
Proteomics-Based Analysis of Protein Complexes in Pluripotent Stem Cells and Cancer Biology


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
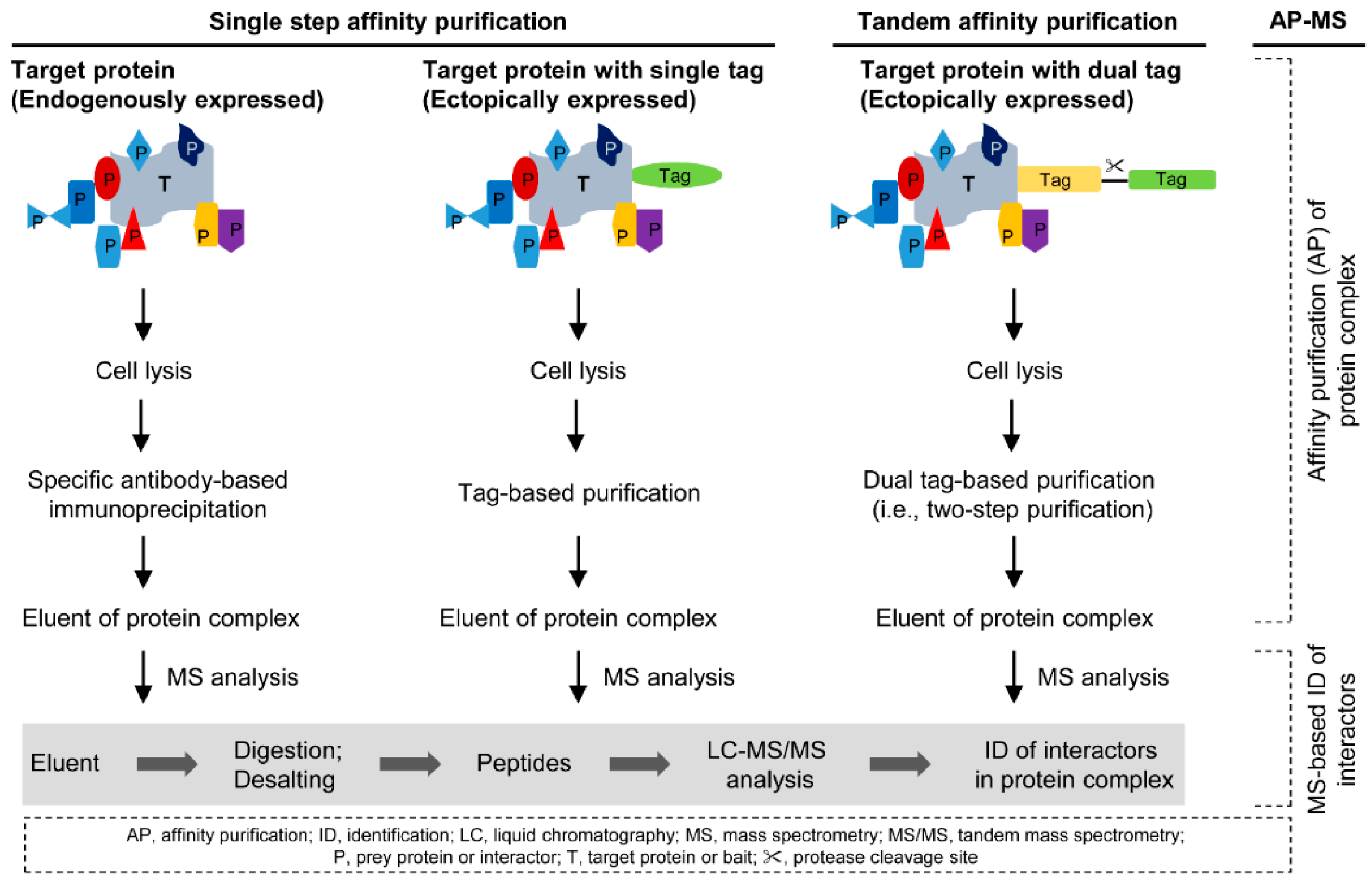
2. Affinity Purification of Protein Complexes
2.1. Single Step Affinity Purification
2.2. Tandem Affinity Purification
3. Basics of Mass Spectrometry (MS) Analysis
3.1. Sample Preparation and MS Analysis
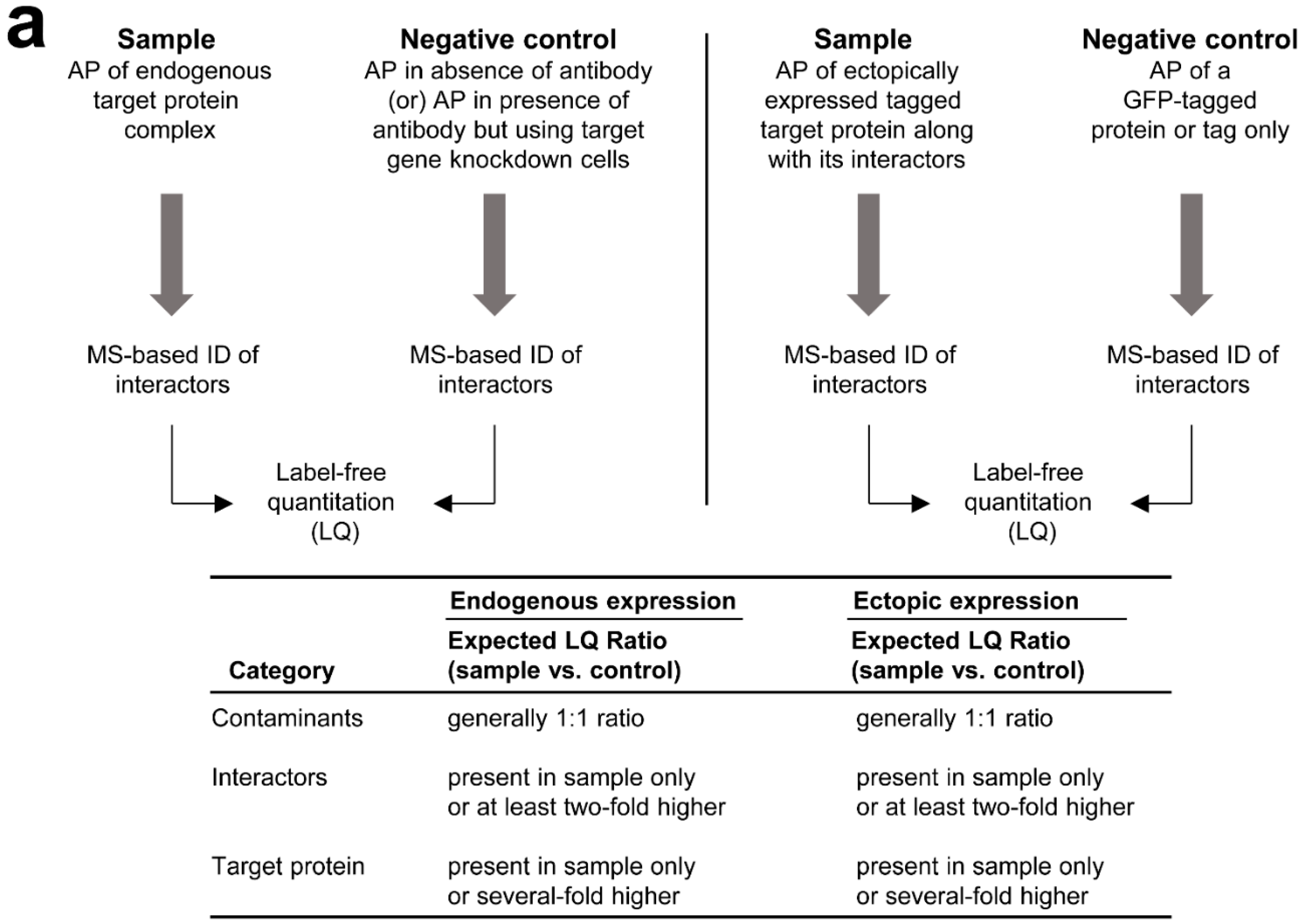
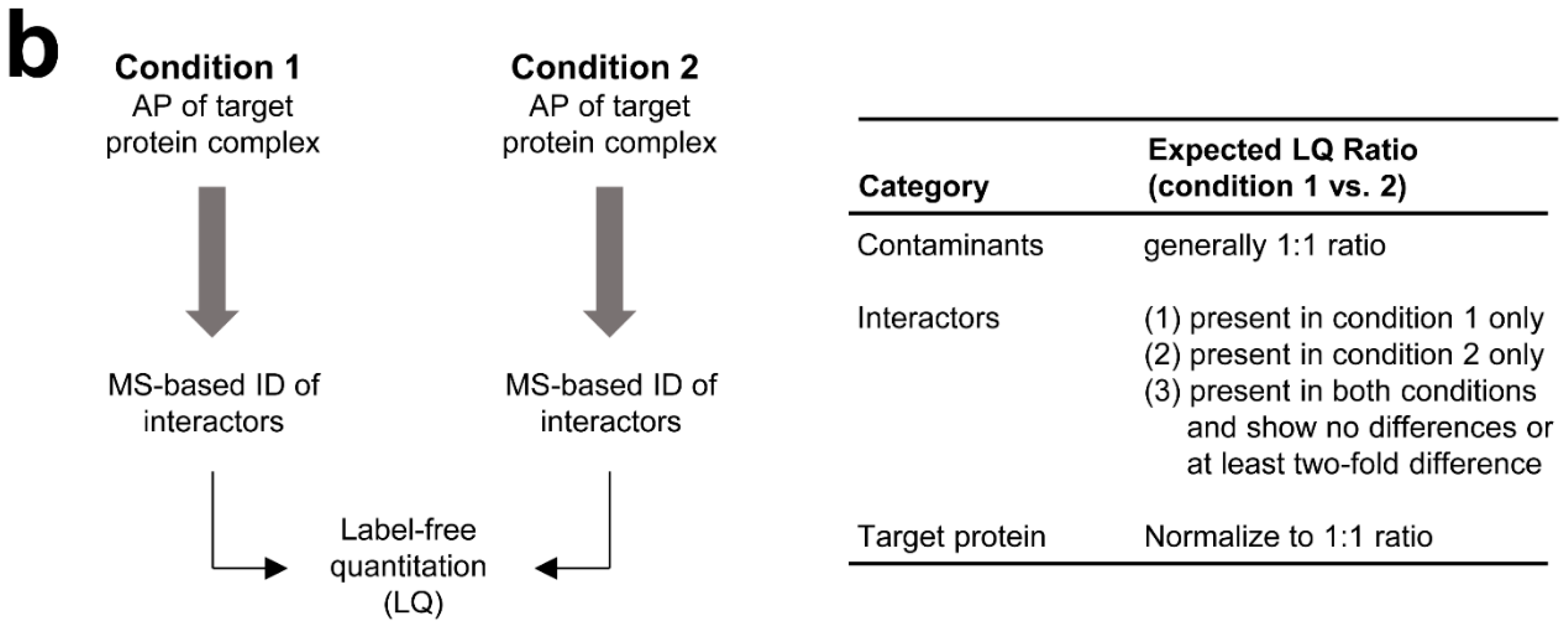
3.2. MS-Based Label-Free Quantitative Analysis
4. Resource Databases of Protein Interactions
5. The Examples of Protein Complexes Identified in Pluripotent Stem Cell Biology
6. The Examples of Protein Complexes Identified in Cancer Cell Biology
7. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ashcroft, A.E. Recent developments in electrospray ionisation mass spectrometry: Noncovalently bound protein complexes. Nat. Prod. Rep. 2005, 22, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Loo, J.A. Studying noncovalent protein complexes by electrospray ionization mass spectrometry. Mass Spectrom. Rev. 1997, 16, 1–23. [Google Scholar] [CrossRef]
- Bauer, A.; Kuster, B. Affinity purification-mass spectrometry. Powerful tools for the characterization of protein complexes. Eur. J. Biochem. 2003, 270, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Gingras, A.C.; Gstaiger, M.; Raught, B.; Aebersold, R. Analysis of protein complexes using mass spectrometry. Nat. Rev. Mol. Cell Biol. 2007, 8, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.A.; Zhang, Z.; Emili, A. Experimental and computational procedures for the assessment of protein complexes on a genome-wide scale. Chem. Rev. 2007, 107, 3585–3600. [Google Scholar] [CrossRef] [PubMed]
- Walzthoeni, T.; Leitner, A.; Stengel, F.; Aebersold, R. Mass spectrometry supported determination of protein complex structure. Curr. Opin. Struct. Biol. 2013, 23, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Fields, S.; Song, O. A novel genetic system to detect protein–protein interactions. Nature 1989, 340, 245–246. [Google Scholar] [CrossRef] [PubMed]
- Stynen, B.; Tournu, H.; Tavernier, J.; van Dijck, P. Diversity in genetic in vivo methods for protein-protein interaction studies: From the yeast two-hybrid system to the mammalian split-luciferase system. Microbiol. Mol. Biol. Rev. 2012, 76, 331–382. [Google Scholar] [CrossRef] [PubMed]
- Hamdi, A.; Colas, P. Yeast two-hybrid methods and their applications in drug discovery. Trends Pharmacol. Sci. 2012, 33, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Batalao, A.; Zhou, H.; Zhu, L. Mammalian two-hybrid system: A complementary approach to the yeast two-hybrid system. Biotechniques 1997, 22, 350–352. [Google Scholar] [PubMed]
- Westermarck, J.; Ivaska, J.; Corthals, G.L. Identification of protein interactions involved in cellular signaling. Mol. Cell. Proteom. 2013, 12, 1752–1763. [Google Scholar] [CrossRef] [PubMed]
