
Identification of Heat Shock Transcription Factor Genes Involved in Thermotolerance of Octoploid Cultivated Strawberry


Abstract
:1. Introduction
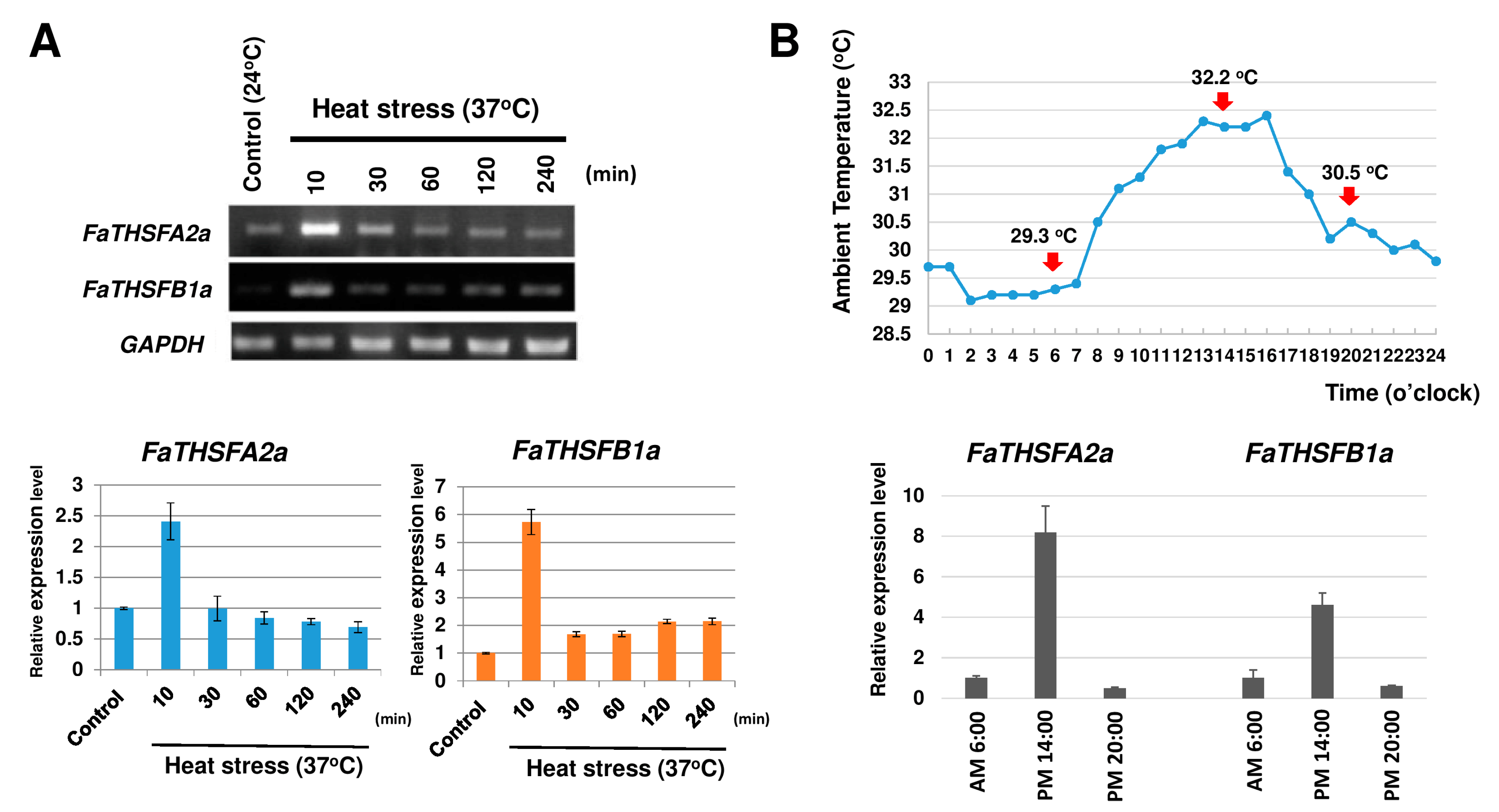
2. Results and Discussion
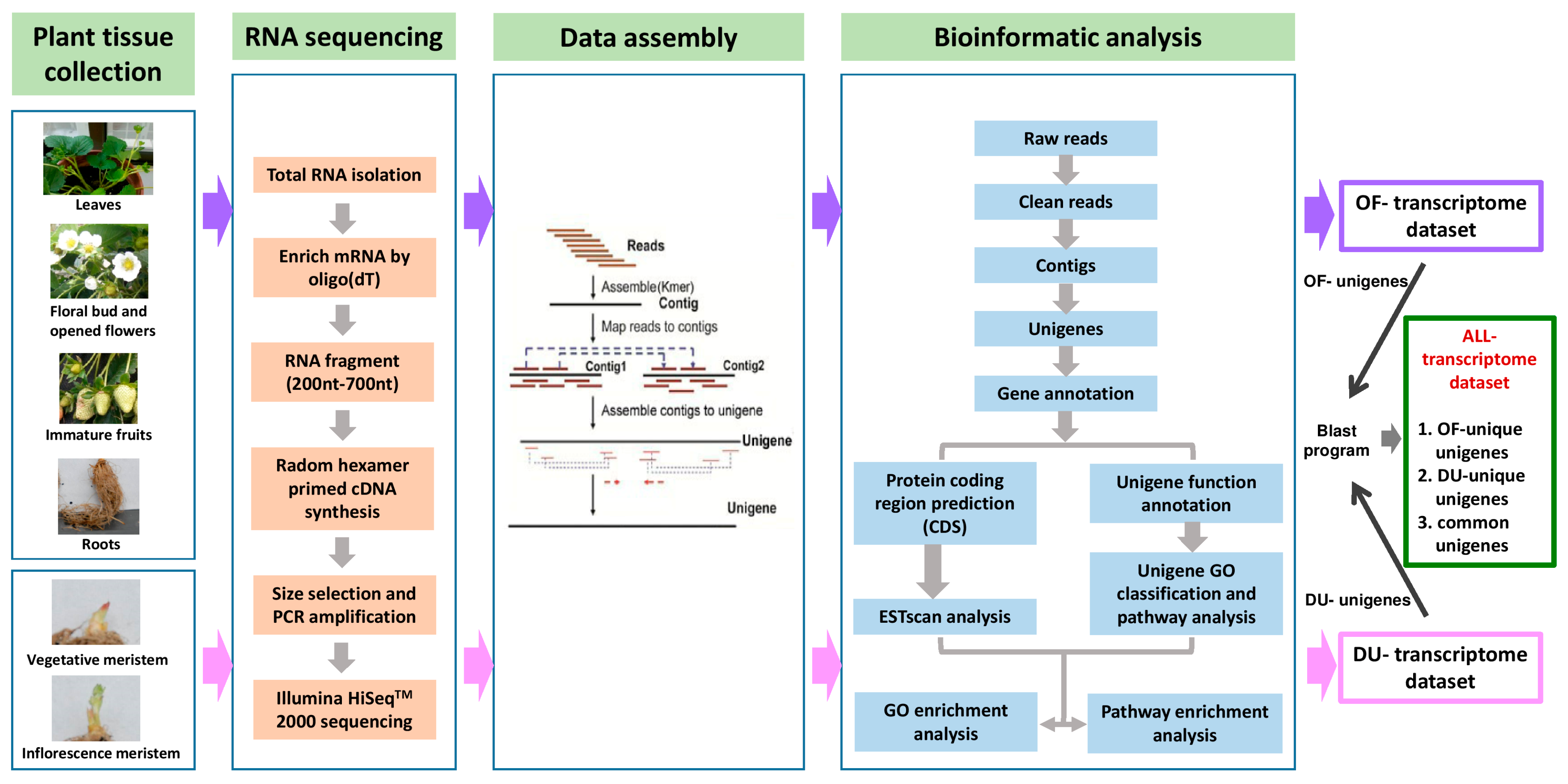
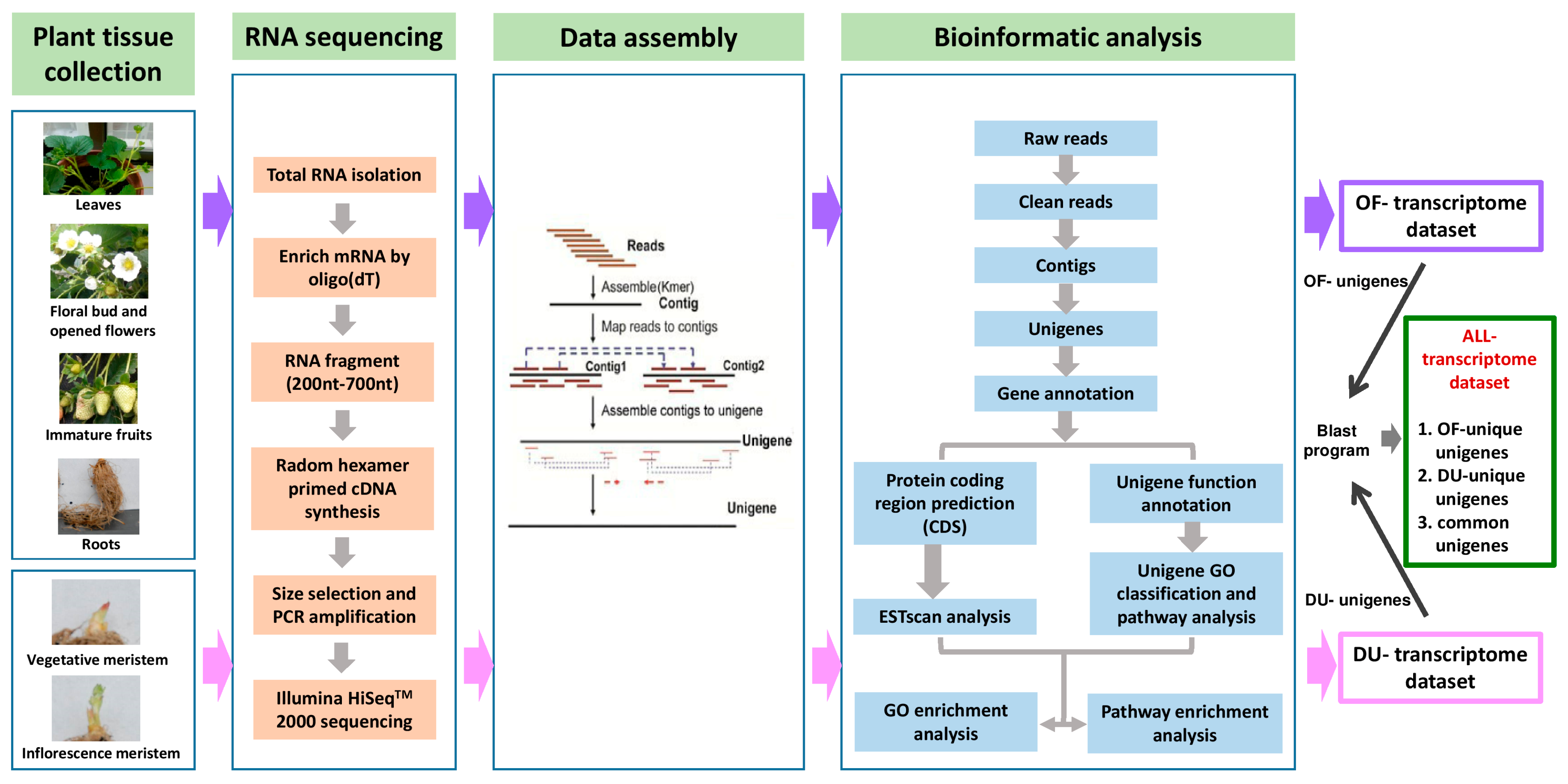
2.1. Illumina Sequencing, De Novo Assembly, and Functional Annotation of the Transcriptome
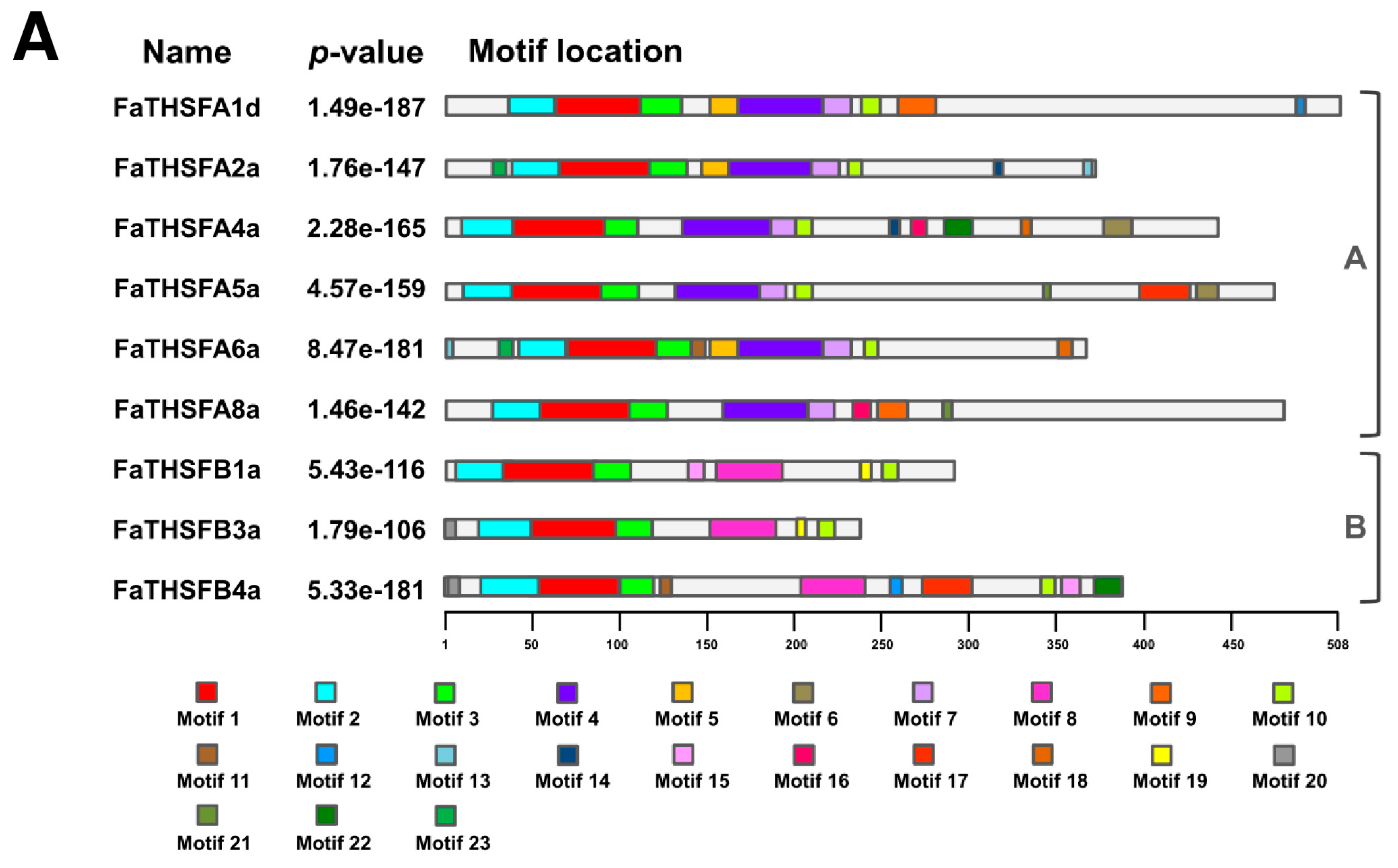
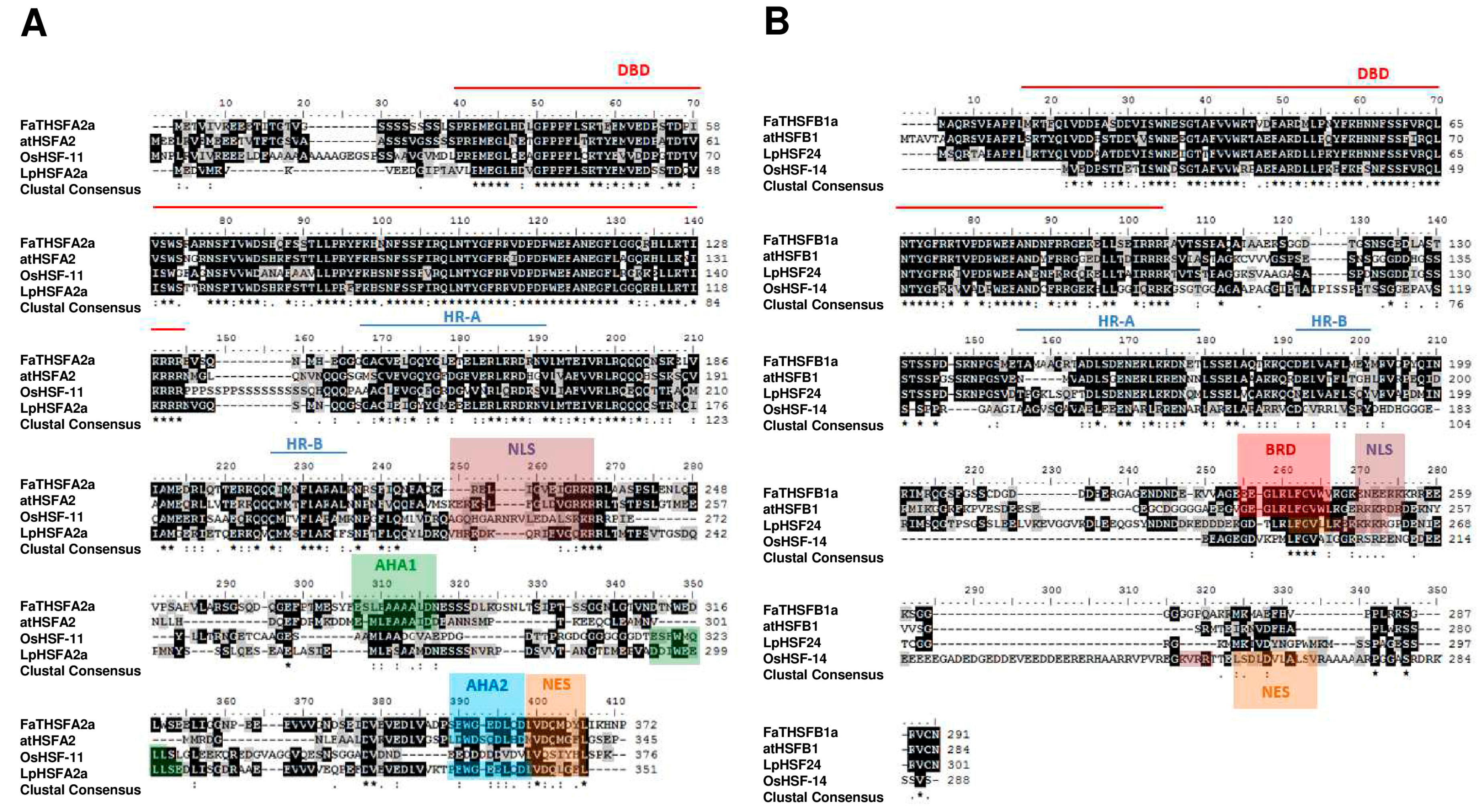
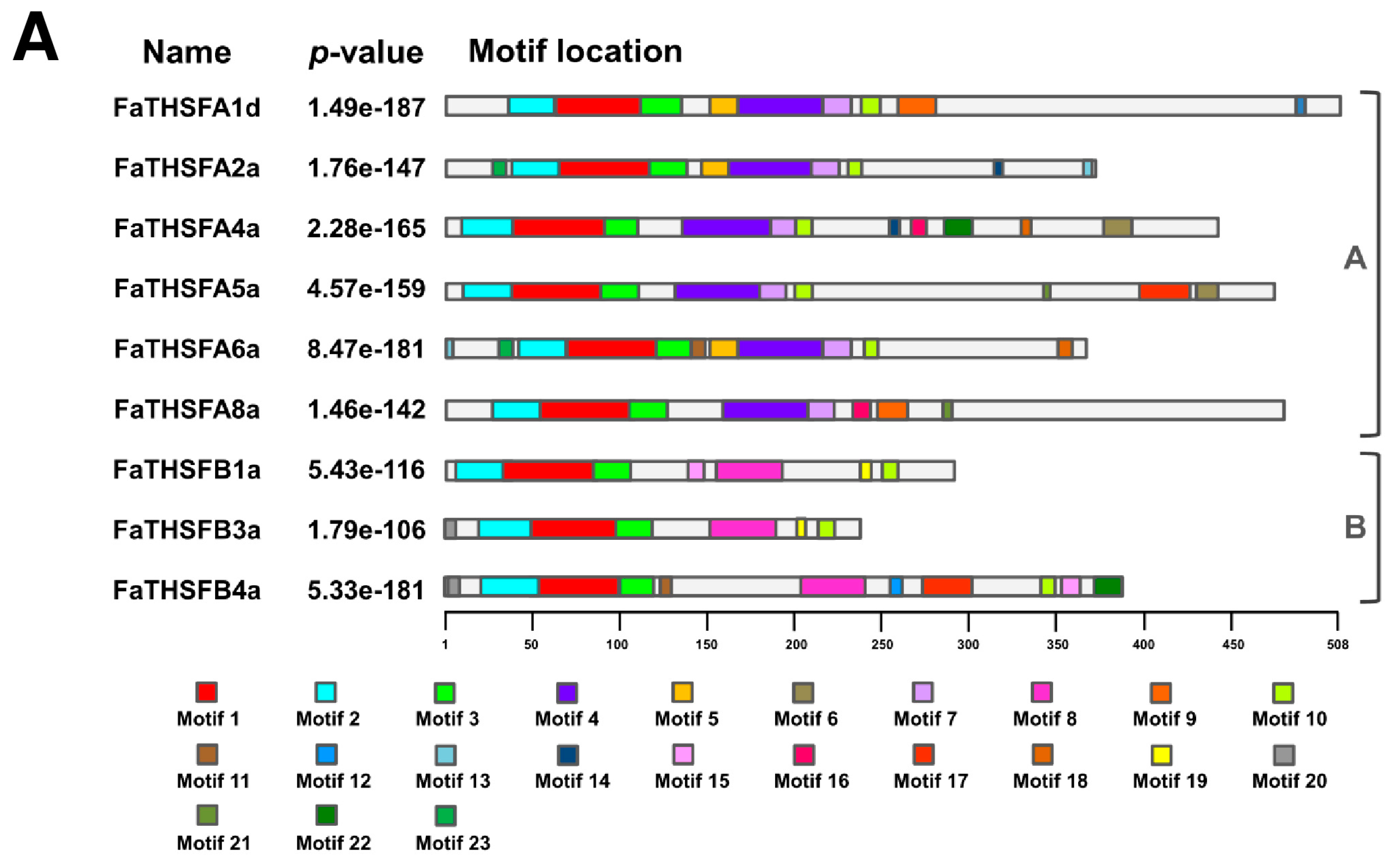
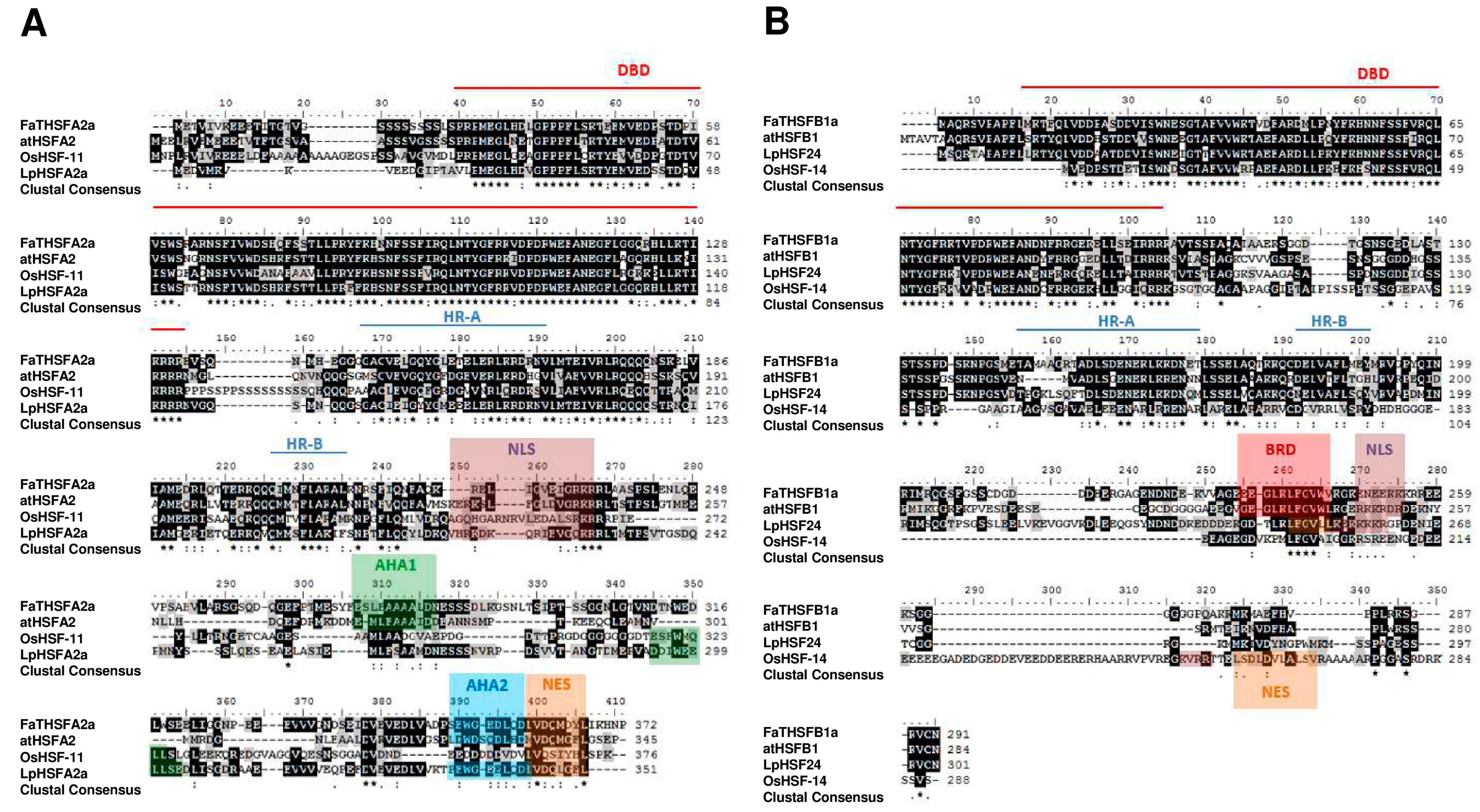
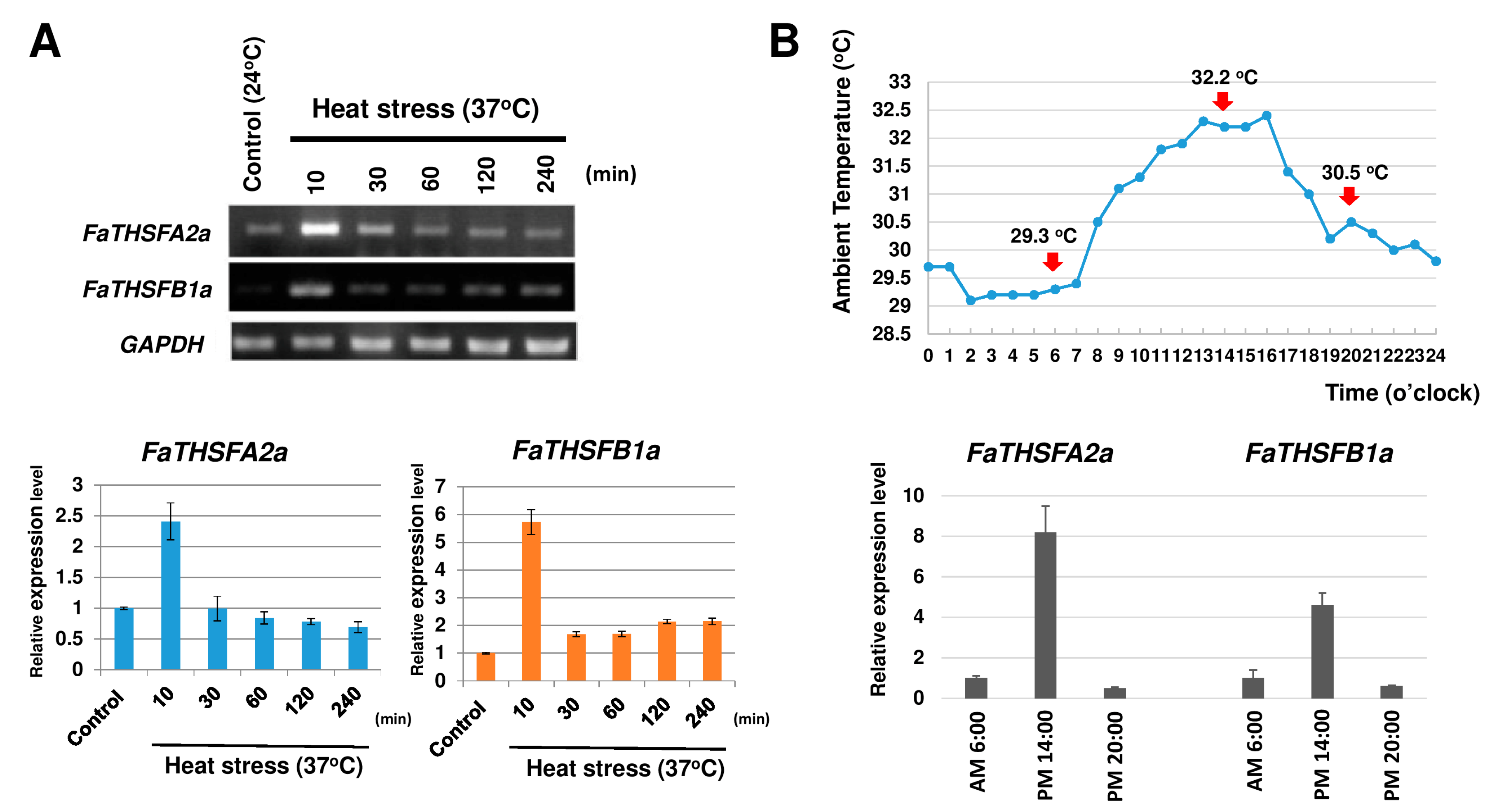
2.2. Identifying Nine FaTHSF Genes with Full-Length cDNA Sequences in F. × ananassa Duch. cv. Toyonoka
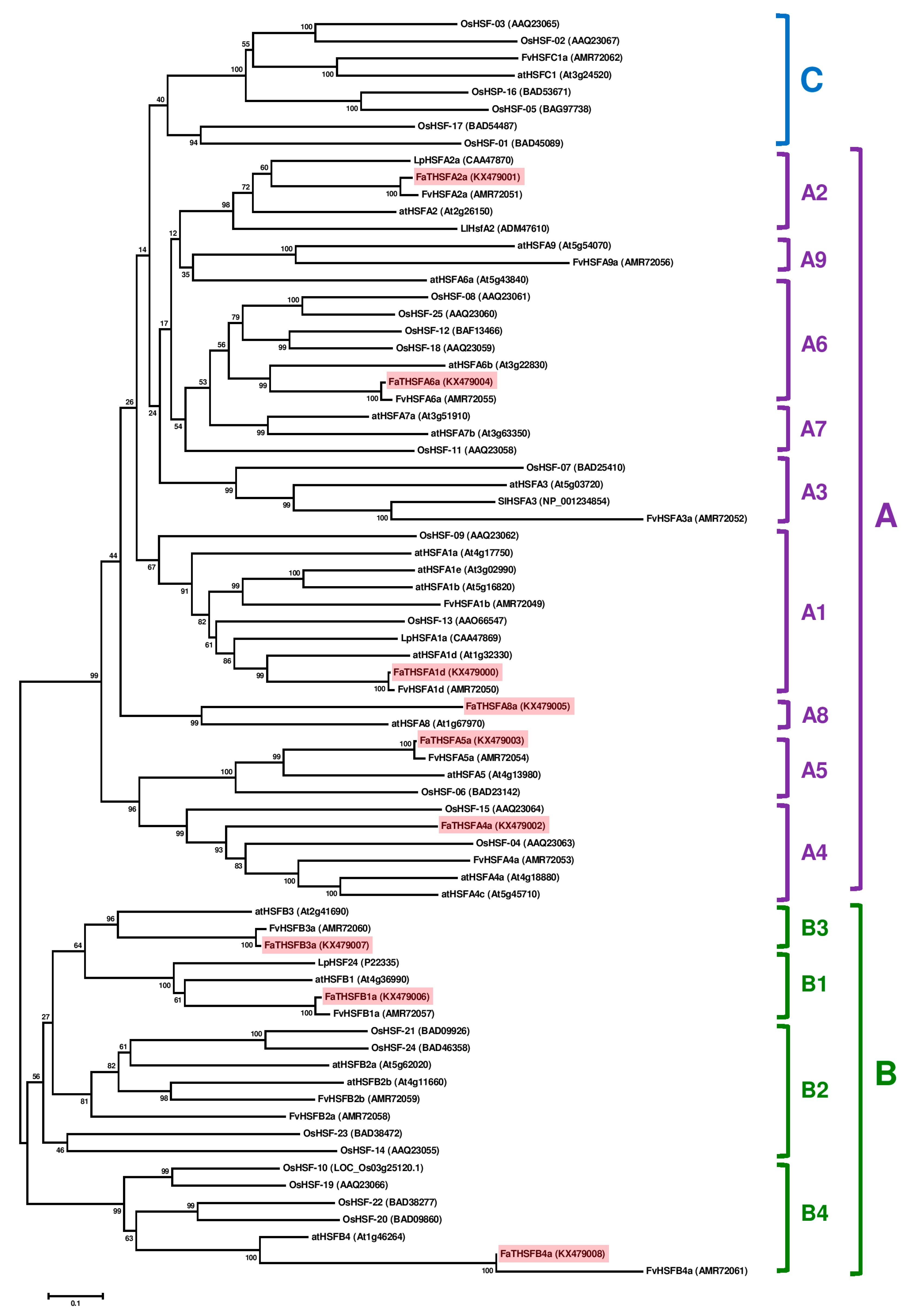
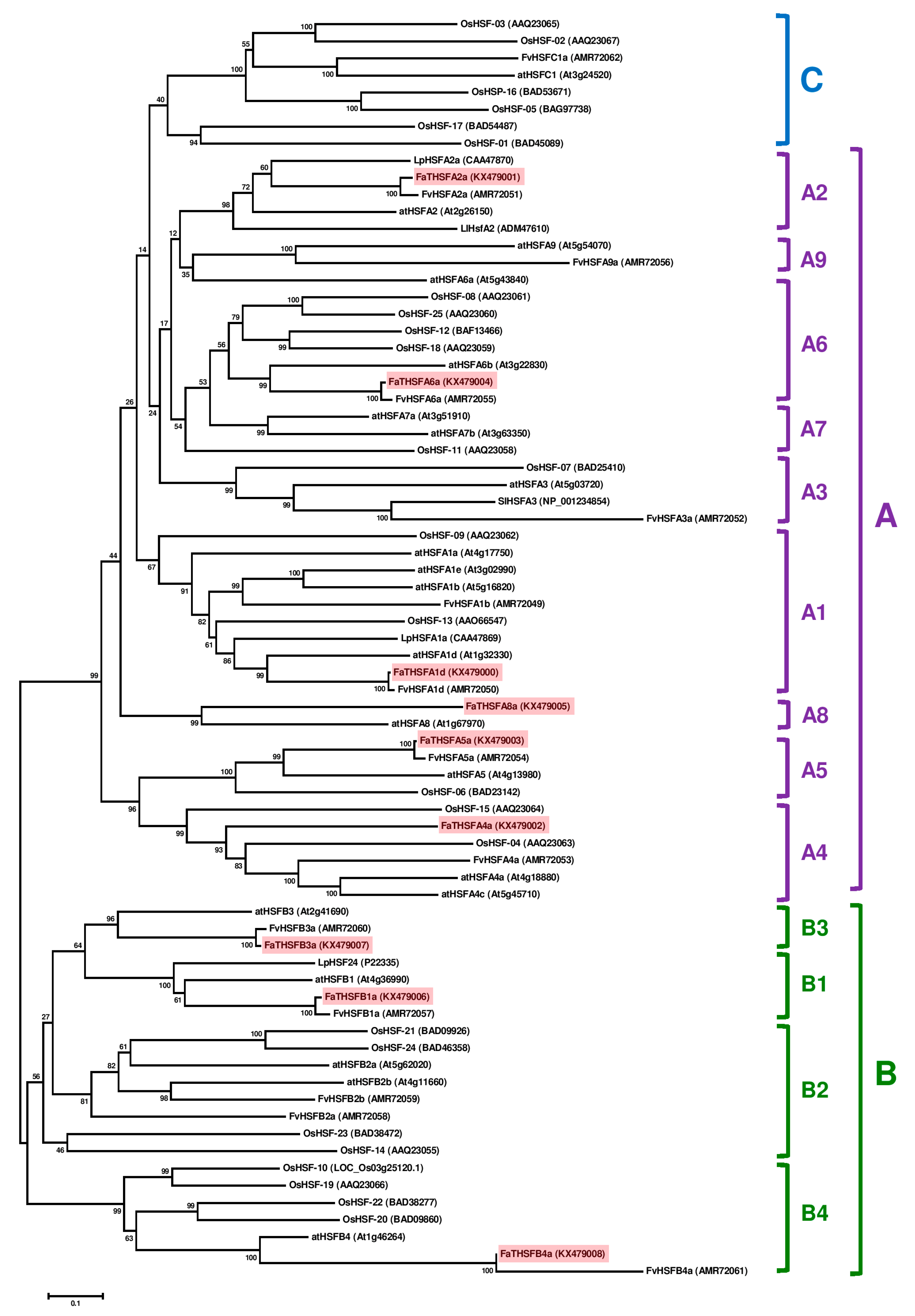
2.3. Classifying FaTHSFA2a and FaTHSFB1a into A2 and B1 HSFs, Respectively, by Phylogenic Analysis
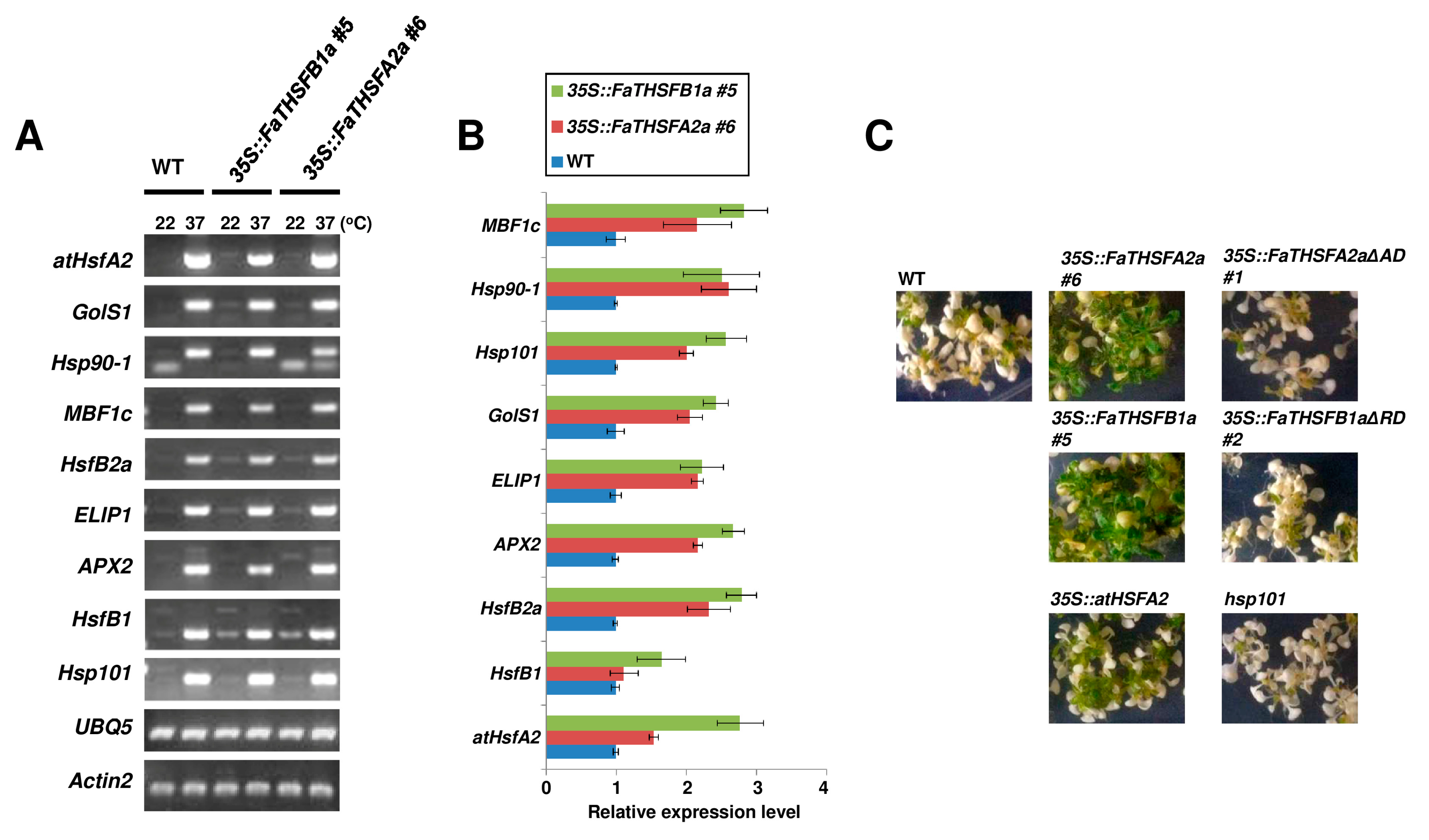
2.4. Enhanced Thermotolerance in FaTHSFA2a- and FaTHSFB1a-Overexpressed Transgenic Arabidopsis Plants
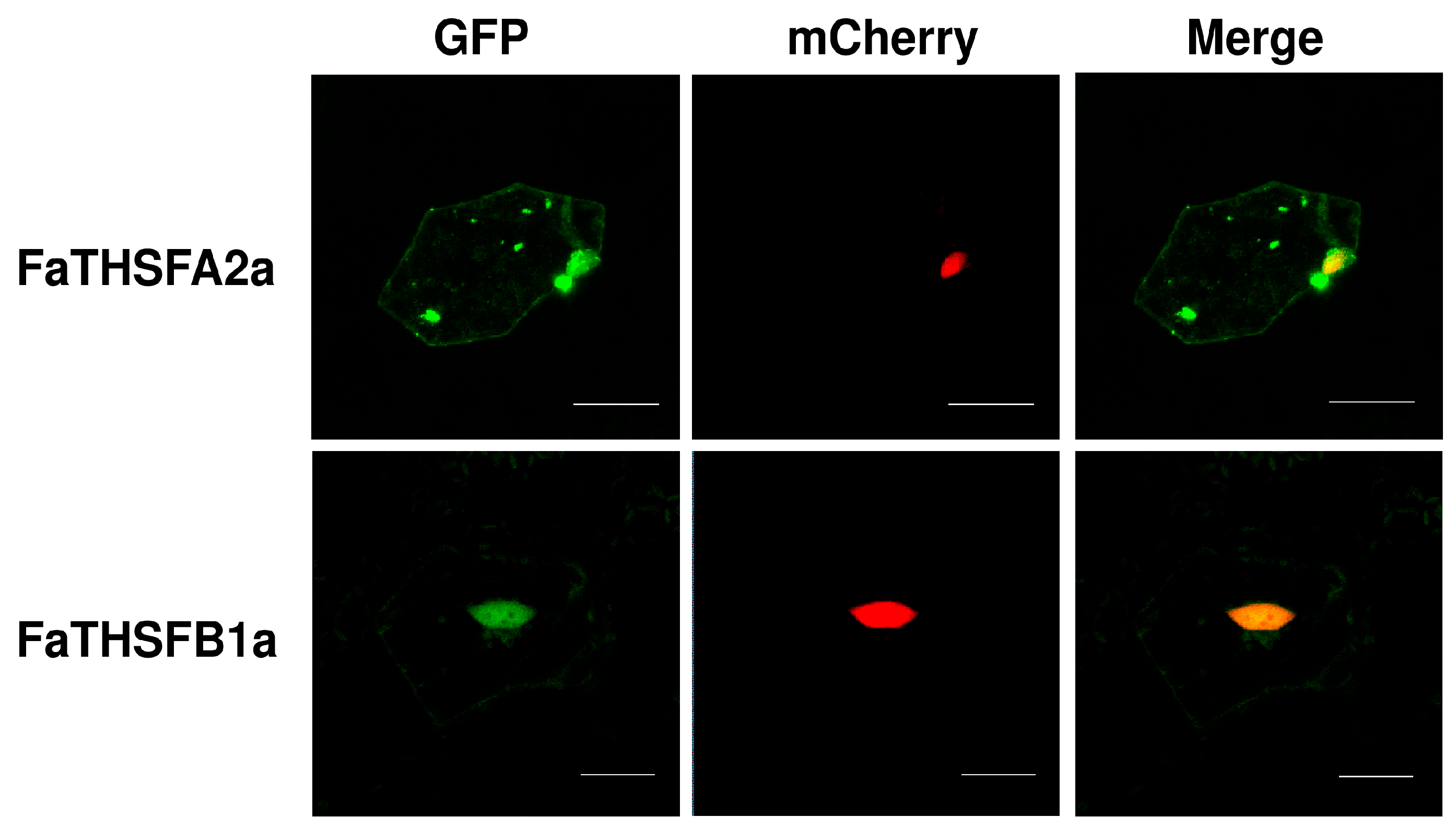
2.5. Nuclear Localization of FaTHSFA2a and FaTHSFB1a Suggesting Their Roles as Transcription Factors
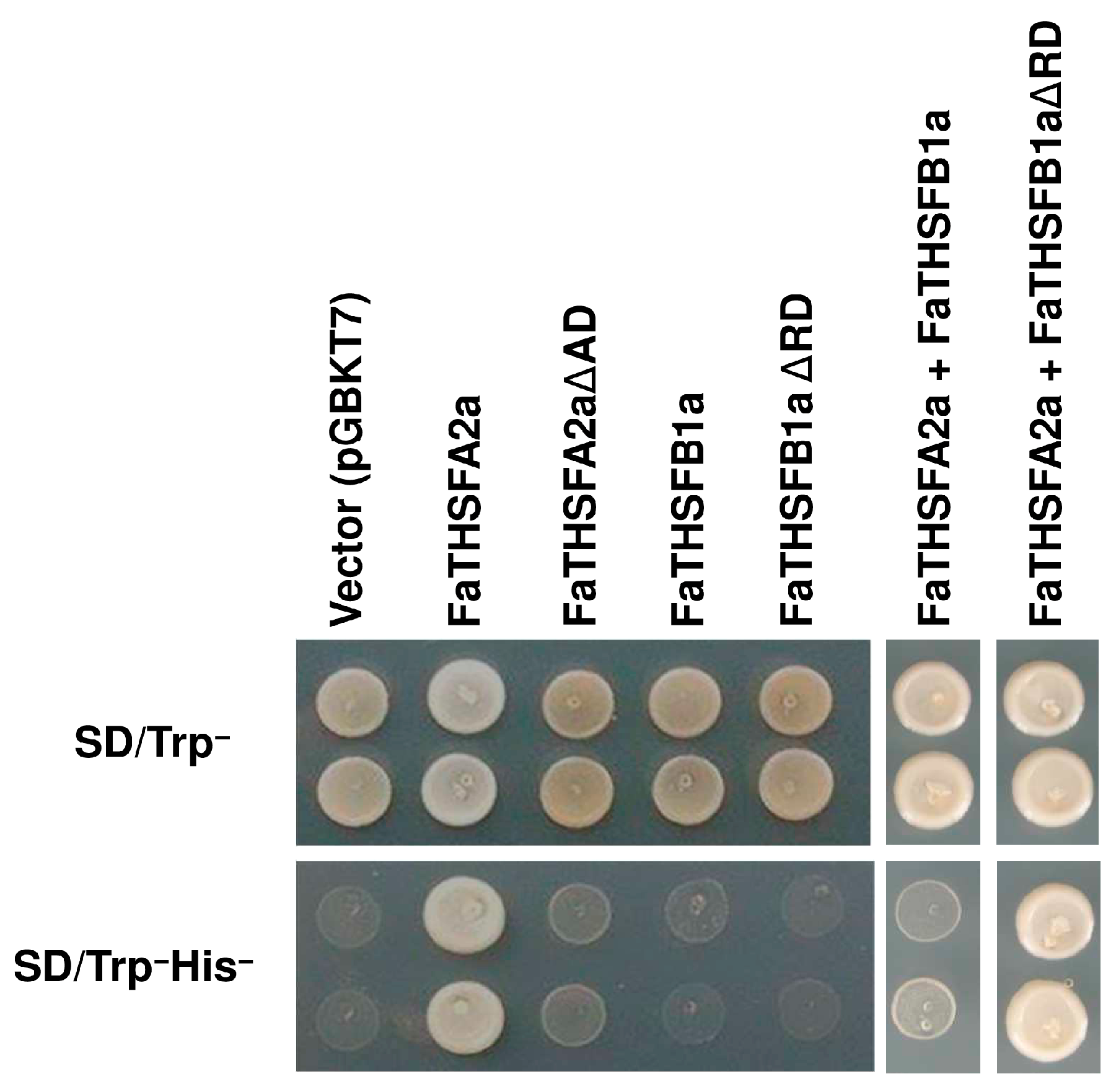
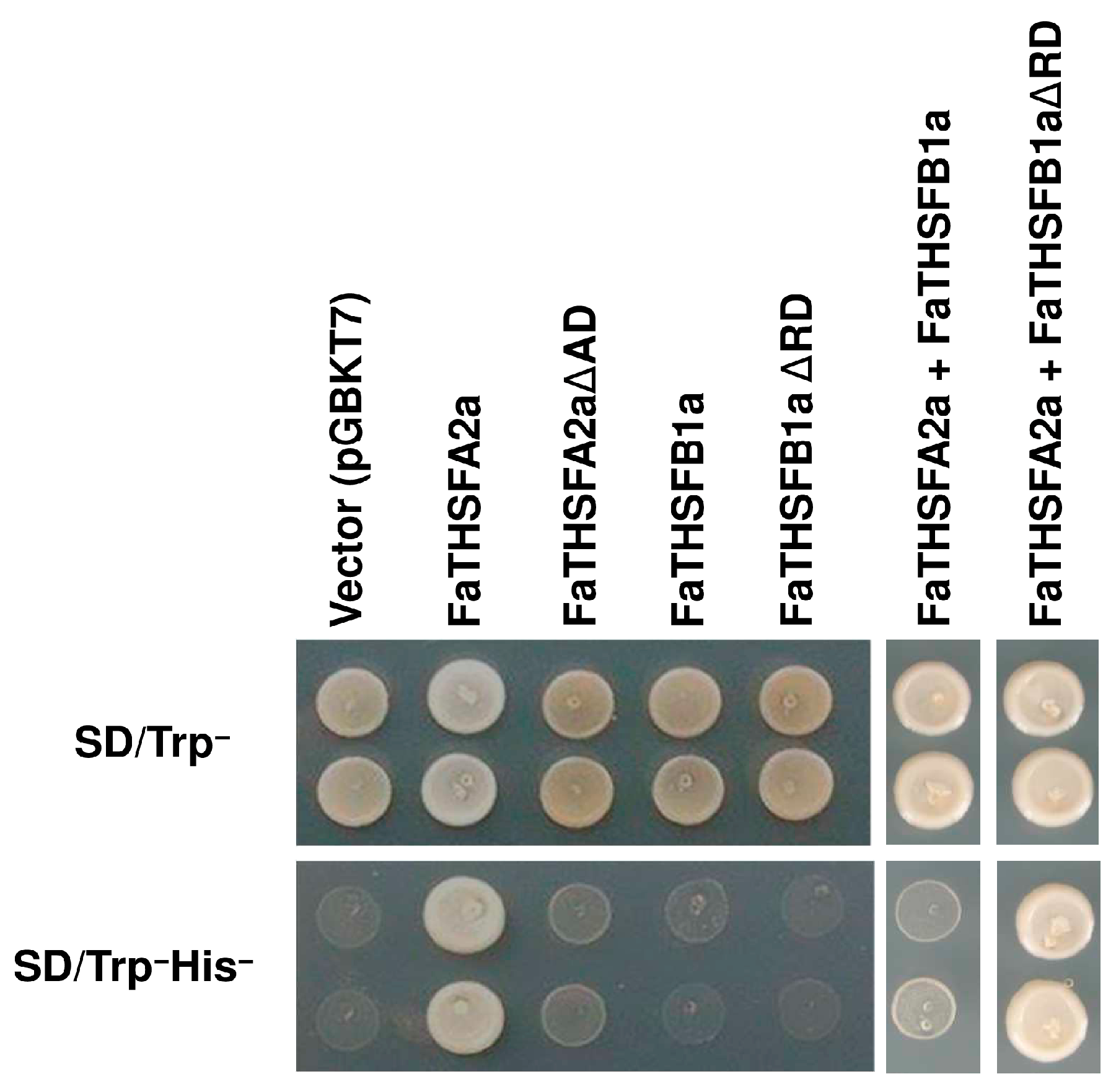
2.6. FaTHSFA2a Exhibited Trans-Activation Function, Whereas FaTHSFB1a Showed Trans-Repression Activity
3. Materials and Methods
3.1. Plant Materials and RNA Extraction
3.2. cDNA Preparation, Sequencing and De Novo Assembly
3.3. Functional Annotation and Classification
3.4. Reverse Transcription Polymerase Chain Reaction (RT-PCR) and Real-Time RT-PCR Analysis
3.5. Construction of 35S::FaTHSFA2a, 35S::FaTHSFA2aΔAD, 35S::FaTHSFB1a and 35S::FaTHSFB1aΔRD Recombinant Plasmids
3.6. Arabidopsis thaliana Transformation and Phenotypic Analysis
3.7. Heat Stress Assays
3.8. Subcellular Localization
3.9. Transcriptional Activation or Repression Activity in Yeast
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Schlenker, W.; Roberts, M.J. Nonlinear temperature effects indicate severe damages to U.S. crop yields under climate change. Proc. Natl. Acad. Sci. USA 2009, 106, 15594–15598. [Google Scholar] [CrossRef] [PubMed]
- Lobell, D.B.; Schlenker, W.; Costa-Roberts, J. Climate trends and global crop production since 1980. Science 2011, 333, 616–620. [Google Scholar] [CrossRef] [PubMed]
