syndrome-related mutations dysregulate alternative pre-mRNA splicing. Mol. Cell. Biol. 2015, 35, 3071–3082. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.N.; Lee, M.; Loh, T.J.; Choi, S.W.; Oh, H.K.; Moon, H.; Cho, S.; Hong, S.E.; Kim, D.H.; Sheng, Z.; et al. Exon 9 skipping of apoptotic caspase-2 pre-mRNA is promoted by SRSF3 through interaction with exon 8. Biochim. Biophys. Acta 2014, 1839, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Park, R.Y.; Chen, J.K.; Kim, J.; Jeong, S.; Ohn, T. Splicing factor SRSF3 represses the translation of programmed cell death 4 mRNA by associating with the 5′-UTR region. Cell Death Differ. 2014, 21, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, K.; Akaike, Y.; Masuda, K.; Kuwano, Y.; Nishida, K.; Yamagishi, N.; Kajita, K.; Tanahashi, T.; Rokutan, K. Downregulation of serine/arginine-rich splicing factor 3 induces G1 cell cycle arrest and apoptosis in colon cancer cells. Oncogene 2014, 33, 1407–1417. [Google Scholar]
- Shen, X.; Li, J.; Liao, W.; Wang, J.; Chen, H.; Yao, Y.; Liu, H.; Ding, K. microRNA-149 targets caspase-2 in glioma progression. Oncotarget 2016, 7, 26388–26399. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jiang, D.; Zhang, Q.; Liu, X.; Cai, Z. Ubiquitin-specific protease 4 inhibits breast cancer cell growth through the upregulation of PDCD4. Int. J. Mol. Med. 2016, 38, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, V.; Jordan, P. Posttranscriptional regulation of splicing factor SRSF1 and its role in cancer cell biology. BioMed Res. Int. 2015, 2015, 287048. [Google Scholar] [CrossRef] [PubMed]
- Anczuków, O.; Akerman, M.; Cléry, A.; Wu, J.; Shen, C.; Shirole, N.H.; Raimer, A.; Sun, S.; Jensen, M.A.; Hua, Y.; et al. SRSF1-regulated alternative splicing in breast cancer. Mol. Cell 2015, 60, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Malakar, P.; Chartarifsky, L.; Hija, A.; Leibowitz, G.; Glaser, B.; Dor, Y.; Karni, R. Insulin receptor alternative splicing is regulated by insulin signaling and modulates β cell survival. Sci. Rep. 2016, 16, 31222. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.C.; Tarn, W.Y.; Hsieh, W.K. Emerging role for RNA binding motif protein 4 in the development of brown adipocytes. Biochim. Biophys. Acta 2014, 1843, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, A.; Chander, P.; Howe, P.H. Heterogeneous nuclear ribonucleoproteins (hnRNPs) in cellular processes: Focus on hnRNP E1’s multifunctional regulatory roles. RNA 2010, 16, 1449–1462. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, M.; Hornbaker, M.J.; Zhang, X.; Hu, P.; Bueso-Ramos, C.; Post, S.M. Aberrant hnRNP K expression: All roads lead to cancer. Cell Cycle 2016, 15, 1552–1557. [Google Scholar] [CrossRef] [PubMed]
- Jean-Philippe, J.; Paz, S.; Caputi, M. HnRNP A1: The Swiss army knife of gene expression. Int. J. Mol. Sci. 2013, 14, 18999–19024. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.k.; Lee, E.; Jang, H.N.; Lee, J.; Moon, H.; Sheng, Z.; Jun, Y.; Loh, T.J.; Cho, S.; Zhou, J.; et al. HnRNP A1 contacts exon 5 to promote exon 6 inclusion of apoptotic Fas gene. Apoptosis 2013, 18, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Revil, T.; Pelletier, J.; Toutant, J.; Cloutier, A.; Chabot, B. Heterogeneous nuclear ribonucleoprotein K represses the production of pro-apoptotic Bcl-xS splice isoform. J. Biol. Chem. 2009, 284, 21458–21467. [Google Scholar] [CrossRef] [PubMed]