- Banks, C.A.; Kong, S.E.; Washburn, M.P. Affinity purification of protein complexes for analysis by multidimensional protein identification technology. Protein Expr. Purif. 2012, 86, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Miteva, Y.V.; Budayeva, H.G.; Cristea, I.M. Proteomics-based methods for discovery, quantification, and validation of protein–protein interactions. Anal. Chem. 2013, 85, 749–768. [Google Scholar] [CrossRef] [PubMed]
- Oeffinger, M. Two steps forward—One step back: Advances in affinity purification mass spectrometry of macromolecular complexes. Proteomics 2012, 12, 1591–1608. [Google Scholar] [CrossRef] [PubMed]
- Figeys, D.; McBroom, L.D.; Moran, M.F. Mass spectrometry for the study of protein-protein interactions. Methods 2001, 24, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Rigaut, G.; Shevchenko, A.; Rutz, B.; Wilm, M.; Mann, M.; Seraphin, B. A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 1999, 17, 1030–1032. [Google Scholar] [CrossRef] [PubMed]
- Kaboord, B.; Perr, M. Isolation of proteins and protein complexes by immunoprecipitation. Methods Mol. Biol. 2008, 424, 349–364. [Google Scholar] [PubMed]
- Keilhauer, E.C.; Hein, M.Y.; Mann, M. Accurate protein complex retrieval by affinity enrichment mass spectrometry (AE-MS) rather than affinity purification mass spectrometry (AP-MS). Mol. Cell. Proteom. 2015, 14, 120–135. [Google Scholar] [CrossRef] [PubMed]
- Rees, J.S.; Lilley, K.S.; Jackson, A.P. Silac-ipac: A quantitative method for distinguishing genuine from non-specific components of protein complexes by parallel affinity capture. J. Proteom. 2015, 115, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Paul, F.E.; Hosp, F.; Selbach, M. Analyzing protein-protein interactions by quantitative mass spectrometry. Methods 2011, 54, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Li, G.; Liang, S. Several affinity tags commonly used in chromatographic purification. J. Anal. Methods Chem. 2013, 2013, 581093. [Google Scholar] [CrossRef] [PubMed]
- Hofener, M.; Heinzlmeir, S.; Kuster, B.; Sewald, N. Probing SH2-domains using inhibitor affinity purification (IAP). Proteome Sci. 2014, 12, 41. [Google Scholar] [CrossRef] [PubMed]
- Rodina, A.; Taldone, T.; Kang, Y.; Patel, P.D.; Koren, J., 3rd; Yan, P.; DaGama Gomes, E.M.; Yang, C.; Patel, M.R.; Shrestha, L.; et al. Affinity purification probes of potential use to investigate the endogenous HSP70 interactome in cancer. ACS Chem. Biol. 2014, 9, 1698–1705. [Google Scholar] [CrossRef] [PubMed]
- Burckstummer, T.; Bennett, K.L.; Preradovic, A.; Schutze, G.; Hantschel, O.; Superti-Furga, G.; Bauch, A. An efficient tandem affinity purification procedure for interaction proteomics in mammalian cells. Nat. Methods 2006, 3, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Li, Y. The tandem affinity purification technology: An overview. Biotechnol. Lett. 2011, 33, 1487–1499. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.O.; Choudhary, J.S. Mapping multiprotein complexes by affinity purification and mass spectrometry. Curr. Opin. Biotechnol. 2008, 19, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Puig, O.; Caspary, F.; Rigaut, G.; Rutz, B.; Bouveret, E.; Bragado-Nilsson, E.; Wilm, M.; Seraphin, B. The tandem affinity purification (TAP) method: A general procedure of protein complex purification. Methods 2001, 24, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Puig, O.; Rutz, B.; Luukkonen, B.G.; Kandels-Lewis, S.; Bragado-Nilsson, E.; Seraphin, B. New constructs and strategies for efficient PCR-based gene manipulations in yeast. Yeast 1998, 14, 1139–1146. [Google Scholar] [CrossRef]
