
From Clinical Standards to Translating Next-Generation Sequencing Research into Patient Care Improvement for Hepatobiliary and Pancreatic Cancers

 and
and
Abstract
:1. Introduction
2. Clinical Standards
2.1. Modern Adjuvant and Neoadjuvant Treatment
2.2. Limitations of Current Therapeutic Interventions
2.3. Underway Phase III RCTs
3. NEXT-Generation Sequencing and Tumor Heterogeneity
3.1. Targeted Next-Generation Sequencing
3.2. Whole-Exome and Whole-Genome Sequencing
3.3. Confirmation of Known Cancer Driver Genes by NGS Supporting Clinical Implications
3.4. Inter-Patient Heterogeneity and Personalized Therapeutic Approach
4. Future Perspectives
5. Conclusions
Author Contributions
Conflicts of Interest
References
- Shendure, J.; Ji, H. Next-generation DNA sequencing. Nat. Biotechnol. 2008, 26, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar]
- Stamatoyannopoulos, J.A. What does our genome encode? Genome Res. 2012, 22, 1602–1611. [Google Scholar] [CrossRef] [PubMed]
- Aronson, S.J.; Rehm, H.L. Building the foundation for genomics in precision medicine. Nature 2015, 526, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.; Noone, A.; Krapcho, M.; Miller, D.; Bishop, K.; Altekruse, S.; Kosary, C.; Yu, M.; Ruhl, J.; Tatalovich, Z.; et al. SEER Cancer Statistics Review, 1975–2013. Available online: https://seer.cancer.gov/csr/1975_2013/ (accessed on 18 October 2016).
- Ku, C.S.; Roukos, D.H. From next-generation sequencing to nanopore sequencing technology: Paving the way to personalized genomic medicine. Expert Rev. Med. Dev. 2013, 10, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, A.; Furuta, M.; Totoki, Y.; Tsunoda, T.; Kato, M.; Shiraishi, Y.; Tanaka, H.; Taniguchi, H.; Kawakami, Y.; Ueno, M.; et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat. Genet. 2016, 48, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, 359–386. [Google Scholar] [CrossRef] [PubMed]
- European Association For The Study Of The Liver; European Organisation For Research and Treatment Of Cancer. EASL-EORTC clinical practice guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2012, 56, 908–943. [Google Scholar]
- Al-Hawary, M.M.; Francis, I.R.; Chari, S.T.; Fishman, E.K.; Hough, D.M.; Lu, D.S.; Macari, M.; Megibow, A.J.; Miller, F.H.; Mortele, K.J.; et al. Pancreatic ductal adenocarcinoma radiology reporting template: Consensus statement of the society of abdominal radiology and the american pancreatic association. Gastroenterology 2014, 146, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Sherman, M. American Association for the Study of Liver Diseases. Management of hepatocellular carcinoma: An update. Hepatology 2011, 53, 1020–1022. [Google Scholar] [CrossRef] [PubMed]
- Brugge, W.R.; de Witt, J.; Klapman, J.B.; Ashfaq, R.; Shidham, V.; Chhieng, D.; Kwon, R.; Baloch, Z.; Zarka, M.; Staerkel, G. Techniques for cytologic sampling of pancreatic and bile duct lesions: The Papanicolaou Society of Cytopathology Guidelines. CytoJournal 2014, 11, 2. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; He, R.; Li, Y.M.; Cao, G.; Ma, Q.Y.; Yang, W.B. Endoscopic ultrasonography for tumor node staging and vascular invasion in pancreatic cancer: A meta-analysis. Dig. Surg. 2014, 31, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, H.; Fan, C.Y.; Kloke, J.; Khalid, A.; McGrath, K.; Landsittel, D.; Papachristou, G.I. Performance characteristics of endoscopic ultrasound in the staging of pancreatic cancer: A meta-analysis. J. Pancreas 2013, 14, 484–497. [Google Scholar]
- Callery, M.P.; Chang, K.J.; Fishman, E.K.; Talamonti, M.S.; William Traverso, L.; Linehan, D.C. Pretreatment assessment of resectable and borderline resectable pancreatic cancer: Expert consensus statement. Ann. Surg. Oncol. 2009, 16, 1727–1733. [Google Scholar] [CrossRef] [PubMed]
- Natsuizaka, M.; Omura, T.; Akaike, T.; Kuwata, Y.; Yamazaki, K.; Sato, T.; Karino, Y.; Toyota, J.; Suga, T.; Asaka, M. Clinical features of hepatocellular carcinoma with extrahepatic metastases. J. Gastroenterol. Hepatol. 2005, 20, 1781–1787. [Google Scholar] [CrossRef] [PubMed]
- Kamath, P.S.; Wiesner, R.H.; Malinchoc, M.; Kremers, W.; Therneau, T.M.; Kosberg, C.L.; d’Amico, G.; Dickson, E.R.; Kim, W.R. A model to predict survival in patients with end-stage liver disease. Hepatology 2001, 33, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Johnson, P.J.; Berhane, S.; Kagebayashi, C.; Satomura, S.; Teng, M.; Reeves, H.L.; O’Beirne, J.; Fox, R.; Skowronska, A.; Palmer, D.; et al. Assessment of liver function in patients with hepatocellular carcinoma: A new evidence-based approach-the ALBI grade. J. Clin. Oncol. 2015, 33, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Torzilli, G.; Belghiti, J.; Kokudo, N.; Takayama, T.; Capussotti, L.; Nuzzo, G.; Vauthey, J.N.; Choti, M.A.; de Santibanes, E.; Donadon, M.; et al. A snapshot of the effective indications and results of surgery for hepatocellular carcinoma in tertiary referral centers: Is it adherent to the EASL/AASLD recommendations?: An observational study of the HCC East-West study group. Ann. Surg. 2013, 257, 929–937. [Google Scholar] [CrossRef] [PubMed]
- NCCN Clinical Practice Guidelines in Oncology. Pancreatic Adenocarcinoma, Version 2. 2016. Available online: https://www.nccn.org/professionals/physician_gls/f_guidelines.asp (accessed on 5 October 2016).
- NCCN Clinical Practice Guidelines in Oncology. Hepatobiliary Cancers, Version 2. 2016. Available online: https://www.nccn.org/professionals/physician_gls/f_guidelines.asp (accessed on 5 October 2016).
- American Cancer Society. American Joint Committee on Cancer Staging and End Results Reporting. In Manual for Staging of Cancer; American Joint Committee: Chicago, IL, USA, 1977. [Google Scholar]
- Hermanek, P.; Sobin, L.H. TNM Classification of Malignant Tumours. In UICC International Union against Cancer, 4th, Fully Revised ed.; Springer: Berlin/Heidelberg, Germany, 1987. [Google Scholar]
- Bruix, J.; Llovet, J.M. Prognostic prediction and treatment strategy in hepatocellular carcinoma. Hepatology 2002, 35, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Truty, M.J.; Vauthey, J.N. Surgical resection of high-risk hepatocellular carcinoma: Patient selection, preoperative considerations, and operative technique. Ann. Surg. Oncol. 2010, 17, 1219–1225. [Google Scholar] [CrossRef] [PubMed]
- Mazzaferro, V.; Regalia, E.; Doci, R.; Andreola, S.; Pulvirenti, A.; Bozzetti, F.; Montalto, F.; Ammatuna, M.; Morabito, A.; Gennari, L. Liver transplantation for the treatment of small hepatocellular carcinomas in patients with cirrhosis. N. Engl. J. Med. 1996, 334, 693–699. [Google Scholar] [CrossRef] [PubMed]
- D’Angelica, M.; Dalal, K.M.; de Matteo, R.P.; Fong, Y.; Blumgart, L.H.; Jarnagin, W.R. Analysis of the extent of resection for adenocarcinoma of the gallbladder. Ann. Surg. Oncol. 2009, 16, 806–816. [Google Scholar] [CrossRef] [PubMed]
- De Jong, M.C.; Nathan, H.; Sotiropoulos, G.C.; Paul, A.; Alexandrescu, S.; Marques, H.; Pulitano, C.; Barroso, E.; Clary, B.M.; Aldrighetti, L.; et al. Intrahepatic cholangiocarcinoma: An international multi-institutional analysis of prognostic factors and lymph node assessment. J. Clin. Oncol. 2011, 29, 3140–3145. [Google Scholar] [CrossRef] [PubMed]
- Nakeeb, A.; Lillemoe, K.D.; Grosfeld, J.L. Surgical techniques for pancreatic cancer. Minerva Chir. 2004, 59, 151–163. [Google Scholar] [PubMed]
- Verslype, C.; Rosmorduc, O.; Rougier, P.; Group, E.G.W. Hepatocellular carcinoma: ESMO-ESDO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2012, 23, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Ono, T.; Yamanoi, A.; Nazmy El Assal, O.; Kohno, H.; Nagasue, N. Adjuvant chemotherapy after resection of hepatocellular carcinoma causes deterioration of long-term prognosis in cirrhotic patients: Metaanalysis of three randomized controlled trials. Cancer 2001, 91, 2378–2385. [Google Scholar] [CrossRef]
- Samuel, M.; Chow, P.K.; Chan Shih-Yen, E.; Machin, D.; Soo, K.C. Neoadjuvant and adjuvant therapy for surgical resection of hepatocellular carcinoma. Cochrane Database Syst. Rev. 2009, 1, CD001199. [Google Scholar]
- Horgan, A.M.; Amir, E.; Walter, T.; Knox, J.J. Adjuvant therapy in the treatment of biliary tract cancer: A systematic review and meta-analysis. J. Clin. Oncol. 2012, 30, 1934–1940. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, O.K.; Crane, C.H. Palliative and postoperative radiotherapy in biliary tract cancer. Surg. Oncol. Clin. N. Am. 2002, 11, 941–954. [Google Scholar] [CrossRef]
- Hezel, A.F.; Zhu, A.X. Systemic therapy for biliary tract cancers. Oncologist 2008, 13, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Le Scodan, R.; Mornex, F.; Girard, N.; Mercier, C.; Valette, P.J.; Ychou, M.; Bibeau, F.; Roy, P.; Scoazec, J.Y.; Partensky, C. Preoperative chemoradiation in potentially resectable pancreatic adenocarcinoma: Feasibility, treatment effect evaluation and prognostic factors, analysis of the SFRO-FFCD 9704 trial and literature review. Ann. Oncol. 2009, 20, 1387–1396. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.B.; Varadhachary, G.R.; Crane, C.H.; Sun, C.C.; Lee, J.E.; Pisters, P.W.; Vauthey, J.N.; Wang, H.; Cleary, K.R.; Staerkel, G.A.; et al. Preoperative gemcitabine-based chemoradiation for patients with resectable adenocarcinoma of the pancreatic head. J. Clin. Oncol. 2008, 26, 3496–3502. [Google Scholar] [CrossRef] [PubMed]
