
Autocrine and Paracrine Mechanisms Promoting Chemoresistance in Cholangiocarcinoma

 , , ,
, , ,
Abstract
:
1. Introduction
2. Mechanisms of Chemoresistance
Mechanisms of Chemoresistance Depending upon Evasion from Apoptosis
3. Tumor Reactive Stroma
3.1. Cancer-Associated Fibroblasts
3.2. Tumor-Associated Macrophages
3.3. Endothelial Cells
3.4. Cancer Stem Cells
4. Main Signals Released within the TRS Promoting CCA Chemoresistance
4.1. Interleukin-6 (IL-6) Family
4.2. Platelet-Derived Growth Factor
4.3. Wnt/β-Catenin
4.4. Hippo Pathway
4.5. Notch
4.6. Hedgehog
5. Concluding Remarks and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Blechacz, B.; Gores, G.J. Cholangiocarcinoma: Advances in pathogenesis, diagnosis, and treatment. Hepatology 2008, 48, 308–321. [Google Scholar] [CrossRef] [PubMed]
- Gatto, M.; Bragazzi, M.C.; Semeraro, R.; Napoli, C.; Gentile, R.; Torrice, A.; Gaudio, E.; Alvaro, D. Cholangiocarcinoma: Update and future perspectives. Dig. Liver Dis. 2010, 42, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Floreani, A.; Lisiero, M.; Baldovin, T.; Baldo, V. Epidemiological aspects of biliary tree tumors in a region of northern Italy: Emerging trends and sex-based differences. Eur. J. Gastroenterol. Hepatol. 2013, 25, 1347–1451. [Google Scholar] [CrossRef] [PubMed]
- Bragazzi, M.; Cardinale, V.; Carpino, G.; Venere, R.; Semeraro, R.; Gentile, R.; Gaudio, E.; Alvaro, D. Cholangiocarcinoma: Epidemiology and risk factors. Transl. Gastrointest. Cancer 2011, 1, 21–32. [Google Scholar]
- Wolpin, B.M.; Mayer, R.J. A step forward in the treatment of advanced biliary tract cancer. N. Engl. J. Med. 2010, 362, 1335–1337. [Google Scholar] [CrossRef] [PubMed]
- Razumilava, N.; Gores, G.J. Classification, diagnosis, and management of cholangiocarcinoma. Clin. Gastroenterol. Hepatol. 2013, 11, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Bridgewater, J.; Galle, P.R.; Khan, S.A.; Llovet, J.M.; Park, J.W.; Patel, T.; Pawlik, T.M.; Gores, G.J. Guidelines for the diagnosis and management of intrahepatic cholangiocarcinoma. J. Hepatol. 2014, 60, 1268–1289. [Google Scholar] [CrossRef] [PubMed]
- Zabron, A.; Edwards, R.J.; Khan, S.A. The challenge of cholangiocarcinoma: Dissecting the molecular mechanisms of an insidious cancer. Dis. Model. Mech. 2013, 6, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Marin, J.J.; Lozano, E.; Briz, O.; Al-Abdulla, R.; Serrano, M.A.; Macias, R.I. Molecular Bases of Chemoresistance In Cholangiocarcinoma. Curr. Drug Targets 2015, in press. [Google Scholar] [CrossRef]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. ABC-02 Trial Investigators. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.T.; Li, Z.L.; He, Z.X.; Qiu, J.X.; Zhou, S.F. Molecular mechanisms for tumour resistance to chemotherapy. Clin. Exp. Pharmacol. Physiol. 2016, 43, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L.; et al. Expert consensus document: Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Strazzabosco, M.; Fabris, L.; Spirli, C. Pathophysiology of cholangiopathies. J. Clin. Gastroenterol. 2005, 39, S90–S102. [Google Scholar] [CrossRef] [PubMed]
- Franke, R.M.; Scherkenbach, L.A.; Sparreboom, A. Pharmacogenetics of the organic anion transporting polypeptide 1A2. Pharmacogenomics 2009, 10, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Wlcek, K.; Svoboda, M.; Riha, J.; Zakaria, S.; Olszewski, U.; Dvorak, Z.; Sellner, F.; Ellinger, I.; Jäger, W.; Thalhammer, T. The analysis of organic anion transporting polypeptide (OATP) mRNA and proteinpatterns in primary and metastatic liver cancer. Cancer Biol. Ther. 2011, 11, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Herraez, E.; Lozano, E.; Macias, R.I.; Vaquero, J.; Bujanda, L.; Banales, J.M.; Marin, J.J.; Briz, O. Expression of SLC22A1 variants may affect the response of hepatocellular carcinoma and cholangiocarcinoma to sorafenib. Hepatology 2013, 58, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- Marin, J.J. Plasma membrane transporters in modern liver pharmacology. Scientifica 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Namwat, N.; Amimanan, P.; Loilome, W.; Jearanaikoon, P.; Sripa, B.; Bhudhisawasdi, V.; Tassaneeyakul, W. Characterization of 5-fluorouracil resistant cholangiocarcinoma cell lines. Chemotherapy 2008, 54, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.J.; dos Santos, D.J.; Ferreira, M.J. P-glycoprotein and membrane roles in multidrug resistance. Future Med. Chem. 2015, 7, 929–946. [Google Scholar] [CrossRef] [PubMed]
- Eaton, J.E.; Talwalkar, J.A.; Lazaridis, K.N.; Gores, G.J.; Lindor, K.D. Pathogenesis of primary sclerosing cholangitis and advances in diagnosis and management. Gastroenterology 2013, 145, 521–536. [Google Scholar] [CrossRef] [PubMed]
- Perez, J.; Bardin, C.; Rigal, C.; Anthony, B.; Rousseau, R.; Dutour, A. Anti-MDR1 siRNA restores chemosensitivity in chemoresistant breast carcinoma and osteosarcoma cell lines. Anticancer Res. 2011, 31, 2813–2820. [Google Scholar] [PubMed]
- Hahnvajanawong, C.; Chaiyagool, J.; Seubwai, W.; Bhudhisawasdi, V.; Namwat, N.; Khuntikeo, N.; Sripa, B.; Pugkhem, A.; Tassaneeyakul, W. Orotate phosphoribosyl transferase mRNA expression and the response of cholangiocarcinoma to 5-fluorouracil. World J. Gastroenterol. 2012, 18, 3955–3961. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, T.; Takayama, T.; Miyanishi, K.; Nobuoka, A.; Hayashi, T.; Abe, T.; Kato, J.; Sakon, K.; Naniwa, Y.; Tanabe, H.; et al. Reversal of multiple drug resistance in cholangiocarcinoma by the glutathione S-transferase-pi-specific inhibitor O1-hexadecyl-γ-glutamyl-S-benzylcysteinyl-d-phenylglycine ethylester. J. Pharmacol. Exp. Ther. 2003, 306, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Jansen, W.J.; Kolfschoten, G.M.; Erkelens, C.A.; Van Ark-Otte, J.; Pinedo, H.M.; Boven, E. Anti-tumor activity of CPT-11 in experimental human ovarian cancer and human soft-tissue sarcoma. Int. J. Cancer 1997, 73, 891–896. [Google Scholar] [CrossRef]