- Forler, D.; Kocher, T.; Rode, M.; Gentzel, M.; Izaurralde, E.; Wilm, M. An efficient protein complex purification method for functional proteomics in higher eukaryotes. Nat. Biotechnol. 2003, 21, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Poser, I.; Sarov, M.; Hutchins, J.R.; Heriche, J.K.; Toyoda, Y.; Pozniakovsky, A.; Weigl, D.; Nitzsche, A.; Hegemann, B.; Bird, A.W.; et al. BAC transgeneomics: A high-throughput method for exploration of protein function in mammals. Nat. Methods 2008, 5, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Hubner, N.C.; Bird, A.W.; Cox, J.; Splettstoesser, B.; Bandilla, P.; Poser, I.; Hyman, A.; Mann, M. Quantitative proteomics combined with BAC transgeneomics reveals in vivo protein interactions. J. Cell Biol. 2010, 189, 739–754. [Google Scholar] [CrossRef] [PubMed]
- Haura, E.B.; Muller, A.; Breitwieser, F.P.; Li, J.; Grebien, F.; Colinge, J.; Bennett, K.L. Using itraq combined with tandem affinity purification to enhance low-abundance proteins associated with somatically mutated EGFR core complexes in lung cancer. J. Proteome Res. 2011, 10, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Lavallee-Adam, M.; Rousseau, J.; Domecq, C.; Bouchard, A.; Forget, D.; Faubert, D.; Blanchette, M.; Coulombe, B. Discovery of cell compartment specific protein–protein interactions using affinity purification combined with tandem mass spectrometry. J. Proteome Res. 2013, 12, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Zhu, H. Tube-gel digestion: A novel proteomic approach for high throughput analysis of membrane proteins. Mol. Cell. Proteom. 2005, 4, 1948–1958. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, A.; Tomas, H.; Havlis, J.; Olsen, J.V.; Mann, M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2006, 1, 2856–2860. [Google Scholar] [CrossRef] [PubMed]
- Sudhir, P.R.; Chen, C.H.; Pavana Kumari, M.; Wang, M.J.; Tsou, C.C.; Sung, T.Y.; Chen, J.Y.; Chen, C.H. Label-free quantitative proteomics and N-glycoproteomics analysis of KRAS-activated human bronchial epithelial cells. Mol. Cell. Proteom. 2012, 11, 901–915. [Google Scholar] [CrossRef] [PubMed]
- Sudhir, P.R.; Hsu, C.L.; Wang, M.J.; Wang, Y.T.; Chen, Y.J.; Sung, T.Y.; Hsu, W.L.; Yang, U.C.; Chen, J.Y. Phosphoproteomics identifies oncogenic RAS signaling targets and their involvement in lung adenocarcinomas. PLoS ONE 2011, 6, e20199. [Google Scholar] [CrossRef] [PubMed]