- Varadhachary, G.R.; Wolff, R.A.; Crane, C.H.; Sun, C.C.; Lee, J.E.; Pisters, P.W.; Vauthey, J.N.; Abdalla, E.; Wang, H.; Staerkel, G.A.; et al. Preoperative gemcitabine and cisplatin followed by gemcitabine-based chemoradiation for resectable adenocarcinoma of the pancreatic head. J. Clin. Oncol. 2008, 26, 3487–3495. [Google Scholar] [CrossRef] [PubMed]
- Regine, W.F.; Winter, K.A.; Abrams, R.A.; Safran, H.; Hoffman, J.P.; Konski, A.; Benson, A.B.; Macdonald, J.S.; Kudrimoti, M.R.; Fromm, M.L.; et al. Fluorouracil vs. gemcitabine chemotherapy before and after fluorouracil-based chemoradiation following resection of pancreatic adenocarcinoma: A randomized controlled trial. JAMA 2008, 299, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Neoptolemos, J.P.; Stocken, D.D.; Bassi, C.; Ghaneh, P.; Cunningham, D.; Goldstein, D.; Padbury, R.; Moore, M.J.; Gallinger, S.; Mariette, C.; et al. Adjuvant chemotherapy with fluorouracil plus folinic acid vs. gemcitabine following pancreatic cancer resection: A randomized controlled trial. JAMA 2010, 304, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Van Laethem, J.L.; Hammel, P.; Mornex, F.; Azria, D.; van Tienhoven, G.; Vergauwe, P.; Peeters, M.; Polus, M.; Praet, M.; Mauer, M.; et al. Adjuvant gemcitabine alone versus gemcitabine-based chemoradiotherapy after curative resection for pancreatic cancer: A randomized EORTC-40013–22012/FFCD-9203/GERCOR phase II study. J. Clin. Oncol. 2010, 28, 4450–4456. [Google Scholar] [CrossRef] [PubMed]
- Oettle, H.; Neuhaus, P.; Hochhaus, A.; Hartmann, J.T.; Gellert, K.; Ridwelski, K.; Niedergethmann, M.; Zulke, C.; Fahlke, J.; Arning, M.B.; et al. Adjuvant chemotherapy with gemcitabine and long-term outcomes among patients with resected pancreatic cancer: The CONKO-001 randomized trial. JAMA 2013, 310, 1473–1481. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.M.; Ngan, H.; Tso, W.K.; Liu, C.L.; Lam, C.M.; Poon, R.T.; Fan, S.T.; Wong, J. Randomized controlled trial of transarterial lipiodol chemoembolization for unresectable hepatocellular carcinoma. Hepatology 2002, 35, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Real, M.I.; Montana, X.; Planas, R.; Coll, S.; Aponte, J.; Ayuso, C.; Sala, M.; Muchart, J.; Sola, R.; et al. Arterial embolisation or chemoembolisation versus symptomatic treatment in patients with unresectable hepatocellular carcinoma: A randomised controlled trial. Lancet 2002, 359, 1734–1739. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.L.; Kang, Y.K.; Chen, Z.; Tsao, C.J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Adnane, L.; Newell, P.; Villanueva, A.; Llovet, J.M.; Lynch, M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol. Cancer Ther. 2008, 7, 3129–3140. [Google Scholar] [CrossRef] [PubMed]
- Keating, G.M.; Santoro, A. Sorafenib: A review of its use in advanced hepatocellular carcinoma. Drugs 2009, 69, 223–240. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Siebels, M.; Negrier, S.; Chevreau, C.; Solska, E.; Desai, A.A.; et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Brose, M.S.; Nutting, C.M.; Jarzab, B.; Elisei, R.; Siena, S.; Bastholt, L.; de la Fouchardiere, C.; Pacini, F.; Paschke, R.; Shong, Y.K.; et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: A randomised, double-blind, phase 3 trial. Lancet 2014, 384, 319–328. [Google Scholar] [CrossRef]
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef] [PubMed]
- Fabian, M.A.; Biggs, W.H., 3rd; Treiber, D.K.; Atteridge, C.E.; Azimioara, M.D.; Benedetti, M.G.; Carter, T.A.; Ciceri, P.; Edeen, P.T.; Floyd, M.; et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005, 23, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Mosquera, C.; Maglic, D.; Zervos, E.E. Molecular targeted therapy for pancreatic adenocarcinoma: A review of completed and ongoing late phase clinical trials. Cancer Genet. 2016, 209, 567–581. [Google Scholar] [CrossRef] [PubMed]
- Huguet, F.; Hammel, P.; Vernerey, D.; Goldstein, D.; Laethem, J.L.V.; Glimelius, B.; Spry, N.; Paget-Bailly, S.; Bonnetain, F.; Louvet, C. Impact of chemoradiotherapy (CRT) on local control and time without treatment in patients with locally advanced pancreatic cancer (LAPC) included in the international phase III LAP 07 study. Pancreatology 2014, 14, 6. [Google Scholar]
- Hammel, P.; Huguet, F.; van Laethem, J.L.; Goldstein, D.; Glimelius, B.; Artru, P.; Borbath, I.; Bouche, O.; Shannon, J.; Andre, T.; et al. Effect of Chemoradiotherapy vs. Chemotherapy on Survival in Patients with Locally Advanced Pancreatic Cancer Controlled after 4 Months of Gemcitabine with or without Erlotinib: The LAP07 Randomized Clinical Trial. JAMA 2016, 315, 1844–1853. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society. Survival Statistics for Bile Duct Cancers. Available online: http://www.cancer.org/cancer/bileductcancer/detailedguide/bile-duct-cancer-survival-by-stage (accessed on 14 September 2016).
- Horner, M.; Ries, L.; Krapcho, M.; Neyman, N.; Aminou, R.; Howlader, N.; Altekruse, S.; Feuer, E.; Huang, L.; Mariotto, A.; et al. SEER Cancer Statistics Review, 1975–2006. Available online: https://seer.cancer.gov/csr/1975_2006/ (accessed on 18 October 2016).
- Ries, L.; Eisner, M.; Kosary, C.; Hankey, B.; Miller, B.; Clegg, L.; Edwards, B. SEER Cancer Statistics Review, 1973–1999. Available online: https://seer.cancer.gov/csr/1973_1999/ (accessed on 18 October 2016).
- Tabrizian, P.; Jibara, G.; Shrager, B.; Schwartz, M.; Roayaie, S. Recurrence of hepatocellular cancer after resection: Patterns, treatments, and prognosis. Ann. Surg. 2015, 261, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Leung, J.Y.; Zhu, A.X.; Gordon, F.D.; Pratt, D.S.; Mithoefer, A.; Garrigan, K.; Terella, A.; Hertl, M.; Cosimi, A.B.; Chung, R.T. Liver transplantation outcomes for early-stage hepatocellular carcinoma: Results of a multicenter study. Liver Transplant. 2004, 10, 1343–1354. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.A.; Cleary, S.P.; Wei, A.C.; Yang, I.; Taylor, B.R.; Hemming, A.W.; Langer, B.; Grant, D.R.; Greig, P.D.; Gallinger, S. Recurrence after liver resection for hepatocellular carcinoma: Risk factors, treatment, and outcomes. Surgery 2007, 141, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Lang, H.; Sotiropoulos, G.C.; Brokalaki, E.I.; Schmitz, K.J.; Bertona, C.; Meyer, G.; Frilling, A.; Paul, A.; Malago, M.; Broelsch, C.E. Survival and recurrence rates after resection for hepatocellular carcinoma in noncirrhotic livers. J. Am. Coll. Surg. 2007, 205, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.Y.; Patt, C.H.; Geschwind, J.F.; Thuluvath, P.J. The outcome of liver transplantation in patients with hepatocellular carcinoma in the United States between 1988 and 2001: 5-Year survival has improved significantly with time. J. Clin. Oncol. 2003, 21, 4329–4335. [Google Scholar] [CrossRef] [PubMed]
- Roayaie, S.; Schwartz, J.D.; Sung, M.W.; Emre, S.H.; Miller, C.M.; Gondolesi, G.E.; Krieger, N.R.; Schwartz, M.E. Recurrence of hepatocellular carcinoma after liver transplant: Patterns and prognosis. Liver Transplant. 2004, 10, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, T.; Uesaka, K.; Mihara, K.; Sasaki, K.; Kanemoto, H.; Mizuno, T.; Okamura, Y. Margin status, recurrence pattern, and prognosis after resection of pancreatic cancer. Surgery 2013, 154, 1078–1086. [Google Scholar] [CrossRef] [PubMed]
- Endo, I.; Gonen, M.; Yopp, A.C.; Dalal, K.M.; Zhou, Q.; Klimstra, D.; d’Angelica, M.; deMatteo, R.P.; Fong, Y.; Schwartz, L.; et al. Intrahepatic cholangiocarcinoma: Rising frequency, improved survival, and determinants of outcome after resection. Ann. Surg. 2008, 248, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Tamandl, D.; Herberger, B.; Gruenberger, B.; Puhalla, H.; Klinger, M.; Gruenberger, T. Influence of hepatic resection margin on recurrence and survival in intrahepatic cholangiocarcinoma. Ann. Surg. Oncol. 2008, 15, 2787–2794. [Google Scholar] [CrossRef] [PubMed]
- Sapisochin, G.; Fidelman, N.; Roberts, J.P.; Yao, F.Y. Mixed hepatocellular cholangiocarcinoma and intrahepatic cholangiocarcinoma in patients undergoing transplantation for hepatocellular carcinoma. Liver Transplant. 2011, 17, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.C.; Petrowsky, H.; Kaldas, F.M.; Farmer, D.G.; Durazo, F.A.; Finn, R.S.; Saab, S.; Han, S.H.; Lee, P.; Markovic, D.; et al. Predictive index for tumor recurrence after liver transplantation for locally advanced intrahepatic and hilar cholangiocarcinoma. J. Am. Coll. Surg. 2011, 212, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.M.; Ryu, J.K.; Lee, S.H.; Yoo, J.W.; Park, J.K.; Kim, Y.T.; Jang, J.Y.; Kim, S.W.; Kang, G.H.; Yoon, Y.B. Recurrence and prognostic factors of ampullary carcinoma after radical resection: Comparison with distal extrahepatic cholangiocarcinoma. Ann. Surg. Oncol. 2007, 14, 3195–3201. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Miwa, S.; Nakata, T.; Miyagawa, S. Disease recurrence patterns after R0 resection of hilar cholangiocarcinoma. Br. J. Surg. 2010, 97, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Jarnagin, W.R.; Ruo, L.; Little, S.A.; Klimstra, D.; d’Angelica, M.; de Matteo, R.P.; Wagman, R.; Blumgart, L.H.; Fong, Y. Patterns of initial disease recurrence after resection of gallbladder carcinoma and hilar cholangiocarcinoma: Implications for adjuvant therapeutic strategies. Cancer 2003, 98, 1689–1700. [Google Scholar] [CrossRef] [PubMed]