- Paronetto, M.P.; Achsel, T.; Massiello, A.; Chalfant, C.E.; Sette, C. The RNA-binding protein Sam68 modulates the alternative splicing of Bcl-x. J. Cell Biol. 2007, 176, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Eder, S.; Lamkowski, A.; Priller, M.; Port, M.; Steinestel, K. Radiosensitization and downregulation of heterogeneous nuclear ribonucleoprotein K (hnRNP K) upon inhibition of mitogen/extracellular signal-regulated kinase (MEK) in malignant melanoma cells. Oncotarget 2015, 6, 17178–17191. [Google Scholar] [CrossRef] [PubMed]
- Calabretta, S.; Bielli, P.; Passacantilli, I.; Pilozzi, E.; Fendrich, V.; Capurso, G.; Fave, G.D.; Sette, C. Modulation of PKM alternative splicing by PTBP1 promotes gemcitabine resistance in pancreatic cancer cells. Oncogene 2016, 35, 2031–2039. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Placzek, W.J. PTBP1 modulation of MCL1 expression regulates cellular apoptosis induced by antitubulin chemotherapeutics. Cell Death Differ. 2016, 23, 1681–1690. [Google Scholar] [CrossRef] [PubMed]
- Stefl, R.; Skrisovska, L.; Allain, F.H. RNA sequence- and shape-dependent recognition by proteins in the ribonucleoprotein particle. EMBO Rep. 2005, 6, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.J.; Du, Y.W.; Hao, Y.Q.; Su, Z.Z.; Zhang, L.; Zhao, L.J.; Zhang, J. RNA-binding motif protein 5 inhibits the proliferation of cigarette smoke-transformed BEAS-2B cells through cell cycle arrest and apoptosis. Oncol. Rep. 2016, 35, 2315–2327. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Bacon, M.L.; Tessier, J.J.; Rintala-Maki, N.D.; Tang, V.; Sutherland, L.C. RBM10 modulates apoptosis and influences TNF-α gene expression. J. Cell Death 2012, 5, 1–19. [Google Scholar] [PubMed]
- Lin, J.C.; Chi, Y.L.; Peng, H.Y.; Lu, Y.H. RBM4-Nova1-SRSF6 splicing cascade modulates the development of brown adipocytes. Biochim. Biophys. Acta 2016, 1859, 1368–1379. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.C.; Lin, C.Y.; Tarn, W.Y.; Li, F.Y. Elevated SRPK1 lessens apoptosis in breast cancer cells through RBM4-regulated splicing events. RNA 2014, 20, 1621–1631. [Google Scholar] [CrossRef] [PubMed]
- Bechara, E.G.; Sebestyén, E.; Bernardis, I.; Eyras, E.; Valcárcel, J. RBM5, 6, and 10 differentially regulate NUMB alternative splicing to control cancer cell proliferation. Mol. Cell 2013, 52, 720–733. [Google Scholar] [CrossRef] [PubMed]
- Fushimi, K.; Ray, P.; Kar, A.; Wang, L.; Sutherland, L.C.; Wu, J.Y. Up-regulation of the proapoptotic caspase 2 splicing isoform by a candidate tumor suppressor, RBM5. Proc. Natl. Acad. Sci. USA 2008, 105, 15708–15713. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Yamamoto, N.; Kimura, M.; Nishio, K.; Yamane, H.; Nakajima, K. RBM10 regulates alternative splicing. FEBS Lett. 2014, 588, 942–947. [Google Scholar] [CrossRef] [PubMed]
- Pece, S.; Confalonieri, S.R.; Romano, P.; di Fiore, P.P. NUMB-ing down cancer by more than just a NOTCH. Biochim. Biophys. Acta 2011, 1815, 26–43. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | AS Region | AS Type | Splicing Regulator | Biological Signatures | Reference |
