
Tamoxifen Resistance: Emerging Molecular Targets

 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Estrogen Receptors (ERα and ERβ)
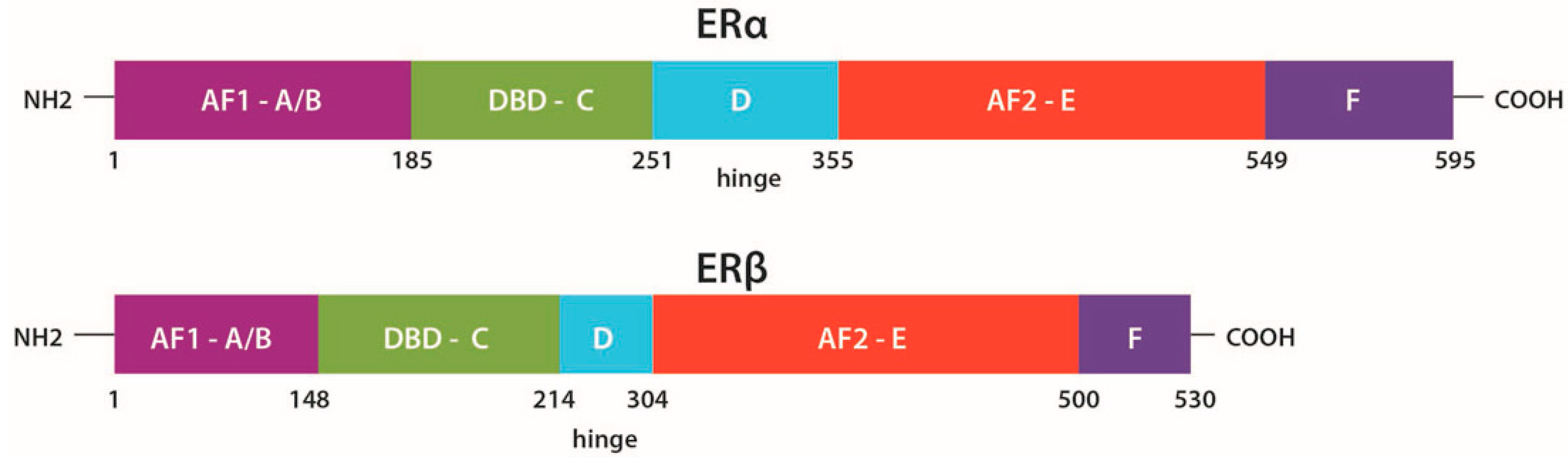
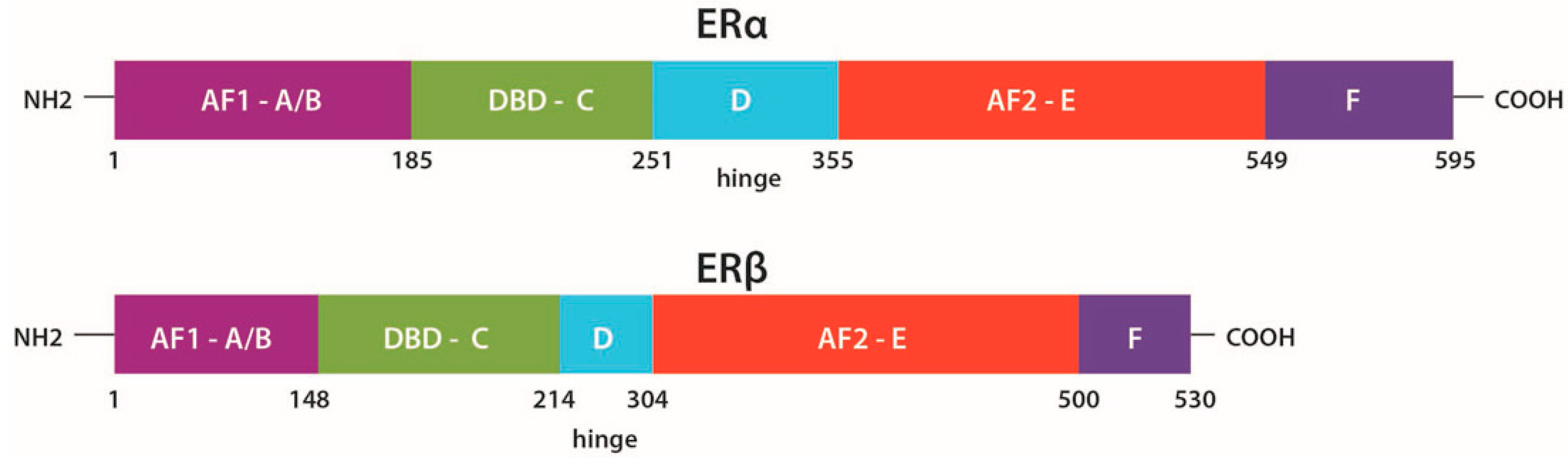
2.1. Structure
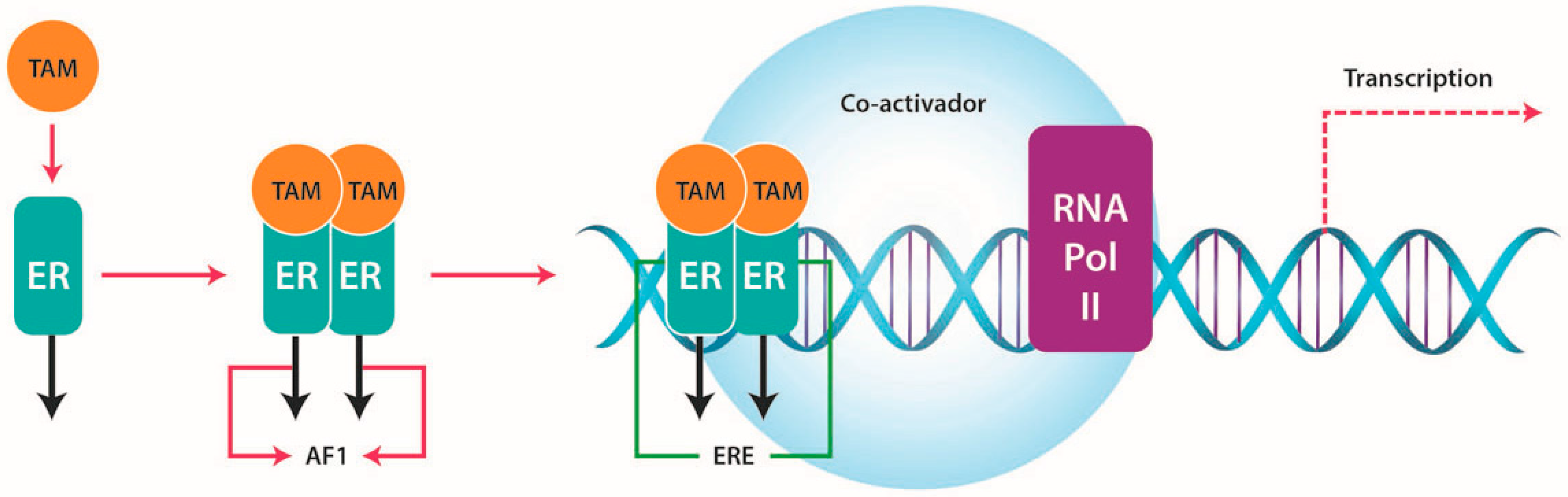
2.2. Function
3. Tamoxifen (TAM)
3.1. Function
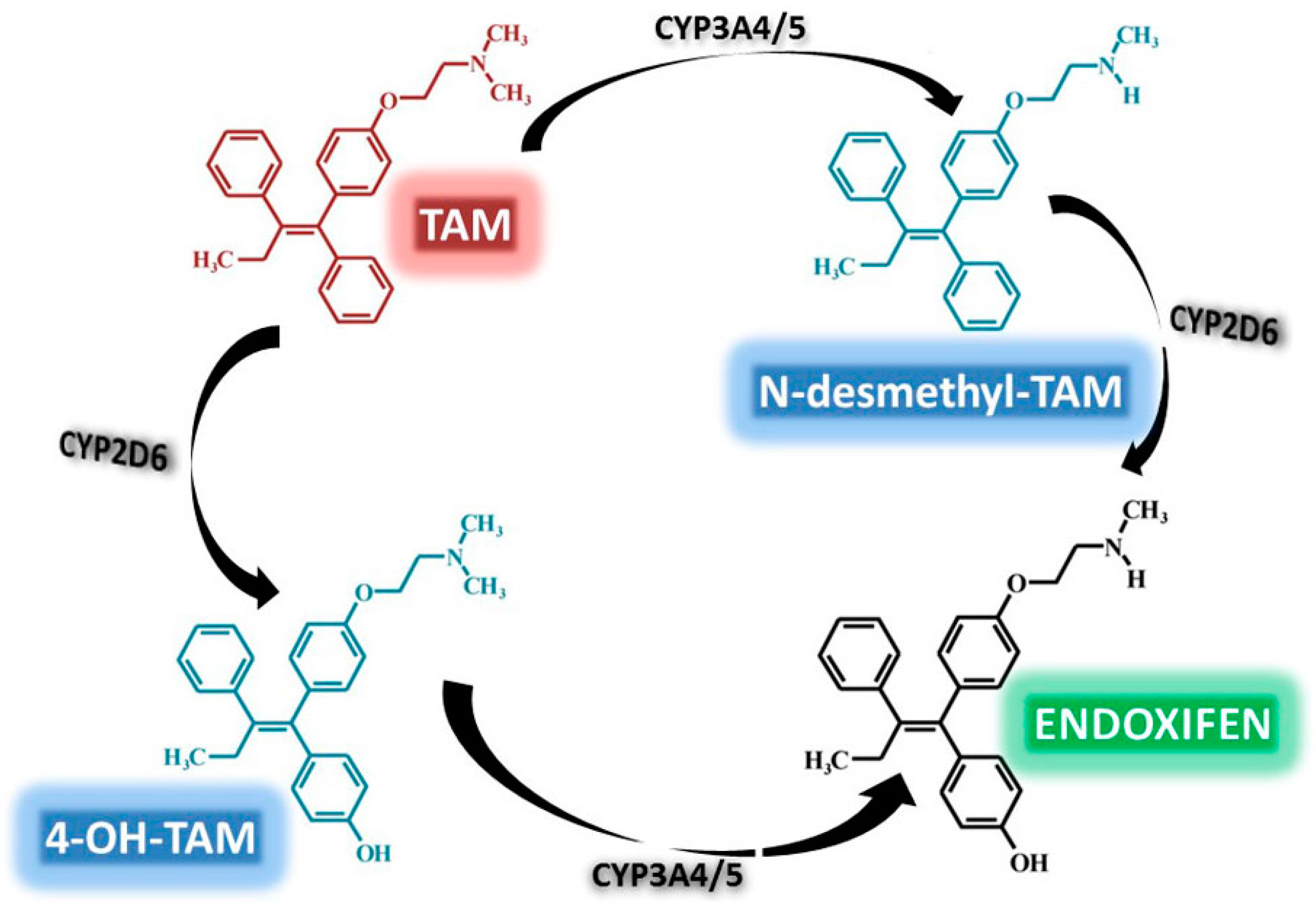
3.2. TAM Metabolism
4. TAM Resistance
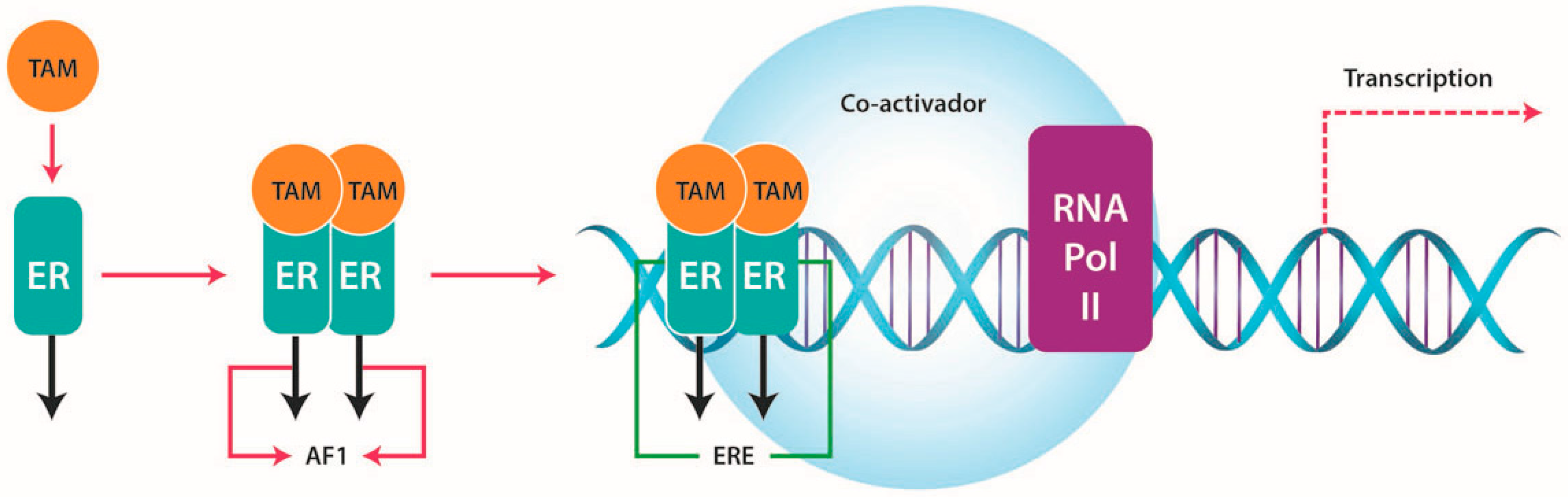
4.1. ERs and TAM Resistance
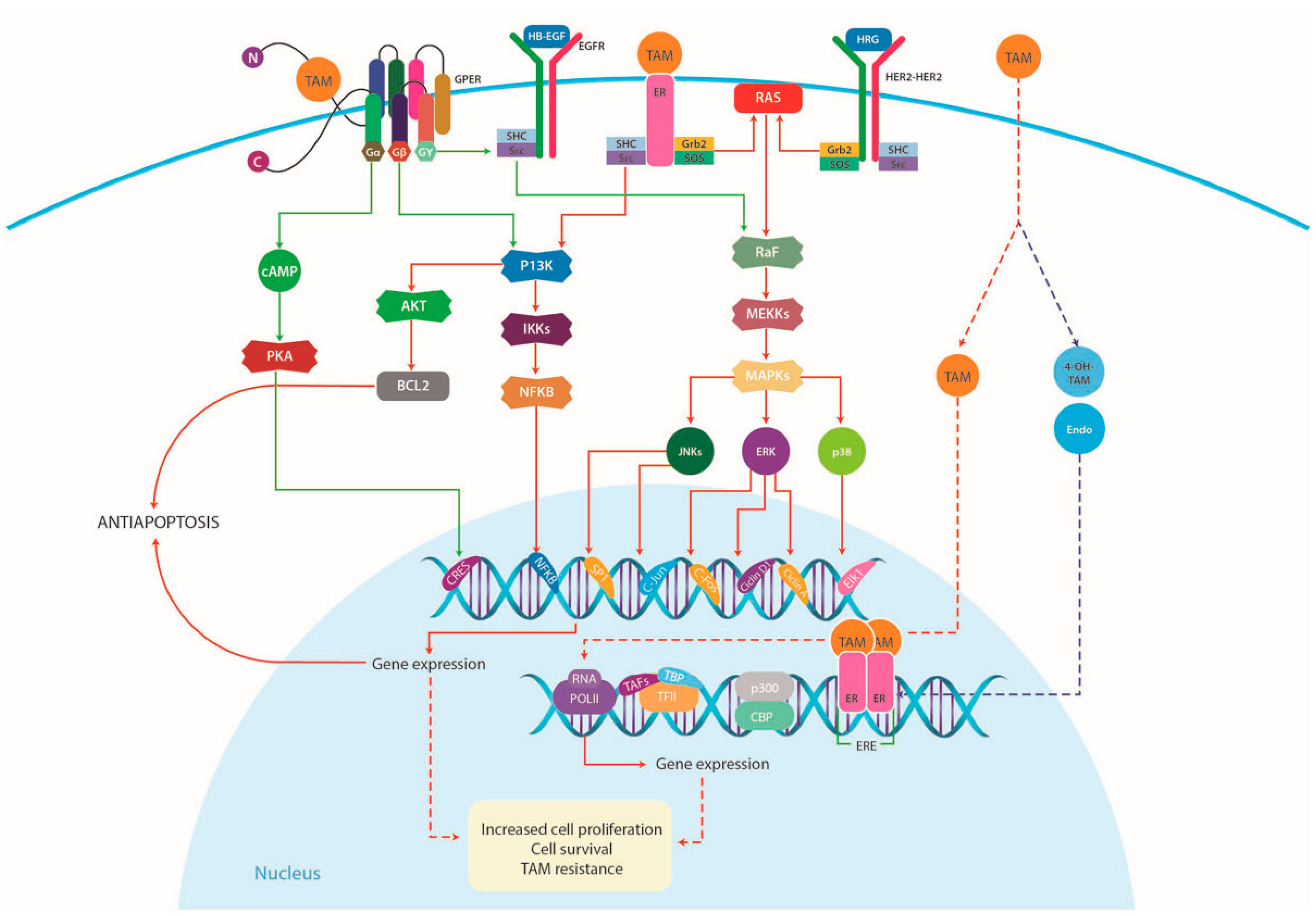
4.1.1. Increased Growth Factor Signaling and Membrane-Associated ERs
4.1.2. Loss of ERα Expression
4.1.3. ERα Mutations
4.2. (G-Protein Coupled Estrogen Receptor) GPER and TAM Resistance
4.2.1. GPER Subcellular Localization
4.2.2. GPER Signaling
4.2.3. GPER in Breast Cancer
4.2.4. GPER in Triple Negative Breast Cancer (TNBC)
4.3. Androgen Receptor (AR) and TAM Resistance
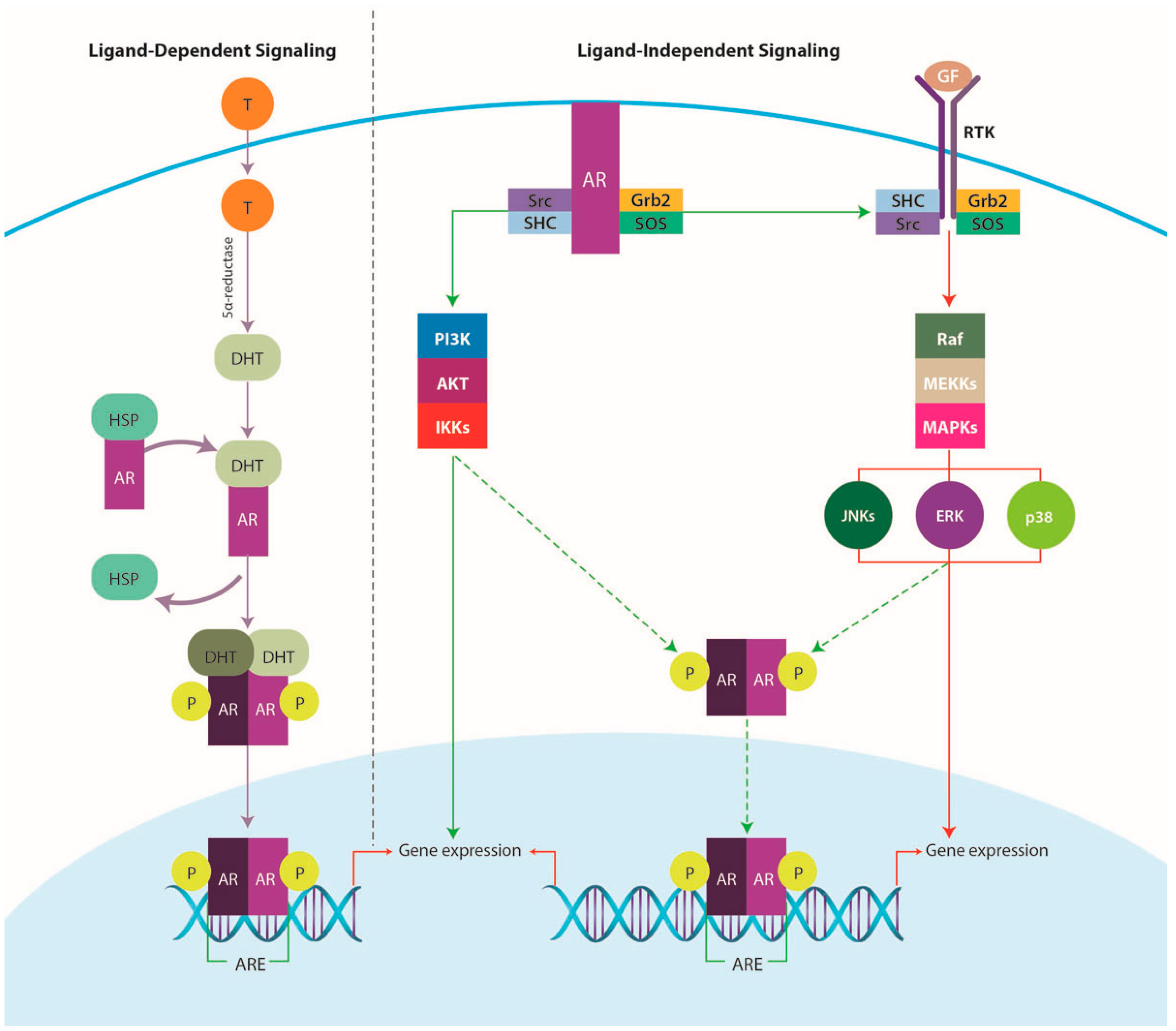
4.3.1. AR Signaling Pathway