- Gores, G.J.; Darwish Murad, S.; Heimbach, J.K.; Rosen, C.B. Liver transplantation for perihilar cholangiocarcinoma. J. Dig. Dis. 2013, 31, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Takada, T.; Amano, H.; Yasuda, H.; Nimura, Y.; Matsushiro, T.; Kato, H.; Nagakawa, T.; Nakayama, T.; Study Group of Surgical Adjuvant Therapy for Carcinomas of the Pancreas and Biliary Tract. Is postoperative adjuvant chemotherapy useful for gallbladder carcinoma? A phase III multicenter prospective randomized controlled trial in patients with resected pancreaticobiliary carcinoma. Cancer 2002, 95, 1685–1695. [Google Scholar] [PubMed]
- Bruix, J.; Takayama, T.; Mazzaferro, V.; Chau, G.Y.; Yang, J.; Kudo, M.; Cai, J.; Poon, R.T.; Han, K.H.; Tak, W.Y.; et al. Adjuvant sorafenib for hepatocellular carcinoma after resection or ablation (STORM): A phase 3, randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2015, 16, 1344–1354. [Google Scholar] [CrossRef]
- Cainap, C.; Qin, S.; Huang, W.T.; Chung, I.J.; Pan, H.; Cheng, Y.; Kudo, M.; Kang, Y.K.; Chen, P.J.; Toh, H.C.; et al. Linifanib versus Sorafenib in patients with advanced hepatocellular carcinoma: Results of a randomized phase III trial. J. Clin. Oncol. 2015, 33, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M.; Han, G.; Finn, R.S.; Poon, R.T.; Blanc, J.F.; Yan, L.; Yang, J.; Lu, L.; Tak, W.Y.; Yu, X.; et al. Brivanib as adjuvant therapy to transarterial chemoembolization in patients with hepatocellular carcinoma: A randomized phase III trial. Hepatology 2014, 60, 1697–1707. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Rosmorduc, O.; Evans, T.R.; Ross, P.J.; Santoro, A.; Carrilho, F.J.; Bruix, J.; Qin, S.; Thuluvath, P.J.; Llovet, J.M.; et al. SEARCH: A phase III, randomized, double-blind, placebo-controlled trial of sorafenib plus erlotinib in patients with advanced hepatocellular carcinoma. J. Clin. Oncol. 2015, 33, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Qin, S.; Ryoo, B.-Y.; Lu, S.-N.; Yen, C.-J.; Feng, Y.-H.; Lim, H.Y.; Izzo, F.; Colombo, M.; Sarker, D.; et al. Phase III randomized study of second line ADI-peg 20 (A) plus best supportive care versus placebo (P) plus best supportive care in patients (pts) with advanced hepatocellular carcinoma (HCC). In Proceedings of the 2016 ASCO Annual Meeting, Chicago, IL, USA, 2016.
- Zhu, A.X.; Park, J.O.; Ryoo, B.Y.; Yen, C.J.; Poon, R.; Pastorelli, D.; Blanc, J.F.; Chung, H.C.; Baron, A.D.; Pfiffer, T.E.; et al. Ramucirumab versus placebo as second-line treatment in patients with advanced hepatocellular carcinoma following first-line therapy with sorafenib (REACH): A randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2015, 16, 859–870. [Google Scholar] [CrossRef]
- Barbare, J.C.; Bouche, O.; Bonnetain, F.; Raoul, J.L.; Rougier, P.; Abergel, A.; Boige, V.; Denis, B.; Blanchi, A.; Pariente, A.; et al. Randomized controlled trial of tamoxifen in advanced hepatocellular carcinoma. J. Clin. Oncol. 2005, 23, 4338–4346. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Decaens, T.; Raoul, J.L.; Boucher, E.; Kudo, M.; Chang, C.; Kang, Y.K.; Assenat, E.; Lim, H.Y.; Boige, V.; et al. Brivanib in patients with advanced hepatocellular carcinoma who were intolerant to sorafenib or for whom sorafenib failed: Results from the randomized phase III BRISK-PS study. J. Clin. Oncol. 2013, 31, 3509–3516. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Lee, J.H.; Lim, Y.S.; Yeon, J.E.; Song, T.J.; Yu, S.J.; Gwak, G.Y.; Kim, K.M.; Kim, Y.J.; Lee, J.W.; et al. Adjuvant immunotherapy with autologous cytokine-induced killer cells for hepatocellular carcinoma. Gastroenterology 2015, 148, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Valle, J.W.; Wasan, H.; Lopes, A.; Backen, A.C.; Palmer, D.H.; Morris, K.; Duggan, M.; Cunningham, D.; Anthoney, D.A.; Corrie, P.; et al. Cediranib or placebo in combination with cisplatin and gemcitabine chemotherapy for patients with advanced biliary tract cancer (ABC-03): A randomised phase 2 trial. Lancet Oncol. 2015, 16, 967–978. [Google Scholar] [CrossRef]
- Middleton, G.; Silcocks, P.; Cox, T.; Valle, J.; Wadsley, J.; Propper, D.; Coxon, F.; Ross, P.; Madhusudan, S.; Roques, T.; et al. Gemcitabine and capecitabine with or without telomerase peptide vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer (TeloVac): An open-label, randomised, phase 3 trial. Lancet Oncol. 2014, 15, 829–840. [Google Scholar] [CrossRef]
- Philip, P.A.; Benedetti, J.; Corless, C.L.; Wong, R.; O’Reilly, E.M.; Flynn, P.J.; Rowland, K.M.; Atkins, J.N.; Mirtsching, B.C.; Rivkin, S.E.; et al. Phase III study comparing gemcitabine plus cetuximab versus gemcitabine in patients with advanced pancreatic adenocarcinoma: Southwest Oncology Group-directed intergroup trial S0205. J. Clin. Oncol. 2010, 28, 3605–3610. [Google Scholar] [CrossRef] [PubMed]
- Biotechnology Events. Available online: http://www.biotechnologyevents.com/node/8843 (accessed on 23 September 2016).
- Van Cutsem, E.; van de Velde, H.; Karasek, P.; Oettle, H.; Vervenne, W.L.; Szawlowski, A.; Schoffski, P.; Post, S.; Verslype, C.; Neumann, H.; et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J. Clin. Oncol. 2004, 22, 1430–1438. [Google Scholar] [CrossRef] [PubMed]
- Ioka, T.; Okusaka, T.; Ohkawa, S.; Boku, N.; Sawaki, A.; Fujii, Y.; Kamei, Y.; Takahashi, S.; Namazu, K.; Umeyama, Y.; et al. Efficacy and safety of axitinib in combination with gemcitabine in advanced pancreatic cancer: Subgroup analyses by region, including Japan, from the global randomized Phase III trial. Jpn. J. Clin. Oncol. 2015, 45, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Kindler, H.L.; Niedzwiecki, D.; Hollis, D.; Sutherland, S.; Schrag, D.; Hurwitz, H.; Innocenti, F.; Mulcahy, M.F.; O’Reilly, E.; Wozniak, T.F.; et al. Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: Phase III trial of the Cancer and Leukemia Group B (CALGB 80303). J. Clin. Oncol. 2010, 28, 3617–3622. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, B.H.; Scott, A.J.; Ma, W.W.; Cohen, S.J.; Aisner, D.L.; Menter, A.R.; Tejani, M.A.; Cho, J.K.; Granfortuna, J.; Coveler, A.L.; et al. A phase II/III randomized study to compare the efficacy and safety of rigosertib plus gemcitabine versus gemcitabine alone in patients with previously untreated metastatic pancreatic cancer. Ann. Oncol. 2016, 27, 1180. [Google Scholar] [CrossRef] [PubMed]
- Gilliam, A.D.; Broome, P.; Topuzov, E.G.; Garin, A.M.; Pulay, I.; Humphreys, J.; Whitehead, A.; Takhar, A.; Rowlands, B.J.; Beckingham, I.J. An international multicenter randomized controlled trial of G17DT in patients with pancreatic cancer. Pancreas 2012, 41, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Yamaue, H.; Tsunoda, T.; Tani, M.; Miyazawa, M.; Yamao, K.; Mizuno, N.; Okusaka, T.; Ueno, H.; Boku, N.; Fukutomi, A.; et al. Randomized phase II/III clinical trial of elpamotide for patients with advanced pancreatic cancer: PEGASUS-PC Study. Cancer Sci. 2015, 106, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Merle, P.; Granito, A.; Huang, Y.-H.; Bodoky, G.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; Gerolami, R.; Masi, G.; et al. Efficacy and safety of regorafenib versus placebo in patients with hepatocellular carcinoma (HCC) progressing on sorafenib: Results of the international, randomized phase 3 RESORCE trial. In Proceedings of ESMO 18th World Congress of Gastrointestinal Cancer, Barcelona, Spain, 29 June–2 July 2016.
- Zhu, A.X.; Galle, P.R.; Kudo, M.; Finn, R.S.; Yang, L.; Abada, P.; Chang, S.-C.; Llovet, J.M. A randomized, double-blind, placebo-controlled phase III study of ramucirumab versus placebo as second-line treatment in patients with hepatocellular carcinoma and elevated baseline alpha-fetoprotein following first-line sorafenib (REACH-2). In Proceedings of the 2016 Gastrointestinal Cancers Symposium, San Francisco, CA, USA, 21–23 January 2016.
- Santoro, A.; Rimassa, L.; Borbath, I.; Daniele, B.; Salvagni, S.; van Laethem, J.L.; van Vlierberghe, H.; Trojan, J.; Kolligs, F.T.; Weiss, A.; et al. Tivantinib for second-line treatment of advanced hepatocellular carcinoma: A randomised, placebo-controlled phase 2 study. Lancet Oncol. 2013, 14, 55–63. [Google Scholar] [CrossRef]
- Santoro, A.; Porta, C.; Rimassa, L.; Borbath, I.; Daniele, B.; Finn, R.S.; Raoul, J.L.; He, R.; Trojan, J.; Peck-Radosavljevic, M.; et al. A Phase 3 Placebo-Controlled Trial with Tivantinib (ARQ 197), in Patients with Second-Line, MET-High, Inoperable Hepatocellular Carcinoma. In Proceedings of the International Liver Cancer Association 9th Annual Conference, Paris, France, 4–6 September 2015.