- Sudhir, P.R.; Kumari, M.P.; Hsu, W.T.; Massiot, J.; Chen, C.H.; Kuo, H.C.; Chen, C.H. Quantitative proteomics of protein complexes and their implications for cell reprograming and pluripotency. J. Proteome Res. 2013, 12, 5878–5890. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Glatter, T.; Ludwig, C.; Ahrne, E.; Aebersold, R.; Heck, A.J.; Schmidt, A. Large-scale quantitative assessment of different in-solution protein digestion protocols reveals superior cleavage efficiency of tandem Lys-C/trypsin proteolysis over trypsin digestion. J. Proteome Res. 2012, 11, 5145–5156. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Ostasiewicz, P.; Dus, K.; Zielinska, D.F.; Gnad, F.; Mann, M. Extensive quantitative remodeling of the proteome between normal colon tissue and adenocarcinoma. Mol. Syst. Biol. 2012, 8, 611. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H. Review of a current role of mass spectrometry for proteome research. Anal. Chim. Acta 2008, 624, 16–36. [Google Scholar] [CrossRef] [PubMed]
- Steen, H.; Mann, M. The ABC’s (and XYZ’s) of peptide sequencing. Nat. Rev. Mol. Cell Biol. 2004, 5, 699–711. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Mazumdar, S. Electrospray ionization mass spectrometry: A technique to access the information beyond the molecular weight of the analyte. Int. J. Anal. Chem. 2012, 2012, 282574. [Google Scholar] [CrossRef] [PubMed]
- Gingras, A.C.; Aebersold, R.; Raught, B. Advances in protein complex analysis using mass spectrometry. J. Physiol. 2005, 563, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Smith, J.W.; Huang, C.M. Mass spectrometry-based label-free quantitative proteomics. J. Biomed. Biotechnol. 2010, 2010, 840518. [Google Scholar] [CrossRef] [PubMed]
- Selbach, M.; Mann, M. Protein interaction screening by quantitative immunoprecipitation combined with knockdown (quick). Nat. Methods 2006, 3, 981–983. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.P.; Ivosev, G.; Couzens, A.L.; Larsen, B.; Taipale, M.; Lin, Z.Y.; Zhong, Q.; Lindquist, S.; Vidal, M.; Aebersold, R.; et al. Mapping differential interactomes by affinity purification coupled with data-independent mass spectrometry acquisition. Nat. Methods 2013, 10, 1239–1245. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, R.M.; Bejide, M.; Trinkle-Mulcahy, L.; Cote, J. Identification of the PRMT1v1 and PRMT1v2 specific interactomes by quantitative mass spectrometry in breast cancer cells. Proteomics 2015, 15, 2187–2197. [Google Scholar] [CrossRef] [PubMed]
- Gunaratne, J.; Goh, M.X.; Swa, H.L.; Lee, F.Y.; Sanford, E.; Wong, L.M.; Hogue, K.A.; Blackstock, W.P.; Okumura, K. Protein interactions of phosphatase and tensin homologue (PTEN) and its cancer-associated G20E mutant compared by using stable isotope labeling by amino acids in cell culture-based parallel affinity purification. J. Biol. Chem. 2011, 286, 18093–18103. [Google Scholar] [CrossRef] [PubMed]
- Collins, B.C.; Gillet, L.C.; Rosenberger, G.; Rost, H.L.; Vichalkovski, A.; Gstaiger, M.; Aebersold, R. Quantifying protein interaction dynamics by swath mass spectrometry: Application to the 14-3-3 system. Nat. Methods 2013, 10, 1246–1253. [Google Scholar] [CrossRef] [PubMed]