|---|---|---|---|---|---|
| Survivin | Exon 3 Exon 2B | Exon skipping Exon skipping | uncharacterized | anti-apoptosis (Survivin ∆Ex3 and 3β) pro-apoptosis (Survivin 2β and 2α) | [49,50,51,52] |
| ERa ERb | Exon 7 Exon 10/11 | Exon skipping Exon skipping | hnRNP G, Tra2b unclear | anti-apoptosis (ERα+7) pro-apoptosis (ERRb2) anti-apoptosis (ERRβsf) | [60,61,62,63] |
| Transient receptor potential melastatin 8 | Exon 5a | Exon skipping | uncharacterized | anti-apoptosis (sTRPM8α) | [65,66,67] |
| Interleukin-15 | Exon 6 | Exon skipping | uncharacterized | pro-apoptosis (IL-15ΔE6) | [69,70] |
| p53 | Exon β/γ | Exon skipping | uncharacterized | pro-apoptosis (p53β variants) | [72,73,74,75,76] |
| Fas | Exon 6 | Exon skipping | TIA-1/TIAR, PTBP1, HuR, hnRNP C | anti-apoptosis (Fas−exon 6) pro-apoptosis (Fas) | [79,80,81,82,83,84,85] |
| c-FLIP | Exon 5 | Alternative 5′ SS | RBM5/10 | pro-apoptosis (c-FLIPL) anti-apoptosis (c-FLIPS) | [87,88] |
| Bcl-x | Exon 2 | Alternative 5′ SS | SRSF1, PTBP1, RBM4, RBM5, RBM10, and RBM11 | anti-apoptosis (Bcl-xL) pro-apoptosis (Bcl-xS) | [91,92,93,94,95,96] |
| Bax | Exon 3 | Exon skipping | uncharacterized | pro-apoptosis (Bax and BaxΔ2) | [99,100] |
| BIM | Exon 3/4 | Mutual selection | SRSF1 | pro-apoptosis (BIM+exon 3) anti-apoptosis (BIM+exon 4) | [101,102,103,104] |
| Splicing Regulator | Specific Candidate | Impact on AS | Biological Signatures | Reference |
|---|---|---|---|---|
| SRSF1 | INSR | Exon 11 inclusion (INSR-B) | anti-apoptosis | [115,116,117] |
| CASC4 | Exon 9 inclusion | anti-apoptosis | ||
| SRSF3 | Casp2 | Exon 9 skipping | anti-apoptosis | [109,110,111,112,113] |
| HIPK2 | Alternative 3′ SS selection (Exon 8) | pro-apoptosis | ||
| PDCD4 | Intron retention (Intron 3) | anti-apoptosis | ||
| HnRNP A1 | Fas | Exon 6 inclusion | anti-apoptosis | [121] |
| HnRNP K | Bcl-x | Authentic 5' SS selection (Bcl-xL) | anti-apoptosis | [122,123,124] |
| HnRNP I | Mcl-1 | Exon 2 inclusion (Mcl-1L) | anti-apoptosis | [126] |
| Bim | Exon 4 inclusion | anti-apoptosis | [101] | |
| RBM4 | Bcl-x | Alternative 5′ SS selection (Bcl-xS) | pro-apoptosis | [95] |
| Mcl-1 | Exon 2 skipping (Mcl-1S) | pro-apoptosis | [131] | |
| RBM5/10 | c-FLIP | Alternative 5′ SS selection | anti-apoptosis (c-FLIPS) | [129] |
| Fas | Exon 6 skipping | anti-apoptosis | [134] | |
| Casp2 | Exon 9 inclusion | pro-apoptosis | [133] | |
| NUMB | Exon 9 inclusion/exclusion | uncharacterized | [132] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, J.-C.; Tsao, M.-F.; Lin, Y.-J. Differential Impacts of Alternative Splicing Networks on Apoptosis. Int. J. Mol. Sci. 2016, 17, 2097. https://doi.org/10.3390/ijms17122097
Lin J-C, Tsao M-F, Lin Y-J. Differential Impacts of Alternative Splicing Networks on Apoptosis. International Journal of Molecular Sciences. 2016; 17(12):2097. https://doi.org/10.3390/ijms17122097
Chicago/Turabian StyleLin, Jung-Chun, Mei-Fen Tsao, and Ying-Ju Lin. 2016. "Differential Impacts of Alternative Splicing Networks on Apoptosis" International Journal of Molecular Sciences 17, no. 12: 2097. https://doi.org/10.3390/ijms17122097