
Neuroprotective Strategy in Retinal Degeneration: Suppressing ER Stress-Induced Cell Death via Inhibition of the mTOR Signal

Abstract
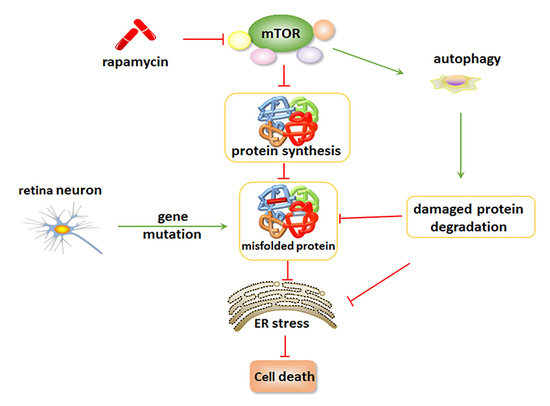
:
1. Retinal Degeneration
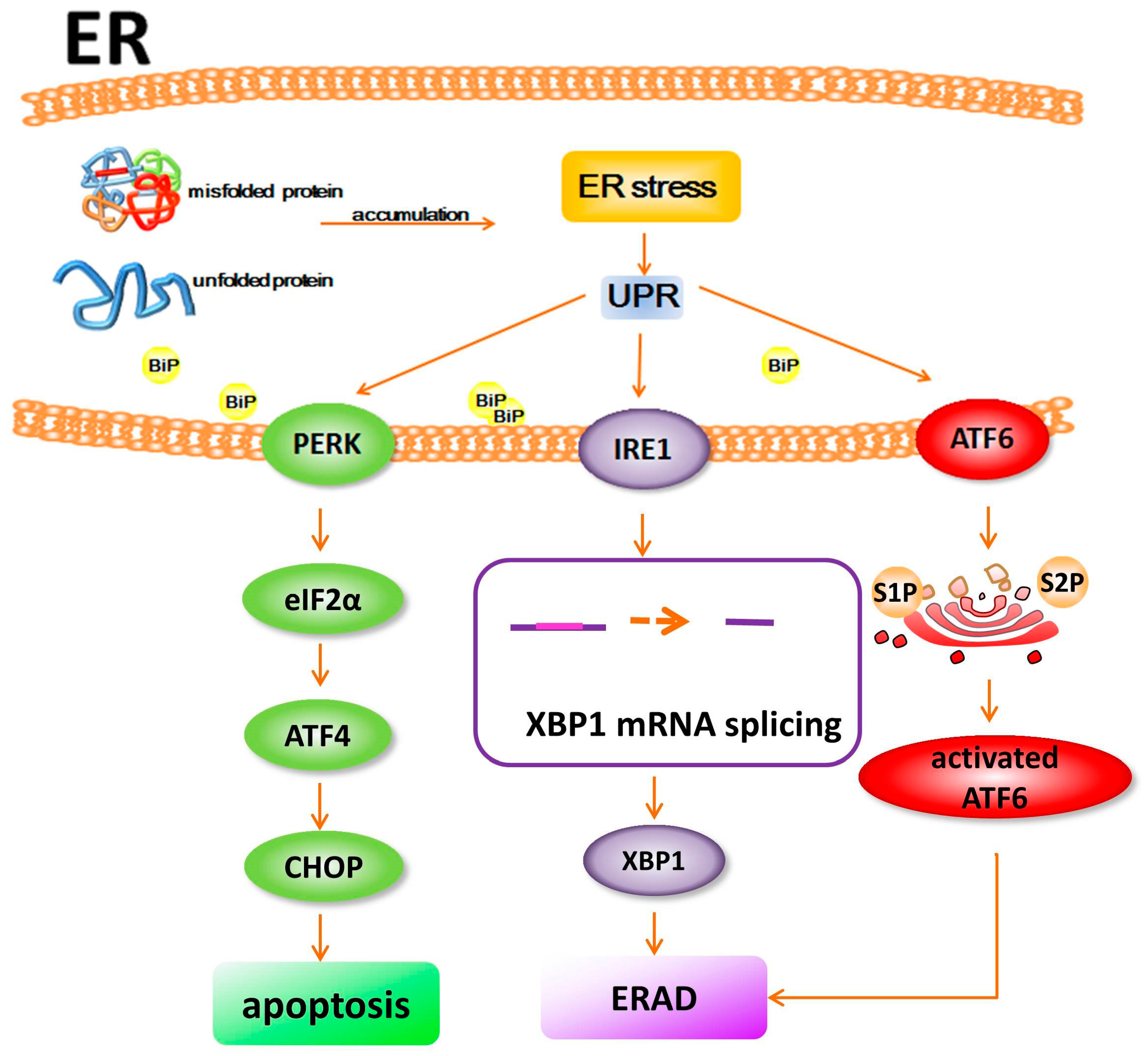
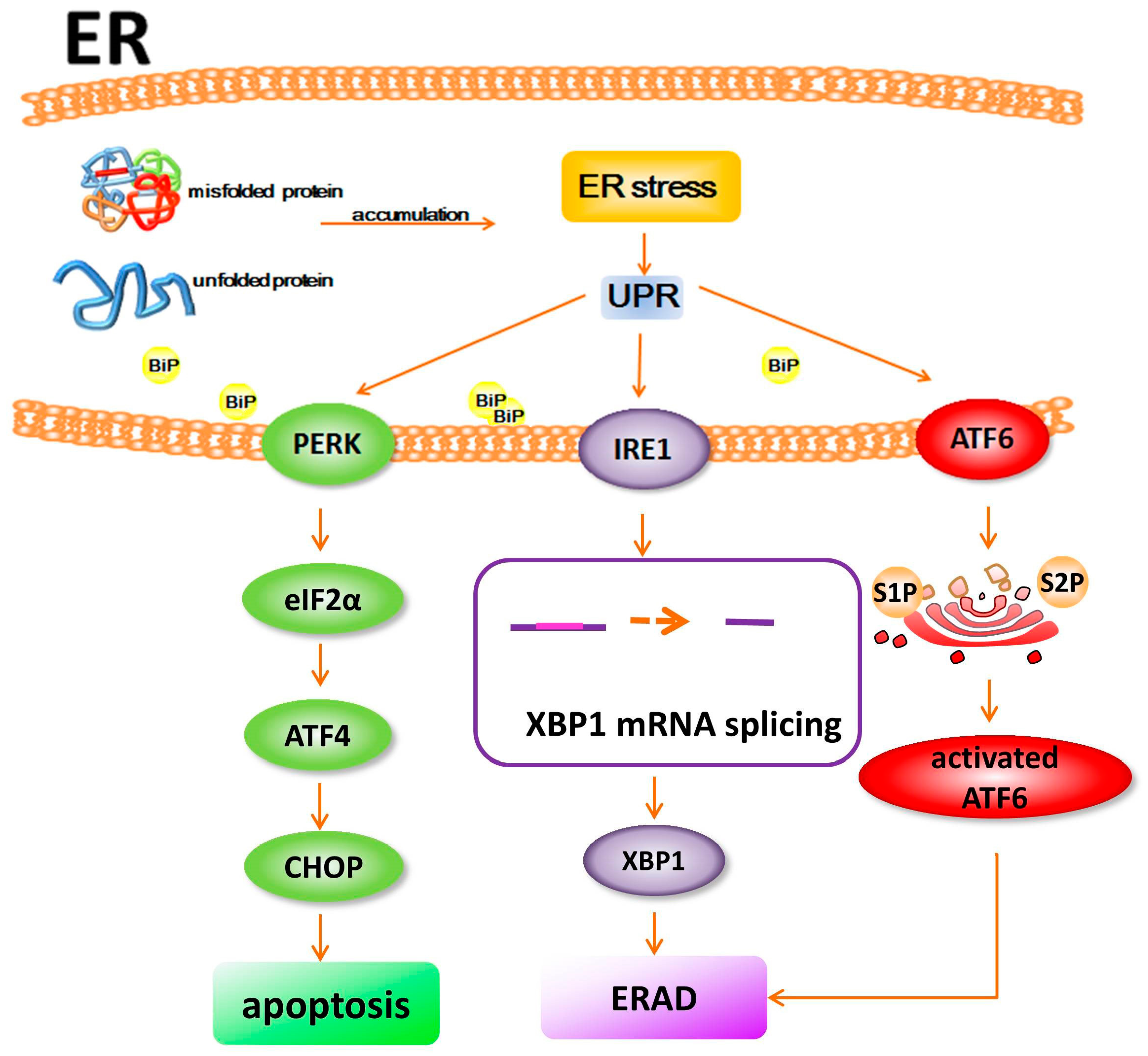
2. Proteostasis and ER Stress
3. Disturbance of Proteostasis and ER Stress in Retinal Degeneration
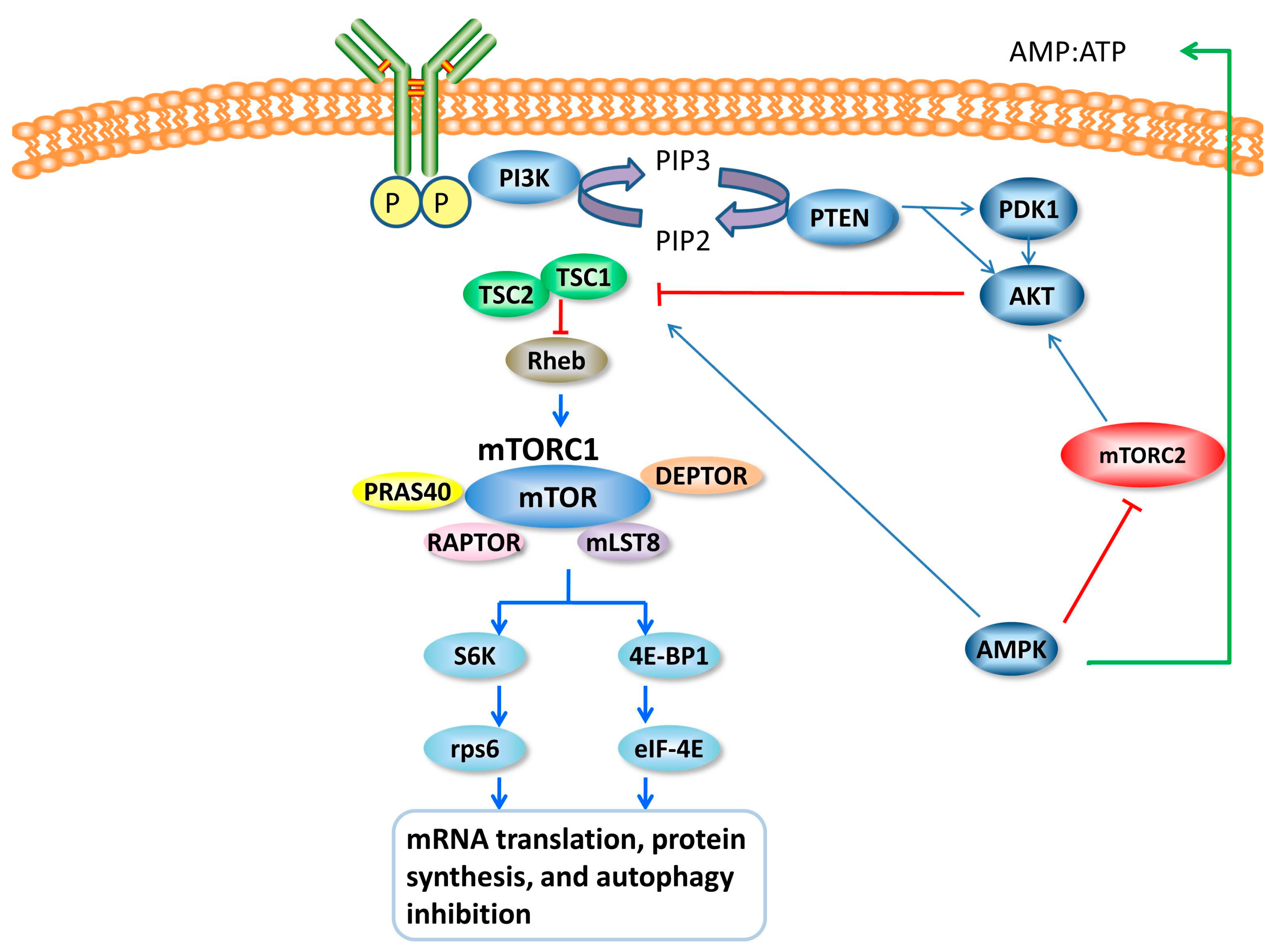
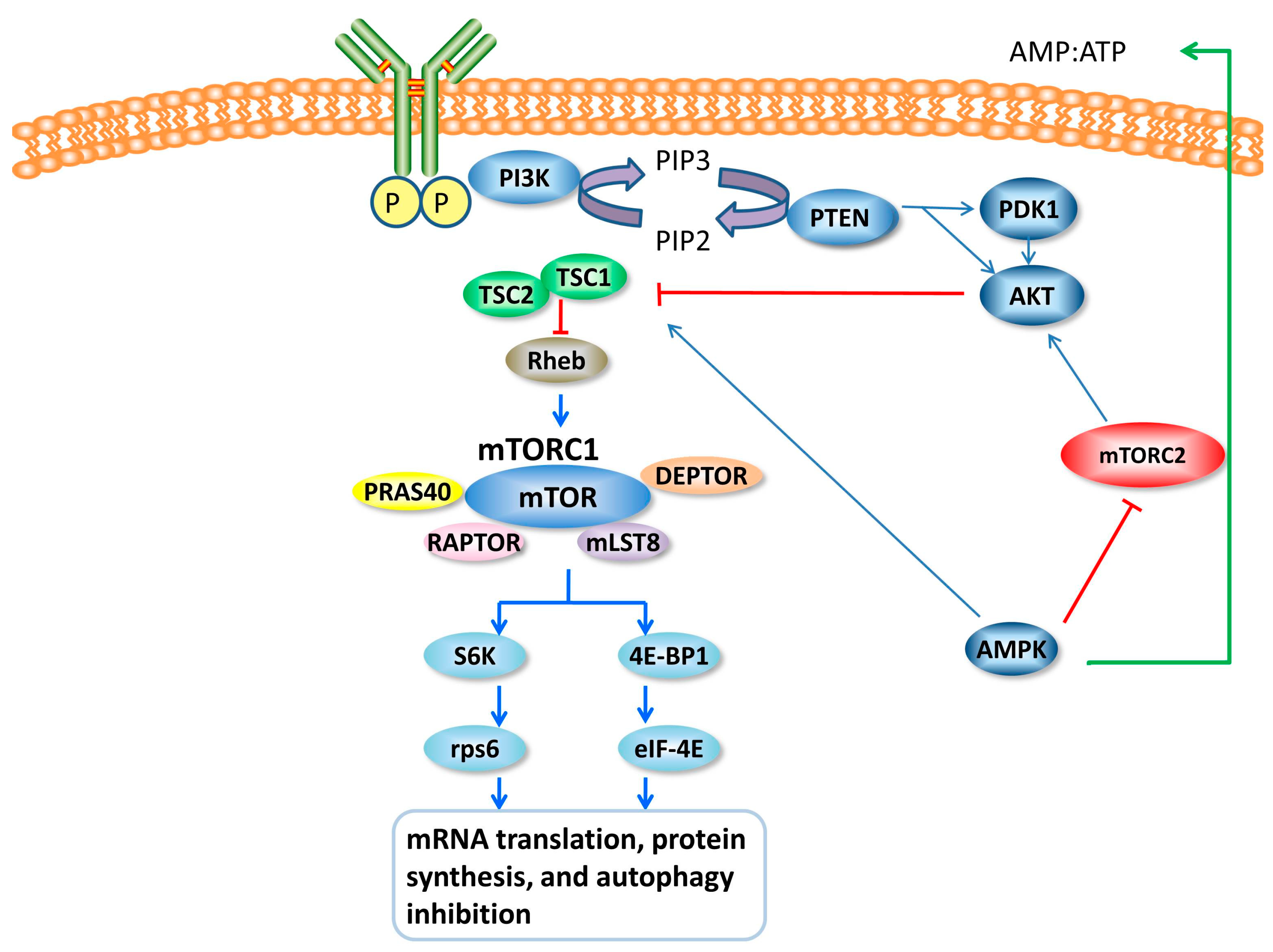
4. mTOR and mTOR Signal
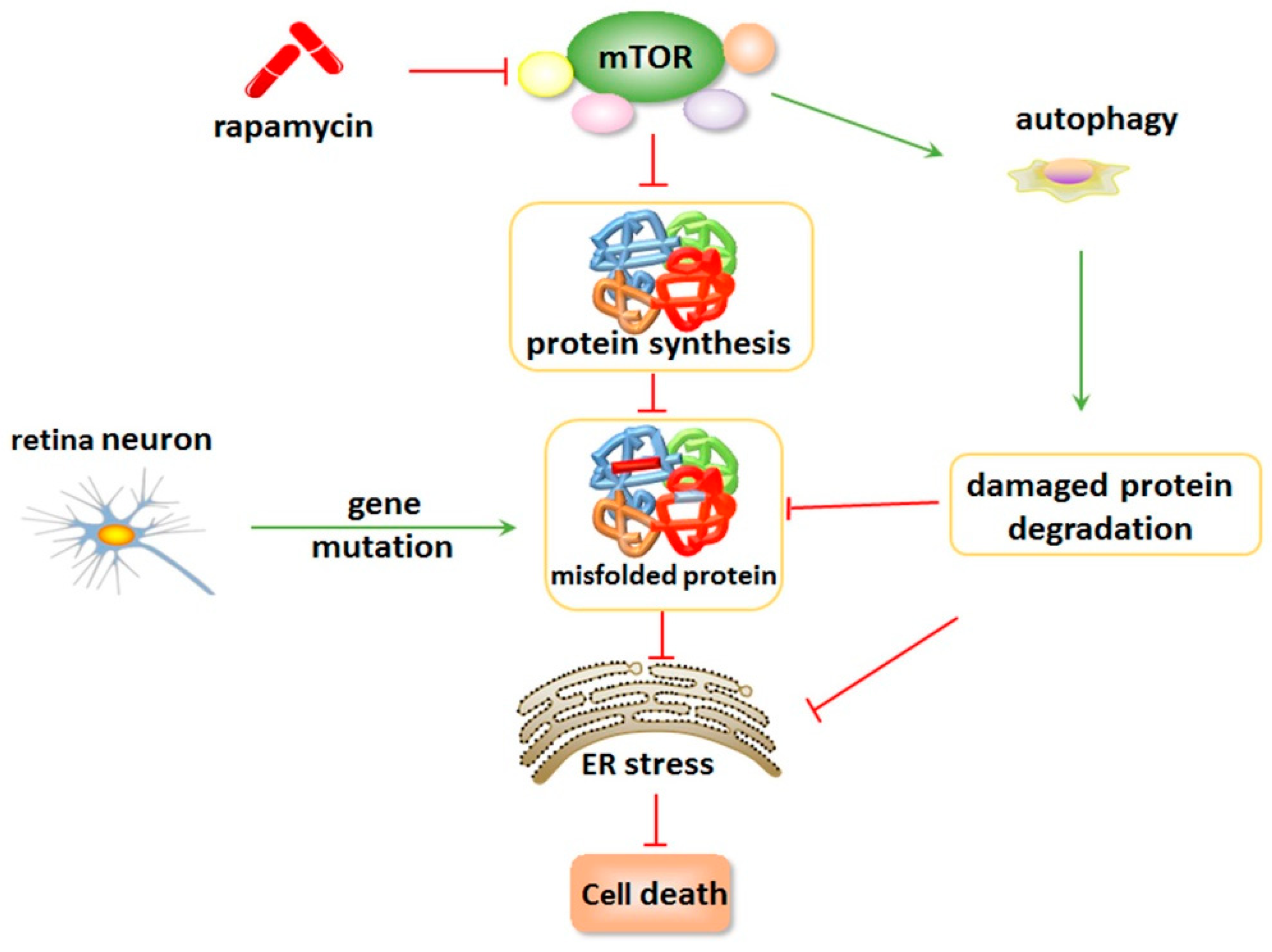
5. Inhibition of mTOR Suppresses ER Stress and Attenuates Retinal Degeneration
6. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| ATF4 | Activating transcription factor 4 |
| ASK1 | Apoptosis signal-regulating kinase 1 |
| ATF6 | Activating transcription factor 6 |
| ATG13 | Autophagy related gene 13 |
| AMP | Adenosine monophosphate |
| AMPK | AMP-dependent kinase |
| AMD | Age-related macular degeneration |
| AAV5 | Adeno-Associated Virus Type 5 |
| AKT1 | AKT serine/threonine kinase 1 |
| adRP | Autosomal dominant RP |
| BiP | Immunoglobulin binding protein |
| cGMP | Cyclic guanosine-mono-phosphate |
| CHOP | C/EBP homologousprotein |
| cKO | Conditional knockout |
| DHRD | Doyne honeycomb retinal dystrophy |
| DEPTOR | Disheveled, Egl-10, and pleckstrin domain-containing mTOR-interacting protein |
| ERAD | ER-associated protein degradation |
| ER | Endoplasmic reticulum |
| ELOVL4 | Elongation of very long chain fatty acids |
| ERK | Extracellular signal related kinase |
| F3 | Fibulin-3 |
| FKBP12 | 12 kDa Protein FK506-binding protein |
| GCL | Ganglion cell layer |
| GRP78 | Glucose-regulated protein 78 |
| GAP | GTPase activating protein |
| HSR | Heat shock response |
| INL | Inner nuclear layer |
| IPL | Inner plexiform layer |
| IRD | Inherited retinal degeneration |