4.3.2. AR and TAM Therapy
4.4. Hedgehog(HH) Signaling Pathway and TAM Resistance
4.4.1. HH Signaling
4.4.2. HH Signaling in Breast Cancer
4.4.3. HH Signaling Crosstalk with Additional Pathways
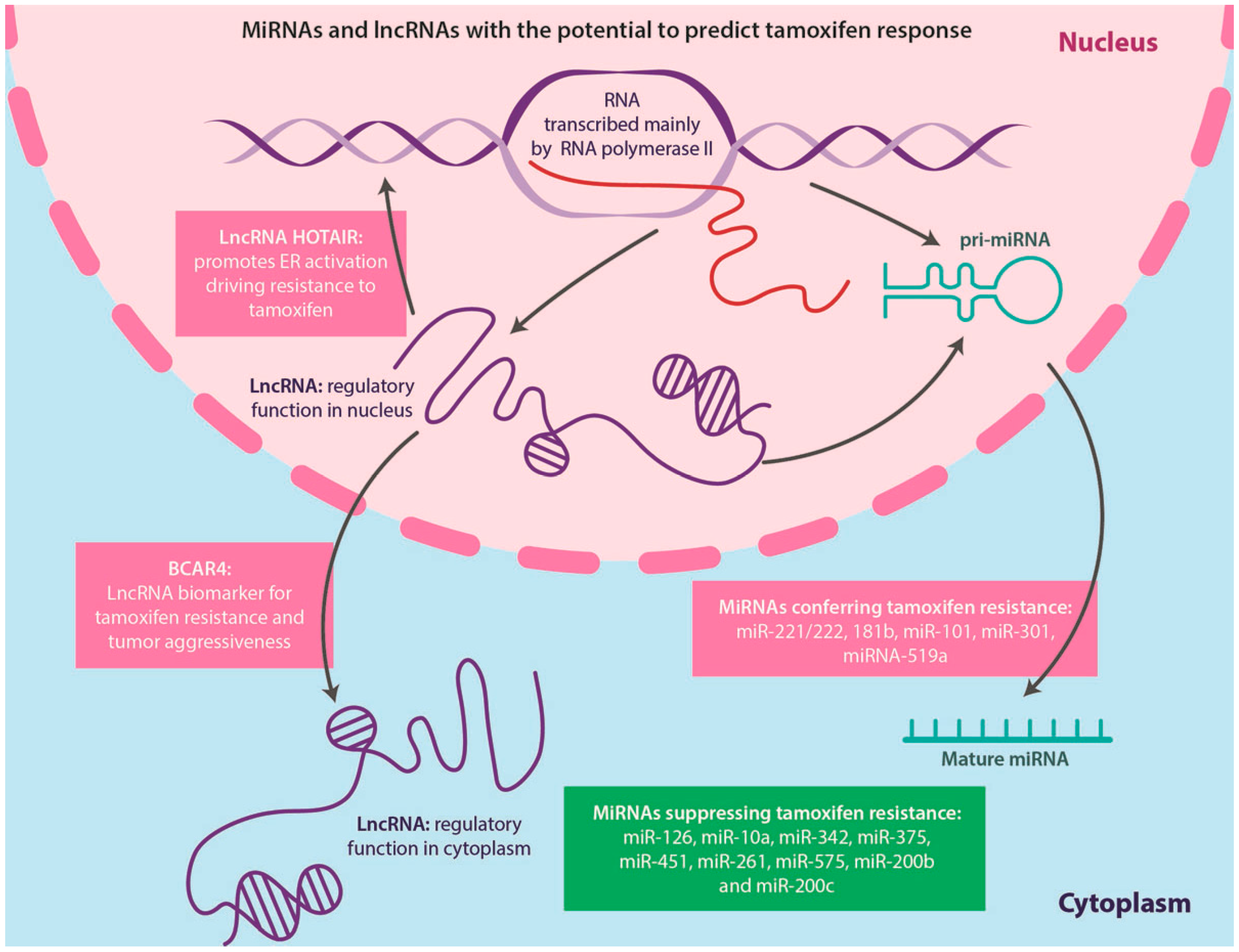
4.5. Non-Coding RNAs and TAM Resistance
4.5.1. miRNAs
4.5.2. LncRNAs
5. Clinical Use of TAM in Combination with Other Agents
5.1. Lower Doses of TAM
5.2. Topical Application of Either TAM or Its Active Metabolites
5.3. Use of Combination Therapies: GPER Inhibitors and TAM
5.4. Use of Combination Therapies: ARs Inhibitors and TAM
5.5. Use of Combination Therapies: HH Inhibitors and TAM
5.6. ncRNAs: Therapeutic Targets in TAM Resistance
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Ervik, M.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Globocan 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC Cancerbase No. 11 [Internet]. Available online: http://globocan.iarc.fr (accessed on 19 February 2016).