- Boyer, J.; McLean, E.G.; Aroori, S.; Wilson, P.; McCulla, A.; Carey, P.D.; Longley, D.B.; Johnston, P.G. Characterization of p53 wild-type and null isogenic colorectal cancer cell lines resistant to 5-fluorouracil, oxaliplatin, and irinotecan. Clin. Cancer Res. 2004, 10, 2158–2167. [Google Scholar] [CrossRef] [PubMed]
- Alvaro, D.; Barbaro, B.; Franchitto, A.; Onori, P.; Glaser, S.S.; Alpini, G.; Francis, H.; Marucci, L.; Sterpetti, P.; Ginanni-Corradini, S.; et al. Estrogens and insulin-like growth factor 1 modulate neoplastic cell growth in human cholangiocarcinoma. Am. J. Pathol. 2006, 169, 877–888. [Google Scholar] [CrossRef] [PubMed]
- Sampson, L.K.; Vickers, S.M.; Ying, W.; Phillips, J.O. Tamoxifen-mediated growth inhibition of human cholangiocarcinoma. Cancer Res. 1997, 57, 1743–1749. [Google Scholar] [PubMed]
- Marzioni, M.; Torrice, A.; Saccomanno, S.; Rychlicki, C.; Agostinelli, L.; Pierantonelli, I.; Rhönnstad, P.; Trozzi, L.; Apelqvist, T.; Gentile, R.; et al. An oestrogen receptor β-selective agonist exerts anti-neoplastic effects in experimental intrahepatic cholangiocarcinoma. Dig. Liver Dis. 2012, 44, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Hector, S.; Bolanowska-Higdon, W.; Zdanowicz, J.; Hitt, S.; Pendyala, L. In vitro studies on the mechanisms of oxaliplatin resistance. Cancer Chemother. Pharmacol. 2001, 48, 398–406. [Google Scholar] [CrossRef]
- Metzger, R.; Leichman, C.G.; Danenberg, K.D.; Danenberg, P.V.; Lenz, H.J.; Hayashi, K.; Groshen, S.; Salonga, D.; Cohen, H.; Laine, L.; et al. ERCC1 mRNA levels complement thymidylate synthase mRNA levels in predicting response and survival for gastric cancer patients receiving combination cisplatin and fluorouracil chemotherapy. J. Clin. Oncol. 1998, 16, 309–316. [Google Scholar] [PubMed]
- Hwang, I.G.; Jang, J.S.; Do, J.H.; Kang, J.H.; Lee, G.W.; Oh, S.Y.; Kwon, H.C.; Jun, H.J.; Lim, H.Y.; Lee, S.; et al. Different relation between ERCC1 overexpression and treatment outcomes of two platinum agents in advanced biliary tract adenocarcinoma patients. Cancer Chemother. Pharmacol. 2011, 68, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Selvakumaran, M.; Pisarcik, D.A.; Bao, R.; Yeung, A.T.; Hamilton, T.C. Enhanced cisplatin cytotoxicity by disturbing the nucleotide excision repair pathway in ovarian cancer cell lines. Cancer Res. 2003, 63, 1311–1316. [Google Scholar] [PubMed]
- Asakawa, H.; Koizumi, H.; Koike, A.; Takahashi, M.; Wu, W.; Iwase, H.; Fukuda, M.; Ohta, T. Prediction of breast cancer sensitivity to neoadjuvant chemotherapy based on status of DNA damage repair proteins. Breast Cancer Res. 2010, 12. [Google Scholar] [CrossRef] [PubMed]
- Obama, K.; Satoh, S.; Hamamoto, R.; Sakai, Y.; Nakamura, Y.; Furukawa, Y. Enhanced expression of RAD51 associating protein-1 is involved in the growth of intrahepatic cholangiocarcinoma cells. Clin. Cancer Res. 2008, 14, 1333–1339. [Google Scholar] [CrossRef] [PubMed]
- Fink, D.; Aebi, S.; Howell, S.B. The role of DNA mismatch repair in drug resistance. Clin. Cancer Res. 1998, 4, 1–6. [Google Scholar] [PubMed]
- Chaney, S.G.; Campbell, S.L.; Temple, B.; Bassett, E.; Wu, Y.; Faldu, M. Protein interactions with platinum-DNA adducts: From structure to function. J. Inorg. Biochem. 2004, 98, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Vogler, M. Targeting BCL2-Proteins for the Treatment of Solid Tumours. Adv. Med. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Harnois, D.M.; Que, F.G.; Celli, A.; LaRusso, N.F.; Gores, G.J. Bcl-2 is overexpressed and alters the threshold for apoptosis in a cholangiocarcinoma cell line. Hepatology 1997, 26, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Fabris, L.; Strazzabosco, M.; Crosby, H.A.; Ballardini, G.; Hubscher, S.G.; Kelly, D.A.; Neuberger, J.M.; Strain, A.J.; Joplin, R. Characterization and isolation of ductular cells coexpressing neural cell adhesion molecule and Bcl-2 from primary cholangiopathies and ductal plate malformations. Am. J. Pathol. 2000, 156, 1599–1612. [Google Scholar] [CrossRef]
- Minagawa, N.; Kruglov, E.A.; Dranoff, J.A.; Robert, M.E.; Gores, G.J.; Nathanson, M.H. The anti-apoptotic protein Mcl-1 inhibits mitochondrial Ca2+ signals. J. Biol. Chem. 2005, 280, 33637–33644. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Cheng, C.; Balasis, M.E.; Liu, Y.; Garner, T.P.; Daniel, K.G.; Li, J.; Qin, Y.; Gavathiotis, E.; Sebti, S.M. Design, synthesis and evaluation of marinopyrrole derivatives as selective inhibitors of Mcl-1 binding to pro-apoptotic Bim and dual Mcl-1/Bcl-xL inhibitors. Eur. J. Med. Chem. 2015, 90, 315–331. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhao, Z.; Wu, K.; Xu, Z.; Liu, K. MCL-1 is the key target of adjuvant chemotherapy to reverse the cisplatin-resistance in NSCLC. Gene 2016, 587, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.; Zhao, J.; Wang, T.; Duan, J.; Zhang, Y.; Deng, X. Role of bile salt in regulating Mcl-1 phosphorylation and chemoresistance in hepatocellular carcinoma cells. Mol. Cancer 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Rampino, N.; Yamamoto, H.; Ionov, Y.; Li, Y.; Sawai, H.; Reed, J.C.; Perucho, M. Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science 1997, 275, 967–969. [Google Scholar] [CrossRef] [PubMed]