| IRE1 | Inositol-requiring protein 1 |
| Ire1α | Inositol-requiring kinase 1 |
| JNK | c-Jun N-terminal kinase |
| LCA | Leber congenital amaurosis |
| ML | Malattia Leventinese |
| mTOR | Mammalian target of rapamycin |
| mTORC1 | mTOR complex 1 |
| mTORC2 | mTOR complex 2 |
| mLST8 | Mammalian lethal with SEC13 protein 8 |
| mSIN1 | Stress-activated MAP kinase-interacting protein 1 |
| ONL | Outer nuclear layer |
| OS | Outer segment |
| OPL | Outer plexiform layer |
| PERK | Protein kinase RNA-like ER kinase |
| PDE6 | Phosphodiesterase |
| PDK1 | 3-Phosphoinositide-dependent protein kinase 1 |
| PIKK | PI3K-kinase-related kinase |
| PI3K | Phosphoinositide 3-kinase |
| PIP2 | Phosphatidylinositol 4,5-bisphosphate |
| PIP3 | Phosphatidylinositol 3,4,5-triphosphate |
| PRAS40 | 40 kDa Pro-rich AKT1 substrate 1 |
| PRR5 | Pro-rich protein 5 |
| PTEN | Phosphatase and tensin homolog deleted on chromosome 10 |
| PN | Photoreceptor neuron |
| p90-RSK | Ribosomal S6 kinase |
| p70S6K | 40S ribosomal protein S6 kinase |
| Rheb | Ras homolog enriched in brain |
| RICTOR | Rapamycin-insensitive companion of mTOR |
| Rh | Rhodopsin |
| RP | Retinitis pigmentosa |
| RPE | Retinal pigment epithelium |
| RAPTOR | Regulatory associated protein of mTOR |
| S6K1 | S6 kinase 1 |
| TSC | Tuberous sclerosis complex |
| TFEB | Transcription factor immunoglobulin E box-binding proteins |
| TOR | Target of rapamycin |
| T17M | Threonine-to-methionine mutation at the 17th residue of rhodopsin |
| UPS | The ubiquitin-proteasome system |
| UPR | The unfolded protein response |
| ULK | UNC-5 like autophagy activating kinase |
| VEGF | Vascular endothelial growth factor |
| WT | Wild-type |
| 4E-BP1 | 4E-binding protein 1 |
| 5′-TOP | 5′-Terminal oligopyrimidine tract |
References