- Santen, R.J.; Yue, W.; Wang, J.P. Estrogen metabolites and breast cancer. Steroids 2015, 99, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Planey, S.L.; Kumar, R.; Arnott, J.A. Estrogen receptors (ERα versus ERβ): Friends or foes in human biology? J. Recept. Signal. Transduct. Res. 2014, 34, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Liehr, J.G. Is estradiol a genotoxic mutagenic carcinogen? Endocr. Rev. 2000, 21, 40–54. [Google Scholar] [PubMed]
- Russo, J.; Fernandez, S.V.; Russo, P.A.; Fernbaugh, R.; Sheriff, F.S.; Lareef, H.M.; Garber, J.; Russo, I.H. 17-β-Estradiol induces transformation and tumorigenesis in human breast epithelial cells. FASEB J. 2006, 20, 1622–1634. [Google Scholar] [CrossRef] [PubMed]
- Santen, R.J. To block estrogen's synthesis or action: That is the question. J. Clin. Endocrinol. Metab. 2002, 87, 3007–3012. [Google Scholar] [PubMed]
- Bocchinfuso, W.P.; Hively, W.P.; Couse, J.F.; Varmus, H.E.; Korach, K.S. A mouse mammary tumor virus-Wnt-1 transgene induces mammary gland hyperplasia and tumorigenesis in mice lacking estrogen receptor-α. Cancer Res. 1999, 59, 1869–1876. [Google Scholar] [PubMed]
- Cheng, S.B.; Graeber, C.T.; Quinn, J.A.; Filardo, E.J. Retrograde transport of the transmembrane estrogen receptor, G-protein-coupled-receptor-30 (GPR30/GPER) from the plasma membrane towards the nucleus. Steroids 2011, 76, 892–896. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Omoto, Y.; Iwase, H.; Yamashita, H.; Toyama, T.; Coombes, R.C.; Filipovic, A.; Warner, M.; Gustafsson, J.A. Differential expression of estrogen receptor α, β1, and β2 in lobular and ductal breast cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 1933–1938. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, G.; Staaf, J.; Vallon-Christersson, J.; Ringner, M.; Holm, K.; Hegardt, C.; Gunnarsson, H.; Fagerholm, R.; Strand, C.; Agnarsson, B.A.; et al. Genomic subtypes of breast cancer identified by array-comparative genomic hybridization display distinct molecular and clinical characteristics. Breast Cancer Res. 2010, 12, R42. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.A.; Muss, H.B.; Thor, A.D.; Dressler, L.; Liu, E.T.; Broadwater, G.; Budman, D.R.; Henderson, I.C.; Barcos, M.; Hayes, D.; et al. Her-2/neu and p53 expression versus tamoxifen resistance in estrogen receptor-positive, node-positive breast cancer. J. Clin. Oncol. 2000, 18, 3471–3479. [Google Scholar] [PubMed]
- Parisot, J.P.; Hu, X.F.; DeLuise, M.; Zalcberg, J.R. Altered expression of the IGF-1 receptor in a tamoxifen-resistant human breast cancer cell line. Br. J. Cancer 1999, 79, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Buzdar, A.; Douma, J.; Davidson, N.; Elledge, R.; Morgan, M.; Smith, R.; Porter, L.; Nabholtz, J.; Xiang, X.; Brady, C. Phase III, multicenter, double-blind, randomized study of letrozole, an aromatase inhibitor, for advanced breast cancer versus megestrol acetate. J. Clin. Oncol. 2001, 19, 3357–3366. [Google Scholar] [PubMed]
- Miller, W.R.; Larionov, A.; Renshaw, L.; Anderson, T.J.; Walker, J.R.; Krause, A.; Sing, T.; Evans, D.B.; Dixon, J.M. Gene expression profiles differentiating between breast cancers clinically responsive or resistant to letrozole. J. Clin. Oncol. 2009, 27, 1382–1387. [Google Scholar] [CrossRef] [PubMed]
- Howell, A.; Dowsett, M. Endocrinology and hormone therapy in breast cancer: Aromatase inhibitors versus antioestrogens. Breast Cancer Res. 2004, 6, 269–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chia, S.; Gradishar, W. Fulvestrant: Expanding the endocrine treatment options for patients with hormone receptor-positive advanced breast cancer. Breast 2008, 17, S16–S21. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.F.; Llombart-Cussac, A.; Rolski, J.; Feltl, D.; Dewar, J.; Macpherson, E.; Lindemann, J.; Ellis, M.J. Activity of fulvestrant 500 mg versus anastrozole 1 mg as first-line treatment for advanced breast cancer: Results from the first study. J. Clin. Oncol. 2009, 27, 4530–4535. [Google Scholar] [CrossRef] [PubMed]
- Fedele, P.; Calvani, N.; Marino, A.; Orlando, L.; Schiavone, P.; Quaranta, A.; Cinieri, S. Targeted agents to reverse resistance to endocrine therapy in metastatic breast cancer: Where are we now and where are we going? Crit. Rev. Oncol. Hematol. 2012, 84, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Nahta, R.; O'Regan, R.M. Therapeutic implications of estrogen receptor signaling in HER2-positive breast cancers. Breast Cancer Res. Treat. 2012, 135, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J.; Massarweh, S.; Huang, S.; Tsimelzon, A.; Hilsenbeck, S.G.; Osborne, C.K.; Shou, J.; Malorni, L.; Schiff, R. Development of resistance to targeted therapies transforms the clinically associated molecular profile subtype of breast tumor xenografts. Cancer Res. 2008, 68, 7493–7501. [Google Scholar] [CrossRef] [PubMed]
- Gee, J.M.; Harper, M.E.; Hutcheson, I.R.; Madden, T.A.; Barrow, D.; Knowlden, J.M.; McClelland, R.A.; Jordan, N.; Wakeling, A.E.; Nicholson, R.I. The antiepidermal growth factor receptor agent gefitinib (ZD1839/Iressa) improves antihormone response and prevents development of resistance in breast cancer in vitro. Endocrinology 2003, 144, 5105–5117. [Google Scholar] [CrossRef] [PubMed]
- Ojo, D.; Wei, F.; Liu, Y.; Wang, E.; Zhang, H.; Lin, X.; Wong, N.; Bane, A.; Tang, D. Factors promoting tamoxifen resistance in breast cancer via stimulating breast cancer stem cell expansion. Curr. Med. Chem. 2015, 22, 2360–2374. [Google Scholar] [CrossRef] [PubMed]
- Menasce, L.P.; White, G.R.; Harrison, C.J.; Boyle, J.M. Localization of the estrogen receptor locus (ESR) to chromosome 6q25.1 by FISH and a simple post-FISH banding technique. Genomics 1993, 17, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Enmark, E.; Pelto-Huikko, M.; Grandien, K.; Lagercrantz, S.; Lagercrantz, J.; Fried, G.; Nordenskjold, M.; Gustafsson, J.A. Human estrogen receptor β-gene structure, chromosomal localization, and expression pattern. J. Clin. Endocrinol. Metab. 1997, 82, 4258–4265. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Butler, R.; Warner, M.; Gustafsson, J.A.; Wilczek, B.; Landgren, B.M. Effects of short-term estradiol and norethindrone acetate treatment on the breasts of normal postmenopausal women. Menopause 2013, 20, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.W.; Perkins, R.; Tong, W.; Hong, H. Versatility or promiscuity: The estrogen receptors, control of ligand selectivity and an update on subtype selective ligands. Int. J. Environ. Res. Public Health 2014, 11, 8709–8742. [Google Scholar] [CrossRef] [PubMed]
- Yager, J.D.; Davidson, N.E. Estrogen carcinogenesis in breast cancer. N. Engl. J. Med. 2006, 354, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Arpino, G.; Wiechmann, L.; Osborne, C.K.; Schiff, R. Crosstalk between the estrogen receptor and the HER tyrosine kinase receptor family: Molecular mechanism and clinical implications for endocrine therapy resistance. Endocr. Rev. 2008, 29, 217–233. [Google Scholar] [CrossRef] [PubMed]
- Acconcia, F.; Marino, M. The effects of 17β-estradiol in cancer are mediated by estrogen receptor signaling at the plasma membrane. Front. Physiol. 2011, 2, 30. [Google Scholar] [CrossRef] [PubMed]
- Janik, M.E.; Belkot, K.; Przybylo, M. Is oestrogen an important player in melanoma progression? Contemp. Oncol. 2014, 18, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Pearce, S.T.; Jordan, V.C. The biological role of estrogen receptors α and β in cancer. Crit. Rev. Oncol. Hematol. 2004, 50, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Chang, M. Dual roles of estrogen metabolism in mammary carcinogenesis. BMB Rep. 2011, 44, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Furth, P.A.; Cabrera, M.C.; Diaz-Cruz, E.S.; Millman, S.; Nakles, R.E. Assessing estrogen signaling aberrations in breast cancer risk using genetically engineered mouse models. Ann. N. Y. Acad. Sci. 2011, 1229, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Kousidou, O.; Berdiaki, A.; Kletsas, D.; Zafiropoulos, A.; Theocharis, A.D.; Tzanakakis, G.N.; Karamanos, N.K. Estradiol-estrogen receptor: A key interplay of the expression of syndecan-2 and metalloproteinase-9 in breast cancer cells. Mol. Oncol. 2008, 2, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Pietras, R.J.; Marquez-Garban, D.C. Membrane-associated estrogen receptor signaling pathways in human cancers. Clin. Cancer Res. 2007, 13, 4672–4676. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y. Microenvironmental regulation of estrogen signals in breast cancer. Breast Cancer 2007, 14, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Nawaz, Z. Molecular biology of estrogen receptor action. In Hormones Brain and Behavior, 2nd ed.; Pfaff, D.W., Arnold, A.P., Fahrbach, S.E., Etgen, A.M., Rubin, R.T., Eds.; Elsevier: Amsterdam, The Netherlands, 2009; p. 31. [Google Scholar]
- Cheskis, B.J. Regulation of cell signalling cascades by steroid hormones. J. Cell. Biochem. 2004, 93, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Song, R.X.; Fan, P.; Yue, W.; Chen, Y.; Santen, R.J. Role of receptor complexes in the extranuclear actions of estrogen receptor α in breast cancer. Endocr. Relat. Cancer 2006, 13, S3–S13. [Google Scholar] [CrossRef] [PubMed]
- Shupnik, M.A. Crosstalk between steroid receptors and the c-Src-receptor tyrosine kinase pathways: Implications for cell proliferation. Oncogene 2004, 23, 7979–7989. [Google Scholar] [CrossRef] [PubMed]