- Golan, T.; Oh, D.-Y.; Reni, M.; Macarulla, T.M.; Tortora, G.; Hall, M.J.; Reinacher-Schick, A.C.; Borg, C.; Hochhauser, D.; Walter, T.; et al. A randomized phase III trial of olaparib maintenance monotherapy in patients (pts) with metastatic pancreatic cancer (mPC) who have a germline BRCA1/2 mutation (gBRCAm). In Proceedings of 2016 ASCO Annual Meeting, Chicago, IL, USA, 3–7 June 2016.
- Biankin, A.V.; Piantadosi, S.; Hollingsworth, S.J. Patient-centric trials for therapeutic development in precision oncology. Nature 2015, 526, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Mirnezami, R.; Nicholson, J.; Darzi, A. Preparing for precision medicine. N. Engl. J. Med. 2012, 366, 489–491. [Google Scholar] [CrossRef] [PubMed]
- Roukos, D.H.; Ku, C.S. Clinical cancer genome and precision medicine. Ann. Surg. Oncol. 2012, 19, 3646–3650. [Google Scholar] [CrossRef] [PubMed]
- Swanton, C.; Soria, J.C.; Bardelli, A.; Biankin, A.; Caldas, C.; Chandarlapaty, S.; de Koning, L.; Dive, C.; Feunteun, J.; Leung, S.Y.; et al. Consensus on precision medicine for metastatic cancers: A report from the MAP conference. Ann. Oncol. 2016, 27, 1443–1448. [Google Scholar] [CrossRef] [PubMed]
- Van Allen, E.M.; Wagle, N.; Stojanov, P.; Perrin, D.L.; Cibulskis, K.; Marlow, S.; Jane-Valbuena, J.; Friedrich, D.C.; Kryukov, G.; Carter, S.L.; et al. Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine. Nat. Med. 2014, 20, 682–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munchel, S.; Hoang, Y.; Zhao, Y.; Cottrell, J.; Klotzle, B.; Godwin, A.K.; Koestler, D.; Beyerlein, P.; Fan, J.B.; Bibikova, M.; et al. Targeted or whole genome sequencing of formalin fixed tissue samples: Potential applications in cancer genomics. Oncotarget 2015, 6, 25943–25961. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.A. Selection and adaptation during metastatic cancer progression. Nature 2013, 501, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Mafficini, A.; Amato, E.; Fassan, M.; Simbolo, M.; Antonello, D.; Vicentini, C.; Scardoni, M.; Bersani, S.; Gottardi, M.; Rusev, B.; et al. Reporting tumor molecular heterogeneity in histopathological diagnosis. PLoS ONE 2014, 9, 104979. [Google Scholar] [CrossRef] [PubMed]
- Froyen, G.; Broekmans, A.; Hillen, F.; Pat, K.; Achten, R.; Mebis, J.; Rummens, J.L.; Willemse, J.; Maes, B. Validation and Application of a Custom-Designed Targeted Next-Generation Sequencing Panel for the Diagnostic Mutational Profiling of Solid Tumors. PLoS ONE 2016, 11, 0154038. [Google Scholar] [CrossRef] [PubMed]
- Roukos, D.H. Trastuzumab and beyond: sequencing cancer genomes and predicting molecular networks. Pharmacogenom. J. 2011, 11, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Hirotsu, Y.; Zheng, T.H.; Amemiya, K.; Mochizuki, H.; Guleng, B.; Omata, M. Targeted and exome sequencing identified somatic mutations in hepatocellular carcinoma. Hepatol. Res. 2016, 46, 1145–1151. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Yin, J.; Dong, R.; Yang, T.; Yuan, L.; Zang, L.; Xu, C.; Peng, B.; Zhao, J.; Du, X. Targeted sequencing of cancer-associated genes in hepatocellular carcinoma using next generation sequencing. Mol. Med. Rep. 2015, 12, 4678–4682. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Kaseb, A.O.; Tsimberidou, A.M.; Wolff, R.A.; Kurzrock, R. Identification of novel therapeutic targets in the PI3K/AKT/mTOR pathway in hepatocellular carcinoma using targeted next generation sequencing. Oncotarget 2014, 5, 3012–3022. [Google Scholar] [CrossRef] [PubMed]
- Kaibori, M.; Sakai, K.; Ishizaki, M.; Matsushima, H.; de Velasco, M.A.; Matsui, K.; Iida, H.; Kitade, H.; Kwon, A.H.; Nagano, H.; et al. Increased FGF19 copy number is frequently detected in hepatocellular carcinoma with a complete response after sorafenib treatment. Oncotarget 2016, 31, 49091–49098. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Takeda, H.; Nishijima, N.; Orito, E.; Joko, K.; Uchida, Y.; Izumi, N.; Nishio, K.; Osaki, Y. Targeted DNA and RNA sequencing of fine-needle biopsy FFPE specimens in patients with unresectable hepatocellular carcinoma treated with sorafenib. Oncotarget 2015, 6, 21636–21644. [Google Scholar] [CrossRef] [PubMed]
- Kawai-Kitahata, F.; Asahina, Y.; Tanaka, S.; Kakinuma, S.; Murakawa, M.; Nitta, S.; Watanabe, T.; Otani, S.; Taniguchi, M.; Goto, F.; et al. Comprehensive analyses of mutations and hepatitis B virus integration in hepatocellular carcinoma with clinicopathological features. J. Gastroenterol. 2016, 51, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Putra, J.; de Abreu, F.B.; Peterson, J.D.; Pipas, J.M.; Mody, K.; Amos, C.I.; Tsongalis, G.J.; Suriawinata, A.A. Molecular profiling of intrahepatic and extrahepatic cholangiocarcinoma using next generation sequencing. Exp. Mol. Pathol. 2015, 99, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.S.; Wang, K.; Gay, L.; Al-Rohil, R.; Rand, J.V.; Jones, D.M.; Lee, H.J.; Sheehan, C.E.; Otto, G.A.; Palmer, G.; et al. New routes to targeted therapy of intrahepatic cholangiocarcinomas revealed by next-generation sequencing. Oncologist 2014, 19, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Yoo, K.H.; Kim, N.K.; Kwon, W.I.; Lee, C.; Kim, S.Y.; Jang, J.; Ahn, J.; Kang, M.; Jang, H.; Kim, S.T.; et al. Genomic Alterations in Biliary Tract Cancer Using Targeted Sequencing. Transl. Oncol. 2016, 9, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Wang, H.E.; Seo, S.Y.; Kim, S.H.; Kim, I.H.; Kim, S.W.; Lee, S.T.; Kim, D.G.; Han, M.K.; Lee, S.O. Cancer related gene alterations can be detected with next-generation sequencing analysis of bile in diffusely infiltrating type cholangiocarcinoma. Exp. Mol. Pathol. 2016, 101, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Pawlik, T.M.; Anders, R.A.; Selaru, F.M.; Streppel, M.M.; Lucas, D.J.; Niknafs, N.; Guthrie, V.B.; Maitra, A.; Argani, P.; et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat. Genet. 2013, 45, 1470–1473. [Google Scholar] [CrossRef] [PubMed]
- Churi, C.R.; Shroff, R.; Wang, Y.; Rashid, A.; Kang, H.C.; Weatherly, J.; Zuo, M.; Zinner, R.; Hong, D.; Meric-Bernstam, F.; et al. Mutation profiling in cholangiocarcinoma: Prognostic and therapeutic implications. PLoS ONE 2014, 9, 115383. [Google Scholar] [CrossRef] [PubMed]
- Simbolo, M.; Fassan, M.; Ruzzenente, A.; Mafficini, A.; Wood, L.D.; Corbo, V.; Melisi, D.; Malleo, G.; Vicentini, C.; Malpeli, G.; et al. Multigene mutational profiling of cholangiocarcinomas identifies actionable molecular subgroups. Oncotarget 2014, 5, 2839–2852. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.; Bekaii-Saab, T.; Jain, A.; Wang, Y.; Kelley, R.K.; Wang, K.; Kang, H.C.; Catenacci, D.; Ali, S.; Krishnan, S.; et al. Biliary cancer: Utility of next-generation sequencing for clinical management. Cancer 2016, 122, 3838–3847. [Google Scholar] [CrossRef] [PubMed]
- Basturk, O.; Tan, M.; Bhanot, U.; Allen, P.; Adsay, V.; Scott, S.N.; Shah, R.; Berger, M.F.; Askan, G.; Dikoglu, E.; et al. The oncocytic subtype is genetically distinct from other pancreatic intraductal papillary mucinous neoplasm subtypes. Modern Pathol. 2016, 29, 1058–1069. [Google Scholar] [CrossRef] [PubMed]
- Pea, A.; Yu, J.; Rezaee, N.; Luchini, C.; He, J.; dal Molin, M.; Griffin, J.F.; Fedor, H.; Fesharakizadeh, S.; Salvia, R.; et al. Targeted DNA Sequencing Reveals Patterns of Local Progression in the Pancreatic Remnant Following Resection of Intraductal Papillary Mucinous Neoplasm (IPMN) of the Pancreas. Ann. Surg. 2016. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, F.C.; Kerr, S.E.; Kipp, B.R.; Voss, J.S.; Minot, D.M.; Tu, Z.J.; Henry, M.R.; Graham, R.P.; Vasmatzis, G.; Cheville, J.C.; et al. Targeted next generation sequencing of endoscopic ultrasound acquired cytology from ampullary and pancreatic adenocarcinoma has the potential to aid patient stratification for optimal therapy selection. Oncotarget 2016, 7, 54526–54536. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.P.; Chesla, D.W.; Chung, M.H. Using next-generation sequencing to determine potential molecularly guided therapy options for patients with resectable pancreatic adenocarcinoma. Am. J. Surg. 2016, 211, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Amato, E.; Molin, M.D.; Mafficini, A.; Yu, J.; Malleo, G.; Rusev, B.; Fassan, M.; Antonello, D.; Sadakari, Y.; Castelli, P.; et al. Targeted next-generation sequencing of cancer genes dissects the molecular profiles of intraductal papillary neoplasms of the pancreas. J. Pathol. 2014, 233, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Chantrill, L.A.; Nagrial, A.M.; Watson, C.; Johns, A.L.; Martyn-Smith, M.; Simpson, S.; Mead, S.; Jones, M.D.; Samra, J.S.; Gill, A.J.; et al. Precision Medicine for Advanced Pancreas Cancer: The Individualized Molecular Pancreatic Cancer Therapy (IMPaCT) Trial. Clin. Cancer Res. 2015, 21, 2029–2037. [Google Scholar] [CrossRef] [PubMed]