- Brzezinski, J.A.; Reh, T.A. Photoreceptor cell fate specification in vertebrates. Development 2015, 142, 3263–3273. [Google Scholar] [CrossRef] [PubMed]
- Silverman, M.S.; Hughes, S.E.; Valentino, T.L.; Liu, Y. Photoreceptor transplantation: Anatomic, electrophysiologic, and behavioral evidence for the functional reconstruction of retinas lacking photoreceptors. Exp. Neurol. 1992, 115, 87–94. [Google Scholar] [CrossRef]
- Wert, K.J.; Lin, J.H.; Tsang, S.H. General pathophysiology in retinal degeneration. Dev. Ophthalmol. 2014, 53, 33–43. [Google Scholar] [PubMed]
- Kolb, H.; Nelson, R. The Organization of Photoreceptor to Bipolar Synapses in the Outer Plexiform Layer; Springer: Houten, The Netherlands, 1995; pp. 273–296. [Google Scholar]
- Schiller, P.H. Parallel information processing channels created in the retina. Proc. Natl. Acad. Sci. USA 2010, 107, 17087–17094. [Google Scholar] [CrossRef] [PubMed]
- Sahel, J.A.; Marazova, K.; Audo, I. Clinical characteristics and current therapies for inherited retinal degenerations. Cold Spring Harb. Perspect. Med. 2014, 5, a017111. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.J.; Bainbridge, J.W.; Ali, R.R. Prospects for retinal gene replacement therapy. Trends Genet. 2009, 25, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.A.; Ali, R.R.; Banin, E.; Branham, K.E.; Flannery, J.G.; Gamm, D.M.; Hauswirth, W.W.; Heckenlively, J.R.; Iannaccone, A.; Jayasundera, K.T.; et al. Advancing therapeutic strategies for inherited retinal degeneration: Recommendations from the Monaciano Symposium. Investig. Ophthalmol. Vis. Sci. 2015, 56, 918–931. [Google Scholar] [CrossRef] [PubMed]