- Allakhverdiev, S.I.; Kreslavski, V.D.; Klimov, V.V.; Los, D.A.; Carpentier, R.; Mohanty, P. Heat stress: An overview of molecular responses in photosynthesis. Photosynth. Res. 2008, 98, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.J.; Yang, K.A.; Hong, J.K.; Choi, J.S.; Yun, D.-J.; Hong, J.C.; Chung, W.S.; Lee, S.Y.; Cho, M.J.; Lim, C.O. Gene expression profiles during heat acclimation in Arabidopsis thaliana suspension-culture cells. J. Plant Res. 2006, 119, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Arkindale, J.; Vierling, E. Core genome responses involved in acclimation to high temperature. Plant Physiol. 2008, 146, 748–761. [Google Scholar] [CrossRef] [PubMed]
- Panchuk, I.I.; Volkov, R.A.; Schöffl, F. Heat stress- and heat shock transcription factor-dependent expression and activity of ascorbate peroxidase in Arabidopsis. Plant Physiol. 2002, 129, 838–853. [Google Scholar] [CrossRef] [PubMed]
- Panikulangara, T.J.; Eggers-Schumacher, G.; Wunderlich, M.; Stransky, H.; Schöffl, F. Galactinol synthase1: A novel heat shock factor target gene responsible for heat-induced synthesis of raffinose family oligosaccharides in Arabidopsis. Plant Physiol. 2004, 136, 3148–3158. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, Y.; Maruyama, K.; Qin, F.; Osakabe, Y.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Dual function of an Arabidopsis transcription factor DREB2A in water-stress-responsive and heat-stress-responsive gene expression. Proc. Natl. Acad. Sci. USA 2006, 103, 18822–18827. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, A.; Yabuta, Y.; Shigeoka, S. Galactinol and raffinose constitute a novel function to protect plants from oxidative damage. Plant Physiol. 2008, 147, 1251–1263. [Google Scholar] [CrossRef] [PubMed]
- Bienz, M.; Pelham, H.R. Mechanisms of heat-shock gene activation in higher eukaryotes. Adv. Genet. 1987, 24, 31–72. [Google Scholar] [PubMed]
- Baniwal, S.K.; Bharti, K.; Chan, K.Y.; Fauth, M.; Ganguli, A.; Kotak, S.; Mishra, S.K.; Nover, L.; Port, M.; Scharf, K.D. Heat stress response in plants: A complex game with chaperons and more. J. Biosci. 2004, 29, 471–487. [Google Scholar] [CrossRef] [PubMed]
- Clos, J.; Westwood, J.T.; Becker, P.B.; Wilson, S.; Lambert, K.; Wu, C. Molecular cloning and expression of a hexameric Drosophila heat shock factor subject to negative regulation. Cell 1990, 63, 1085–1097. [Google Scholar] [CrossRef] [Green Version]