- Fabre, B.; Lambour, T.; Garrigues, L.; Ducoux-Petit, M.; Amalric, F.; Monsarrat, B.; Burlet-Schiltz, O.; Bousquet-Dubouch, M.P. Label-free quantitative proteomics reveals the dynamics of proteasome complexes composition and stoichiometry in a wide range of human cell lines. J. Proteome Res. 2014, 13, 3027–3037. [Google Scholar] [CrossRef] [PubMed]
- Fabre, B.; Lambour, T.; Garrigues, L.; Amalric, F.; Vigneron, N.; Menneteau, T.; Stella, A.; Monsarrat, B.; van den Eynde, B.; Burlet-Schiltz, O.; et al. Deciphering preferential interactions within supramolecular protein complexes: The proteasome case. Mol. Syst. Biol. 2015, 11, 771. [Google Scholar] [CrossRef] [PubMed]
- Fabre, B.; Lambour, T.; Bouyssié, D.; Menneteau, T.; Monsarrat, B.; Burlet-Schiltz, O.; Bousquet-Dubouch, M.P. Comparison of label-free quantification methods for the determination of protein complexes subunits stoichiometry. EuPA Open Proteom. 2014, 4, 82–86. [Google Scholar] [CrossRef]
- Meyer, K.; Selbach, M. Quantitative affinity purification mass spectrometry: A versatile technology to study protein-protein interactions. Front. Genet. 2015, 6, 237. [Google Scholar] [CrossRef] [PubMed]
- Ruepp, A.; Brauner, B.; Dunger-Kaltenbach, I.; Frishman, G.; Montrone, C.; Stransky, M.; Waegele, B.; Schmidt, T.; Doudieu, O.N.; Stumpflen, V.; et al. Corum: The comprehensive resource of mammalian protein complexes. Nucleic Acids. Res. 2008, 36, D646–D650. [Google Scholar] [CrossRef] [PubMed]
- Ruepp, A.; Waegele, B.; Lechner, M.; Brauner, B.; Dunger-Kaltenbach, I.; Fobo, G.; Frishman, G.; Montrone, C.; Mewes, H.W. Corum: The comprehensive resource of mammalian protein complexes—2009. Nucleic Acids Res. 2010, 38, D497–D501. [Google Scholar] [CrossRef] [PubMed]
- Bader, G.D.; Betel, D.; Hogue, C.W. Bind: The biomolecular interaction network database. Nucleic Acids Res. 2003, 31, 248–250. [Google Scholar] [CrossRef] [PubMed]
- Salwinski, L.; Miller, C.S.; Smith, A.J.; Pettit, F.K.; Bowie, J.U.; Eisenberg, D. The database of interacting proteins: 2004 update. Nucleic Acids Res. 2004, 32, D449–D451. [Google Scholar] [CrossRef] [PubMed]
- Keshava Prasad, T.S.; Goel, R.; Kandasamy, K.; Keerthikumar, S.; Kumar, S.; Mathivanan, S.; Telikicherla, D.; Raju, R.; Shafreen, B.; Venugopal, A.; et al. Human protein reference database—2009 update. Nucleic Acids Res. 2009, 37, D767–D772. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Park, B.; Kim, H.; Hong, J.; Park, J. Hpid: The human protein interaction database. Bioinformatics 2004, 20, 2466–2470. [Google Scholar] [CrossRef] [PubMed]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; del-Toro, N.; et al. The mintact project—Intact as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014, 42, D358–D363. [Google Scholar] [CrossRef] [PubMed]
- Chatr-aryamontri, A.; Ceol, A.; Palazzi, L.M.; Nardelli, G.; Schneider, M.V.; Castagnoli, L.; Cesareni, G. Mint: The molecular interaction database. Nucleic Acids Res. 2007, 35, D572–D574. [Google Scholar] [CrossRef] [PubMed]