- Travis, G.H. Mechanisms of cell death in the inherited retinal degenerations. Am. J. Hum. Genet. 1998, 62, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Sin, O.; Nollen, E.A. Regulation of protein homeostasis in neurodegenerative diseases: The role of coding and non-coding genes. Cell. Mol. Life Sci. 2015, 72, 4027–4047. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Matsuda, N. Proteostasis and neurodegeneration: The roles of proteasomal degradation and autophagy. Biochim. Biophys. Acta 2014, 1843, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Castelao, B.; Ruiz-Rivas, C.; Castano, J.G. A critical appraisal of quantitative studies of protein degradation in the framework of cellular proteostasis. Biochem. Res. Int. 2012, 2012, 823597. [Google Scholar] [CrossRef] [PubMed]
- Jing, G.; Wang, J.J.; Zhang, S.X. ER stress and apoptosis: A new mechanism for retinal cell death. Exp. Diabetes Res. 2012, 2012, 589589. [Google Scholar] [CrossRef] [PubMed]
- Crippa, V.; D'Agostino, V.G.; Cristofani, R.; Rusmini, P.; Cicardi, M.E.; Messi, E.; Loffredo, R.; Pancher, M.; Piccolella, M.; Galbiati, M.; et al. Transcriptional induction of the heat shock protein B8 mediates the clearance of misfolded proteins responsible for motor neuron diseases. Sci. Rep. 2016, 6, 22827. [Google Scholar] [CrossRef] [PubMed]
- Schuldt, A. Protein metabolism: A channel for ERAD. Nat. Rev. Mol. Cell Biol. 2014, 15, 2. [Google Scholar] [CrossRef] [PubMed]
- Appenzeller-Herzog, C.; Hall, M.N. Bidirectional crosstalk between endoplasmic reticulum stress and mTOR signaling. Trends Cell Biol. 2012, 22, 274–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deldicque, L. Endoplasmic reticulum stress in human skeletal muscle: Any contribution to sarcopenia? Front. Physiol. 2013, 4, 236. [Google Scholar] [CrossRef] [PubMed]
- Shibata, Y.; Voeltz, G.K.; Rapoport, T.A. Rough sheets and smooth tubules. Cell 2006, 126, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Hiramatsu, N.; Chiang, W.C.; Kurt, T.D.; Sigurdson, C.J.; Lin, J.H. Multiple Mechanisms of unfolded protein response-induced cell death. Am. J. Pathol. 2015, 185, 1800–1808. [Google Scholar] [CrossRef] [PubMed]