- Kymionis, G.D.; Dimitrakakis, C.E.; Konstadoulakis, M.M.; Arzimanoglou, I.; Leandros, E.; Chalkiadakis, G.; Keramopoulos, A.; Michalas, S. Can expression of apoptosis genes, bcl-2 and bax, predict survival and responsiveness to chemotherapy in node-negative breast cancer patients? J. Surg. Res. 2001, 99, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Sjöström, J.; Blomqvist, C.; von Boguslawski, K.; Bengtsson, N.O.; Mjaaland, I.; Malmström, P.; Ostenstadt, B.; Wist, E.; Valvere, V.; Takayama, S.; et al. The predictive value of bcl-2, bax, bcl-xL, bag-1, fas, and fasL for chemotherapy response in advanced breast cancer. Clin. Cancer Res. 2002, 8, 811–816. [Google Scholar] [PubMed]
- Paradiso, A.; Simone, G.; Lena, M.D.; Leone, B.; Vallejo, C.; Lacava, J.; Dellapasqua, S.; Daidone, M.G.; Costa, A. Expression of apoptosis-related markers and clinical outcome in patients with advanced colorectal cancer. Br. J. Cancer 2001, 84, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.; Min, J.K.; Lee, J.W.; Kim, D.G.; Hong, H.J. Acquisition of chemoresistance in intrahepatic cholangiocarcinoma cells by activation of AKT and extracellular signal-regulated kinase (ERK)1/2. Biochem. Biophys. Res. Commun. 2011, 405, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Bunz, F.; Hwang, P.M.; Torrance, C.; Waldman, T.; Zhang, Y.; Dillehay, L.; Williams, J.; Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J. Clin. Investig. 1999, 104, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Thomas, H.C.; Toledano, M.B.; Cox, I.J.; Taylor-Robinson, S.D. p53 Mutations in human cholangiocarcinoma: A review. Liver Int. 2005, 25, 704–716. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.Q.; Jiang, X.; Fraser, M.; Li, M.; Dan, H.C.; Sun, M.; Tsang, B.K. Role of X-linked inhibitor of apoptosis protein in chemoresistance in ovarian cancer: Possible involvement of the phosphoinositide-3 kinase/Akt pathway. Drug Resist. Updat. 2002, 5, 131–146. [Google Scholar] [CrossRef]
- Tamm, I.; Wang, Y.; Sausville, E.; Scudiero, D.A.; Vigna, N.; Oltersdorf, T.; Reed, J.C. IAP-family protein survivin inhibits caspase activity and apoptosis induced by Fas (CD95), Bax, caspases, and anticancer drugs. Cancer Res. 1998, 58, 5315–5320. [Google Scholar] [PubMed]
- Zaffaroni, N.; Pennati, M.; Colella, G.; Perego, P.; Supino, R.; Gatti, L.; Pilotti, S.; Zunino, F.; Daidone, M.G. Expression of the anti-apoptotic gene survivin correlates with taxol resistance in human ovarian cancer. Cell. Mol. Life Sci. 2002, 59, 1406–1412. [Google Scholar] [CrossRef] [PubMed]
- Kato, J.; Kuwabara, Y.; Mitani, M.; Shinoda, N.; Sato, A.; Toyama, T.; Mitsui, A.; Nishiwaki, T.; Moriyama, S.; Kudo, J.; et al. Expression of survivin in esophageal cancer: Correlation with the prognosis and response to chemotherapy. Int. J. Cancer 2001, 95, 92–95. [Google Scholar] [CrossRef]
- Martinez-Becerra, P.; Vaquero, J.; Romero, M.R.; Lozano, E.; Anadon, C.; Macias, R.I.; Serrano, M.A.; Grañé-Boladeras, N.; Muñoz-Bellvis, L.; Alvarez, L.; et al. No correlation between the expression of FXR and genes involved in multidrug resistance phenotype of primary liver tumors. Mol. Pharm. 2012, 9, 1693–1704. [Google Scholar] [CrossRef] [PubMed]
- Wehrkamp, C.J.; Gutwein, A.R.; Natarajan, S.K.; Phillippi, M.A.; Mott, J.L. XIAP antagonist embelin inhibited proliferation of cholangiocarcinoma cells. PLoS ONE 2014, 9, e90238. [Google Scholar] [CrossRef] [PubMed]
- Ueno, Y.; Ishii, M.; Yahagi, K.; Mano, Y.; Kisara, N.; Nakamura, N.; Shimosegawa, T.; Toyota, T.; Nagata, S. Fas-mediated cholangiopathy in the murine model of graft versus host disease. Hepatology 2000, 31, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Alvaro, D.; Alpini, G.; Onori, P.; Perego, L.; Svegliati-Baroni, G.; Franchitto, A.; Baiocchi, L.; Glaser, S.S.; Le Sage, G.; Folli, F.; et al. Estrogens stimulate proliferation of intrahepatic biliary epithelium in rats. Gastroenterology 2000, 119, 1681–1691. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Kojima, Y.; Ikejima, K.; Harada, K.; Yamashina, S.; Okumura, K.; Aoyama, T.; Frese, S.; Ikeda, H.; Haynes, N.M.; et al. Death receptor 5 mediated-apoptosis contributes to cholestatic liver disease. Proc. Natl. Acad. Sci. USA 2008, 105, 10895–10900. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Shirley, S.; Dufour, F. Death receptors as targets in cancer. Br. J. Pharmacol. 2013, 169, 1723–1744. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; McDonald, E.R., 3rd; Dicker, D.T.; El-Deiry, W.S. Deficient tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptor transport to the cell surface in human colon cancer cells selected for resistance to TRAIL-induced apoptosis. J. Biol. Chem. 2004, 279, 35829–35839. [Google Scholar] [CrossRef] [PubMed]
- Wise, J.F.; Berkova, Z.; Mathur, R.; Zhu, H.; Braun, F.K.; Tao, R.H.; Sabichi, A.L.; Ao, X.; Maeng, H.; Samaniego, F. Nucleolin inhibits Fas ligand binding and suppresses Fas-mediated apoptosis in vivo via a surface nucleolin-Fas complex. Blood 2013, 121, 4729–4739. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Tumor-necrosis-factor-related apoptosis-inducing ligand (TRAIL). Adv. Exp. Med. Biol. 2014, 818, 167–180. [Google Scholar] [PubMed]
- Longley, D.B.; Wilson, T.R.; McEwan, M.; Allen, W.L.; McDermott, U.; Galligan, L.; Johnston, P.G. c-FLIP inhibits chemotherapy-induced colorectal cancer cell death. Oncogene 2006, 25, 838–848. [Google Scholar] [CrossRef] [PubMed]
- Okano, H.; Shiraki, K.; Inoue, H.; Kawakita, T.; Yamanaka, T.; Deguchi, M.; Sugimoto, K.; Sakai, T.; Ohmori, S.; Fujikawa, K.; et al. Cellular FLICE/caspase-8-inhibitory protein as a principal regulator of cell death and survival in human hepatocellular carcinoma. Lab. Investig. 2003, 83, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, T.F.; Samal, A.B.; Bedwell, G.J.; Chen, Y.; Saad, J.S. Structural and biophysical characterization of the interactions between the death domain of Fas receptor and calmodulin. J. Biol. Chem. 2013, 288, 21898–21908. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Karin, M. NF-kappaB in cancer: A marked target. Semin. Cancer Biol. 2003, 13, 107–114. [Google Scholar] [CrossRef]