- Le Tourneau, C.; Delord, J.P.; Goncalves, A.; Gavoille, C.; Dubot, C.; Isambert, N.; Campone, M.; Tredan, O.; Massiani, M.A.; Mauborgne, C.; et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): A multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015, 16, 1324–1334. [Google Scholar] [CrossRef]
- Valero, V., 3rd; Saunders, T.J.; He, J.; Weiss, M.J.; Cameron, J.L.; Dholakia, A.; Wild, A.T.; Shin, E.J.; Khashab, M.A.; O’Broin-Lennon, A.M.; et al. Reliable Detection of Somatic Mutations in Fine Needle Aspirates of Pancreatic Cancer with Next-generation Sequencing: Implications for Surgical Management. Ann. Surg. 2016, 263, 153–161. [Google Scholar] [PubMed]
- De Biase, D.; Visani, M.; Baccarini, P.; Polifemo, A.M.; Maimone, A.; Fornelli, A.; Giuliani, A.; Zanini, N.; Fabbri, C.; Pession, A.; et al. Next generation sequencing improves the accuracy of KRAS mutation analysis in endoscopic ultrasound fine needle aspiration pancreatic lesions. PLoS ONE 2014, 9, 87651. [Google Scholar] [CrossRef] [PubMed]
- Sibinga Mulder, B.G.; Mieog, J.S.; Handgraaf, H.J.; Farina Sarasqueta, A.; Vasen, H.F.; Potjer, T.P.; Swijnenburg, R.J.; Luelmo, S.A.; Feshtali, S.; Inderson, A.; et al. Targeted next-generation sequencing of FNA-derived DNA in pancreatic cancer. J. Clin. Pathol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Roukos, D.H. Crossroad between linear and nonlinear transcription concepts in the discovery of next-generation sequencing systems-based anticancer therapies. Drug Discov. Today 2016, 21, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Dong, R.; Jing, Y.; Xu, D.; Wang, Q.; Chen, L.; Li, Q.; Huang, Y.; Zhang, Y.; Zhang, Z.; et al. Exome sequencing of hepatoblastoma reveals novel mutations and cancer genes in the Wnt pathway and ubiquitin ligase complex. Hepatology 2014, 60, 1686–1696. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Deng, Q.; Wang, Q.; Li, K.Y.; Dai, J.H.; Li, N.; Zhu, Z.D.; Zhou, B.; Liu, X.Y.; Liu, R.F.; et al. Exome sequencing of hepatitis B virus-associated hepatocellular carcinoma. Nat. Genet. 2012, 44, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Ki Kim, S.; Ueda, Y.; Hatano, E.; Kakiuchi, N.; Takeda, H.; Goto, T.; Shimizu, T.; Yoshida, K.; Ikura, Y.; Shiraishi, Y.; et al. TERT promoter mutations and chromosome 8p loss are characteristic of nonalcoholic fatty liver disease-related hepatocellular carcinoma. Int. J. Cancer 2016, 139, 2512–2518. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Yonescu, R.; Offerhaus, G.J.; Klimstra, D.S.; Maitra, A.; Eshleman, J.R.; Herman, J.G.; Poh, W.; Pelosof, L.; Wolfgang, C.L.; et al. Whole-exome sequencing of pancreatic neoplasms with acinar differentiation. J. Pathol. 2014, 232, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Guichard, C.; Amaddeo, G.; Imbeaud, S.; Ladeiro, Y.; Pelletier, L.; Maad, I.B.; Calderaro, J.; Bioulac-Sage, P.; Letexier, M.; Degos, F.; et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 2012, 44, 694–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dal Molin, M.; Zhang, M.; de Wilde, R.F.; Ottenhof, N.A.; Rezaee, N.; Wolfgang, C.L.; Blackford, A.; Vogelstein, B.; Kinzler, K.W.; Papadopoulos, N.; et al. Very Long-term Survival Following Resection for Pancreatic Cancer Is Not Explained by Commonly Mutated Genes: Results of Whole-Exome Sequencing Analysis. Clin. Cancer Res. 2015, 21, 1944–1950. [Google Scholar] [CrossRef] [PubMed]
- Cornella, H.; Alsinet, C.; Sayols, S.; Zhang, Z.; Hao, K.; Cabellos, L.; Hoshida, Y.; Villanueva, A.; Thung, S.; Ward, S.C.; et al. Unique genomic profile of fibrolamellar hepatocellular carcinoma. Gastroenterology 2015, 148, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Cleary, S.P.; Jeck, W.R.; Zhao, X.; Chen, K.; Selitsky, S.R.; Savich, G.L.; Tan, T.X.; Wu, M.C.; Getz, G.; Lawrence, M.S.; et al. Identification of driver genes in hepatocellular carcinoma by exome sequencing. Hepatology 2013, 58, 1693–1702. [Google Scholar] [CrossRef] [PubMed]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sausen, M.; Phallen, J.; Adleff, V.; Jones, S.; Leary, R.J.; Barrett, M.T.; Anagnostou, V.; Parpart-Li, S.; Murphy, D.; Kay Li, Q.; et al. Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat. Commun. 2015, 6, 7686. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Losic, B.; Moeini, A.; Cabellos, L.; Hao, K.; Revill, K.; Bonal, D.; Miltiadous, O.; Zhang, Z.; Hoshida, Y.; et al. Massive parallel sequencing uncovers actionable FGFR2-PPHLN1 fusion and ARAF mutations in intrahepatic cholangiocarcinoma. Nat. Commun. 2015, 6, 6087. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.M.; Jang, S.J.; Shim, J.H.; Kim, D.; Hong, S.M.; Sung, C.O.; Baek, D.; Haq, F.; Ansari, A.A.; Lee, S.Y.; et al. Genomic portrait of resectable hepatocellular carcinomas: Implications of RB1 and FGF19 aberrations for patient stratification. Hepatology 2014, 60, 1972–1982. [Google Scholar] [CrossRef] [PubMed]
- Schulze, K.; Imbeaud, S.; Letouze, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Totoki, Y.; Tatsuno, K.; Covington, K.R.; Ueda, H.; Creighton, C.J.; Kato, M.; Tsuji, S.; Donehower, L.A.; Slagle, B.L.; Nakamura, H.; et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat. Genet. 2014, 46, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.S.; Craig, D.W.; Carpten, J.; Borad, M.J.; Demeure, M.J.; Weiss, G.J.; Izatt, T.; Sinari, S.; Christoforides, A.; Aldrich, J.; et al. Genome-wide characterization of pancreatic adenocarcinoma patients using next generation sequencing. PLoS ONE 2012, 7, 43192. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, L.; Lee, J.; Park, C.K.; Mao, M.; Shi, Y.; Gong, Z.; Zheng, H.; Li, Y.; Zhao, Y.; Wang, G.; et al. Whole-genome sequencing of matched primary and metastatic hepatocellular carcinomas. BMC Med. Genom. 2014, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Darcy, D.G.; Chiaroni-Clarke, R.; Murphy, J.M.; Honeyman, J.N.; Bhanot, U.; LaQuaglia, M.P.; Simon, S.M. The genomic landscape of fibrolamellar hepatocellular carcinoma: Whole genome sequencing of ten patients. Oncotarget 2015, 6, 755–770. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, Y.; Fujimoto, A.; Furuta, M.; Tanaka, H.; Chiba, K.; Boroevich, K.A.; Abe, T.; Kawakami, Y.; Ueno, M.; Gotoh, K.; et al. Integrated analysis of whole genome and transcriptome sequencing reveals diverse transcriptomic aberrations driven by somatic genomic changes in liver cancers. PLoS ONE 2014, 9, 114263. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, A.; Totoki, Y.; Abe, T.; Boroevich, K.A.; Hosoda, F.; Nguyen, H.H.; Aoki, M.; Hosono, N.; Kubo, M.; Miya, F.; et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat. Genet. 2012, 44, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Jhunjhunwala, S.; Jiang, Z.; Stawiski, E.W.; Gnad, F.; Liu, J.; Mayba, O.; Du, P.; Diao, J.; Johnson, S.; Wong, K.F.; et al. Diverse modes of genomic alteration in hepatocellular carcinoma. Genome Biol. 2014, 15, 36. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.K.; Zheng, H.; Li, S.; Chen, R.; Liu, X.; Li, Y.; Lee, N.P.; Lee, W.H.; Ariyaratne, P.N.; Tennakoon, C.; et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012, 44, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, A.; Furuta, M.; Shiraishi, Y.; Gotoh, K.; Kawakami, Y.; Arihiro, K.; Nakamura, T.; Ueno, M.; Ariizumi, S.; Nguyen, H.H.; et al. Whole-genome mutational landscape of liver cancers displaying biliary phenotype reveals hepatitis impact and molecular diversity. Nat. Commun. 2015, 6, 6120. [Google Scholar] [CrossRef] [PubMed]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Sundin, T.; Hentosh, P. InTERTesting association between telomerase, mTOR and phytochemicals. Expert Rev. Mol. Med. 2012, 14, 8. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, M. A second chance for telomerase reverse transcriptase in anticancer immunotherapy. Nat. Rev. 2016. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A. A new era for cancer immunotherapy based on the genes that encode cancer antigens. Immunity 1999, 10, 281–287. [Google Scholar] [CrossRef]