- Muoio, D.M.; Newgard, C.B. Biomedicine. Insulin resistance takes a trip through the ER. Science. 2004, 306, 425–426. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef] [PubMed]
- Perri, E.R.; Thomas, C.J.; Parakh, S.; Spencer, D.M.; Atkin, J.D. The Unfolded protein response and the role of protein disulfide isomerase in neurodegeneration. Front. Cell Dev. Biol. 2015, 3, 80. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Chabre, M.; le Maire, M. Monomeric G-protein-coupled receptor as a functional unit. Biochemistry 2005, 44, 9395–9403. [Google Scholar] [CrossRef] [PubMed]
- Powers, E.T.; Morimoto, R.I.; Dillin, A.; Kelly, J.W.; Balch, W.E. Biological and chemical approaches to diseases of proteostasis deficiency. Annu. Rev. Biochem. 2009, 78, 959–991. [Google Scholar] [CrossRef] [PubMed]
- Mendes, C.S.; Levet, C.; Chatelain, G.; Dourlen, P.; Fouillet, A.; Dichtel-Danjoy, M.L.; Gambis, A.; Ryoo, H.D.; Steller, H.; Mollereau, B. ER stress protects from retinal degeneration. EMBO J. 2009, 28, 1296–1307. [Google Scholar] [CrossRef] [PubMed]
- Athanasiou, D.; Aguila, M.; Bevilacqua, D.; Novoselov, S.S.; Parfitt, D.A.; Cheetham, M.E. The cell stress machinery and retinal degeneration. FEBS Lett. 2013, 587, 2008–2017. [Google Scholar] [CrossRef] [PubMed]
- Bhootada, Y.; Kotla, P.; Zolotukhin, S.; Gorbatyuk, O.; Bebok, Z.; Athar, M.; Gorbatyuk, M. Limited ATF4 expression in degenerating retinas with ongoing ER stress promotes photoreceptor survival in a mouse model of autosomal dominant retinitis pigmentosa. PLoS ONE 2016, 11, e0154779. [Google Scholar] [CrossRef] [PubMed]
- RetNet: Retinal Information Network. Available online: https://sph.uth.edu/Retnet (accessed on 18 January 2017).