- Kato, T.; Duffey, D.C.; Ondrey, F.G.; Dong, G.; Chen, Z.; Cook, J.A.; Mitchell, J.B.; Van Waes, C. Cisplatin and radiation sensitivity in human head and neck squamous carcinomas are independently modulated by glutathione and transcription factor NF-κB. Head Neck 2000, 22, 748–759. [Google Scholar] [CrossRef]
- Arlt, A.; Gehrz, A.; Müerköster, S.; Vorndamm, J.; Kruse, M.L.; Fölsch, U.R.; Schäfer, H. Role of NF-κB and Akt/PI3K in the resistance of pancreatic carcinoma cell lines against gemcitabine-induced cell death. Oncogene 2003, 22, 3243–3251. [Google Scholar] [CrossRef] [PubMed]
- Gores, G.J. Cholangiocarcinoma: Current concepts and insights. Hepatology 2003, 37, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Cadamuro, M.; Morton, S.D.; Strazzabosco, M.; Fabris, L. Unveiling the Role of Tumor Reactive Stroma in Cholangiocarcinoma: An Opportunity for New Therapeutic Strategies. Transl. Gastrointest. Cancer 2013, 2, 130–144. [Google Scholar]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Duluc, D.; Delneste, Y.; Tan, F.; Moles, M.P.; Grimaud, L.; Lenoir, J.; Preisser, L.; Anegon, I.; Catala, L.; Ifrah, N.; et al. Tumor-associated leukemia inhibitory factor and IL-6 skew monocyte differentiation into tumor-associated macrophage-like cells. Blood 2007, 110, 4319–4330. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [PubMed]
- Schober, M.; Jesenofsky, R.; Faissner, R.; Weidenauer, C.; Hagmann, W.; Michl, P.; Heuchel, R.L.; Haas, S.L.; Löhr, J.M. Desmoplasia and chemoresistance in pancreatic cancer. Cancers 2014, 6, 2137–2154. [Google Scholar] [CrossRef] [PubMed]
- Velaei, K.; Samadi, N.; Barazvan, B.; Soleimani Rad, J. Tumor microenvironment-mediated chemoresistance in breast cancer. Breast 2016, 30, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Jayson, G.C.; Kohn, E.C.; Kitchener, H.C.; Ledermann, J.A. Ovarian cancer. Lancet 2014, 384, 1376–1388. [Google Scholar] [CrossRef]
- Zhang, W.; Meng, Y.; Liu, N.; Wen, X.F.; Yang, T. Insights into Chemoresistance of Prostate Cancer. Int. J. Biol. Sci. 2015, 11, 1160–1170. [Google Scholar] [CrossRef] [PubMed]
- Goubran, H.A.; Kotb, R.R.; Stakiw, J.; Emara, M.E.; Burnouf, T. Regulation of tumor growth and metastasis: The role of tumor microenvironment. Cancer Growth Metastasis 2014, 7, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Sakai, R. Direct Interaction between Carcinoma Cells and Cancer Associated Fibroblasts for the Regulation of Cancer Invasion. Cancers 2015, 7, 2054–2062. [Google Scholar] [CrossRef] [PubMed]
- McCarroll, J.A.; Naim, S.; Sharbeen, G.; Russia, N.; Lee, J.; Kavallaris, M.; Goldstein, D.; Phillips, P.A. Role of pancreatic stellate cells in chemoresistance in pancreatic cancer. Front. Physiol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Chi, J.Y.; Hsiao, Y.W.; Li, C.F.; Lo, Y.C.; Lin, Z.Y.; Hong, J.Y.; Liu, Y.M.; Han, X.; Wang, S.M.; Chen, B.K.; et al. Targeting chemotherapy-induced PTX3 in tumor stroma to prevent the progression of drug-resistant cancers. Oncotarget 2015, 6, 23987–24001. [Google Scholar] [CrossRef] [PubMed]
- Chu, A.S.; Diaz, R.; Hui, J.J.; Yanger, K.; Zong, Y.; Alpini, G.; Stanger, B.Z.; Wells, R.G. Lineage tracing demonstrates no evidence of cholangiocyte epithelial-to-mesenchymal transition in murine models of hepatic fibrosis. Hepatology 2011, 53, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Brenner, D.A. Is it the end of the line for the EMT? Hepatology 2011, 53, 1433–1435. [Google Scholar] [CrossRef] [PubMed]
- Cadamuro, M.; Nardo, G.; Indraccolo, S.; Dall’olmo, L.; Sambado, L.; Moserle, L.; Franceschet, I.; Colledan, M.; Massani, M.; Stecca, T.; et al. Platelet-derived growth factor-D and Rho GTPases regulate recruitment of cancer-associated fibroblasts in cholangiocarcinoma. Hepatology 2013, 58, 1042–1053. [Google Scholar] [CrossRef] [PubMed]
- Fabris, L.; Strazzabosco, M. Epithelial-mesenchymal interactions in biliary diseases. Semin. Liver Dis. 2011, 31, 11–32. [Google Scholar] [CrossRef] [PubMed]
- Clapéron, A.; Mergey, M.; Aoudjehane, L.; Ho-Bouldoires, T.H.; Wendum, D.; Prignon, A.; Merabtene, F.; Firrincieli, D.; Desbois-Mouthon, C.; Scatton, O.; et al. Hepatic myofibroblasts promote the progression of human cholangiocarcinoma through activation of epidermal growth factor receptor. Hepatology 2013, 58, 2001–2011. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.; Gores, G.J. Pathogenesis, diagnosis, and management of cholangiocarcinoma. Gastroenterology 2013, 145, 1215–1229. [Google Scholar] [CrossRef] [PubMed]
- Utispan, K.; Sonongbua, J.; Thuwajit, P.; Chau-In, S.; Pairojkul, C.; Wongkham, S.; Thuwajit, C. Periostin activates integrin α5β1 through a PI3K/AKT-dependent pathway in invasion of cholangiocarcinoma. Int. J. Oncol. 2012, 41, 1110–1118. [Google Scholar] [PubMed]
- Sirica, A.E.; Almenara, J.A.; Li, C. Periostin in intrahepatic cholangiocarcinoma: Pathobiological insights and clinical implications. Exp. Mol. Pathol. 2014, 97, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Hasita, H.; Komohara, Y.; Okabe, H.; Masuda, T.; Ohnishi, K.; Lei, X.F.; Beppu, T.; Baba, H.; Takeya, M. Significance of alternatively activated macrophages in patients with intrahepatic cholangiocarcinoma. Cancer Sci. 2010, 101, 1913–1919. [Google Scholar] [CrossRef] [PubMed]