- Levin, E.R. Integration of the extranuclear and nuclear actions of estrogen. Mol. Endocrinol. 2005, 19, 1951–1959. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Liu, M.; Luo, H.; Wu, C.; Tang, X.; Tang, S.; Hu, P.; Yan, Y.; Wang, Z.; Tu, G. GPER mediates enhanced cell viability and motility via non-genomic signaling induced by 17β-estradiol in triple-negative breast cancer cells. J. Steroid. Biochem. Mol. Biol. 2014, 143, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Mo, Z.; Liu, M.; Yang, F.; Luo, H.; Li, Z.; Tu, G.; Yang, G. GPR30 as an initiator of tamoxifen resistance in hormone-dependent breast cancer. Breast Cancer Res. 2013, 15, R114. [Google Scholar] [CrossRef] [PubMed]
- Prossnitz, E.R.; Arterburn, J.B.; Smith, H.O.; Oprea, T.I.; Sklar, L.A.; Hathaway, H.J. Estrogen signaling through the transmembrane G protein-coupled receptor GPR30. Annu. Rev. Physiol. 2008, 70, 165–190. [Google Scholar] [CrossRef] [PubMed]
- Lattrich, C.; Juhasz-Boess, I.; Ortmann, O.; Treeck, O. Detection of an elevated HER2 expression in MCF-7 breast cancer cells overexpressing estrogen receptor β1. Oncol. Rep. 2008, 19, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.S.; Symmans, W.F.; Pusztai, L.; Hortobagyi, G.N. Breast cancer biomarkers. Adv. Clin. Chem. 2005, 40, 99–125. [Google Scholar] [PubMed]
- Sturgeon, C.M.; Duffy, M.J.; Stenman, U.H.; Lilja, H.; Brunner, N.; Chan, D.W.; Babaian, R.; Bast, R.C., Jr.; Dowell, B.; Esteva, F.J.; et al. National academy of clinical biochemistry laboratory medicine practice guidelines for use of tumor markers in testicular, prostate, colorectal, breast, and ovarian cancers. Clin. Chem. 2008, 54, e11–e79. [Google Scholar] [CrossRef] [PubMed]
- Maximov, P.Y.; McDaniel, R.E.; Fernandes, D.J.; Bhatta, P.; Korostyshevskiy, V.R.; Curpan, R.F.; Jordan, V.C. Pharmacological relevance of endoxifen in a laboratory simulation of breast cancer in postmenopausal patients. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.K. Tamoxifen in the treatment of breast cancer. N. Engl. J. Med. 1998, 339, 1609–1618. [Google Scholar] [PubMed]
- Massarweh, S.; Osborne, C.K.; Creighton, C.J.; Qin, L.; Tsimelzon, A.; Huang, S.; Weiss, H.; Rimawi, M.; Schiff, R. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res. 2008, 68, 826–833. [Google Scholar] [CrossRef] [PubMed]
- Howell, A.; Osborne, C.K.; Morris, C.; Wakeling, A.E. ICI 182,780 (Faslodex): Development of a novel, “pure” antiestrogen. Cancer 2000, 89, 817–825. [Google Scholar] [CrossRef]
- Sestak, I. Preventative therapies for healthy women at high risk of breast cancer. Cancer Manag. Res. 2014, 6, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Mandlekar, S.; Kong, A.N. Mechanisms of tamoxifen-induced apoptosis. Apoptosis 2001, 6, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Salami, S.; Karami-Tehrani, F. Biochemical studies of apoptosis induced by tamoxifen in estrogen receptor positive and negative breast cancer cell lines. Clin. Biochem. 2003, 36, 247–253. [Google Scholar] [CrossRef]
- Freedman, O.C.; Fletcher, G.G.; Gandhi, S.; Mates, M.; Dent, S.F.; Trudeau, M.E.; Eisen, A. Adjuvant endocrine therapy for early breast cancer: A systematic review of the evidence for the 2014 cancer care ontario systemic therapy guideline. Curr. Oncol. 2015, 22, S95–S113. [Google Scholar] [CrossRef] [PubMed]
- Notas, G.; Pelekanou, V.; Kampa, M.; Alexakis, K.; Sfakianakis, S.; Laliotis, A.; Askoxilakis, J.; Tsentelierou, E.; Tzardi, M.; Tsapis, A.; et al. Tamoxifen induces a pluripotency signature in breast cancer cells and human tumors. Mol. Oncol. 2015, 9, 1744–1759. [Google Scholar] [CrossRef] [PubMed]
- Ariazi, E.A.; Cunliffe, H.E.; Lewis-Wambi, J.S.; Slifker, M.J.; Willis, A.L.; Ramos, P.; Tapia, C.; Kim, H.R.; Yerrum, S.; Sharma, C.G.; et al. Estrogen induces apoptosis in estrogen deprivation-resistant breast cancer through stress responses as identified by global gene expression across time. Proc. Natl. Acad. Sci. USA 2011, 108, 18879–18886. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, A.; Okada, T.; Shibutani, S.; Sonoda, E.; Hochegger, H.; Nishigori, C.; Miyachi, Y.; Takeda, S.; Yamazoe, M. Extensive chromosomal breaks are induced by tamoxifen and estrogen in DNA repair-deficient cells. Cancer Res. 2004, 64, 3144–3147. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.L.; Kern, F.G. Growth factor signal transduction and hormone independence in breast cancer. Adv. Oncobiology 1999, 2, 69. [Google Scholar]
- Cronin-Fenton, D.P.; Damkier, P.; Lash, T.L. Metabolism and transport of tamoxifen in relation to its effectiveness: New perspectives on an ongoing controversy. Future Oncol. 2014, 10, 107–122. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.D.; Zuo, H.; Lee, K.H.; Trebley, J.P.; Rae, J.M.; Weatherman, R.V.; Desta, Z.; Flockhart, D.A.; Skaar, T.C. Pharmacological characterization of 4-hydroxy-N-desmethyl tamoxifen, a novel active metabolite of tamoxifen. Breast Cancer Res. Treat. 2004, 85, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Stearns, V.; Johnson, M.D.; Rae, J.M.; Morocho, A.; Novielli, A.; Bhargava, P.; Hayes, D.F.; Desta, Z.; Flockhart, D.A. Active tamoxifen metabolite plasma concentrations after coadministration of tamoxifen and the selective serotonin reuptake inhibitor paroxetine. J. Natl. Cancer Inst. 2003, 95, 1758–1764. [Google Scholar] [CrossRef] [PubMed]
- Jager, N.G.; Linn, S.C.; Schellens, J.H.; Beijnen, J.H. Tailored tamoxifen treatment for breast cancer patients: A perspective. Clin. Breast Cancer 2015, 15, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Prossnitz, E.R.; Barton, M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat. Rev. Endocrinol. 2011, 7, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Subramaniam, M.; Grygo, S.B.; Sun, Z.; Negron, V.; Lingle, W.L.; Goetz, M.P.; Ingle, J.N.; Spelsberg, T.C.; Hawse, J.R. Estrogen receptor-β sensitizes breast cancer cells to the anti-estrogenic actions of endoxifen. Breast Cancer Res. 2011, 13, R27. [Google Scholar] [CrossRef] [PubMed]
- De Souza, J.A.; Olopade, O.I. CYP2D6 genotyping and tamoxifen: An unfinished story in the quest for personalized medicine. Semin. Oncol. 2011, 38, 263–273. [Google Scholar] [CrossRef] [PubMed]
- International Breast Cancer Study Group; Colleoni, M.; Gelber, S.; Goldhirsch, A.; Aebi, S.; Castiglione-Gertsch, M.; Price, K.N.; Coates, A.S.; Gelber, R.D. Tamoxifen after adjuvant chemotherapy for premenopausal women with lymph node-positive breast cancer: International breast cancer study group trial 13–93. J. Clin. Oncol. 2006, 24, 1332–1341. [Google Scholar] [CrossRef] [PubMed]
- Pietras, R.J.; Arboleda, J.; Reese, D.M.; Wongvipat, N.; Pegram, M.D.; Ramos, L.; Gorman, C.M.; Parker, M.G.; Sliwkowski, M.X.; Slamon, D.J. HER-2 tyrosine kinase pathway targets estrogen receptor and promotes hormone-independent growth in human breast cancer cells. Oncogene 1995, 10, 2435–2446. [Google Scholar] [PubMed]
- Girault, I.; Bieche, I.; Lidereau, R. Role of estrogen receptor α transcriptional coregulators in tamoxifen resistance in breast cancer. Maturitas 2006, 54, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Nass, N.; Kalinski, T. Tamoxifen resistance: From cell culture experiments towards novel biomarkers. Pathol. Res. Pract. 2015, 211, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Pan, X.; Zhang, X.T.; Guo, Y.M.; Wang, Z.Y.; Gong, Y.; Wang, M. Downregulation of ER-α36 expression sensitizes HER2 overexpressing breast cancer cells to tamoxifen. Am. J. Cancer Res. 2015, 5, 530–544. [Google Scholar] [PubMed]
- Gago, F.E.; Fanelli, M.A.; Ciocca, D.R. Co-expression of steroid hormone receptors (estrogen receptor α and/or progesterone receptors) and Her2/neu (c-erbB-2) in breast cancer: Clinical outcome following tamoxifen-based adjuvant therapy. J. Steroid Biochem. Mol. Biol. 2006, 98, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Droog, M.; Beelen, K.; Linn, S.; Zwart, W. Tamoxifen resistance: From bench to bedside. Eur. J. Pharmacol. 2013, 717, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Early Breast Cancer Trialists' Collaborative Group; Davies, C.; Godwin, J.; Gray, R.; Clarke, M.; Cutter, D.; Darby, S.; McGale, P.; Pan, H.C.; Taylor, C.; et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: Patient-level meta-analysis of randomised trials. Lancet 2011, 378, 771–784. [Google Scholar] [CrossRef]
- Early Breast Cancer Trialists’ Collaborative Group. Systemic treatment of early breast cancer by hormonal, cytotoxic, or immune therapy: 133 randomised trials involving 31,000 recurrences and 24,000 deaths among 75,000 women. Lancet 1992, 339, 71–85. [Google Scholar]
- Early Breast Cancer Trialists’ Collaborative Group. Tamoxifen for early breast cancer: An overview of the randomised trials. Lancet 1998, 351, 1451–1467. [Google Scholar]
- Gruvberger-Saal, S.K.; Bendahl, P.O.; Saal, L.H.; Laakso, M.; Hegardt, C.; Eden, P.; Peterson, C.; Malmstrom, P.; Isola, J.; Borg, A.; et al. Estrogen receptor β expression is associated with tamoxifen response in ERα-negative breast carcinoma. Clin. Cancer Res. 2007, 13, 1987–1994. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Zakharov, M.N.; Khan, S.H.; Miki, R.; Jang, H.; Toraldo, G.; Singh, R.; Bhasin, S.; Jasuja, R. The dynamic structure of the estrogen receptor. J. Amino Acids 2011, 2011, 812540. [Google Scholar] [CrossRef] [PubMed]