- Snel, B.; Lehmann, G.; Bork, P.; Huynen, M.A. String: A web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res. 2000, 28, 3442–3444. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. String v10: Protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Lage, K.; Karlberg, E.O.; Storling, Z.M.; Olason, P.I.; Pedersen, A.G.; Rigina, O.; Hinsby, A.M.; Tumer, Z.; Pociot, F.; Tommerup, N.; et al. A human phenome-interactome network of protein complexes implicated in genetic disorders. Nat. Biotechnol. 2007, 25, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Stingele, S.; Stoehr, G.; Peplowska, K.; Cox, J.; Mann, M.; Storchova, Z. Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells. Mol. Syst. Biol. 2012, 8, 608. [Google Scholar] [CrossRef] [PubMed]
- Dejosez, M.; Zwaka, T.P. Pluripotency and nuclear reprogramming. Annu. Rev. Biochem. 2012, 81, 737–765. [Google Scholar] [CrossRef] [PubMed]
- Na, J.; Plews, J.; Li, J.; Wongtrakoongate, P.; Tuuri, T.; Feki, A.; Andrews, P.W.; Unger, C. Molecular mechanisms of pluripotency and reprogramming. Stem Cell Res. Ther. 2010, 1, 33. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Amato, P.; Sparman, M.; Gutierrez, N.M.; Tippner-Hedges, R.; Ma, H.; Kang, E.; Fulati, A.; Lee, H.S.; Sritanaudomchai, H.; et al. Human embryonic stem cells derived by somatic cell nuclear transfer. Cell 2013, 153, 1228–1238. [Google Scholar] [CrossRef] [PubMed]
- Stadtfeld, M.; Hochedlinger, K. Induced pluripotency: History, mechanisms, and applications. Genes Dev. 2010, 24, 2239–2263. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wang, J. The extended pluripotency protein interactome and its links to reprogramming. Curr. Opin. Genet. Dev. 2014, 28, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Pardo, M.; Lang, B.; Yu, L.; Prosser, H.; Bradley, A.; Babu, M.M.; Choudhary, J. An expanded oct4 interaction network: Implications for stem cell biology, development, and disease. Cell Stem Cell 2010, 6, 382–395. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, D.L.; Snoek, T.; Mullin, N.P.; Yates, A.; Bezstarosti, K.; Demmers, J.; Chambers, I.; Poot, R.A. An Oct4-centered protein interaction network in embryonic stem cells. Cell Stem Cell 2010, 6, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Xu, H.; Faiola, F.; Ma’ayan, A.; Wang, J. Oct4 links multiple epigenetic pathways to the pluripotency network. Cell Res. 2012, 22, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Cox, J.L.; Gilmore, J.M.; Ormsbee, B.D.; Mallanna, S.K.; Washburn, M.P.; Rizzino, A. Determination of protein interactome of transcription factor Sox2 in embryonic stem cells engineered for inducible expression of four reprogramming factors. J. Biol. Chem. 2012, 287, 11384–11397. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Rao, S.; Chu, J.; Shen, X.; Levasseur, D.N.; Theunissen, T.W.; Orkin, S.H. A protein interaction network for pluripotency of embryonic stem cells. Nature 2006, 444, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Wan, M.; Zhang, Y.; Gu, P.; Xin, H.; Jung, S.Y.; Qin, J.; Wong, J.; Cooney, A.J.; Liu, D.; et al. Nanog and oct4 associate with unique transcriptional repression complexes in embryonic stem cells. Nat. Cell Biol. 2008, 10, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Woo, A.J.; Chu, J.; Snow, J.W.; Fujiwara, Y.; Kim, C.G.; Cantor, A.B.; Orkin, S.H. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell 2010, 143, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Emani, M.R.; Narva, E.; Stubb, A.; Chakroborty, D.; Viitala, M.; Rokka, A.; Rahkonen, N.; Moulder, R.; Denessiouk, K.; Trokovic, R.; et al. The L1TD1 protein interactome reveals the importance of post-transcriptional regulation in human pluripotency. Stem Cell Rep. 2015, 4, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.; Ronan, J.L.; Wu, J.; Staahl, B.T.; Chen, L.; Kuo, A.; Lessard, J.; Nesvizhskii, A.I.; Ranish, J.; Crabtree, G.R. An embryonic stem cell chromatin remodeling complex, esBAF, is essential for embryonic stem cell self-renewal and pluripotency. Proc. Natl. Acad. Sci. USA 2009, 106, 5181–5186. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.; Lufkin, T. Building a pluripotency protein interaction network for embryonic stem cells. In Embryonic Stem Cells—Basic Biology to Bioengineering; Kallos, M., Ed.; InTech: West Palm Beach, FL, USA, 2011; pp. 305–320. [Google Scholar]
- Gagliardi, A.; Mullin, N.P.; Ying Tan, Z.; Colby, D.; Kousa, A.I.; Halbritter, F.; Weiss, J.T.; Felker, A.; Bezstarosti, K.; Favaro, R.; et al. A direct physical interaction between nanog and Sox2 regulates embryonic stem cell self-renewal. EMBO J. 2013, 32, 2231–2247. [Google Scholar] [CrossRef] [PubMed]
- Foerster, S.; Kacprowski, T.; Dhople, V.M.; Hammer, E.; Herzog, S.; Saafan, H.; Bien-Moller, S.; Albrecht, M.; Volker, U.; Ritter, C.A. Characterization of the EGFR interactome reveals associated protein complex networks and intracellular receptor dynamics. Proteomics 2013, 13, 3131–3144. [Google Scholar] [CrossRef] [PubMed]
- Saldanha, R.G.; Molloy, M.P.; Bdeir, K.; Cines, D.B.; Song, X.; Uitto, P.M.; Weinreb, P.H.; Violette, S.M.; Baker, M.S. Proteomic identification of lynchpin urokinase plasminogen activator receptor protein interactions associated with epithelial cancer malignancy. J. Proteome Res. 2007, 6, 1016–1028. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Shang, Y. Estrogen and cancer. Annu. Rev. Physiol. 2013, 75, 225–240. [Google Scholar] [CrossRef] [PubMed]
- Tarallo, R.; Bamundo, A.; Nassa, G.; Nola, E.; Paris, O.; Ambrosino, C.; Facchiano, A.; Baumann, M.; Nyman, T.A.; Weisz, A. Identification of proteins associated with ligand-activated estrogen receptor alpha in human breast cancer cell nuclei by tandem affinity purification and nano LC-MS/MS. Proteomics 2011, 11, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Nassa, G.; Tarallo, R.; Ambrosino, C.; Bamundo, A.; Ferraro, L.; Paris, O.; Ravo, M.; Guzzi, P.H.; Cannataro, M.; Baumann, M.; et al. A large set of estrogen receptor β-interacting proteins identified by tandem affinity purification in hormone-responsive human breast cancer cell nuclei. Proteomics 2011, 11, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Stellato, C.; Nassa, G.; Tarallo, R.; Giurato, G.; Ravo, M.; Rizzo, F.; Marchese, G.; Alexandrova, E.; Cordella, A.; Baumann, M.; et al. Identification of cytoplasmic proteins interacting with unliganded estrogen receptor alpha and β in human breast cancer cells. Proteomics 2015, 15, 1801–1807. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Bedford, M.T. Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 2013, 13, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Le Naour, F.; Andre, M.; Greco, C.; Billard, M.; Sordat, B.; Emile, J.F.; Lanza, F.; Boucheix, C.; Rubinstein, E. Profiling of the tetraspanin web of human colon cancer cells. Mol. Cell. Proteom. 2006, 5, 845–857. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Hao, Y.; Du, Z.; Wang, Z.; Ewing, R.M. Identifying novel protein complexes in cancer cells using epitope-tagging of endogenous human genes and affinity-purification mass spectrometry. J. Proteome Res. 2012, 11, 5630–5641. [Google Scholar] [CrossRef] [PubMed]
- Koch, H.B.; Zhang, R.; Verdoodt, B.; Bailey, A.; Zhang, C.D.; Yates, J.R., 3rd; Menssen, A.; Hermeking, H. Large-scale identification of c-MYC-associated proteins using a combined TAP/MudPIT approach. Cell Cycle 2007, 6, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Weina, K.; Utikal, J. Sox2 and cancer: Current research and its implications in the clinic. Clin. Transl. Med. 2014, 3, 19. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.L.; Wilder, P.J.; Gilmore, J.M.; Wuebben, E.L.; Washburn, M.P.; Rizzino, A. The Sox2-interactome in brain cancer cells identifies the requirement of MSI2 and USP9X for the growth of brain tumor cells. PLoS ONE 2013, 8, e62857. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, F.; Guo, L.; Wang, Y.; Zhang, P.; Wang, R.; Zhang, N.; Chen, R. Interactome analysis reveals that C1QBP (complement component 1, q subcomponent binding protein) is associated with cancer cell chemotaxis and metastasis. Mol. Cell. Proteom. 2013, 12, 3199–3209. [Google Scholar] [CrossRef] [PubMed]
- Kaake, R.M.; Wang, X.; Huang, L. Profiling of protein interaction networks of protein complexes using affinity purification and quantitative mass spectrometry. Mol. Cell. Proteom. 2010, 9, 1650–1665. [Google Scholar] [CrossRef] [PubMed]
- Dunham, I.; Kundaje, A.; Aldred, S.F.; Collins, P.J.; Davis, C.A.; Doyle, F.; Epstein, C.B.; Frietze, S.; Harrow, J.; Kaul, R. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]



© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sudhir, P.-R.; Chen, C.-H. Proteomics-Based Analysis of Protein Complexes in Pluripotent Stem Cells and Cancer Biology. Int. J. Mol. Sci. 2016, 17, 432. https://doi.org/10.3390/ijms17030432
Sudhir P-R, Chen C-H. Proteomics-Based Analysis of Protein Complexes in Pluripotent Stem Cells and Cancer Biology. International Journal of Molecular Sciences. 2016; 17(3):432. https://doi.org/10.3390/ijms17030432
Chicago/Turabian StyleSudhir, Putty-Reddy, and Chung-Hsuan Chen. 2016. "Proteomics-Based Analysis of Protein Complexes in Pluripotent Stem Cells and Cancer Biology" International Journal of Molecular Sciences 17, no. 3: 432. https://doi.org/10.3390/ijms17030432