- Treuter, E.; Nover, L.; Ohme, K.; Scharf, K.D. Promoter specificity and deletion analysis of three heat stress transcription factors of tomato. Mol. Gen. Genet. 1993, 240, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Nover, L.; Scharf, K.D.; Gagliardi, D.; Vergne, P.; Czarnecka-Verner, E.; Gurley, W.B. The Hsf world: Classification and properties of plant heat stress transcription factors. Cell Stress Chaperones 1996, 1, 215–223. [Google Scholar] [CrossRef]
- Bailey, T.L.; Gribskov, M. Combining evidence using p-values: Application to sequence homology searches. Bioinformatics 1998, 14, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Döring, P.; Treuter, E.; Kistner, C.; Lyck, R.; Chen, A.; Nover, L. The role of AHA motifs in the activator function of tomato heat stress transcription factors HsfA1 and HsfA2. Plant Cell 2000, 12, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Scharf, K. D.; Berberich, T.; Ebersberger, I.; Nover, L. The plant heat stress transcription factor (Hsf) family: Structure, function and evolution. Biochim. Biophys. Acta 2012, 1819, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.J.; Ye, C.J.; Lu, H.Y. Cloning of GmHSFA1 gene and its overexpression leading to enhancement of heat tolerance in transgenic soybean. Acta Genet. Sin. 2006, 28, 1411–1420. [Google Scholar]
- Morimoto, R.I. Regulation of the heat shock transcriptional response: Cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 1998, 12, 3788–3796. [Google Scholar] [CrossRef] [PubMed]
- Nakai, A. New aspects in the vertebrate heat shock factor system: Hsf3 and Hsf4. Cell Stress Chaperones 1999, 4, 86–93. [Google Scholar] [CrossRef]
- Kotak, S.; Port, M.; Ganguli, A.; Bicker, F.; von Koskull-Doring, P. Characterization of C-terminal domains of Arabidopsis heat stress transcription factors (Hsfs) and identification of a new signature combination of plant class A Hsfs with AHA and NES motifs essential for activator function and intracellular localization. Plant J. 2004, 39, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Xing, H.; Wilkerson, D.C.; Mayhew, C.N.; Lubert, E.J.; Skaggs, H.S.; Goodson, M.L.; Hong, Y.; Park-Sarge, O.-K.; Sarge, K.D. Mechanism of hsp70i gene bookmarking. Science 2005, 307, 421–423. [Google Scholar] [CrossRef] [PubMed]
- Nover, L.; Bharti, K.; Doring, P.; Mishra, S.K.; Ganguli, A.; Scharf, K.D. Arabidopsis and the heat stress transcription factor world: How many heat stress transcription factors do we need? Cell Stress Chaperones 2001, 6, 177–189. [Google Scholar] [CrossRef]
- Schultheiss, J.; Kunert, O.; Gase, U.; Scharf, K.D.; Nover, L.; Ruterjans, H. Solution structure of the DNA-binding domain of the tomato heat-stress transcription factor HSF24. Eur. J. Biochem. 1996, 236, 911–921. [Google Scholar] [CrossRef] [PubMed]