- Shou, J.; Massarweh, S.; Osborne, C.K.; Wakeling, A.E.; Ali, S.; Weiss, H.; Schiff, R. Mechanisms of tamoxifen resistance: Increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J. Natl. Cancer Inst. 2004, 96, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.W.; McNally, C.; Nickbarg, E.; Komm, B.S.; Cheskis, B.J. Estrogen receptor-interacting protein that modulates its nongenomic activity-crosstalk with Src/Erk phosphorylation cascade. Proc. Natl. Acad. Sci. USA 2002, 99, 14783–14788. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, B.; Mackey, J.R.; Clemens, M.R.; Bapsy, P.P.; Vaid, A.; Wardley, A.; Tjulandin, S.; Jahn, M.; Lehle, M.; Feyereislova, A.; et al. Trastuzumab plus anastrozole versus anastrozole alone for the treatment of postmenopausal women with human epidermal growth factor receptor 2-positive, hormone receptor-positive metastatic breast cancer: Results from the randomized phase III TAnDEM study. J. Clin. Oncol. 2009, 27, 5529–5537. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Phillips, D.L.; Ferguson, A.T.; Nelson, W.G.; Herman, J.G.; Davidson, N.E. Synergistic activation of functional estrogen receptor (ER)-α by DNA methyltransferase and histone deacetylase inhibition in human ER-α-negative breast cancer cells. Cancer Res. 2001, 61, 7025–7029. [Google Scholar] [PubMed]
- Parl, F.F. Multiple mechanisms of estrogen receptor gene repression contribute to ER-negative breast cancer. Pharm. J. 2003, 3, 251–253. [Google Scholar] [CrossRef] [PubMed]
- Fuqua, S.A.; Wiltschke, C.; Zhang, Q.X.; Borg, A.; Castles, C.G.; Friedrichs, W.E.; Hopp, T.; Hilsenbeck, S.; Mohsin, S.; O’Connell, P.; et al. A hypersensitive estrogen receptor-α mutation in premalignant breast lesions. Cancer Res. 2000, 60, 4026–4029. [Google Scholar] [PubMed]
- Johnston, S.R.; Saccani-Jotti, G.; Smith, I.E.; Salter, J.; Newby, J.; Coppen, M.; Ebbs, S.R.; Dowsett, M. Changes in estrogen receptor, progesterone receptor, and pS2 expression in tamoxifen-resistant human breast cancer. Cancer Res. 1995, 55, 3331–3338. [Google Scholar] [PubMed]
- Gutierrez, M.C.; Detre, S.; Johnston, S.; Mohsin, S.K.; Shou, J.; Allred, D.C.; Schiff, R.; Osborne, C.K.; Dowsett, M. Molecular changes in tamoxifen-resistant breast cancer: Relationship between estrogen receptor, HER-2, and p38 mitogen-activated protein kinase. J. Clin. Oncol. 2005, 23, 2469–2476. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.K.; Pippen, J.; Jones, S.E.; Parker, L.M.; Ellis, M.; Come, S.; Gertler, S.Z.; May, J.T.; Burton, G.; Dimery, I.; et al. Double-blind, randomized trial comparing the efficacy and tolerability of fulvestrant versus anastrozole in postmenopausal women with advanced breast cancer progressing on prior endocrine therapy: Results of a North American trial. J. Clin. Oncol. 2002, 20, 3386–3395. [Google Scholar] [CrossRef] [PubMed]
- Howell, A.; Pippen, J.; Elledge, R.M.; Mauriac, L.; Vergote, I.; Jones, S.E.; Come, S.E.; Osborne, C.K.; Robertson, J.F. Fulvestrant versus anastrozole for the treatment of advanced breast carcinoma: A prospectively planned combined survival analysis of two multicenter trials. Cancer 2005, 104, 236–239. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, S.; Rasouli, M.; Nouri, M.; Kalbasi, S. Association of estrogen receptor-α A908G (K303R) mutation with breast cancer risk. Int. J. Clin. Exp. Med. 2013, 6, 39–49. [Google Scholar] [PubMed]
- Conway, K.; Parrish, E.; Edmiston, S.N.; Tolbert, D.; Tse, C.K.; Geradts, J.; Livasy, C.A.; Singh, H.; Newman, B.; Millikan, R.C. The estrogen receptor-α A908G (K303R) mutation occurs at a low frequency in invasive breast tumors: Results from a population-based study. Breast Cancer Res. 2005, 7, R871–R880. [Google Scholar] [CrossRef] [PubMed]
- Ghimenti, C.; Mello-Grand, M.; Regolo, L.; Zambelli, A.; Chiorino, G. Absence of the K303R estrogen receptor α mutation in breast cancer patients exhibiting different responses to aromatase inhibitor anastrozole neoadjuvant treatment. Exp. Ther. Med. 2010, 1, 939–942. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.P.; O'Neill, P.A.; Innes, H.; Sibson, D.R. Hypersensitive K303R oestrogen receptor-α variant not found in invasive carcinomas. Breast Cancer Res. 2005, 7, R113–R118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toy, W.; Shen, Y.; Won, H.; Green, B.; Sakr, R.A.; Will, M.; Li, Z.; Gala, K.; Fanning, S.; King, T.A.; et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat. Genet. 2013, 45, 1439–1445. [Google Scholar] [CrossRef] [PubMed]
- Merenbakh-Lamin, K.; Ben-Baruch, N.; Yeheskel, A.; Dvir, A.; Soussan-Gutman, L.; Jeselsohn, R.; Yelensky, R.; Brown, M.; Miller, V.A.; Sarid, D.; et al. D538G mutation in estrogen receptor-α: A novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res. 2013, 73, 6856–6864. [Google Scholar] [CrossRef] [PubMed]
- Jeselsohn, R.; Yelensky, R.; Buchwalter, G.; Frampton, G.; Meric-Bernstam, F.; Gonzalez-Angulo, A.M.; Ferrer-Lozano, J.; Perez-Fidalgo, J.A.; Cristofanilli, M.; Gomez, H.; et al. Emergence of constitutively active estrogen receptor-α mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin. Cancer Res. 2014, 20, 1757–1767. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Andres, G.; Kramer, K.; Kundranda, M.N.; Alvarez, R.H.; Klimant, E.; Parikh, A.R.; Tan, B.; Staren, E.D.; Markman, M. Incidence and clinical significance of ESR1 mutations in heavily pretreated metastatic breast cancer patients. Onco Targets Ther. 2015, 8, 3323–3328. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.R.; Wu, Y.M.; Vats, P.; Su, F.; Lonigro, R.J.; Cao, X.; Kalyana-Sundaram, S.; Wang, R.; Ning, Y.; Hodges, L.; et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet. 2013, 45, 1446–1451. [Google Scholar] [CrossRef] [PubMed]
- Prossnitz, E.R.; Oprea, T.I.; Sklar, L.A.; Arterburn, J.B. The ins and outs of GPR30: A transmembrane estrogen receptor. J. Steroid Biochem. Mol. Biol. 2008, 109, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Arias-Pulido, H.; Royce, M.; Gong, Y.; Joste, N.; Lomo, L.; Lee, S.J.; Chaher, N.; Verschraegen, C.; Lara, J.; Prossnitz, E.R.; et al. GPR30 and estrogen receptor expression: New insights into hormone dependence of inflammatory breast cancer. Breast Cancer Res. Treat. 2010, 123, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Samartzis, E.P.; Noske, A.; Meisel, A.; Varga, Z.; Fink, D.; Imesch, P. The G protein-coupled estrogen receptor (GPER) is expressed in two different subcellular localizations reflecting distinct tumor properties in breast cancer. PLoS ONE 2014, 9, e83296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ignatov, A.; Ignatov, T.; Weissenborn, C.; Eggemann, H.; Bischoff, J.; Semczuk, A.; Roessner, A.; Costa, S.D.; Kalinski, T. G-protein-coupled estrogen receptor GPR30 and tamoxifen resistance in breast cancer. Breast Cancer Res. Treat. 2011, 128, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J.; Quinn, J.A.; Bland, K.I.; Frackelton, A.R., Jr. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol. Endocrinol. 2000, 14, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Revankar, C.M.; Cimino, D.F.; Sklar, L.A.; Arterburn, J.B.; Prossnitz, E.R. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 2005, 307, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Girgert, R.; Emons, G.; Grundker, C. Inactivation of GPR30 reduces growth of triple-negative breast cancer cells: Possible application in targeted therapy. Breast Cancer Res. Treat. 2012, 134, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Zhang, Z.; Liao, H.; Wu, L.; Wu, X.; Zhou, D.; Xi, X.; Zhu, Y.; Feng, Y. Nuclear estrogen receptor-mediated Notch signaling and GPR30-mediated PI3K/AKT signaling in the regulation of endometrial cancer cell proliferation. Oncol. Rep. 2012, 27, 504–510. [Google Scholar] [PubMed]
- Thomas, P.; Pang, Y.; Filardo, E.J.; Dong, J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology 2005, 146, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J.; Quinn, J.A.; Frackelton, A.R., Jr.; Bland, K.I. Estrogen action via the G protein-coupled receptor, GPR30: Stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol. Endocrinol. 2002, 16, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Vera-Ramirez, L.; Sanchez-Rovira, P.; Ramirez-Tortosa, M.C.; Ramirez-Tortosa, C.L.; Granados-Principal, S.; Lorente, J.A.; Quiles, J.L. Free radicals in breast carcinogenesis, breast cancer progression and cancer stem cells: Biological bases to develop oxidative-based therapies. Crit. Rev. Oncol. Hematol. 2011, 80, 347–368. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.; Liehr, J.G. Estrogen, DNA damage and mutations. Mutat. Res. 1999, 424, 107–115. [Google Scholar] [CrossRef]
- Lappano, R.; Pisano, A.; Maggiolini, M. GPER function in breast cancer: An overview. Front. Endocrinol. 2014, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Pupo, M.; Pisano, A.; Abonante, S.; Maggiolini, M.; Musti, A.M. GPER activates Notch signaling in breast cancer cells and cancer-associated fibroblasts (CAFs). Int. J. Biochem. Cell Biol. 2014, 46, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Q.; Russo, J. ERα-negative and triple negative breast cancer: Molecular features and potential therapeutic approaches. Biochim. Biophys. Acta 2009, 1796, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Girgert, R.; Emons, G.; Grundker, C. Inhibition of GPR30 by estriol prevents growth stimulation of triple-negative breast cancer cells by 17β-estradiol. BMC Cancer 2014, 14, 935. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Li, X.; Blanchard, A.; Bramwell, V.H.; Pritchard, K.I.; Tu, D.; Shepherd, L.; Myal, Y.; Penner, C.; Watson, P.H.; et al. Expression of both estrogen receptor-β1 (ER-β1) and its co-regulator steroid receptor RNA activator protein (SRAP) are predictive for benefit from tamoxifen therapy in patients with estrogen receptor-alpha (ER-α)-negative early breast cancer (EBC). Ann. Oncol. 2013, 24, 1986–1993. [Google Scholar] [CrossRef] [PubMed]
- Brys, M. Androgens and androgen receptor: Do they play a role in breast cancer? Med. Sci. Monit. 2000, 6, 433–438. [Google Scholar] [PubMed]