- Dallas, P.B.; Pacchione, S.; Wilsker, D.; Bowrin, V.; Kobayashi, R.; Moran, E. The human SWI-SNF complex protein p270 is an ARID family member with non-sequence-specific DNA binding activity. Mol. Cell. Biol. 2000, 20, 3137–3146. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Schones, D.E.; Cui, K.; Ybarra, R.; Northrup, D.; Tang, Q.; Gattinoni, L.; Restifo, N.P.; Huang, S.; Zhao, K. Regulation of nucleosome landscape and transcription factor targeting at tissue-specific enhancers by BRG1. Genome Res. 2011, 21, 1650–1658. [Google Scholar] [CrossRef] [PubMed]
- Kassabov, S.R.; Zhang, B.; Persinger, J.; Bartholomew, B. SWI/SNF unwraps, slides, and rewraps the nucleosome. Mol. Cell 2003, 11, 391–403. [Google Scholar] [CrossRef]
- Tolstorukov, M.Y.; Sansam, C.G.; Lu, P.; Koellhoffer, E.C.; Helming, K.C.; Alver, B.H.; Tillman, E.J.; Evans, J.A.; Wilson, B.G.; Park, P.J.; et al. Swi/Snf chromatin remodeling/tumor suppressor complex establishes nucleosome occupancy at target promoters. Proc. Natl. Acad. Sci. USA 2013, 110, 10165–10170. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Ui, A.; Kanno, S.; Ogiwara, H.; Nagase, T.; Kohno, T.; Yasui, A. SWI/SNF factors required for cellular resistance to DNA damage include ARID1A and ARID1B and show interdependent protein stability. Cancer Res. 2014, 74, 2465–2475. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhao, H.; Zhang, X.; Wood, L.D.; Anders, R.A.; Choti, M.A.; Pawlik, T.M.; Daniel, H.D.; Kannangai, R.; Offerhaus, G.J.; et al. Inactivating mutations of the chromatin remodeling gene ARID2 in hepatocellular carcinoma. Nat. Genet. 2011, 43, 828–829. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Giacomini, C.P.; Matsukuma, K.; Karikari, C.A.; Bashyam, M.D.; Hidalgo, M.; Maitra, A.; Pollack, J.R. Convergent structural alterations define SWItch/Sucrose NonFermentable (SWI/SNF) chromatin remodeler as a central tumor suppressive complex in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Varela, I.; Tarpey, P.; Raine, K.; Huang, D.; Ong, C.K.; Stephens, P.; Davies, H.; Jones, D.; Lin, M.L.; Teague, J.; et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 2011, 469, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Versteege, I.; Sevenet, N.; Lange, J.; Rousseau-Merck, M.F.; Ambros, P.; Handgretinger, R.; Aurias, A.; Delattre, O. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 1998, 394, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.S.; Kalloger, S.E.; et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N. Engl. J. Med. 2010, 363, 1532–4153. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Pollack, J.R. The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLoS ONE 2013, 8, 55119. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Kan, J.; Yuen, S.T.; Shi, S.T.; Chu, K.M.; Law, S.; Chan, T.L.; Kan, Z.; Chan, A.S.; Tsui, W.Y.; et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat. Genet. 2011, 43, 1219–1223. [Google Scholar] [CrossRef] [PubMed]
- Bitler, B.G.; Aird, K.M.; Garipov, A.; Li, H.; Amatangelo, M.; Kossenkov, A.V.; Schultz, D.C.; Liu, Q.; Shih Ie, M.; Conejo-Garcia, J.R.; et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat. Med. 2015, 21, 231–238. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Zhu, Z.; Johnson, C.; Stoops, J.; Eaker, A.E.; Bowen, W.; de Frances, M.C. PIK3IP1, a negative regulator of PI3K, suppresses the development of hepatocellular carcinoma. Cancer Res. 2008, 68, 5591–5598. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.G.; Wang, X.; Shen, X.; McKenna, E.S.; Lemieux, M.E.; Cho, Y.J.; Koellhoffer, E.C.; Pomeroy, S.L.; Orkin, S.H.; Roberts, C.W. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 2010, 18, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Fillmore, C.M.; Xu, C.; Desai, P.T.; Berry, J.M.; Rowbotham, S.P.; Lin, Y.J.; Zhang, H.; Marquez, V.E.; Hammerman, P.S.; Wong, K.K.; et al. EZH2 inhibition sensitizes BRG1 and EGFR mutant lung tumours to TopoII inhibitors. Nature 2015, 520, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Roberts, C.W. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Helming, K.C.; Wang, X.; Wilson, B.G.; Vazquez, F.; Haswell, J.R.; Manchester, H.E.; Kim, Y.; Kryukov, G.V.; Ghandi, M.; Aguirre, A.J.; et al. ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nat. Med. 2014, 20, 251–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze, K.; Nault, J.C.; Villanueva, A. Genetic profiling of hepatocellular carcinoma using next-generation sequencing. J. Hepatol. 2016, 65, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas. National Cancer Institute, National Human Genome Research Institute. Available online: https://cancergenome.nih.gov/ (accessed on 1 October 2016).
- International Cancer Genome Consortium. Available online: http://icgc.org/ (accessed on 1 October 2016).
- Roychowdhury, S.; Chinnaiyan, A.M. Translating cancer genomes and transcriptomes for precision oncology. CA Cancer J. Clin. 2016, 66, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Xue, R.; Li, R.; Guo, H.; Guo, L.; Su, Z.; Ni, X.; Qi, L.; Zhang, T.; Li, Q.; Zhang, Z.; et al. Variable Intra-Tumor Genomic Heterogeneity of Multiple Lesions in Patients With Hepatocellular Carcinoma. Gastroenterology 2016, 150, 998–1008. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Wang, Z.C.; Duan, M.; Lin, Y.H.; Zhou, X.Y.; Worthley, D.L.; Wang, X.Y.; Niu, G.; Xia, Y.; Deng, M.; et al. Cell Culture System for Analysis of Genetic Heterogeneity Within Hepatocellular Carcinomas and Response to Pharmacologic Agents. Gastroenterology 2016. accepted. [Google Scholar] [CrossRef] [PubMed]
- Eirew, P.; Steif, A.; Khattra, J.; Ha, G.; Yap, D.; Farahani, H.; Gelmon, K.; Chia, S.; Mar, C.; Wan, A.; et al. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature 2015, 518, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Murtaza, M.; Dawson, S.J.; Tsui, D.W.; Gale, D.; Forshew, T.; Piskorz, A.M.; Parkinson, C.; Chin, S.F.; Kingsbury, Z.; Wong, A.S.; et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013, 497, 108–112. [Google Scholar] [CrossRef] [PubMed]


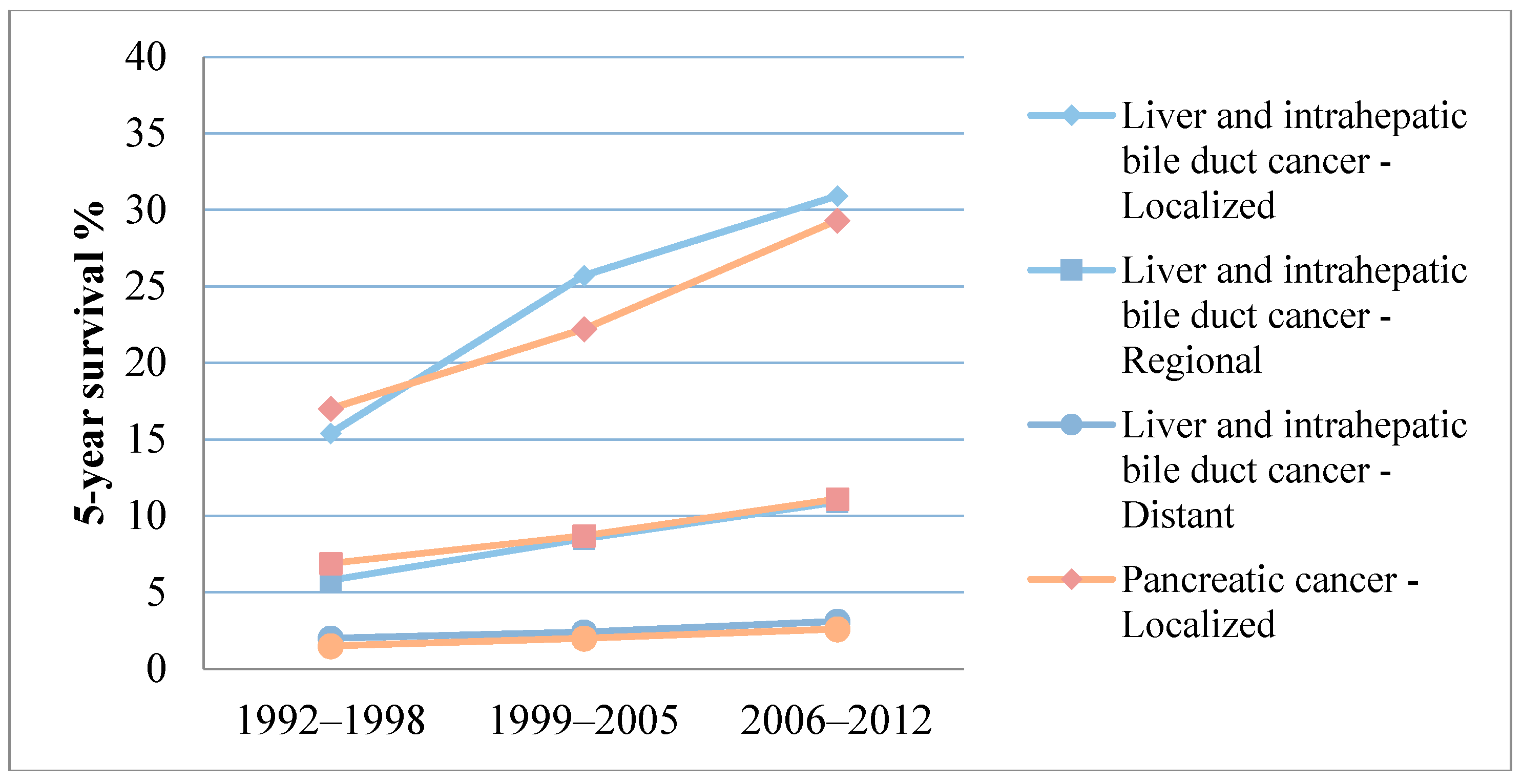{kind=link}
| Cancer Type | Localized | Regional | Distant | Unstaged | Overall |
|---|---|---|---|---|---|
| Liver and intahepatic bile duct cancer | 30.9% [7] | 10.9% [7] | 3.1% [7] | 6.1% [7] | 17.5% [7] |
| Intrahepatic bile duct cancer | 15% [62] | 6% [62] | 2% [62] | N/A | N/A |
| Extrahepatic bile duct cancer | 30% [62] | 24% [62] | 2% [62] | N/A | N/A |
| Pancreatic cancer | 29.3% [7] | 11.1% [7] | 2.6% [7] | 4.9% [7] | 7.7% [7] |
| Type of Cancer | Treatment | Recurrence Rate (%) |
|---|---|---|
| HCC | Hepatectomy | 51% [67]; 54% [65]; 63% [68] |
| HCC | OLT | 7.6% [69]; 15.3% [66]; 18.3% [70] |
| PDA | Pancreatectomy and adjuvant chemotherapy | 75%–85% [71]; 81%–93% (Phase III RCT) [48] |
| ICC | Resection | 62.2% [72]; 70% [73] |
| Mixed HCC-CC/ICC Hilar/ICC | OLT | 60% [74]; 38% [75] |
| ECC | ||
| Distal | Resection | 39% [76] |
| Hilar | Resection, OLT | 53% [77]; 68% [78]; After OLT: 20% [79] |
| GBC | Resection, resection and adjuvant chemotherapy | 66% [78]; 81.4% [80] |
| N | Setting | Intervention | Results | Reference/Clinicaltrials.gov Identifier |
|---|---|---|---|---|
| 1114 | Adjuvant HCC | Sorafenib vs. Placebo | RFS HR = 0.940; [95% CI, 0.780–1134]; one-sided p = 0.26 | STORM trial [81] |
| 1075 | Advanced HCC First-line | Sunitinib vs. Sorafenib | Terminated based on a higher incidence of serious adverse events in the sunitinib and on failure to demonstrate superiority or non-inferiority to sorafenib | NCT00699374 |
| 1035 | Advanced HCC First-line | Linifanib vs. Sorafenib | OS HR = 1.046; [95% CI, 0.896–1.221] | [82] |
| 870 | Intermediate Unresectable HCC | Brivanib vs. Placebo after TACE | HR = 0.90 [95% CI, 0.66–1.23]; log-rank p = 0.5280 | [83] |
| 720 | Advanced HCC First-line | Sorafenib + Erlotinib vs. Sorafenib + Placebo | OS 9.5 vs. 8.5 months, HR = 0.929; p = 0.408 | SEARCH trial [84] |
| 635 | Advanced HCC Second-line | ADI-PEG 20 vs. Placebo | OS 7.8 vs. 7.4 months; HR = 1.022 [95% CI, 0.847–1.233]; p = 0.884 PFS 2.6 vs. 2.6 months; HR = 1.175 [95% CI, 0.964–1.432]; p = 0.075 | [85] |
| 565 | Advanced HCC Second-line | Ramucirumab vs. Placebo after Sorafenib | 9.2 vs. 7.6 months; HR = 0.87 [95% CI, 0.72–1.05]; p = 0.14 HR = 0.674; p = 0.0059 with baseline AFP ≥ 400 ng/mL | REACH trial [86] |
| 420 | Advanced HCC | Tamoxifen + SOC vs. SOC alone | OS 4.8 [95% CI, 3.6–6] vs. 4.0 months [95% CI, 3.5–4.5] | [87] |
| 395 | Advanced HCC Second-line | Brivanib vs. Placebo | OS 9.4 vs. 8.2 months; HR = 0.89 [95.8% CI, 0.69–1.15]; p = 0.3307 | BRISK PS trial [88] |
| 230 | Adjuvant HCC | CIK vs. Placebo | RFS 44.0 vs. 30.0 months; HR = 0.63; [95% CI, 0.43–0.94]; p = 0.010 OS HR = 0.21 [95% CI, 0.06–0.75]; p = 0.008 | [89] |