- Parfitt, D.A.; Cheetham, M.E. Targeting the proteostasis network in rhodopsin retinitis pigmentosa. Adv. Exp. Med. Biol. 2016, 854, 479–484. [Google Scholar] [PubMed]
- Ferrington, D.A.; Sinha, D.; Kaarniranta, K. Defects in retinal pigment epithelial cell proteolysis and the pathology associated with age-related macular degeneration. Prog. Retin. Eye Res. 2016, 51, 69–89. [Google Scholar] [CrossRef] [PubMed]
- Daiger, S.P.; Bowne, S.J.; Sullivan, L.S. Perspective on genes and mutations causing retinitis pigmentosa. Arch. Ophthalmol. 2007, 125, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Galy, A.; Roux, M.J.; Sahel, J.A.; Leveillard, T.; Giangrande, A. Rhodopsin maturation defects induce photoreceptor death by apoptosis: A fly model for RhodopsinPro23His human retinitis pigmentosa. Hum. Mol. Genet. 2005, 14, 2547–2557. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Li, H.; Yasumura, D.; Cohen, H.R.; Zhang, C.; Panning, B.; Shokat, K.M.; Lavail, M.M.; Walter, P. IRE1 signaling affects cell fate during the unfolded protein response. Science 2007, 318, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Gorbatyuk, M.S.; Knox, T.; LaVail, M.M.; Gorbatyuk, O.S.; Noorwez, S.M.; Hauswirth, W.W.; Lin, J.H.; Muzyczka, N.; Lewin, A.S. Restoration of visual function in P23H rhodopsin transgenic rats by gene delivery of BiP/Grp78. Proc. Natl. Acad. Sci. USA 2010, 107, 5961–5966. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, S.; Bhootada, Y.; Gorbatyuk, O.; Gorbatyuk, M. Caspase-7 ablation modulates UPR, reprograms TRAF2-JNK apoptosis and protects T17M rhodopsin mice from severe retinal degeneration. Cell Death Dis. 2013, 4, e528. [Google Scholar] [CrossRef] [PubMed]
- Chiang, W.C.; Messah, C.; Lin, J.H. IRE1 directs proteasomal and lysosomal degradation of misfolded rhodopsin. Mol. Biol. Cell 2012, 23, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Chiang, W.C.; Kroeger, H.; Sakami, S.; Messah, C.; Yasumura, D.; Matthes, M.T.; Coppinger, J.A.; Palczewski, K.; LaVail, M.M.; Lin, J.H. Robust endoplasmic reticulum-associated degradation of rhodopsin precedes retinal degeneration. Mol. Neurobiol. 2015, 52, 679–695. [Google Scholar] [CrossRef] [PubMed]
- Karan, G.; Yang, Z.; Howes, K.; Zhao, Y.; Chen, Y.; Cameron, D.J.; Lin, Y.; Pearson, E.; Zhang, K. Loss of ER retention and sequestration of the wild-type ELOVL4 by Stargardt disease dominant negative mutants. Mol. Vis. 2005, 11, 657–664. [Google Scholar] [PubMed]
- Tran, H.V.; Moret, E.; Vaclavik, V.; Marcelli, F.; Abitbol, M.M.; Munier, F.L.; Schorderet, D.F. Swiss family with dominant Stargardt Disease caused by a recurrent mutation in the ELOVL4 gene. Klin. Monbl. Augenheilkd. 2016, 233, 475–477. [Google Scholar] [CrossRef] [PubMed]
- Mandal, N.A.; Tran, J.T.; Zheng, L.; Wilkerson, J.L.; Brush, R.S.; McRae, J.; Agbaga, M.P.; Zhang, K.; Petrukhin, K.; Ayyagari, R.; et al. In vivo effect of mutant ELOVL4 on the expression and function of wild-type ELOVL4. Investig. Ophthalmol. Vis. Sci. 2014, 55, 2705–2713. [Google Scholar] [CrossRef] [PubMed]
- Karan, G.; Lillo, C.; Yang, Z.; Cameron, D.J.; Locke, K.G.; Zhao, Y.; Thirumalaichary, S.; Li, C.; Birch, D.G.; Vollmer-Snarr, H.R.; et al. Lipofuscin accumulation, abnormal electrophysiology, and photoreceptor degeneration in mutant ELOVL4 transgenic mice: A model for macular degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 4164–4169. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, A.; Meire, F.; Hoyng, C.B.; Vink, C.; van Regemorter, N.; Karan, G.; Yang, Z.; Cremers, F.P.; Zhang, K. A novel mutation in the ELOVL4 gene causes autosomal dominant Stargardt-like macular dystrophy. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4263–4267. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Kniazeva, M.; Han, M.; Li, W.; Yu, Z.; Yang, Z.; Li, Y.; Metzker, M.L.; Allikmets, R.; Zack, D.J.; et al. A 5-bp deletion in ELOVL4 is associated with two related forms of autosomal dominant macular dystrophy. Nat. Genet. 2001, 27, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Logan, S.; Agbaga, M.P.; Chan, M.D.; Kabir, N.; Mandal, N.A.; Brush, R.S.; Anderson, R.E. Deciphering mutant ELOVL4 activity in autosomal-dominant Stargardt macular dystrophy. Proc. Natl. Acad. Sci. USA 2013, 110, 5446–5451. [Google Scholar] [CrossRef] [PubMed]
- Bowes, C.; Li, T.; Danciger, M.; Baxter, L.C.; Applebury, M.L.; Farber, D.B. Retinal degeneration in the rd mouse is caused by a defect in the β subunit of rod cGMP-phosphodiesterase. Nature 1990, 347, 677–680. [Google Scholar] [CrossRef] [PubMed]
- Pittler, S.J.; Baehr, W. Identification of a nonsense mutation in the rod photoreceptor cGMP phosphodiesterase β-subunit gene of the rd mouse. Proc. Natl. Acad. Sci. USA 1991, 88, 8322–8326. [Google Scholar] [CrossRef] [PubMed]
- Ma, E.Y.; Lewis, A.; Barabas, P.; Stearns, G.; Suzuki, S.; Krizaj, D.; Brockerhoff, S.E. Loss of Pde6 reduces cell body Ca2+ transients within photoreceptors. Cell Death Dis. 2013, 4, e797. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.P.; Wu, L.M.; Guo, X.J.; Tso, M.O. Activation of endoplasmic reticulum stress in degenerating photoreceptors of the rd1 mouse. Investig. Ophthalmol. Vis. Sci. 2007, 48, 5191–5198. [Google Scholar] [CrossRef] [PubMed]