- Raggi, C.; Correnti, M.; Sica, A.; Andersen, J.B.; Cardinale, V.; Alvaro, D.; Chiorino, G.; Forti, E.; Glaser, S.; Alpini, G.; et al. Cholangiocarcinoma stem-like subset shapes tumor-initiating niche by educating associated macrophages. J. Hepatol. 2017, 66, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, L.; Cadamuro, M.; Spirlì, C.; Fiorotto, R.; Lecchi, S.; Morell, C.M.; Popov, Y.; Scirpo, R.; de Matteis, M.; Amenduni, M.; et al. Macrophage recruitment by fibrocystin-defective biliary epithelial cells promotes portal fibrosis in congenital hepatic fibrosis. Hepatology 2016, 63, 965–982. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.E.; Pollard, J.W. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006, 66, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Schito, L.; Rey, S.; Tafani, M.; Zhang, H.; Wong, C.C.; Russo, A.; Russo, M.A.; Semenza, G.L. Hypoxia-inducible factor 1-dependent expression of platelet-derived growth factor B promotes lymphatic metastasis of hypoxic breast cancer cells. Proc. Natl. Acad. Sci. USA 2012, 109, E2707–E2716. [Google Scholar] [CrossRef] [PubMed]
- Karnezis, T.; Shayan, R.; Caesar, C.; Roufail, S.; Harris, N.C.; Ardipradja, K.; Zhang, Y.F.; Williams, S.P.; Farnsworth, R.H.; Chai, M.G.; et al. VEGF-D promotes tumor metastasis by regulating prostaglandins produced by the collecting lymphatic endothelium. Cancer Cell 2012, 21, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.T.; Hung, W.C. Inhibition of proliferation, sprouting, tube formation and Tie2 signaling of lymphatic endothelial cells by the histone deacetylase inhibitor SAHA. Oncol. Rep. 2013, 30, 961–967. [Google Scholar] [PubMed]
- Morine, Y.; Shimada, M.; Utsunomiya, T.; Imura, S.; Ikemoto, T.; Mori, H.; Hanaoka, J.; Kanamoto, M.; Iwahashi, S.; Miyake, H. Hypoxia inducible factor expression in intrahepatic cholangiocarcinoma. Hepatogastroenterology 2011, 58, 1439–1444. [Google Scholar] [CrossRef] [PubMed]
- Le Calvé, B.; Griveau, A.; Vindrieux, D.; Maréchal, R.; Wiel, C.; Svrcek, M.; Gout, J.; Azzi, L.; Payen, L.; Cros, J.; et al. Lysyl oxidase family activity promotes resistance of pancreatic ductal adenocarcinoma to chemotherapy by limiting the intratumoral anticancer drug distribution. Oncotarget 2016, 7, 32100–32112. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T.; Jung, A.; Spaderna, S.; Hlubek, F.; Kirchner, T. Opinion: Migrating cancer stem cells—An integrated concept of malignant tumour progression. Nat. Rev. Cancer 2005, 5, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Reim, F.; Dombrowski, Y.; Ritter, C.; Buttmann, M.; Häusler, S.; Ossadnik, M.; Krockenberger, M.; Beier, D.; Beier, C.P.; Dietl, J.; et al. Immunoselection of breast and ovarian cancer cells with trastuzumab and natural killer cells: Selective escape of CD44high/CD24low/HER2low breast cancer stem cells. Cancer Res. 2009, 69, 8058–8066. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, T.; Wauthier, E.; Dinh, T.A.; Selitsky, S.R.; Reyna-Neyra, A.; Carpino, G.; Levine, R.; Cardinale, V.; Klimstra, D.; Gaudio, E.; et al. Model of fibrolamellar hepatocellular carcinomas reveals striking enrichment in cancer stem cells. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, V.; Renzi, A.; Carpino, G.; Torrice, A.; Bragazzi, M.C.; Giuliante, F.; DeRose, A.M.; Fraveto, A.; Onori, P.; Napoletano, C.; et al. Profiles of cancer stem cell subpopulations in cholangiocarcinomas. Am. J. Pathol. 2015, 185, 1724–1739. [Google Scholar] [CrossRef] [PubMed]
- Alison, M.R. Liver stem cells: Implications for hepatocarcinogenesis. Stem Cell Rev. 2005, 1, 253–260. [Google Scholar] [CrossRef]
- Haraguchi, N.; Ishii, H.; Mimori, K.; Tanaka, F.; Ohkuma, M.; Kim, H.M.; Akita, H.; Takiuchi, D.; Hatano, H.; Nagano, H.; et al. CD13 is a therapeutic target in human liver cancer stem cells. J. Clin. Investig. 2010, 120, 3326–3339. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.K.; Castilho, A.; Cheung, V.C.; Tang, K.H.; Ma, S.; Ng, I.O. CD24+ liver tumor-initiating cells drive self-renewal and tumor initiation through STAT3-mediated NANOG regulation. Cell Stem Cell 2011, 9, 50–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keeratichamroen, S.; Leelawat, K.; Thongtawee, T.; Narong, S.; Aegem, U.; Tujinda, S.; Praditphol, N.; Tohtong, R. Expression of CD24 in cholangiocarcinoma cells is associated with disease progression and reduced patient survival. Int. J. Oncol. 2011, 39, 873–881. [Google Scholar] [PubMed]
- Leelawat, K.; Keeratichamroen, S.; Leelawat, S.; Tohtong, R. CD24 induces the invasion of cholangiocarcinoma cells by upregulating CXCR4 and increasing the phosphorylation of ERK1/2. Oncol. Lett. 2013, 6, 1439–1446. [Google Scholar] [CrossRef] [PubMed]
- Takaishi, S.; Okumura, T.; Tu, S.; Wang, S.S.; Shibata, W.; Vigneshwaran, R.; Gordon, S.A.; Shimada, Y.; Wang, T.C. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells 2009, 27, 1006–1020. [Google Scholar] [PubMed]
- Gu, M.J.; Jang, B.I. Clinicopathologic significance of Sox2, CD44 and CD44v6 expression in intrahepatic cholangiocarcinoma. Pathol. Oncol. Res. 2014, 20, 655–660. [Google Scholar] [PubMed]
- Yang, Z.F.; Ho, D.W.; Ng, M.N.; Lau, C.K.; Yu, W.C.; Ngai, P.; Chu, P.W.; Lam, C.T.; Poon, R.T.; Fan, S.T. Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell 2008, 13, 153–166. [Google Scholar] [PubMed]
- Sukowati, C.H.; Anfuso, B.; Torre, G.; Francalanci, P.; Crocè, L.S.; Tiribelli, C. The expression of CD90/Thy-1 in hepatocellular carcinoma: An in vivo and in vitro study. PLoS ONE 2013, 8, e76830. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xiao, J.; Jiang, J.; Qin, R. CD133 and ALDH may be the molecular markers of cholangiocarcinoma stem cells. Int. J. Cancer 2011, 128, 1996–1997. [Google Scholar] [CrossRef] [PubMed]