- Czarnecka-Verner, E.; Yuan, C.X.; Scharf, K.D.; Englich, G.; Gurley, W.B. Plants contain a novel multi-member class of heat shock factors without transcriptional activator potential. Plant Mol. Biol. 2000, 43, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Ohme-Takagi, M. A novel group of transcriptional repressors in Arabidopsis. Plant Cell Physiol. 2009, 50, 970–975. [Google Scholar] [CrossRef] [PubMed]
- Fragkostefanakis, S.; Röth, S.; Schleiff, E.; Scharf, K.D. Prospects of engineering thermotolerance in crops through modulation of heat stress transcription factor and heat shock protein networks. Plant Cell Environ. 2015, 38, 1881–1895. [Google Scholar] [CrossRef] [PubMed]
- Schramm, F.; Larkindale, J.; Kiehlmann, E.; Ganguli, A.; Englich, G.; Vierling, E.; von Koskull-Döring, P. A cascade of transcription factor DREB2A and heat stress transcription factor HsfA3 regulates the heat stress response of Arabidopsis. Plant J. 2008, 53, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Fujita, Y.; Sayama, H.; Kidokoro, S.; Maruyama, K.; Mizoi, J.; Shinozaki, K.; Yamaguchi-Shinozaki, K. AREB1, AREB2, and ABF3 are master transcription factors that cooperatively regulate ABRE-dependent ABA signaling involved in drought stress tolerance and require ABA for full activation. Plant J. 2010, 61, 672–685. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.M.; Kim, D.W.; Woo, M.S.; Jeong, H.S.; Son, Y.S.; Akhter, S.; Choi, G.J.; Bahk, J.D. Functional characterization of Arabidopsis HsfA6a as a heat-shock transcription factor under high salinity and dehydration conditions. Plant Cell Environ. 2014, 37, 1202–1222. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.K.; Wu, J.; Ji, Q.; Wang, C.; Luo, L.; Yuan, Y.; Wang, Y.; Wang, J. Genome-wide analysis of heat shock transcription factor families in rice and Arabidopsis. J. Genet. Genom. 2008, 35, 105–118. [Google Scholar] [CrossRef]
- Guo, M.; Lu, J.P.; Zhai, Y.F.; Chai, W.G.; Gong, Z.H.; Lu, M.H. Genome-wide analysis, expression profile of heat shock factor gene family (CaHsfs) and characterisation of CaHsfA2 in pepper (Capsicum annuum L.). BMC Plant Biol. 2015, 15, 151–170. [Google Scholar] [CrossRef] [PubMed]