- Rakha, E.A.; El-Sayed, M.E.; Green, A.R.; Lee, A.H.; Robertson, J.F.; Ellis, I.O. Prognostic markers in triple-negative breast cancer. Cancer 2007, 109, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Koo, J.; Park, H.S.; Kim, J.H.; Choi, S.Y.; Lee, J.H.; Park, B.W.; Lee, K.S. Expression of androgen receptors in primary breast cancer. Ann. Oncol. 2010, 21, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Castellano, I.; Allia, E.; Accortanzo, V.; Vandone, A.M.; Chiusa, L.; Arisio, R.; Durando, A.; Donadio, M.; Bussolati, G.; Coates, A.S.; et al. Androgen receptor expression is a significant prognostic factor in estrogen receptor positive breast cancers. Breast Cancer Res. Treat. 2010, 124, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Tsang, J.Y.; Ni, Y.B.; Chan, S.K.; Shao, M.M.; Law, B.K.; Tan, P.H.; Tse, G.M. Androgen receptor expression shows distinctive significance in ER positive and negative breast cancers. Ann. Surg. Oncol. 2014, 21, 2218–2228. [Google Scholar] [CrossRef] [PubMed]
- Witzel, I.; Graeser, M.; Karn, T.; Schmidt, M.; Wirtz, R.; Schutze, D.; Rausch, A.; Janicke, F.; Milde-Langosch, K.; Muller, V. Androgen receptor expression is a predictive marker in chemotherapy-treated patients with endocrine receptor-positive primary breast cancers. J. Cancer Res. Clin. Oncol. 2013, 139, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Saltzman, A.; Yeh, S.; Young, W.; Keller, E.; Lee, H.J.; Wang, C.; Mizokami, A. Androgen receptor: An overview. Crit. Rev. Eukaryot. Gene Expr. 1995, 5, 97–125. [Google Scholar] [CrossRef] [PubMed]
- Higa, G.M.; Fell, R.G. Sex hormone receptor repertoire in breast cancer. Int. J. Breast Cancer 2013, 2013, 284036. [Google Scholar] [CrossRef] [PubMed]
- Heinlein, C.A.; Chang, C. The roles of androgen receptors and androgen-binding proteins in nongenomic androgen actions. Mol. Endocrinol. 2002, 16, 2181–2187. [Google Scholar] [CrossRef] [PubMed]
- Tarulli, G.A.; Butler, L.M.; Tilley, W.D.; Hickey, T.E. Bringing androgens up a Notch in breast cancer. Endocr. Relat. Cancer 2014, 21, T183–T202. [Google Scholar] [CrossRef] [PubMed]
- Cuenca-Lopez, M.D.; Montero, J.C.; Morales, J.C.; Prat, A.; Pandiella, A.; Ocana, A. Phospho-kinase profile of triple negative breast cancer and androgen receptor signaling. BMC Cancer 2014, 14, 302. [Google Scholar] [CrossRef] [PubMed]
- De Amicis, F.; Thirugnansampanthan, J.; Cui, Y.; Selever, J.; Beyer, A.; Parra, I.; Weigel, N.L.; Herynk, M.H.; Tsimelzon, A.; Lewis, M.T.; et al. Androgen receptor overexpression induces tamoxifen resistance in human breast cancer cells. Breast Cancer Res. Treat. 2010, 121, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ciupek, A.; Rechoum, Y.; Gu, G.; Gelsomino, L.; Beyer, A.R.; Brusco, L.; Covington, K.R.; Tsimelzon, A.; Fuqua, S.A. Androgen receptor promotes tamoxifen agonist activity by activation of EGFR in ERα-positive breast cancer. Breast Cancer Res. Treat. 2015, 154, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Cochrane, D.R.; Bernales, S.; Jacobsen, B.M.; Cittelly, D.M.; Howe, E.N.; D'Amato, N.C.; Spoelstra, N.S.; Edgerton, S.M.; Jean, A.; Guerrero, J.; et al. Role of the androgen receptor in breast cancer and preclinical analysis of enzalutamide. Breast Cancer Res. 2014, 16, R7. [Google Scholar] [CrossRef] [PubMed]
- Carreno, G.; Del Casar, J.M.; Corte, M.D.; Gonzalez, L.O.; Bongera, M.; Merino, A.M.; Juan, G.; Obregon, R.; Martinez, E.; Vizoso, F.J. Local recurrence after mastectomy for breast cancer: Analysis of clinicopathological, biological and prognostic characteristics. Breast Cancer Res. Treat. 2007, 102, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, B.; Lu, Y.; Teng, K.Y.; Nuovo, G.; Li, X.; Shapiro, C.L.; Majumder, S. Hedgehog signaling is a novel therapeutic target in tamoxifen-resistant breast cancer aberrantly activated by PI3K/AKT pathway. Cancer Res. 2012, 72, 5048–5059. [Google Scholar] [CrossRef] [PubMed]
- Matevossian, A.; Resh, M.D. Hedgehog Acyltransferase as a target in estrogen receptor positive, HER2 amplified, and tamoxifen resistant breast cancer cells. Mol. Cancer 2015, 14, 72. [Google Scholar] [CrossRef] [PubMed]
- Chamoun, Z.; Mann, R.K.; Nellen, D.; von Kessler, D.P.; Bellotto, M.; Beachy, P.A.; Basler, K. Skinny hedgehog, an acyltransferase required for palmitoylation and activity of the hedgehog signal. Science 2001, 293, 2080–2084. [Google Scholar] [CrossRef] [PubMed]
- Micchelli, C.A.; The, I.; Selva, E.; Mogila, V.; Perrimon, N. Rasp, a putative transmembrane acyltransferase, is required for Hedgehog signaling. Development 2002, 129, 843–851. [Google Scholar] [PubMed]
- Villegas, V.E.; Rondon-Lagos, M.; Annaratone, L.; Castellano, I.; Grismaldo, A.; Sapino, A.; Zaphiropoulos, P.G. Tamoxifen treatment of breast cancer cells: Impact on Hedgehog/GLI1 signaling. Int. J. Mol. Sci. 2016, 17, 308. [Google Scholar] [CrossRef] [PubMed]
- O'Toole, S.A.; Machalek, D.A.; Shearer, R.F.; Millar, E.K.; Nair, R.; Schofield, P.; McLeod, D.; Cooper, C.L.; McNeil, C.M.; McFarland, A.; et al. Hedgehog overexpression is associated with stromal interactions and predicts for poor outcome in breast cancer. Cancer Res. 2011, 71, 4002–4014. [Google Scholar] [CrossRef] [PubMed]
- Rubin, L.L.; de Sauvage, F.J. Targeting the Hedgehog pathway in cancer. Nat. Rev. Drug Discov. 2006, 5, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Petrova, R.; Joyner, A.L. Roles for Hedgehog signaling in adult organ homeostasis and repair. Development 2014, 141, 3445–3457. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Martelotto, L.G.; Peifer, M.; Sos, M.L.; Karnezis, A.N.; Mahjoub, M.R.; Bernard, K.; Conklin, J.F.; Szczepny, A.; Yuan, J.; et al. A crucial requirement for Hedgehog signaling in small cell lung cancer. Nat. Med. 2011, 17, 1504–1508. [Google Scholar] [CrossRef] [PubMed]
- Benvenuto, M.; Masuelli, L.; de Smaele, E.; Fantini, M.; Mattera, R.; Cucchi, D.; Bonanno, E.; di Stefano, E.; Frajese, G.V.; Orlandi, A.; et al. In vitro and in vivo inhibition of breast cancer cell growth by targeting the Hedgehog/GLI pathway with SMO (GDC-0449) or GLI (GANT-61) inhibitors. Oncotarget 2016, 7, 9250–9270. [Google Scholar] [PubMed]
- Li, X.; Wang, Z.; Ma, Q.; Xu, Q.; Liu, H.; Duan, W.; Lei, J.; Ma, J.; Wang, X.; Lv, S.; et al. Sonic hedgehog paracrine signaling activates stromal cells to promote perineural invasion in pancreatic cancer. Clin. Cancer Res. 2014, 20, 4326–4338. [Google Scholar] [CrossRef] [PubMed]
- Suzman, D.L.; Antonarakis, E.S. Clinical implications of hedgehog pathway signaling in prostate cancer. Cancers (Basel) 2015, 7, 1983–1993. [Google Scholar] [CrossRef] [PubMed]
- Varjosalo, M.; Taipale, J. Hedgehog: Functions and mechanisms. Genes Dev. 2008, 22, 2454–2472. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, D.; Stone, D.M.; Brush, J.; Ryan, A.; Armanini, M.; Frantz, G.; Rosenthal, A.; de Sauvage, F.J. Characterization of two patched receptors for the vertebrate hedgehog protein family. Proc. Natl. Acad. Sci. USA 1998, 95, 13630–13634. [Google Scholar] [CrossRef] [PubMed]
- Ruch, J.M.; Kim, E.J. Hedgehog signaling pathway and cancer therapeutics: Progress to date. Drugs 2013, 73, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Aberger, F.; Ruiz, I.A.A. Context-dependent signal integration by the GLI code: The oncogenic load, pathways, modifiers and implications for cancer therapy. Semin. Cell Dev. Biol. 2014, 33, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Stecca, B.; Ruiz, I.A.A. Context-dependent regulation of the GLI code in cancer by HEDGEHOG and NON-HEDGEHOG signals. J. Mol. Cell. Biol. 2010, 2, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Teglund, S.; Toftgard, R. Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim. Biophys. Acta 2010, 1805, 181–208. [Google Scholar] [CrossRef] [PubMed]
- Hatsell, S.; Frost, A.R. Hedgehog signaling in mammary gland development and breast cancer. J. Mammary Gland Biol. Neoplasia 2007, 12, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Hui, M.; Cazet, A.; Nair, R.; Watkins, D.N.; O'Toole, S.A.; Swarbrick, A. The Hedgehog signalling pathway in breast development, carcinogenesis and cancer therapy. Breast Cancer Res. 2013, 15, 203. [Google Scholar] [CrossRef] [PubMed]
- Brennan, D.; Chen, X.; Cheng, L.; Mahoney, M.; Riobo, N.A. Noncanonical Hedgehog signaling. Vitam Horm. 2012, 88, 55–72. [Google Scholar] [PubMed]
- Kasper, M.; Jaks, V.; Fiaschi, M.; Toftgard, R. Hedgehog signalling in breast cancer. Carcinogenesis 2009, 30, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Kubo, M.; Nakamura, M.; Tasaki, A.; Yamanaka, N.; Nakashima, H.; Nomura, M.; Kuroki, S.; Katano, M. Hedgehog signaling pathway is a new therapeutic target for patients with breast cancer. Cancer Res. 2004, 64, 6071–6074. [Google Scholar] [CrossRef] [PubMed]
- Flemban, A.; Qualtrough, D. The potential role of hedgehog signaling in the luminal/basal phenotype of breast epithelia and in breast cancer invasion and metastasis. Cancers (Basel) 2015, 7, 1863–1884. [Google Scholar] [CrossRef] [PubMed]