| 124 * | Advanced BDC | Cis/Gem + Cediranib vs. Cis/Gem + Placebo | PFS HR = 0.93 [95% CI, 0.65–1.35]; p = 0.72 | ABC-03 trial [90] |
| N | Setting | Intervention | Results | Reference |
|---|---|---|---|---|
| 1062 | Advanced PDA First-line | Arm I: Chemotherapy alone Arm II: Chemotherapy with sequential GV1001 (telomerase peptide vaccine) Arm III: Chemotherapy with concurrent GV1001 | Sequential chemoimmunotherapy group OS HR = 1.19 [98.25% CI, 0.97–1.48]; p = 0.05 Concurrent chemoimmunotherapy group OS HR = 1.05 [98.25% CI, 0.85–1.29]; p = 0.64 | TeloVac trial [91] |
| 745 | Locally Advanced PDA First-line | Gemcitabine + Cetuximab vs. Gemcitabine alone | OS HR = 1.06 [95% CI, 0.91–1.23]; p = 0.23, one-sided | Southwest Oncology Group-directed intergroup trial S0205 [92] |
| 722 | Adjuvant PDA | Algenpantucel-L (HAPa) Immunotherapy + SOC vs. SOC alone | Study completed No statistically significant difference on preliminary report OS 27.3 vs. 30.4 months | IMPRESS trial [93] |
| 688 | Advanced PDA First-line | Gemcitabine + Tipifarnib (R115777) vs. Gemcitabine + Placebo | OS HR = 1.03 [95% CI, 0.86–1.23]; stratified log-rank p = 0.75 | [94] |
| 632 | Advanced PDA First-line | Gemcitabine + AG-013736 (Axitinib) vs. Gemcitabine + Placebo | OS HR = 1.014 [95% CI, 0.786–1.309]; p = 0.5436 | [95] |
| 602 | Advanced PDA First-line | Gemcitabine + Bevacizumab vs. Gemcitabine plus Placebo | OS HR = 1.044 [95% CI, 0.88 to 1.24]; p = 0.95 | CALGB 80303 trial [96] |
| 160 * | Metastatic PDA First-line | Rigosertib (ON 01910.Na) + Gemcitabine vs. Gemcitabine alone | OS HR = 1.24 [95% CI, 0.85–1.81] | [97] |
| 154 | Advanced PDA First-line | G17DT immunogen vs. Placebo | Mortality HR = 0.75 [95% CI, 0.51–1.10]; p = 0.138 | [98] |
| 153 * | Advanced PDA First-line | Elpamotide + Gemcitabine vs. Placebo + Gemcitabine | OS HR = 0.87 [95% CI, 0.486–1.557]; Harrington-Fleming p-value = 0.918; log-rank p-value = 0.897 | [99] |
| N | Findings | Clinical Implications | Reference |
|---|---|---|---|
| 9 tNGS; 1 WES | Mutations were observed in TP53 and CTNNB1 genes in 5/9 tumors | Larger studies are required | [116] |
| 12 | TP53 mutations in 5/12 patients, RUNX1 in 3/12 and other less frequent mutations | Larger studies required | [117] |
| 14 (advanced-metastatic) | Mutations identified in several well-known genes and pathways | Larger studies required | [118] |
| 45 patients (pts) treated with sorafenib (tNGS and CN assay; 6 CR, 39 non-CR) | FGFR mutations in 5/45 FGF19 copy number gain was detected more frequently among CR cases (2/6 vs. 2/39; p = 0.024) | Larger studies are required to evaluate potential clinical utility of CN gain for FGF19 as a predictive biomarker to sorafenib | [119] |
| 46 pts treated with sorafenib | Average number of detected oncogene mutations differed significantly between the PD and non-PD groups (p = 0.0446) | Targeted sequencing could predict response to sorafenib | [120] |
| 104 | Most frequent mutations: TERT (65%, associated with HCV infection), TP53 (38%, associated with HBV infection), CTNNB1 (30%, associated with absence of HBV infection) | TERT promoter mutations are related to poor prognosis Results may influence diagnostic and therapeutic strategies | [121] |
| N | Findings | Clinical Implications | Reference |
|---|---|---|---|
| 11 (3 ICC, 8 ECC) | Molecular heterogeneity was identified between ICC and ECC | This molecular classification could potentially provide personalized therapeutic implications | [122] |
| 28 | In 71% of cases, at least one potentially actionable alteration was found in known genes | Identification of these novel gene fusions (FGFR2-KIAA1598, FGFR2-BICC1, FGFR2-TACC3, and RABGAP1L-NTRK1) provides potential for personalized treatment | [123] |
| 40 (15 ECC, 10 ICC, 14 GBC, 1 AVC) | More (TP53) or less (NRAS, KRAS, ERBB215, PIK3CA) frequently mutated genes were identified | This is another study confirming the potential utility for umbrella studies | [124] |
| 41 (Diffusely infiltrating type CCA; 24 ERCP bile samples, 17 tumor samples) | tNGS on bile samples was feasible and comparable to tumor tNGS Diffusely infiltrating type CCA was genetically distinct from mass-forming type CCA | Encouraging results provide ground for larger studies to evaluate the reliability of TS on bile samples | [125] |
| 41 (32 ICC, 9 GBC, WES in 2) | Comparison of ICC with GBC revealed these two types are genetically distinct | Further investigation of chromatin remodeling could lead to the development of novel therapies | [126] |
| 75 (55 ICC, 20 ECC; 26 surgical resections, 49 biopsies) | Genetic aberrations were significantly different between ICC and ECC | TS could identify mutated genes-based subgroups of patients with potential prognostic and therapeutic relevance | [127] |
| 153 (70 ICC, 57 ECC, 26 GBC) | IDH1/2 and BAP1 mutations were characteristic of ICC, while KRAS and TP53 were more frequent in ECC and GBC Potentially actionable mutations were identified in 104/153 (68%) | Clinical utility of molecular classification identified by this study requires evaluation by clinical trials | [128] |
| 554 (412 ICCs, 57 ECCs and 85 GBCs) | Most frequently mutated genes: ICC: TP53, CDKN2A/B, KRAS, ARID1A, IDH1 ECC: KRAS, TP53, CDKN2A/B, SMAD4 GBC: TP53, CDKN2A/B, ARID1A, ERBB2 | In the ICC group, TP53, KRAS and FGFR2 mutations can be used as prognostic markers Identification of FGFR mutations in ICC patients could predict therapeutic response to FGFR inhibitors (BGJ398, pazopanib, dovitinib, TAS-120) tNGS can be utilized for clinical benefit and for designing umbrella and basket studies | [129] |
| N | Findings | Clinical Implications | Reference |
|---|---|---|---|
| 11 (oncocytic subtype IPMN; 11 TS, 2 WGS) | Typical oncocytic subtype IPMNs did not have KRAS or GNAS mutations and only one had both RNF43 and PIK3R1 mutations; ARHGAP26, ASXL1, EPHA8, and ERBB4 genes were mutated in more than one sample | Larger studies are required to explore the genetic profile and biologic behavior of the oncocytic subtype of IPMN | [130] |
| 23 (IPMN) | Identification of distinct mechanisms for the development of cancer in patients with IPMN using tNGS | Potential stratification and surveillance of patients based on the risk for pancreatic cancer | [131] |
| TS on FNA samples from 29 pts (25 PDA, 4 AVC) | Most frequent mutations identified: KRAS (93%), TP53 (72%), SMAD4 (31%), and GNAS (10%) Feasibility, reliability and concordance of FNA as compared to tumor samples for tNGS analysis | FNA-based tNGS analysis enables biomarker-based patient selection for clinical trials | [132] |
| 30 (PDA) | Substantial mutational heterogeneity (73%) | tNGS shapes the development of targeted therapy for pancreatic cancer | [133] |
| 52 (48 IPMNs, 4 ITPNs) | GNAS was mutated in 38/48 (79%) IPMNs, KRAS in 24/48 (50%) both in 18/48 (37.5%); Other mutations were less frequent | Identification of mutations in cyst fluid could enhance diagnosis and prognostic stratification of pancreatic cystic neoplasms | [134] |
| 76 (PDA) | 22 candidate cases have been identified (14 KRAS wild-type, 5 HER2 amplifications, 2 mutations in BRCA2 and 1 ATM mutation) | The availability of drugs targeting these mutated or amplified genes (cetuximab, transtuzumab) enables basket design of clinical trials | [135] |
| Cancer Type | N | Findings | Clinical Implications | Reference |
|---|---|---|---|---|
| HB | 6 | 21 mutated genes, including mutations in the WNT pathway | Larger studies are required to explore the mutational background of HB | [141] |
| HCC (HBV positive) | 10 (110 samples, including PVTTs and intrahepatic metastases) | ARID1A was mutated in 14 of 110 samples (13%) ARID1A loss-of-function mutations may be crucial for HCC invasion and metastasis | ARID1A is a potential novel biomarker for treatment and prognosis | [142] † |
| NAFLD-related HCC | 10 (11 samples, WES, TS, CNV studies) | 12 genes were frequently mutated including novel genes (FGA, SYNE1) | Larger studies are required to confirm the validity of novel genes | [143] |
| PDA (acinar differentiation) | 23 | Potentially targetable mutations in well-known genes (BRCA2, PALB2, ATM, BAP1, BRAF and JAK1) were identified in 1/3 of patients | This study supports the conduction of umbrella studies | [144] |
| HCC | 24 WES (NGS); 125 CNA in total with CGH array analysis | New recurrent mutations in ARID1A, RPS6KA3, NFE2L2 and IRF2 Inactivation of chromatin remodelers was frequent and was associated with alcohol Wnt/b-catenin pathway promotes tumorigenesis through both oxidative stress metabolism and MAPK pathways | Association of environmental risk factors with specific gene mutations could improve screening and early diagnosis | [145] |