- Qiao, X.; Li, M.; Li, L.; Yin, H.; Wu, J.; Zhang, S. Genome-wide identification and comparative analysis of the heat shock transcription factor family in Chinese white pear (Pyrus bretschneideri) and five other Rosaceae species. BMC Plant Biol. 2015, 15, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Xue, G.P.; Sadat, S.; Drenth, J.; McIntyre, C.L. The heat shock factor family from Triticum aestivum in response to heat and other major abiotic stresses and their role in regulation of heat shock protein genes. J. Exp. Bot. 2014, 65, 539–557. [Google Scholar] [CrossRef] [PubMed]
- Shulaev, V.; Korban, S.S.; Sosinski, B.; Abbott, A.G.; Aldwinckle, H.S.; Folta, K.M.; Iezzoni, A.; Main, D.; Aru’s, P.; Dandekar, A.M.; et al. Multiple models for Rosaceae genomics. Plant Physiol. 2008, 147, 985–1003. [Google Scholar] [CrossRef] [PubMed]
- Shulaev, V.; Sargent, D.J.; Crowhurst, R.N.; Mockler, T.C.; Folkerts, O.; Delcher, A.L.; Jaiswal, P.; Mockaitis, K.; Liston, A.; Mane, S.P.; et al. The genome of woodland strawberry (Fragaria vesca). Nat. Genet. 2011, 43, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Folta, K.M.; Clancy, M.A.; Chamala, S.; Dhingra, A.; Brunings, A.M.; Gomide, L.; Kulathinal, R.J.; Peres, N.A.; Davis, T.M.; Barbazuk, W.B. A Transcript accounting from diverse tissues of a cultivated strawberry. Plant Genome 2010, 3, 90–105. [Google Scholar] [CrossRef]
- Rousseau-Gueutin, M.; Lerceteau-Köhler, E.; Barrot, L.; Sargent, D.J.; Monfort, A.; Simpson, D.; Arús, P.; Guérin, G.; Denoyes-Rothan, B. Comparative genetic mapping between octoploid and diploid Fragaria species reveals a high level of colinearity between their genomes and the essentially disomic behavior of the cultivated octoploid strawberry. Genetics 2008, 179, 2045–2060. [Google Scholar] [CrossRef] [PubMed]
- Folta, K.M.; Davis, T.M. Strawberry genes and genomics. Crit. Rev. Plant Sci. 2006, 25, 399–415. [Google Scholar] [CrossRef]
- Mezzetti, B.; Costantini, E. Strawberry (Fragaria x ananassa). Methods Mol. Biol. 2006, 344, 287–295. [Google Scholar] [PubMed]
- Oosumi, T.; Gruszewski, H.A.; Blischak, L.A.; Baxter, A.J.; Wadl, P.A.; Shuman, J.L.; Veilleux, R.E.; Shulaev, V. High-efficiency transformation of the diploid strawberry (Fragaria vesca) for functional genomics. Planta 2006, 223, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Xie, W.F.; Zhang, L.; Valpuesta, V.; Ye, Z.W.; Gao, Q.H.; Duan, K. Auxin biosynthesis by the YUCCA6 flavin monooxygenase gene in woodland strawberry. J. Integr. Plant Biol. 2014, 56, 350–363. [Google Scholar] [CrossRef] [PubMed]