- Shimada, M.; Sugimoto, K.; Iwahashi, S.; Utsunomiya, T.; Morine, Y.; Imura, S.; Ikemoto, T. CD133 expression is a potential prognostic indicator in intrahepatic cholangiocarcinoma. J. Gastroenterol. 2010, 45, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; He, F.; Liu, H.; Zhu, J.; Liu, Y.; Yin, Z.; Wang, L.; Guo, Y.; Wang, Z.; Yan, Q.; et al. CD133: A potential indicator for differentiation and prognosis of human cholangiocarcinoma. BMC Cancer 2011, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Xiao, J.; Shen, M.; Yahong, Y.; Tian, R.; Zhu, F.; Jiang, J.; Du, Z.; Hu, J.; Liu, W.; et al. Isolation and characterization of tumorigenic extrahepatic cholangiocarcinoma cells with stem cell-like properties. Int. J. Cancer 2011, 128, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Govaere, O.; Wouters, J.; Petz, M.; Vandewynckel, Y.P.; Van den Eynde, K.; Van den Broeck, A.; Verhulst, S.; Dollé, L.; Gremeaux, L.; Ceulemans, A.; et al. Laminin-332 sustains chemoresistance and quiescence as part of the human hepatic cancer stem cell niche. J. Hepatol. 2016, 64, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Cavalloni, G.; Peraldo-Neia, C.; Varamo, C.; Casorzo, L.; Dell’Aglio, C.; Bernabei, P.; Chiorino, G.; Aglietta, M.; Leone, F. Establishment and characterization of a human intrahepatic cholangiocarcinoma cell line derived from an Italian patient. Tumour Biol. 2016, 37, 4041–4052. [Google Scholar] [CrossRef] [PubMed]
- Bourguignon, L.Y.; Wong, G.; Earle, C.; Chen, L. Hyaluronan-CD44v3 interaction with Oct4-Sox2-Nanog promotes miR-302 expression leading to self-renewal, clonal formation, and cisplatin resistance in cancer stem cells from head and neck squamous cell carcinoma. J. Biol. Chem. 2012, 287, 32800–32824. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Werneburg, N.W.; Bronk, S.F.; Kaufmann, S.H.; Gores, G.J. Interleukin-6 contributes to Mcl-1 up-regulation and TRAIL resistance via an Akt-signaling pathway in cholangiocarcinoma cells. Gastroenterology 2005, 128, 2054–2065. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Yamagiwa, Y.; Ueno, Y.; Patel, T. Over-expression of interleukin-6 enhances cell survival and transformed cell growth in human malignant cholangiocytes. J. Hepatol. 2006, 44, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Tadlock, L.; Gores, G.J.; Patel, T. Inhibition of interleukin 6-mediated mitogen-activated protein kinase activation attenuates growth of a cholangiocarcinoma cell line. Hepatology 1999, 30, 1128–1133. [Google Scholar] [CrossRef] [PubMed]
- Trouillas, M.; Saucourt, C.; Guillotin, B.; Gauthereau, X.; Taupin, J.L.; Moreau, J.F.; Boeuf, H. The LIF cytokine: Towards adulthood. Eur. Cytokine Netw. 2009, 20, 51–62. [Google Scholar] [PubMed]
- Mathieu, M.E.; Saucourt, C.; Mournetas, V.; Gauthereau, X.; Thézé, N.; Praloran, V.; Thiébaud, P.; Bœuf, H. LIF-dependent signaling: New pieces in the Lego. Stem Cell Rev. 2012, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Aghajanova, L. Leukemia inhibitory factor and human embryo implantation. Ann. N. Y. Acad. Sci. 2004, 1034, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Mahic, M.; Kalland, M.E.; Aandahl, E.M.; Torgersen, K.M.; Taskén, K. Human naturally occurring and adaptive regulatory T cells secrete high levels of leukaemia inhibitory factor upon activation. Scand. J. Immunol. 2008, 68, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Sims, N.A.; Johnson, R.W. Leukemia inhibitory factor: A paracrine mediator of bone metabolism. Growth Factors 2012, 30, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Oskowitz, A.Z.; Lu, J.; Penfornis, P.; Ylostalo, J.; McBride, J.; Flemington, E.K.; Prockop, D.J.; Pochampally, R. Human multipotent stromal cells from bone marrow and microRNA: Regulation of differentiation and leukemia inhibitory factor expression. Proc. Natl. Acad. Sci. USA 2008, 105, 18372–18377. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, P.C.; Behrmann, I.; Müller-Newen, G.; Schaper, F.; Graeve, L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem. J. 1998, 334, 297–314. [Google Scholar] [CrossRef] [PubMed]
- Giese, B.; Roderburg, C.; Sommerauer, M.; Wortmann, S.B.; Metz, S.; Heinrich, P.C.; Müller-Newen, G. Dimerization of the cytokine receptors gp130 and LIFR analysed in single cells. J. Cell Sci. 2005, 118, 5129–5140. [Google Scholar] [CrossRef] [PubMed]
- Zouein, F.A.; Kurdi, M.; Booz, G.W. LIF and the heart: Just another brick in the wall? Eur. Cytokine Netw. 2013, 24, 11–19. [Google Scholar] [PubMed]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Müller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Morton, S.D.; Cadamuro, M.; Brivio, S.; Vismara, M.; Stecca, T.; Massani, M.; Bassi, N.; Furlanetto, A.; Joplin, R.E.; Floreani, A.; et al. Leukemia inhibitory factor protects cholangiocarcinoma cells from drug-induced apoptosis via a PI3K/AKT-dependent Mcl-1 activation. Oncotarget 2015, 6, 26052–26064. [Google Scholar] [CrossRef] [PubMed]
- Kellokumpu-Lehtinen, P.; Talpaz, M.; Harris, D.; Van, Q.; Kurzrock, R.; Estrov, Z. Leukemia-inhibitory factor stimulates breast, kidney and prostate cancer cell proliferation by paracrine and autocrine pathways. Int. J. Cancer 1996, 66, 515–519. [Google Scholar] [CrossRef]
- Kamohara, H.; Sakamoto, K.; Ishiko, T.; Masuda, Y.; Abe, T.; Ogawa, M. Leukemia inhibitory factor induces apoptosis and proliferation of human carcinoma cells through different oncogene pathways. Int. J. Cancer 1997, 72, 687–695. [Google Scholar] [CrossRef]
- Andrae, J.; Gallini, R.; Betsholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008, 22, 1276–1312. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Westermark, B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol. Rev. 1999, 79, 1283–1316. [Google Scholar] [PubMed]