| PDA from VLTSs (≥10 years) | 35 (8 WES, 27 TS) | Frequently mutated genes were identified (KRAS, TP53, RNF43, CDKN2A, and SMAD4) Combined WES and TS data showed no significant difference between VLTSs and patients unselected for survival | Validity of these data must be investigated by larger studies | [146] |
| FLC | 78 (48 WES + TES, 58 whole-transcriptome, 41 SNP arrays) | Identification of 3 molecular classes: proliferation with altered mTOR pathway, inflammation with altered cytokine production genes and unannotated | Larger studies are required to confirm the validity of the developed prognostic 8-gene expression signature (PEAR1, KRTAP, KLRD1, OSBPL8, RPL32, SLC26A11, RGS11 and RAPGEF1) | [147] |
| HCC | 87 | Substantial genetic heterogeneity NFE2L2-KEAP1 and MLL pathways were recurrently mutated | Further larger WES studies are needed for completing the cancer driver genes catalog and developing individualized therapy | [148] |
| PDA | 99 with early stage (I and II; WES and CNA) | Substantial genetic heterogeneity 8 novel mutated genes: EPC1 and ARID2 (chromatin modification), ATM (DNA damage repair), ZIM2, MAP2K4, NALCN, SLC16A4 and MAGEA6 | The novel mutated genes identified could potentially be used as therapeutic targets but validation is required by larger studies | [149] |
| PDA | 101 (24 WES and 77 TS) | Mutated chromatin regulating genes MLL, MLL2, MLL3, ARID1A were associated with improved survival Detection of ctDNA was associated with predictable recurrence 6.5 months before occurrence | These genes may have prognostic significance and ctDNA could potentially be used as a biomarker to predict recurrence | [150] |
| PDA | 109 | Identification of multiple novel mutated genes in PDA, with select genes harboring prognostic significance KRAS mutations were observed in >90% of cases ARID1A was a marker of poorer outcome RBM10 mutation was associated with longer survival BRAF and PIK3CA mutations expand the spectrum of oncogenic drivers | PDA is a complex cancer and WES can provide insight on pathogenesis, diagnosis and therapeutic management of these tumors | [151] |
| ICC | 135 (7 fresh frozen samples, 107 FFPE, 21 FFPE mixed HCC-ICC; WES in 8, WGS in 1) | Chromosomal translocation t(10;12)(q26;q12) leads to FGFR2–PPHLN1 fusion; it is successfully inhibited by a selective FGFR2 inhibitor in vitro | Novel fusion event (FGFR2–PPHLN1) could provide therapeutic benefit Most CCA patients harbor potentially targetable molecular alterations | [152] |
| HCC | 231 (WES and CNA) | Mutated RB1 was a predictor of recurrence and poor survival after HCC resection | RB1 mutations could be used as a prognostic molecular biomarker for resectable HCC | [153] |
| HCC | 243 | 28% of the tumors featured genetic alterations targeted by FDA-approved drugs and 3 groups of genes were associated with risk factors: CTNNB1 (alcohol), TP53 (HBV) and AXINI | Association of environmental risk factors with specific genes provides new potential for HCC prevention and early-stage diagnosis | [154] |
| HCC | 503 (452 WES) * | TERT alterations were identified in 68% of the patients AXIN1 was more frequently mutated in HBV-positive and ARID1A in non-virus cases Druggable kinase alterations were rarely found (<2%) | Mutations in genes coding for metabolic enzymes, chromatin remodelers and mTOR pathway could provide diagnostic and therapeutic potential | [155] ‡ |
| Cancer Type | N | Findings | Clinical Implications | Reference |
|---|---|---|---|---|
| PDA | 3 | KRAS signaling pathway was the most heavily impacted pathway | Larger WGS studies are required for assessing clinical utility | [156] |
| HCC with pulmonary metastasis | 4 | Somatic SNVs, SVs and CNAs were similar between primary and metastatic tumors | Larger studies with multiple biopsies are required to investigate similarities and differences between primary and metastatic tumors | [157] |
| FLC | 10 | Few coding, somatic mutations, no recurrent SVs Molecular differentiation from HCC | This study supports further research on the DNAJB1-PRKACA fusion protein for potential diagnostic and therapeutic clinical implementation | [158] |
| HCC | 22 * | TERT alterations were identified in 68% of the patients AXIN1 was more frequently mutated in HBV-positive and ARID1A in non-virus cases Druggable kinase alterations were rarely found (<2%) | Mutations in genes coding for metabolic enzymes, chromatin remodelers and mTOR pathway could provide diagnostic and therapeutic potential | [155] |
| HBV-related HCC | 22 (WGS and RNA seq.) | Mutations, including non-coding alterations and SVs and virus integrations can create diverse transcriptomic aberrations | Integrative analysis of WGS and RNA-Seq is crucial for understanding the importance of comprehensive GA identification, shaping new diagnostic and therapeutic avenues | [159] |
| HCC | 27 (25 HBV- or HCV-related) | In the two multicentric tumors, WGS analysis suggested origins from independent mutations Chromatin regulation genes (ARID1A, ARID1B, ARID2, MLL, MLL3) were mutated in approximately 50% of the tumors Frequent integration of HBV DNA in TERT locus | GAs and carcinogenesis can be influenced by the etiological background (viral hepatitis) Further elucidation on the molecular background of HCC is required to achieve significant clinical benefit | [160] |
| HCC | 42 (WGS, WES and whole-transcriptome seq.) | More (TP53, CTNNB1 and AXIN1) or less (BAP1 and IDH1) frequent mutations and a novel deletion in CTNNB1 were identified; LAMA2 was a predictor of recurrence and poor survival | Identification of GAs and virus-associated genomic changes provide new predictive and therapeutic potential | [161] |
| HCC | 88 (81 HBV positive) | HBV integration is more frequent in the tumors (86.4%) than in adjacent liver tissues (30.7%) Recurrent HBV integration in TERT, MLL4 and CCNE1 genes, with upregulated gene expression | The number of HBV integrations is associated with survival and could have prognostic significance | [162] |
| HCC/LCB | 90 (30 LCB, 60 HCC) | LCBs feature recurrent mutations in TERT promoter, chromatin regulators (BAP1, PBRM1 and ARID2), a synapse organization gene (PCLO), IDH genes and KRAS KRAS and IDH mutations were more frequent in hepatitis-negative LCBs and are associated with poor disease-free survival | Chronic hepatitis has a major impact on the mutational status of liver cancer | [163] |
| PDA | 100 (WGS and CNV analysis) | Identification of altered genes (TP53, SMAD4, CDKN2A, ARID1A and ROBO2), novel gene mutations (KDM6A and PREX2) and frequent targetable gene mutations (ERBB2, MET, FGFR1, CDK6, PIK3R3 and PIK3CA) | KDM6A and PREX2 are potential biomarkers and therapeutic targets | [164] |
| HCC, ICC | 300 (268 HCC, 24 ICC, 8 combined HCC/ICC) | Mutations related to liver carcinogenesis and recurrently mutated coding and noncoding regions were identified Known (CDKN2A, CCND1, APC, and TERT) and novel (ASH1L, NCOR1, and MACROD2) cancer-related genes were identified in SV analysis | WGS is crucial for detection of cancer driver genes Association of risk factors (smoking, HCV, HBV, alcohol) with specific mutations can predict tumorigenesis and provide prognostic potential | [10] |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kyrochristos, I.D.; Glantzounis, G.K.; Ziogas, D.E.; Gizas, I.; Schizas, D.; Lykoudis, E.G.; Felekouras, E.; Machairas, A.; Katsios, C.; Liakakos, T.; et al. From Clinical Standards to Translating Next-Generation Sequencing Research into Patient Care Improvement for Hepatobiliary and Pancreatic Cancers. Int. J. Mol. Sci. 2017, 18, 180. https://doi.org/10.3390/ijms18010180
Kyrochristos ID, Glantzounis GK, Ziogas DE, Gizas I, Schizas D, Lykoudis EG, Felekouras E, Machairas A, Katsios C, Liakakos T, et al. From Clinical Standards to Translating Next-Generation Sequencing Research into Patient Care Improvement for Hepatobiliary and Pancreatic Cancers. International Journal of Molecular Sciences. 2017; 18(1):180. https://doi.org/10.3390/ijms18010180
Chicago/Turabian StyleKyrochristos, Ioannis D., Georgios K. Glantzounis, Demosthenes E. Ziogas, Ioannis Gizas, Dimitrios Schizas, Efstathios G. Lykoudis, Evangelos Felekouras, Anastasios Machairas, Christos Katsios, Theodoros Liakakos, and et al. 2017. "From Clinical Standards to Translating Next-Generation Sequencing Research into Patient Care Improvement for Hepatobiliary and Pancreatic Cancers" International Journal of Molecular Sciences 18, no. 1: 180. https://doi.org/10.3390/ijms18010180