- Griciuc, A.; Aron, L.; Roux, M.J.; Klein, R.; Giangrande, A.; Ueffing, M. Inactivation of VCP/ter94 suppresses retinal pathology caused by misfolded rhodopsin in Drosophila. PLoS Genet. 2010, 6, e1001075. [Google Scholar] [CrossRef] [PubMed]
- Trapani, I.; Banfi, S.; Simonelli, F.; Surace, E.M.; Auricchio, A. Gene therapy of inherited retinal degenerations: Prospects and challenges. Hum. Gene Ther. 2015, 26, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Hulleman, J.D. Malattia Leventinese/Doyne honeycomb retinal dystrophy: Similarities to age-related macular degeneration and potential therapies. Adv. Exp. Med. Biol. 2016, 854, 153–158. [Google Scholar] [PubMed]
- Griciuc, A.; Aron, L.; Piccoli, G.; Ueffing, M. Clearance of Rhodopsin(P23H) aggregates requires the ERAD effector VCP. Biochim. Biophys. Acta 2010, 1803, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Tam, B.M.; Moritz, O.L. Characterization of rhodopsin P23H-induced retinal degeneration in a Xenopus laevis model of retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3234–3241. [Google Scholar] [CrossRef] [PubMed]
- Hulleman, J.D.; Kelly, J.W. Genetic ablation of N-linked glycosylation reveals two key folding pathways for R345W fibulin-3, a secreted protein associated with retinal degeneration. FASEB J. 2015, 29, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Schroder, M. Endoplasmic reticulum stress responses. Cell. Mol. Life Sci. 2008, 65, 862–894. [Google Scholar] [CrossRef] [PubMed]
- Lopez, E.; Berna-Erro, A.; Lopez, J.J.; Granados, M.P.; Bermejo, N.; Brull, J.M.; Salido, G.M.; Rosado, J.A.; Redondo, P.C. Role of mTOR1 and mTOR2 complexes in MEG-01 cell physiology. Thromb. Haemost. 2015, 114, 969–981. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.S.; Choi, C.S. The role of amino acid-induced mammalian target of rapamycin complex 1 (mTORC1) signaling in insulin resistance. Exp. Mol. Med. 2016, 48, e201. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Wang, X.; Proud, C.G. mTOR inhibitors in cancer therapy. F1000Research 2016. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.D.; Chen, X.W.; Kaplan, R.E.; Saltiel, A.R.; Walker, C.L.; Reiner, D.J.; Der, C.J. Ral and Rheb GTPase activating proteins integrate mTOR and GTPase signaling in aging, autophagy, and tumor cell invasion. Mol. Cell 2014, 53, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Lei, C.; Fan, J.; Wang, J. miR-18a promotes cell proliferation of esophageal squamous cell carcinoma cells by increasing cylin D1 via regulating PTEN-PI3K-AKT-mTOR signaling axis. Biochem. Biophys. Res. Commun. 2016, 477, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Li, C.M.; Narayanan, R.; Lu, Y.; Hurh, E.; Coss, C.C.; Barrett, C.M.; Miller, D.D.; Dalton, J.T. 2-Arylthiazolidine-4-carboxylic acid amides (ATCAA) target dual pathways in cancer cells: 5′-AMP-activated protein kinase (AMPK)/mTOR and PI3K/Akt/mTOR pathways. Int. J. Oncol. 2010, 37, 1023–1030. [Google Scholar] [PubMed]
- Santos, R.X.; Correia, S.C.; Cardoso, S.; Carvalho, C.; Santos, M.S.; Moreira, P.I. Effects of rapamycin and TOR on aging and memory: Implications for Alzheimer's disease. J. Neurochem. 2011, 117, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Banaszynski, L.A.; Liu, C.W.; Wandless, T.J. Characterization of the FKBP.rapamycin.FRB ternary complex. J. Am. Chem. Soc. 2005, 127, 4715–4721. [Google Scholar] [CrossRef] [PubMed]
- Rogina, B.; Helfand, S.L. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc. Natl. Acad. Sci. USA 2004, 101, 15998–16003. [Google Scholar] [CrossRef] [PubMed]
- Von Walden, F.; Liu, C.; Aurigemma, N.; Nader, G.A. mTOR signaling regulates myotube hypertrophy by modulating protein synthesis, rDNA transcription and chromatin remodeling. Am. J. Physiol. Cell Physiol. 2016, 311, C663–C672. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, F.; Li, M.; Gao, X.; Wang, Y.; Wang, D.; Ma, X.; Ma, T.; Gu, S. The antidepressant-like effect of alarin is related to TrkB-mTOR signaling and synaptic plasticity. Behav. Brain Res. 2016, 313, 158–171. [Google Scholar] [CrossRef] [PubMed]
- Rivera Rivera, A.; Castillo-Pichardo, L.; Gerena, Y.; Dharmawardhane, S. Anti-Breast Cancer Potential of Quercetin via the Akt/AMPK/Mammalian Target of Rapamycin (mTOR) Signaling Cascade. PLoS ONE 2016, 11, e0157251. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Kahaar, E.; Kabakchiev, M.; Hartmann, B.; Wieland, E.; Shipkova, M. Performance of a phosphoflow assay to determine phosphorylation of S6 ribosomal protein as a pharmacodynamic read out for mTOR inhibition. Clin. Biochem. 2016, 49, 1181–1187. [Google Scholar] [CrossRef] [PubMed]