- Pick, T.; Jaskiewicz, M.; Peterhänsel, C.; Conrath, U. Heat shock factor HsfB1 primes gene transcription and systemic acquired resistance in Arabidopsis. Plant Physiol. 2012, 159, 52–55. [Google Scholar] [CrossRef] [PubMed]
- Chan-Schaminet, K.Y.; Baniwal, S.K.; Bublak, D.; Nover, L.; Scharf, K.D. Specific interaction between tomato HsfA1 and HsfA2 creates hetero-oligomeric superactivator complexes for synergistic activation of heat stress gene expression. J. Biol. Chem. 2009, 284, 20848–20857. [Google Scholar] [CrossRef] [PubMed]
- Schramm, F.; Ganguli, A.; Kiehlmann, E.; Englich, G.; Walch, D.; von Koskull-Döring, P. The heat stress transcription factor HsfA2 serves as a regulatory amplifier of a subset of genes in the heat stress response in Arabidopsis. Plant Mol. Biol. 2006, 60, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Mittal, D.; Chakrabarti, S.; Sarkar, A.; Singh, A.; Grover, A. Heat shock factor gene family in rice: Genomic organization and transcript expression profiling in response to high temperature, low temperature and oxidative stresses. Plant Physiol. Biochem. 2009, 47, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Busch, W.; Birke, H.; Kemmerling, B.; Nürnberger, T.; Schöffl, F. Heat shock factors HsfB1 and HsfB2b are involved in the regulation of Pdf1.2 expression and pathogen resistance in Arabidopsis. Mol. Plant 2009, 2, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Mitsuda, N.; Ohme-Takagi, M. Arabidopsis HsfB1 and HsfB2b act as repressors of the expression of heat-inducible Hsfs but positively regulate the acquired thermotolerance. Plant Physiol. 2011, 157, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Von Koskull-Döring, P.; Scharf, K.D.; Nover, L. The diversity of plant heat stress transcription factors. Trends Plant Sci. 2007, 12, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Han, Y.-T.; Wei, W.; Li, Y.-J.; Zhang, K.; Gao, Y.-R.; Zhao, F.-L.; Feng, J.-Y. Identification, isolation, and expression analysis of heat shock transcription factors in the diploid woodland strawberry Fragaria vesca. Front. Plant Sci. 2015, 6, 736–751. [Google Scholar] [CrossRef] [PubMed]
- Busch, W.; Wunderlich, M.; Schőffl, F. Identification of novel heat shock factor-dependent genes and biochemical pathways in Arabidopsis thaliana. Plant J. 2005, 41, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, A.; Yabuta, Y.; Yoshida, E.; Maruta, T.; Yoshimura, K.; Shigeoka, S. Arabidopsis heat shock transcription factor A2 as a key regulator in response to several types of environmental stress. Plant J. 2006, 48, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Charng, Y.Y.; Liu, H.C.; Liu, N.Y.; Chi, W.T.; Wang, C.N.; Chang, S.H.; Wang, T.T. A heat-inducible transcription factor, HsfA2, is required for extension of acquired thermotolerance in Arabidopsis. Plant Physiol. 