- Jeng, K.S.; Sheen, I.S.; Jeng, W.J.; Yu, M.C.; Hsiau, H.I.; Chang, F.Y. High expression of Sonic Hedgehog signaling pathway genes indicates a risk of recurrence of breast carcinoma. Onco Targets Ther. 2013, 7, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Noman, A.S.; Uddin, M.; Rahman, M.Z.; Nayeem, M.J.; Alam, S.S.; Khatun, Z.; Wahiduzzaman, M.; Sultana, A.; Rahman, M.L.; Ali, M.Y.; et al. Overexpression of Sonic Hedgehog in the triple negative breast cancer: Clinicopathological characteristics of high burden breast cancer patients from bangladesh. Sci. Rep. 2016, 6, 18830. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Wang, L.H.; Wen, Y.Y.; Song, M.; Li, B.L.; Chen, X.L.; Xu, M.; An, S.X.; Zhao, J.; Lu, Y.Y.; et al. Expression and regulation mechanisms of Sonic Hedgehog in breast cancer. Cancer Sci. 2010, 101, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Gao, H.F.; Wang, Y.; Liu, F.; Tian, X.F.; Zhang, Y. Overexpression of GLI1 in cancer interstitial tissues predicts early relapse after radical operation of breast cancer. Chin. J. Cancer Res. 2012, 24, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Inaguma, S.; Riku, M.; Ito, H.; Tsunoda, T.; Ikeda, H.; Kasai, K. Gli1 orchestrates CXCR4/CXC7 signaling to enhance migration and metastasis of breast cancer cells. Oncotarget 2015, 6, 33648–33657. [Google Scholar] [PubMed]
- Xuan, Y.; Lin, Z. Expression of indian Hedgehog signaling molecules in breast cancer. J. Cancer Res. Clin. Oncol. 2009, 135, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Im, S.; Choi, H.J.; Yoo, C.; Jung, J.H.; Jeon, Y.W.; Suh, Y.J.; Kang, C.S. Hedgehog related protein expression in breast cancer: Gli-2 is associated with poor overall survival. Korean J. Pathol. 2013, 47, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Qu, Y.; Jin, Y.; Yu, Y.; Deng, N.; Wawrowsky, K.; Zhang, X.; Li, N.; Bose, S.; Wang, Q.; et al. FOXC1 Activates Smoothened-independent Hedgehog Signaling in Basal-like Breast Cancer. Cell Rep. 2015, 13, 1046–1058. [Google Scholar] [CrossRef] [PubMed]
- Okolowsky, N.; Furth, P.A.; Hamel, P.A. Oestrogen receptor-α regulates non-canonical Hedgehog-signalling in the mammary gland. Dev. Biol. 2014, 391, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Koga, K.; Nakamura, M.; Nakashima, H.; Akiyoshi, T.; Kubo, M.; Sato, N.; Kuroki, S.; Nomura, M.; Tanaka, M.; Katano, M. Novel link between estrogen receptor α and hedgehog pathway in breast cancer. Anticancer Res. 2008, 28, 731–740. [Google Scholar] [PubMed]
- Che, J.; Zhang, F.Z.; Zhao, C.Q.; Hu, X.D.; Fan, S.J. Cyclopamine is a novel Hedgehog signaling inhibitor with significant anti-proliferative, anti-invasive and anti-estrogenic potency in human breast cancer cells. Oncol. Lett. 2013, 5, 1417–1421. [Google Scholar] [PubMed]
- Malhotra, G.K.; Zhao, X.; Band, H.; Band, V. Shared signaling pathways in normal and breast cancer stem cells. J. Carcinog. 2011, 10, 38. [Google Scholar] [PubMed]
- Johnson, R.W.; Nguyen, M.P.; Padalecki, S.S.; Grubbs, B.G.; Merkel, A.R.; Oyajobi, B.O.; Matrisian, L.M.; Mundy, G.R.; Sterling, J.A. TGF-β promotion of Gli2-induced expression of parathyroid hormone-related protein, an important osteolytic factor in bone metastasis, is independent of canonical Hedgehog signaling. Cancer Res. 2011, 71, 822–831. [Google Scholar] [CrossRef] [PubMed]
- Javelaud, D.; Alexaki, V.I.; Dennler, S.; Mohammad, K.S.; Guise, T.A.; Mauviel, A. TGF-β/SMAD/GLI2 signaling axis in cancer progression and metastasis. Cancer Res. 2011, 71, 5606–5610. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Katoh, M. Integrative genomic analyses on GLI1: Positive regulation of GLI1 by Hedgehog-GLI, TGFβ-Smads, and RTK-PI3K-AKT signals, and negative regulation of GLI1 by Notch-CSL-HES/HEY, and GPCR-Gs-PKA signals. Int. J. Oncol. 2009, 35, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.W.; Merkel, A.R.; Page, J.M.; Ruppender, N.S.; Guelcher, S.A.; Sterling, J.A. Wnt signaling induces gene expression of factors associated with bone destruction in lung and breast cancer. Clin. Exp. Metastasis 2014, 31, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Mimeault, M.; Batra, S.K. Frequent deregulations in the Hedgehog signaling network and cross-talks with the epidermal growth factor receptor pathway involved in cancer progression and targeted therapies. Pharmacol. Rev. 2010, 62, 497–524. [Google Scholar] [CrossRef] [PubMed]
- Androutsellis-Theotokis, A.; Leker, R.R.; Soldner, F.; Hoeppner, D.J.; Ravin, R.; Poser, S.W.; Rueger, M.A.; Bae, S.K.; Kittappa, R.; McKay, R.D. Notch signalling regulates stem cell numbers in vitro and in vivo. Nature 2006, 442, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Liu, M.; Gonzalez-Perez, R.R. Role of Notch and its oncogenic signaling crosstalk in breast cancer. Biochim. Biophysi. Acta 2011, 1815, 197–213. [Google Scholar] [CrossRef] [PubMed]
- Van Bakel, H.; Nislow, C.; Blencowe, B.J.; Hughes, T.R. Most “dark matter” transcripts are associated with known genes. PLoS Biol. 2010, 8, e1000371. [Google Scholar] [CrossRef] [PubMed]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet. 2006, 15, R17–R29. [Google Scholar] [CrossRef] [PubMed]
- Huttenhofer, A.; Schattner, P.; Polacek, N. Non-coding rnas: Hope or hype? Trends Genet. 2005, 21, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Taft, R.J.; Pang, K.C.; Mercer, T.R.; Dinger, M.; Mattick, J.S. Non-coding RNAs: regulators of disease. J. Pathol. 2010, 220, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Kapranov, P.; Cheng, J.; Dike, S.; Nix, D.A.; Duttagupta, R.; Willingham, A.T.; Stadler, P.F.; Hertel, J.; Hackermuller, J.; Hofacker, I.L.; et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 2007, 316, 1484–1488. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.V.; Mattick, J.S. The rise of regulatory RNA. Nat. Rev. Genet. 2014, 15, 423–437. [Google Scholar] [CrossRef] [PubMed]
- Sana, J.; Faltejskova, P.; Svoboda, M.; Slaby, O. Novel classes of non-coding RNAs and cancer. J. Transl. Med. 2012, 10, 103. [Google Scholar] [CrossRef] [PubMed]
- Ferrarelli, L.K. Focus issue: Noncoding RNAs in cancer. Sci. Signal. 2015, 8, eg3. [Google Scholar] [CrossRef] [PubMed]
- Calore, F.; Lovat, F.; Garofalo, M. Non-coding RNAs and cancer. Int. J. Mol. Sci. 2013, 14, 17085–17110. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. Elegans heterochronic gene lin-4 encodes small rnas with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. Elegans. Cell 1993, 75, 855–862. [Google Scholar] [CrossRef]
- Lam, J.K.; Chow, M.Y.; Zhang, Y.; Leung, S.W. siRNA versus miRNA as therapeutics for gene silencing. Mol. Ther. Nucleic Acids 2015, 4, e252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinske, L.C.; Galante, P.A.; Kuo, W.P.; Ohno-Machado, L. A potential role for intragenic miRNAs on their hosts' interactome. BMC Genom. 2010, 11, 533. [Google Scholar] [CrossRef] [PubMed]
- Clark, B.S.; Blackshaw, S. Long non-coding RNA-dependent transcriptional regulation in neuronal development and disease. Front. Genet. 2014, 5, 164. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Su, H.; Liu, C.; Skogerbo, G.; He, H.; He, D.; Zhu, X.; Liu, T.; Zhao, Y.; Chen, R. MicroRNA-encoding long non-coding RNAs. BMC Genom. 2008, 9, 236. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, J.E.; Sunwoo, H.; Spector, D.L. Long noncoding RNAs: Functional surprises from the RNA world. Genes Dev. 2009, 23, 1494–1504. [Google Scholar] [CrossRef] [PubMed]
- Drakaki, A.; Iliopoulos, D. MicroRNA gene networks in oncogenesis. Curr. Genom. 2009, 10, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Van Schooneveld, E.; Wildiers, H.; Vergote, I.; Vermeulen, P.B.; Dirix, L.Y.; van Laere, S.J. Dysregulation of microRNAs in breast cancer and their potential role as prognostic and predictive biomarkers in patient management. Breast Cancer Res. 2015, 17, 21. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.E.; Ghoshal, K.; Ramaswamy, B.; Roy, S.; Datta, J.; Shapiro, C.L.; Jacob, S.; Majumder, S. MicroRNA-221/222 confers tamoxifen resistance in breast cancer by targeting p27kip1. J. Biol. Chem. 2008, 283, 29897–29903. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, M.; Quintavalle, C.; Romano, G.; Croce, C.M.; Condorelli, G. miR221/222 in cancer: Their role in tumor progression and response to therapy. Curr. Mol. Med. 2012, 12, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Roy, S.; Nuovo, G.; Ramaswamy, B.; Miller, T.; Shapiro, C.; Jacob, S.T.; Majumder, S. Anti-microRNA-222 (anti-miR-222) and -181B suppress growth of tamoxifen-resistant xenografts in mouse by targeting TIMP3 protein and modulating mitogenic signal. J. Biol. Chem. 2011, 286, 42292–42302. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, M.; Mo, Y.Y. MicroRNA-145 suppresses cell invasion and metastasis by directly targeting mucin 1. Cancer Res. 2010, 70, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Gerster, K.; Alajez, N.M.; Tsang, J.; Waldron, L.; Pintilie, M.; Hui, A.B.; Sykes, J.; P'ng, C.; Miller, N.; et al. MicroRNA-301 mediates proliferation and invasion in human breast cancer. Cancer Res. 2011, 71, 2926–2937. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.; Shukla, K.; Balwierz, A.; Soons, Z.; Konig, R.; Sahin, O.; Wiemann, S. MicroRNA-519a is a novel oncomir conferring tamoxifen resistance by targeting a network of tumour-suppressor genes in ER+ breast cancer. J. Pathol. 2014, 233, 368–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoppe, R.; Achinger-Kawecka, J.; Winter, S.; Fritz, P.; Lo, W.Y.; Schroth, W.; Brauch, H. Increased expression of miR-126 and miR-10a predict prolonged relapse-free time of primary oestrogen receptor-positive breast cancer following tamoxifen treatment. Eur. J. Cancer 2013, 49, 3598–3608. [Google Scholar] [CrossRef] [PubMed]
- Tavazoie, S.F.; Alarcon, C.; Oskarsson, T.; Padua, D.; Wang, Q.; Bos, P.D.; Gerald, W.L.; Massague, J. Endogenous human microRNAs that suppress breast cancer metastasis. Nature 2008, 451, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhu, T.; Chen, Y.; Mertani, H.C.; Lee, K.O.; Lobie, P.E. Human growth hormone-regulated HOXA1 is a human mammary epithelial oncogene. J. Biol. Chem. 2003, 278, 7580–7590. [Google Scholar] [CrossRef] [PubMed]