- Bahrami, B.F.; Ataie-Kachoie, P.; Pourgholami, M.H.; Morris, D.L. p70 Ribosomal protein S6 kinase (Rps6kb1): An update. J. Clin. Pathol. 2014, 67, 1019–1025. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Patsis, C.; Koromilas, A.E. Stat1 stimulates cap-independent mRNA translation to inhibit cell proliferation and promote survival in response to antitumor drugs. Proc. Natl. Acad. Sci. USA 2015, 112, E2149–E2155. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, B.D.; Zakaria, C.; Jia, J.J.; Graber, T.E.; Svitkin, Y.; Tahmasebi, S.; Healy, D.; Hoang, H.D.; Jensen, J.M.; Diao, I.T.; et al. La-related protein 1 (LARP1) represses terminal oligopyrimidine (TOP) mRNA translation downstream of mTOR complex 1 (mTORC1). J. Biol. Chem. 2015, 290, 15996–16020. [Google Scholar] [CrossRef] [PubMed]
- Alers, S.; Loffler, A.S.; Paasch, F.; Dieterle, A.M.; Keppeler, H.; Lauber, K.; Campbell, D.G.; Fehrenbacher, B.; Schaller, M.; Wesselborg, S.; et al. Atg13 and FIP200 act independently of Ulk1 and Ulk2 in autophagy induction. Autophagy 2011, 7, 1423–1433. [Google Scholar] [CrossRef] [PubMed]
- Loffler, A.S.; Alers, S.; Dieterle, A.M.; Keppeler, H.; Franz-Wachtel, M.; Kundu, M.; Campbell, D.G.; Wesselborg, S.; Alessi, D.R.; Stork, B. Ulk1-mediated phosphorylation of AMPK constitutes a negative regulatory feedback loop. Autophagy 2011, 7, 696–706. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Weber, K.J.; Diwan, A.; Schilling, J.D. Inhibition of mTOR reduces lipotoxic cell death in primary macrophages through an autophagy-independent mechanism. J. Leukoc. Biol. 2016, 100, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Kashyap, M.P.; Tripathi, V.K.; Singh, S.; Garg, G.; Rizvi, S.I. Neuroprotection through rapamycin-induced activation of autophagy and PI3K/Akt1/mTOR/CREB signaling against amyloid-β-induced oxidative stress, synaptic/neurotransmission dysfunction, and neurodegeneration in adult rats. Mol. Neurobiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Mendes, H.F.; Cheetham, M.E. Pharmacological manipulation of gain-of-function and dominant-negative mechanisms in rhodopsin retinitis pigmentosa. Hum. Mol. Genet. 2008, 17, 3043–3054. [Google Scholar] [CrossRef] [PubMed]
- Sizova, O.S.; Shinde, V.M.; Lenox, A.R.; Gorbatyuk, M.S. Modulation of cellular signaling pathways in P23H rhodopsin photoreceptors. Cell. Signal. 2014, 26, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Griciuc, A.; Roux, M.J.; Merl, J.; Giangrande, A.; Hauck, S.M.; Aron, L.; Ueffing, M. Proteomic survey reveals altered energetic patterns and metabolic failure prior to retinal degeneration. J. Neurosci. 2014, 34, 2797–2812. [Google Scholar] [CrossRef] [PubMed]
- Rajala, A.; Tanito, M.; Le, Y.Z.; Kahn, C.R.; Rajala, R.V. Loss of neuroprotective survival signal in mice lacking insulin receptor gene in rod photoreceptor cells. J. Biol. Chem. 2008, 283, 19781–19792. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Anderson, R.E.; Tomita, H.; Adler, R.; Liu, X.; Zack, D.J.; Rajala, R.V. Nonredundant role of Akt2 for neuroprotection of rod photoreceptor cells from light-induced cell death. J. Neurosci. 2007, 27, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Nakahara, T.; Asano, D.; Ushikubo, H.; Mori, A.; Sakamoto, K.; Ishii, K. Preventive effects of rapamycin on inflammation and capillary degeneration in a rat model of NMDA-induced retinal injury. Biol. Pharm. Bull. 2015, 38, 321–324. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, X.; Zhang, J. Bridges between mitochondrial oxidative stress, ER stress and mTOR signaling in pancreatic β cells. Cell. Signal. 2016, 28, 1099–1104. [Google Scholar] [CrossRef] [PubMed]


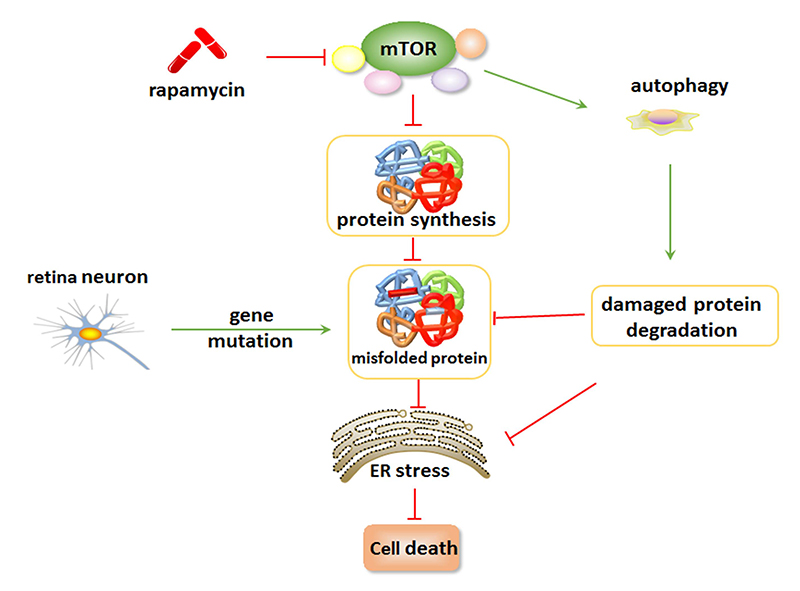{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Animal Model | Mutant Gene | Dysregulated Components | Related Retinal Degeneration | ER Stress Activation | Reference |
|---|---|---|---|---|---|
| Drosophila | RhoP23H | rhodopsin | ADRP | + | [33] |
| Xenopus laevis | RhoP23H | rhodopsin | RP | + | [54] |
| Rats | RhoP23H | rhodopsin | RP | + | [50] |
| Mice | RhoT17M | rhodopsin | ADRP | + | [28] |
| Transfected cell | ELOVL4 | an enzyme involved in the generation of long-chain fatty acids | Stargardt macular dystrophy | + | [39] |
| Rd1 mouse | PDE6-β | a catalytic subunit of a phosphodiesterase, regulating cGMP levels in photoreceptors | RP | + | [49] |
| ARPE-19 cells | R345W | N-Linked glycosylation | Malattia Leventinese and Doyne honeycomb retinal dystrophy | + | [55] |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, B.; Sun, Y.-J.; Liu, S.-Y.; Che, L.; Li, G.-Y. Neuroprotective Strategy in Retinal Degeneration: Suppressing ER Stress-Induced Cell Death via Inhibition of the mTOR Signal. Int. J. Mol. Sci. 2017, 18, 201. https://doi.org/10.3390/ijms18010201
Fan B, Sun Y-J, Liu S-Y, Che L, Li G-Y. Neuroprotective Strategy in Retinal Degeneration: Suppressing ER Stress-Induced Cell Death via Inhibition of the mTOR Signal. International Journal of Molecular Sciences. 2017; 18(1):201. https://doi.org/10.3390/ijms18010201
Chicago/Turabian StyleFan, Bin, Ying-Jian Sun, Shu-Yan Liu, Lin Che, and Guang-Yu Li. 2017. "Neuroprotective Strategy in Retinal Degeneration: Suppressing ER Stress-Induced Cell Death via Inhibition of the mTOR Signal" International Journal of Molecular Sciences 18, no. 1: 201. https://doi.org/10.3390/ijms18010201