2007, 143, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.C.; Liao, H.T.; Charng, Y.Y. The role of class A1 heat shock factors (HSFA1s) in response to heat and other stresses in Arabidopsis. Plant Cell Environ. 2011, 34, 738–751. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa-Yokoi, A.; Nosaka, R.; Hayashi, H.; Tainaka, H.; Maruta, T.; Tamoi, M.; Ikeda, M.; Ohme-Takagi, M.; Yoshimura, K.; Yabuta, Y.; et al. HsfA1d and HsfA1e involved in the transcriptional regulation of HsfA2 function as key regulators for the Hsf signaling network in response to environmental stress. Plant Cell Physiol. 2011, 52, 933–945. [Google Scholar] [CrossRef] [PubMed]
- Czarnecka-Verner, E.; Pan, S.; Salem, T.; Gurley, W.B. Plant class B HSFs inhibit transcription and exhibit affinity for TFIIB and TBP. Plant Mol. Biol. 2004, 56, 57–75. [Google Scholar] [CrossRef] [PubMed]
- Morishita, M.; Mochizuki, T.; Yamakawa, O. Flower induction and selection on earliness of strawberry seedlings by short-day and low night temperature treatment. J. Jpn. Soc. Hortic. Sci. 1993, 61, 857–864. [Google Scholar] [CrossRef]
- Ledesma, N.A.; Nakata, M.; Sugiyama, N. Effect of high temperature stress on the reproductive growth of strawberry cvs. “Nyoho” and “Toyonoka”. Sci. Hortic-Amsterdam 2008, 116, 186–193. [Google Scholar] [CrossRef]
- Chang, S.; Puryear, J.; Cairney, J. A simple and efficient method for isolating RNA from pine trees. Plant Mol. Biol. Rep. 1993, 11, 113–116. [Google Scholar] [CrossRef]
- Li, R.; Zhu, H.; Ruan, J.; Qian, W.; Fang, X.; Shi, Z.; Li, Y.; Li, S.; Shan, G.; Kristiansen, K.; et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010, 20, 265–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iseli, C.; Jongeneel, C.V.; Bucher, P. ESTScan: A program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1999, 138–148. [Google Scholar]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Natale, D.A.; Garkavtsev, I.V.; Tatusova, T.A.; Shankavaram, U.T.; Rao, B.S.; Kiryutin, B.; Galperin, M.Y.; Fedorova, N.D.; Koonin, E.V. The COG database: New developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001, 29, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed]