- Jechlinger, M.; Sommer, A.; Moriggl, R.; Seither, P.; Kraut, N.; Capodiecci, P.; Donovan, M.; Cordon-Cardo, C.; Beug, H.; Grünert, S. Autocrine PDGFR signaling promotes mammary cancer metastasis. J. Clin. Investig. 2006, 116, 1561–1570. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ahmad, A.; Li, Y.; Kong, D.; Azmi, A.S.; Banerjee, S.; Sarkar, F.H. Emerging roles of PDGF-D signaling pathway in tumor development and progression. Biochim. Biophys. Acta 2010, 1806, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Fingas, C.D.; Bronk, S.F.; Werneburg, N.W.; Mott, J.L.; Guicciardi, M.E.; Cazanave, S.C.; Mertens, J.C.; Sirica, A.E.; Gores, G.J. Myofibroblast-derived PDGF-BB promotes Hedgehog survival signaling in cholangiocarcinoma cells. Hepatology 2011, 54, 2076–2688. [Google Scholar] [CrossRef] [PubMed]
- Pietras, K.; Sjöblom, T.; Rubin, K.; Heldin, C.H.; Ostman, A. PDGF receptors as cancer drug targets. Cancer Cell 2003, 3, 439–443. [Google Scholar] [CrossRef]
- Strazzabosco, M.; Fabris, L. Development of the bile ducts: Essentials for the clinical hepatologist. J. Hepatol. 2012, 56, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. Wnt/β-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Spirli, C.; Locatelli, L.; Morell, C.M.; Fiorotto, R.; Morton, S.D.; Cadamuro, M.; Fabris, L.; Strazzabosco, M. Protein kinase A-dependent pSer675-β-catenin, a novel signaling defect in a mouse model of congenital hepatic fibrosis. Hepatology 2013, 58, 1713–1723. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.Y.; Zhang, W.; Zeng, X.; Liu, C.Q. Inhibition of Wnt/β-catenin signaling downregulates P-glycoprotein and reverses multi-drug resistance of cholangiocarcinoma. Cancer Sci. 2013, 104, 1303–1308. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.L.; Shen, D.Y.; Cai, C.F.; Zhang, Q.Y.; Ren, H.Y.; Chen, Q.X. β-escin reverses multidrug resistance through inhibition of the GSK3β/β-catenin pathway in cholangiocarcinoma. World J. Gastroenterol. 2015, 21, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhong, W.; Yuan, J.; Yan, C.; Hu, S.; Tong, Y.; Mao, Y.; Hu, T.; Zhang, B.; Song, G. Involvement of Wnt/β-catenin signaling in the mesenchymal stem cells promote metastatic growth and chemoresistance of cholangiocarcinoma. Oncotarget 2015, 6, 42276–42289. [Google Scholar] [PubMed]
- Okabe, H.; Yang, J.; Sylakowski, K.; Yovchev, M.; Miyagawa, Y.; Nagarajan, S.; Chikina, M.; Thompson, M.; Oertel, M.; Baba, H.; et al. Wnt signaling regulates hepatobiliary repair following cholestatic liver injury in mice. Hepatology 2016, 64, 1652–1666. [Google Scholar] [CrossRef] [PubMed]
- Lamar, J.M.; Stern, P.; Liu, H.; Schindler, J.W.; Jiang, Z.G.; Hynes, R.O. The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc. Natl. Acad. Sci. USA 2012, 109, E2441–E2450. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Cordenonsi, M.; Dupont, S. Molecular pathways: YAP and TAZ take center stage in organ growth and tumorigenesis. Clin. Cancer Res. 2013, 19, 4925–4930. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef] [PubMed]
- Azzolin, L.; Panciera, T.; Soligo, S.; Enzo, E.; Bicciato, S.; Dupont, S.; Bresolin, S.; Frasson, C.; Basso, G.; Guzzardo, V.; et al. YAP/TAZ incorporation in the β-catenin destruction complex orchestrates the Wnt response. Cell 2014, 158, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Yeh, Y.T.; Nguyen, P.; Limqueco, E.; Lopez, J.; Thorossian, S.; Guan, K.L.; Li, Y.J.; Chien, S. Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proc. Natl. Acad. Sci. USA 2016, 113, 11525–11530. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [PubMed]
- Marti, P.; Stein, C.; Blumer, T.; Abraham, Y.; Dill, M.T.; Pikiolek, M.; Orsini, V.; Jurisic, G.; Megel, P.; Makowska, Z.; et al. YAP promotes proliferation, chemoresistance, and angiogenesis in human cholangiocarcinoma through TEAD transcription factors. Hepatology 2015, 62, 1497–1510. [Google Scholar] [CrossRef] [PubMed]
- Morell, C.M.; Fiorotto, R.; Fabris, L.; Strazzabosco, M. Notch signalling beyond liver development: Emerging concepts in liver repair and oncogenesis. Clin. Res. Hepatol. Gastroenterol. 2013, 37, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, I.; Pochampally, R.; Xing, F.; Watabe, K.; Miele, L. Notch signaling: Targeting cancer stem cells and epithelial-to-mesenchymal transition. OncoTargets Ther. 2013, 6, 1249–1259. [Google Scholar] [PubMed]
- Geisler, F.; Strazzabosco, M. Emerging roles of Notch signaling in liver disease. Hepatology 2015, 61, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A.; Alsinet, C.; Yanger, K.; Hoshida, Y.; Zong, Y.; Toffanin, S.; Rodriguez-Carunchio, L.; Solé, M.; Thung, S.; Stanger, B.Z.; et al. Notch signaling is activated in human hepatocellular carcinoma and induces tumor formation in mice. Gastroenterology 2012, 143, 1660–1669. [Google Scholar] [CrossRef] [PubMed]
- Strazzabosco, M.; Fabris, L. Notch signaling in hepatocellular carcinoma: Guilty in association! Gastroenterology 2012, 143, 1430–1434. [Google Scholar] [CrossRef] [PubMed]
- Fan, B.; Malato, Y.; Calvisi, D.F.; Naqvi, S.; Razumilava, N.; Ribback, S.; Gores, G.J.; Dombrowski, F.; Evert, M.; Chen, X.; et al. Cholangiocarcinomas can originate from hepatocytes in mice. J. Clin. Investig. 2012, 122, 2911–2915. [Google Scholar] [CrossRef] [PubMed]
- Zender, S.; Nickeleit, I.; Wuestefeld, T.; Sörensen, I.; Dauch, D.; Bozko, P.; El-Khatib, M.; Geffers, R.; Bektas, H.; Manns, M.P.; et al. A critical role for notch signaling in the formation of cholangiocellular carcinomas. Cancer Cell 2013, 23, 784–795. [Google Scholar] [CrossRef]
- Wu, W.R.; Zhang, R.; Shi, X.D.; Zhu, M.S.; Xu, L.B.; Zeng, H.; Liu, C. Notch1 is overexpressed in human intrahepatic cholangiocarcinoma and is associated with its proliferation, invasiveness and sensitivity to 5-fluorouracil in vitro. Oncol. Rep. 2014, 31, 2515–2524. [Google Scholar] [CrossRef] [PubMed]