- Cittelly, D.M.; Das, P.M.; Spoelstra, N.S.; Edgerton, S.M.; Richer, J.K.; Thor, A.D.; Jones, F.E. Downregulation of miR-342 is associated with tamoxifen resistant breast tumors. Mol. Cancer 2010, 9, 317. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.; Balwierz, A.; Zhang, J.D.; Kublbeck, M.; Pawitan, Y.; Hielscher, T.; Wiemann, S.; Sahin, O. Re-expression of microRNA-375 reverses both tamoxifen resistance and accompanying EMT-like properties in breast cancer. Oncogene 2013, 32, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, A.; Katzenellenbogen, B.S. Tamoxifen downregulation of miR-451 increases 14–3-3zeta and promotes breast cancer cell survival and endocrine resistance. Oncogene 2012, 31, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Hrstka, R.; Nenutil, R.; Fourtouna, A.; Maslon, M.M.; Naughton, C.; Langdon, S.; Murray, E.; Larionov, A.; Petrakova, K.; Muller, P.; et al. The pro-metastatic protein anterior gradient-2 predicts poor prognosis in tamoxifen-treated breast cancers. Oncogene 2010, 29, 4838–4847. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, T.T.; Teng, Y.; Litchfield, L.M.; Muluhngwi, P.; Al-Rayyan, N.; Klinge, C.M. Reduced expression of miR-200 family members contributes to antiestrogen resistance in LY2 human breast cancer cells. PLoS ONE 2013, 8, e62334. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xu, J.; Shi, Y.; Sun, Q.; Zhang, Q.; Guan, X. The novel role of miRNAs for tamoxifen resistance in human breast cancer. Cell. Mol. Life Sci. 2015, 72, 2575–2584. [Google Scholar] [CrossRef] [PubMed]
- Li, C.H.; Chen, Y. Targeting long non-coding RNAs in cancers: Progress and prospects. Int. J. Biochem. Cell Biol. 2013, 45, 1895–1910. [Google Scholar] [CrossRef] [PubMed]
- Orom, U.A.; Derrien, T.; Beringer, M.; Gumireddy, K.; Gardini, A.; Bussotti, G.; Lai, F.; Zytnicki, M.; Notredame, C.; Huang, Q.; et al. Long noncoding RNAs with enhancer-like function in human cells. Cell 2010, 143, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Orom, U.A.; Shiekhattar, R. Noncoding RNAs and enhancers: Complications of a long-distance relationship. Trends Genet. 2011, 27, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Prensner, J.R.; Chinnaiyan, A.M. The emergence of lncRNAs in cancer biology. Cancer Discov. 2011, 1, 391–407. [Google Scholar] [CrossRef] [PubMed]
- Lottin, S.; Adriaenssens, E.; Dupressoir, T.; Berteaux, N.; Montpellier, C.; Coll, J.; Dugimont, T.; Curgy, J.J. Overexpression of an ectopic H19 gene enhances the tumorigenic properties of breast cancer cells. Carcinogenesis 2002, 23, 1885–1895. [Google Scholar] [CrossRef] [PubMed]
- Mourtada-Maarabouni, M.; Pickard, M.R.; Hedge, V.L.; Farzaneh, F.; Williams, G.T. GAS5, a non-protein-coding RNA, controls apoptosis and is downregulated in breast cancer. Oncogene 2009, 28, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Godinho, M.F.; Sieuwerts, A.M.; Look, M.P.; Meijer, D.; Foekens, J.A.; Dorssers, L.C.; van Agthoven, T. Relevance of BCAR4 in tamoxifen resistance and tumour aggressiveness of human breast cancer. Br. J. Cancer 2010, 103, 1284–1291. [Google Scholar] [CrossRef] [PubMed]
- Hayes, E.L.; Lewis-Wambi, J.S. Mechanisms of endocrine resistance in breast cancer: An overview of the proposed roles of noncoding RNA. Breast Cancer Res. 2015, 17, 40. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Malouf, G.G.; Chen, Y.; Zhang, J.; Yao, H.; Valero, V.; Weinstein, J.N.; Spano, J.P.; Meric-Bernstam, F.; Khayat, D.; et al. Comprehensive analysis of long non-coding RNAs in human breast cancer clinical subtypes. Oncotarget 2014, 5, 9864–9876. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Yang, Y.A.; Zhang, A.; Fong, K.W.; Kim, J.; Song, B.; Li, S.; Zhao, J.C.; Yu, J. LncRNA HOTAIR enhances er signaling and confers tamoxifen resistance in breast cancer. Oncogene 2016, 35, 2746–2755. [Google Scholar] [CrossRef] [PubMed]
- Xing, Z.; Lin, A.; Li, C.; Liang, K.; Wang, S.; Liu, Y.; Park, P.K.; Qin, L.; Wei, Y.; Hawke, D.H.; et al. LncRNA directs cooperative epigenetic regulation downstream of chemokine signals. Cell 2014, 159, 1110–1125. [Google Scholar] [CrossRef] [PubMed]
- Barton, S.; Swanton, C. Recent developments in treatment stratification for metastatic breast cancer. Drugs 2011, 71, 2099–2113. [Google Scholar] [CrossRef] [PubMed]
- De Lima, G.R.; Facina, G.; Shida, J.Y.; Chein, M.B.; Tanaka, P.; Dardes, R.C.; Jordan, V.C.; Gebrim, L.H. Effects of low dose tamoxifen on normal breast tissue from premenopausal women. Eur. J. Cancer 2003, 39, 891–898. [Google Scholar] [CrossRef]
- Guerrieri-Gonzaga, A.; Botteri, E.; Lazzeroni, M.; Rotmensz, N.; Goldhirsch, A.; Varricchio, C.; Serrano, D.; Cazzaniga, M.; Bassi, F.; Luini, A.; et al. Low-dose tamoxifen in the treatment of breast ductal intraepithelial neoplasia: Results of a large observational study. Ann. Oncol. 2010, 21, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Maltoni, C.; Minardi, F.; Belpoggi, F.; Pinto, C.; Lenzi, A.; Filippini, F. Experimental results on the chemopreventive and side effects of tamoxifen using a human-equivalent animal model. In The Scientific Bases of Cancer Chemoprevention; Maltoni, C., Soffritti, M., Davis, W., Eds.; Elsevier Science BV: Amsterdam, The Netherlands, 1996; p. 10. [Google Scholar]
- Decensi, A.; Robertson, C.; Viale, G.; Pigatto, F.; Johansson, H.; Kisanga, E.R.; Veronesi, P.; Torrisi, R.; Cazzaniga, M.; Mora, S.; et al. A randomized trial of low-dose tamoxifen on breast cancer proliferation and blood estrogenic biomarkers. J. Natl. Cancer Inst. 2003, 95, 779–790. [Google Scholar] [CrossRef] [PubMed]
- Rouanet, P.; Linares-Cruz, G.; Dravet, F.; Poujol, S.; Gourgou, S.; Simony-Lafontaine, J.; Grenier, J.; Kramar, A.; Girault, J.; Le Nestour, E.; et al. Neoadjuvant percutaneous 4-hydroxytamoxifen decreases breast tumoral cell proliferation: A prospective controlled randomized study comparing three doses of 4-hydroxytamoxifen gel to oral tamoxifen. J. Clin. Oncol. 2005, 23, 2980–2987. [Google Scholar] [CrossRef] [PubMed]
- Mansel, R.; Goyal, A.; Nestour, E.L.; Masini-Eteve, V.; O'Connell, K.; Afimoxifene Breast Pain Research Group. A phase II trial of afimoxifene (4-hydroxytamoxifen gel) for cyclical mastalgia in premenopausal women. Breast Cancer Res. Treat. 2007, 106, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.; Chen, Y.; Lim, E.; Wimberly, H.; Bailey, S.T.; Imai, Y.; Rimm, D.L.; Liu, X.S.; Brown, M. Targeting androgen receptor in estrogen receptor-negative breast cancer. Cancer Cell 2011, 20, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Gucalp, A.; Tolaney, S.; Isakoff, S.J.; Ingle, J.N.; Liu, M.C.; Carey, L.A.; Blackwell, K.; Rugo, H.; Nabell, L.; Forero, A.; et al. Phase II trial of bicalutamide in patients with androgen receptor-positive, estrogen receptor-negative metastatic breast cancer. Clin. Cancer Res. 2013, 19, 5505–5512. [Google Scholar] [CrossRef] [PubMed]
- Rimkus, T.K.; Carpenter, R.L.; Qasem, S.; Chan, M.; Lo, H.W. Targeting the Sonic Hedgehog signaling pathway: Review of Smoothened and GLI inhibitors. Cancers (Basel) 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Dlugosz, A.A.; Talpaz, M. Following the Hedgehog to new cancer therapies. N. Engl. J. Med. 2009, 361, 1202–1205. [Google Scholar] [CrossRef] [PubMed]
- Block, A.M.; Alite, F.; Diaz, A.Z.; Borrowdale, R.W.; Clark, J.I.; Choi, M. Combination trimodality therapy using Vismodegib for basal cell carcinoma of the face. Case Rep. Oncol. Med. 2015, 2015, 827608. [Google Scholar] [CrossRef] [PubMed]
- Di Magno, L.; Coni, S.; di Marcotullio, L.; Canettieri, G. Digging a hole under Hedgehog: Downstream inhibition as an emerging anticancer strategy. Biochim. Biophys. Acta 2015, 1856, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Wang, Y.; Liu, Z.; Sun, Y.; Wang, X.; Wei, G.; Wei, J. Metformin exerts anticancer effects through the inhibition of the Sonic Hedgehog signaling pathway in breast cancer. Int. J. Mol. Med. 2015, 36, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Della Corte, C.M.; Ciaramella, V.; di Mauro, C.; Castellone, M.D.; Papaccio, F.; Fasano, M.; Sasso, F.C.; Martinelli, E.; Troiani, T.; de Vita, F.; et al. Metformin increases antitumor activity of MEK inhibitors through GLI1 downregulation in LKB1 positive human NSCLC cancer cells. Oncotarget 2016, 7, 4265–4278. [Google Scholar] [PubMed]
- Li, Z.; Rana, T.M. Therapeutic targeting of microRNAs: Current status and future challenges. Nat. Rev. Drug Discov. 2014, 13, 622–638. [Google Scholar] [CrossRef] [PubMed]
- Matsui, M.; Corey, D.R. Non-coding RNAs as drug targets. Nat. Rev. Drug Discov. 2016. [Google Scholar] [CrossRef] [PubMed]
- Burnett, J.C.; Rossi, J.J. RNA-based therapeutics: Current progress and future prospects. Chem. Biol. 2012, 19, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.C.; Wood, M.J. Therapeutic targeting of non-coding RNAs. Essays Biochem. 2013, 54, 127–145. [Google Scholar] [CrossRef] [PubMed]




© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rondón-Lagos, M.; Villegas, V.E.; Rangel, N.; Sánchez, M.C.; Zaphiropoulos, P.G. Tamoxifen Resistance: Emerging Molecular Targets. Int. J. Mol. Sci. 2016, 17, 1357. https://doi.org/10.3390/ijms17081357
Rondón-Lagos M, Villegas VE, Rangel N, Sánchez MC, Zaphiropoulos PG. Tamoxifen Resistance: Emerging Molecular Targets. International Journal of Molecular Sciences. 2016; 17(8):1357. https://doi.org/10.3390/ijms17081357
Chicago/Turabian StyleRondón-Lagos, Milena, Victoria E. Villegas, Nelson Rangel, Magda Carolina Sánchez, and Peter G. Zaphiropoulos. 2016. "Tamoxifen Resistance: Emerging Molecular Targets" International Journal of Molecular Sciences 17, no. 8: 1357. https://doi.org/10.3390/ijms17081357