- Clough, S.J.; Bent, A.F. Floral dip: A simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 1998, 16, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, D.; Yamaguchi, K.; Nishiuchi, T. High-level overexpression of the Arabidopsis HsfA2 gene confers not only increased themotolerance but also salt/osmotic stress tolerance and enhanced callus growth. J. Exp. Bot. 2007, 58, 3373–3383. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.T.; Liao, D.C.; Wu, F.H.; Liu, N.T.; Shen, S.C.; Chou, S.J.; Tung, S.Y.; Yang, C.H.; Chan, M.T.; Lin, C.S. Integration of molecular biology tools for identifying promoters and genes abundantly expressed in flowers of Oncidium Gower Ramsey. BMC Plant Biol. 2011, 11, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Karimi, M.; Inze, D.; Depicker, A. GATEWAY vectors for Agrobacterium-mediated plant transformation. Trends Plant Sci. 2002, 7, 193–195. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Total Clean Reads 1 | Total Clean Nucleotides (nt) 1 | Q20 Percentage 2 | N Percentage 3 | GC Percentage 4 |
|---|---|---|---|---|---|
| OF | 47,551,534 | 4,279,638,060 | 95.98% | 0.00% | 48.16% |
| DU | 49,032,098 | 4,412,888,820 | 96.99% | 0.00% | 47.45% |
| Classification | Sample | Total Number | Total Length (nt) | Mean Length (nt) | N50 1 |
|---|---|---|---|---|---|
| Contig | OF | 117,813 | 41,433,873 | 352 | 740 |
| DU | 122,261 | 43,572,190 | 356 | 805 | |
| Unigene | OF | 65,164 | 45,910,155 | 705 | 1218 |
| DU | 72,914 | 50,183,039 | 688 | 1155 | |
| All | 65,768 | 57,422,693 | 873 | 1387 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, W.-Y.; Lin, L.-F.; Jheng, J.-L.; Wang, C.-C.; Yang, J.-H.; Chou, M.-L. Identification of Heat Shock Transcription Factor Genes Involved in Thermotolerance of Octoploid Cultivated Strawberry. Int. J. Mol. Sci. 2016, 17, 2130. https://doi.org/10.3390/ijms17122130
Liao W-Y, Lin L-F, Jheng J-L, Wang C-C, Yang J-H, Chou M-L. Identification of Heat Shock Transcription Factor Genes Involved in Thermotolerance of Octoploid Cultivated Strawberry. International Journal of Molecular Sciences. 2016; 17(12):2130. https://doi.org/10.3390/ijms17122130
Chicago/Turabian StyleLiao, Wan-Yu, Lee-Fong Lin, Jing-Lian Jheng, Chun-Chung Wang, Jui-Hung Yang, and Ming-Lun Chou. 2016. "Identification of Heat Shock Transcription Factor Genes Involved in Thermotolerance of Octoploid Cultivated Strawberry" International Journal of Molecular Sciences 17, no. 12: 2130. https://doi.org/10.3390/ijms17122130