- Omenetti, A.; Diehl, A.M. Hedgehog signaling in cholangiocytes. Curr. Opin. Gastroenterol. 2011, 27, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Riedlinger, D.; Bahra, M.; Boas-Knoop, S.; Lippert, S.; Bradtmöller, M.; Guse, K.; Seehofer, D.; Bova, R.; Sauer, I.M.; Neuhaus, P.; et al. Hedgehog pathway as a potential treatment target in human cholangiocarcinoma. J. Hepatobiliary Pancreat. Sci. 2014, 21, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Kurita, S.; Mott, J.L.; Almada, L.L.; Bronk, S.F.; Werneburg, N.W.; Sun, S.Y.; Roberts, L.R.; Fernandez-Zapico, M.E.; Gores, G.J. GLI3-dependent repression of DR4 mediates hedgehog antagonism of TRAIL-induced apoptosis. Oncogene 2010, 29, 4848–4858. [Google Scholar] [CrossRef] [PubMed]
- Razumilava, N.; Bronk, S.F.; Smoot, R.L.; Fingas, C.D.; Werneburg, N.W.; Roberts, L.R.; Mott, J.L. miR-25 targets TNF-related apoptosis inducing ligand (TRAIL) death receptor-4 and promotes apoptosis resistance in cholangiocarcinoma. Hepatology 2012, 55, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Fingas, C.D.; Mertens, J.C.; Razumilava, N.; Sydor, S.; Bronk, S.F.; Christensen, J.D.; Rizvi, S.H.; Canbay, A.; Treckmann, J.W.; Paul, A.; et al. Polo-like kinase 2 is a mediator of hedgehog survival signaling in cholangiocarcinoma. Hepatology 2013, 58, 1362–1374. [Google Scholar] [CrossRef] [PubMed]
- El Khatib, M.; Kalnytska, A.; Palagani, V.; Kossatz, U.; Manns, M.P.; Malek, N.P.; Wilkens, L.; Plentz, R.R. Inhibition of hedgehog signaling attenuates carcinogenesis in vitro and increases necrosis of cholangiocellular carcinoma. Hepatology 2013, 57, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Hong, I.S.; Lee, H.Y.; Nam, J.S. Cancer stem cells: The “Achille’s heel” of chemo-resistant tumors. Recent Pat. Anticancer Drug Discov. 2015, 10, 2–22. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, S.K.; Roger, E.; Toti, U.; Niu, L.; Ohlfest, J.R.; Panyam, J. CD133-targeted paclitaxel delivery inhibits local tumor recurrence in a mouse model of breast cancer. J. Control. Release 2013, 171, 280–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marangoni, E.; Lecomte, N.; Durand, L.; de Pinieux, G.; Decaudin, D.; Chomienne, C.; Smadja-Joffe, F.; Poupon, M.F. CD44 targeting reduces tumour growth and prevents post-chemotherapy relapse of human breast cancers xenografts. Br. J. Cancer 2009, 100, 918–922. [Google Scholar] [CrossRef] [PubMed]
- Edris, B.; Weiskopf, K.; Volkmer, A.K.; Volkmer, J.P.; Willingham, S.B.; Contreras-Trujillo, H.; Liu, J.; Majeti, R.; West, R.B.; Fletcher, J.A.; et al. Antibody therapy targeting the CD47 protein is effective in a model of aggressive metastatic leiomyosarcoma. Proc. Natl. Acad. Sci. USA 2012, 109, 6656–6661. [Google Scholar] [CrossRef] [PubMed]

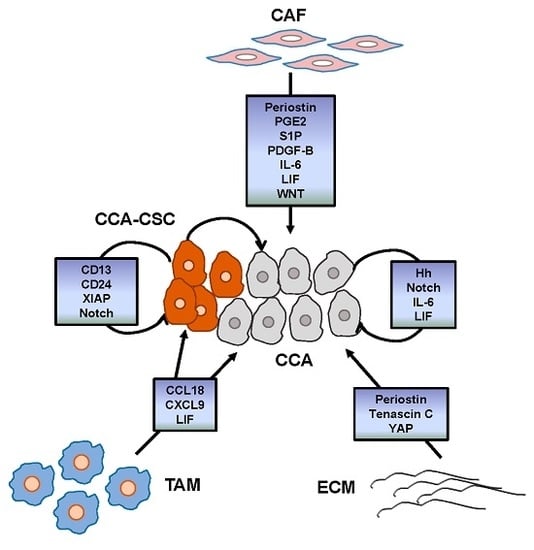
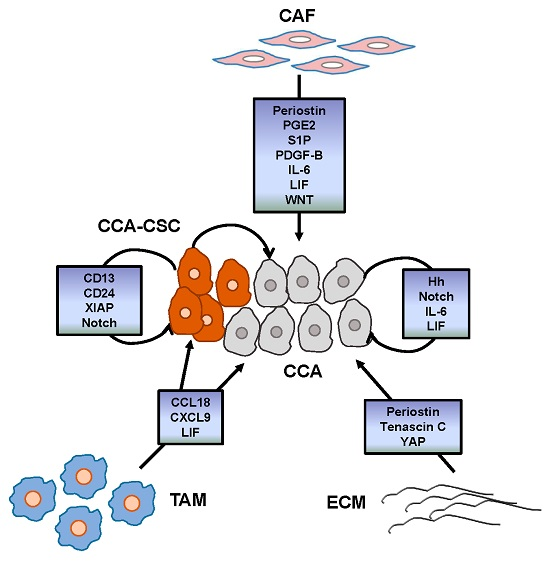{kind=link}
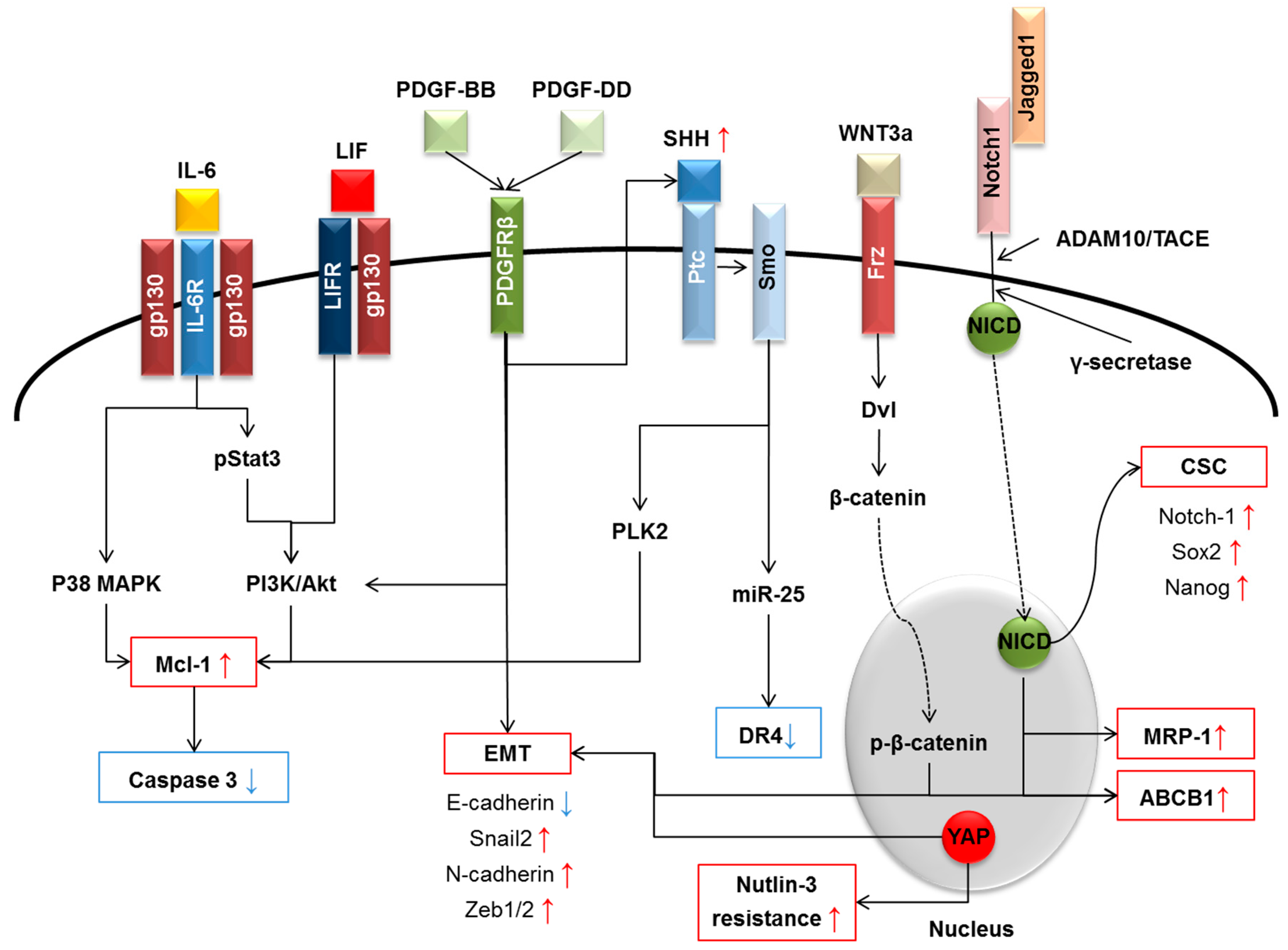
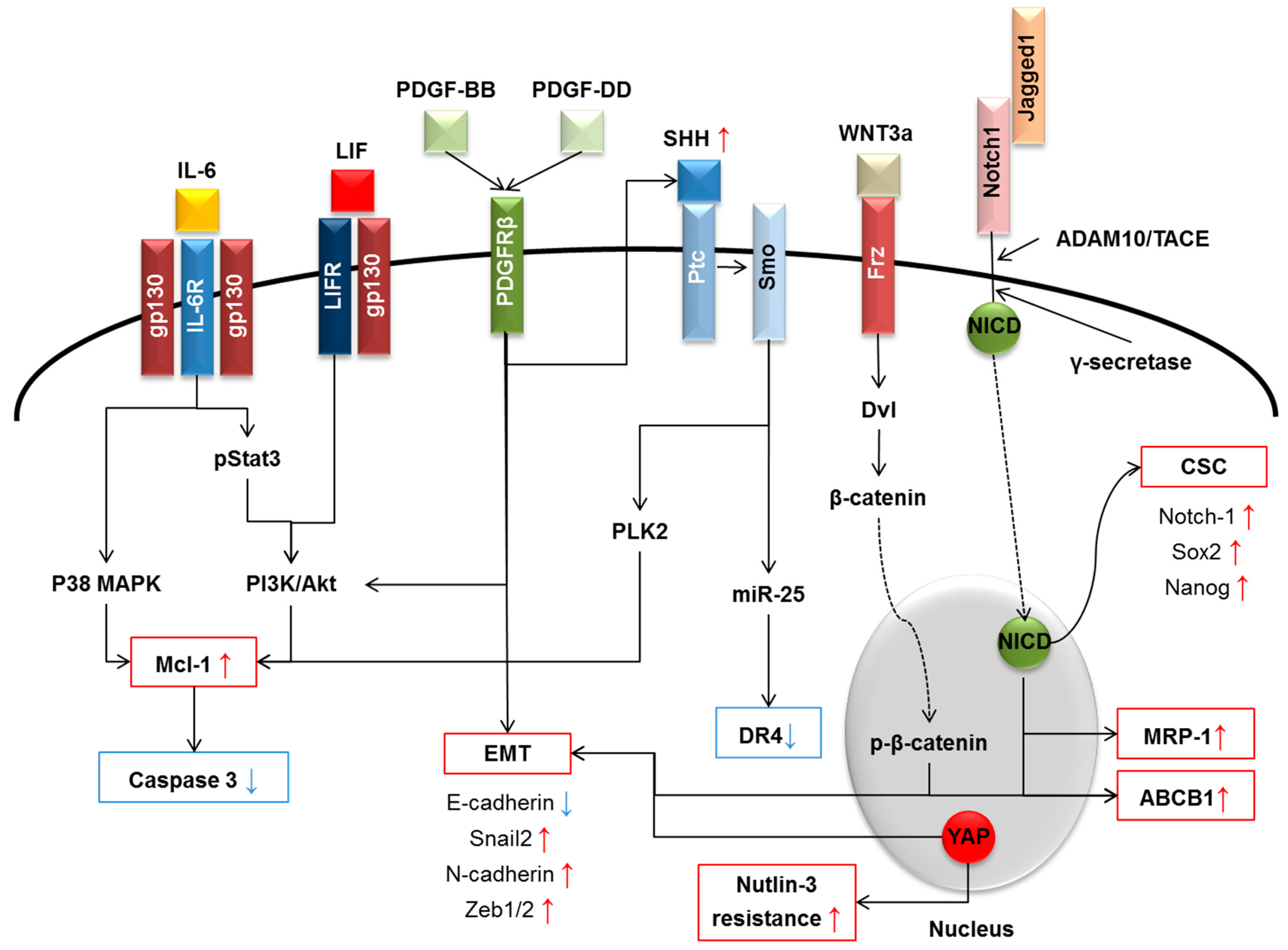{kind=link}
| Biomarker | Molecular Identity | Biological Significance and Relevance | Ref. |
|---|---|---|---|
| CD13 | Transmembrane glycoprotein expressed by granulocytes, monocytes, fibroblasts and some epithelial cells | Escape from drug-induced apoptosis, reported in HCC | [106,108] |
| CD24 | Membrane sialoglycoprotein overexpressed in hematological and epithelial malignancies | Increased cell invasiveness, marker of poor outcome in CCA | [109,110,111] |
| CD44 | Transmembrane hyaluronic acid binding glycoprotein overexpressed in several epithelial cancers | Increased tumorigenicity by synergizing with other peptides | [112,113] |
| CD90 | Phosphatidyl-bound cell surface glycoprotein, expressed by mesenchymal stem cells and by CSC in HCC and CCA | Unknown in CCA, proposed as CSC marker | [106,114,115] |
| CD133 | Transmembrane glycoprotein expressed by hematopoietic stem cells, adult progenitor cells and in fetal liver | Possible marker of poor outcome in CCA | [116,117,118] |
| EpCAM | Adhesion molecule involved in cell-cell interactions, overexpressed in several tumors | Unknown in CCA, proposed as CSC marker | [119] |
| Laminin-332 | Matricellular peptide involved in cell adhesion and metastasization | Preserved stemness of CSC and induced resistance to doxorubicin and sorafenib, in CCA | [120] |
| LGR5 | G protein-coupled receptor expressed by CSC in CCA | Unknown in CCA, proposed as CSC marker | [106] |
| Nanog | Transcription factor regulating developmental features | Involved in self-renewal and differentiation of embryonic stem cells, used as CSC marker | [109,121] |
| Sox2 | Transcription factor regulating developmental features | Involved in stem cell differentiation, correlation with increased lymphatic metastasization and poor outcome in CCA | [113] |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cadamuro, M.; Brivio, S.; Spirli, C.; Joplin, R.E.; Strazzabosco, M.; Fabris, L. Autocrine and Paracrine Mechanisms Promoting Chemoresistance in Cholangiocarcinoma. Int. J. Mol. Sci. 2017, 18, 149. https://doi.org/10.3390/ijms18010149
Cadamuro M, Brivio S, Spirli C, Joplin RE, Strazzabosco M, Fabris L. Autocrine and Paracrine Mechanisms Promoting Chemoresistance in Cholangiocarcinoma. International Journal of Molecular Sciences. 2017; 18(1):149. https://doi.org/10.3390/ijms18010149
Chicago/Turabian StyleCadamuro, Massimiliano, Simone Brivio, Carlo Spirli, Ruth E. Joplin, Mario Strazzabosco, and Luca Fabris. 2017. "Autocrine and Paracrine Mechanisms Promoting Chemoresistance in Cholangiocarcinoma" International Journal of Molecular Sciences 18, no. 1: 149. https://doi.org/10.3390/ijms18010149