
A Systems Biological View of Life-and-Death Decision with Respect to Endoplasmic Reticulum Stress—The Role of PERK Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
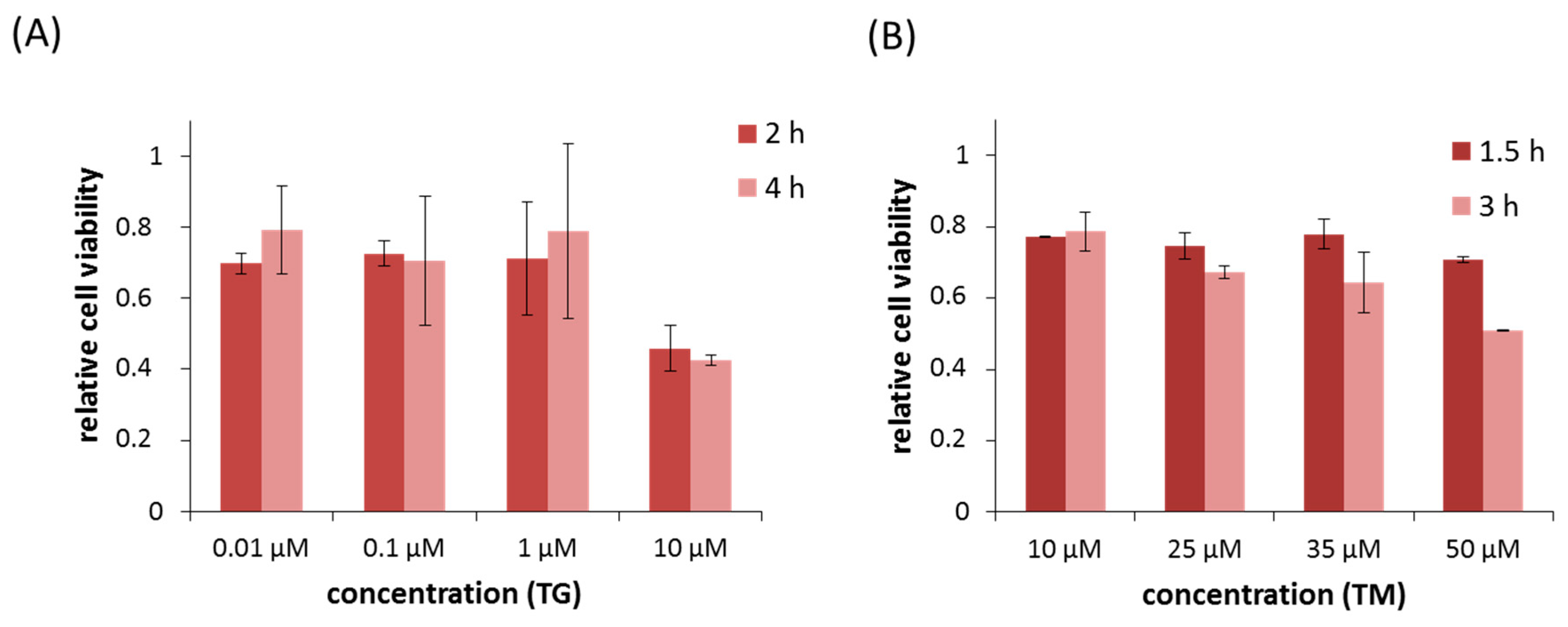
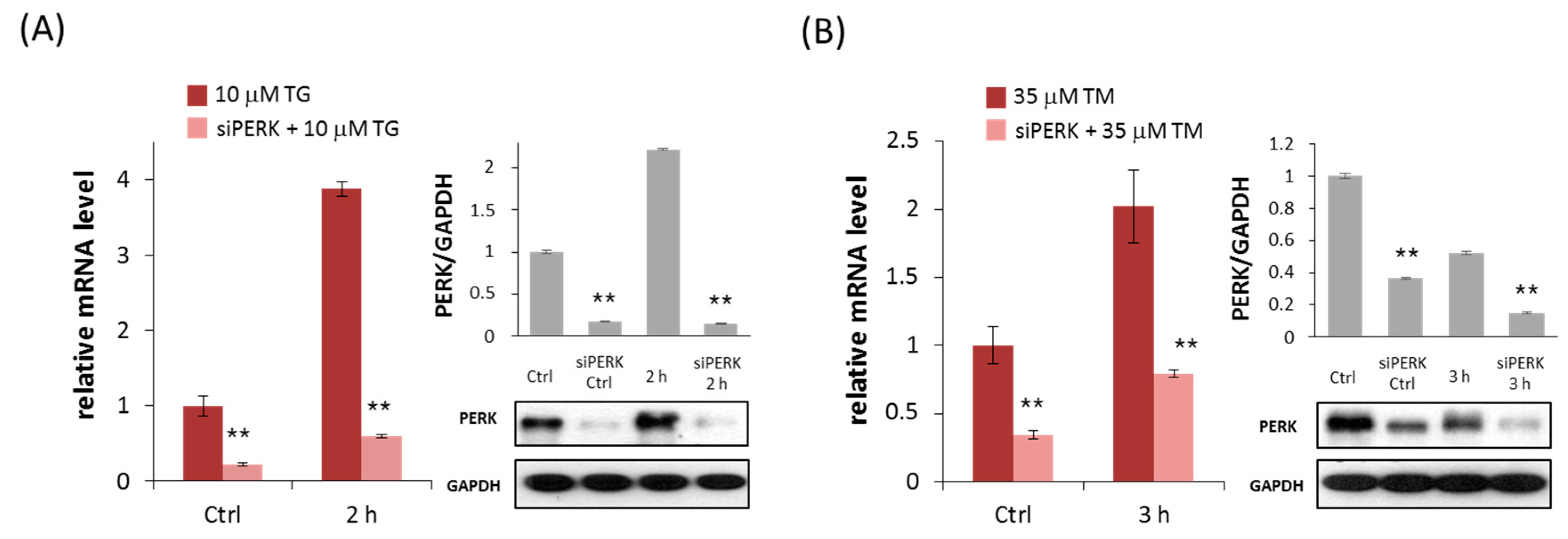
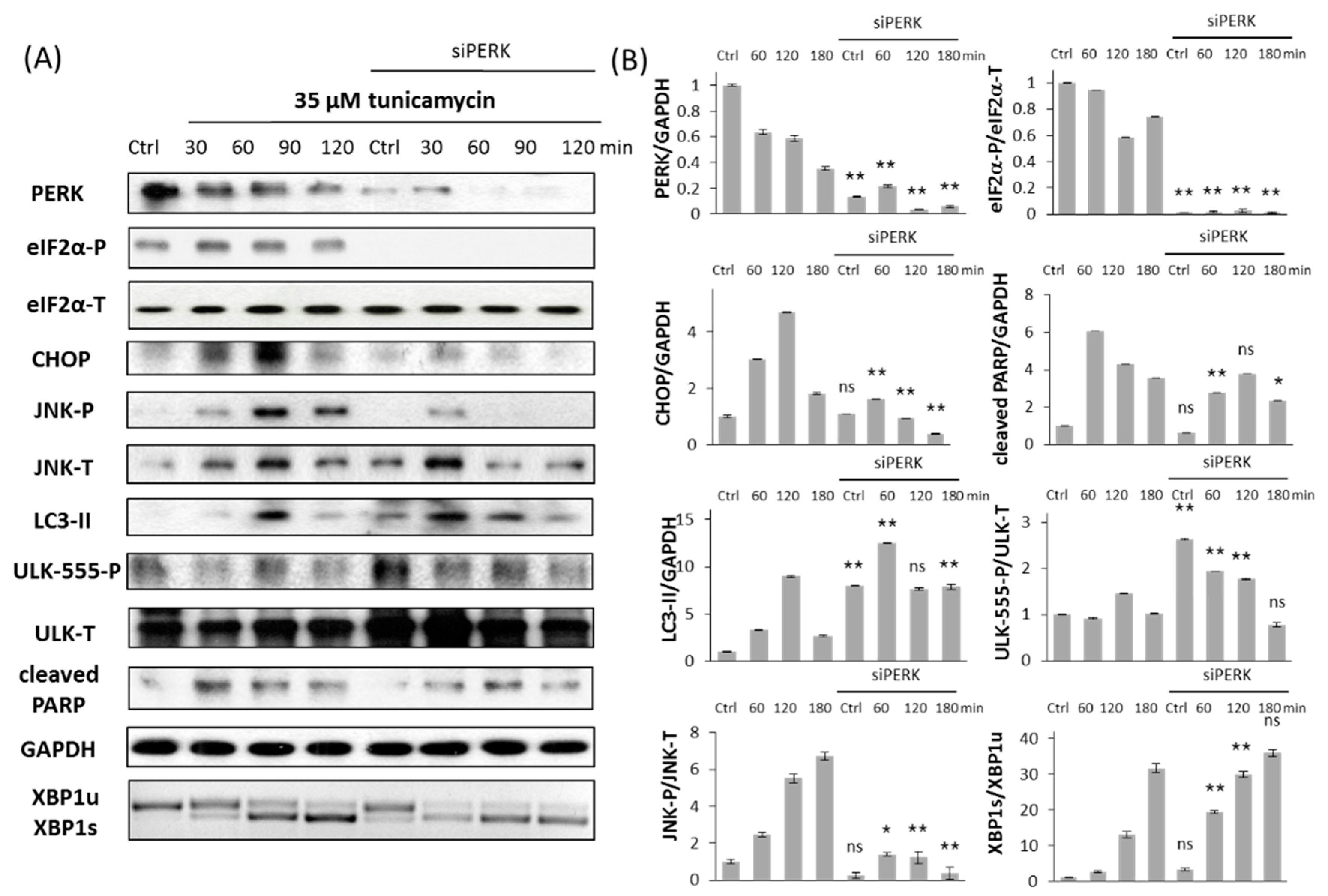
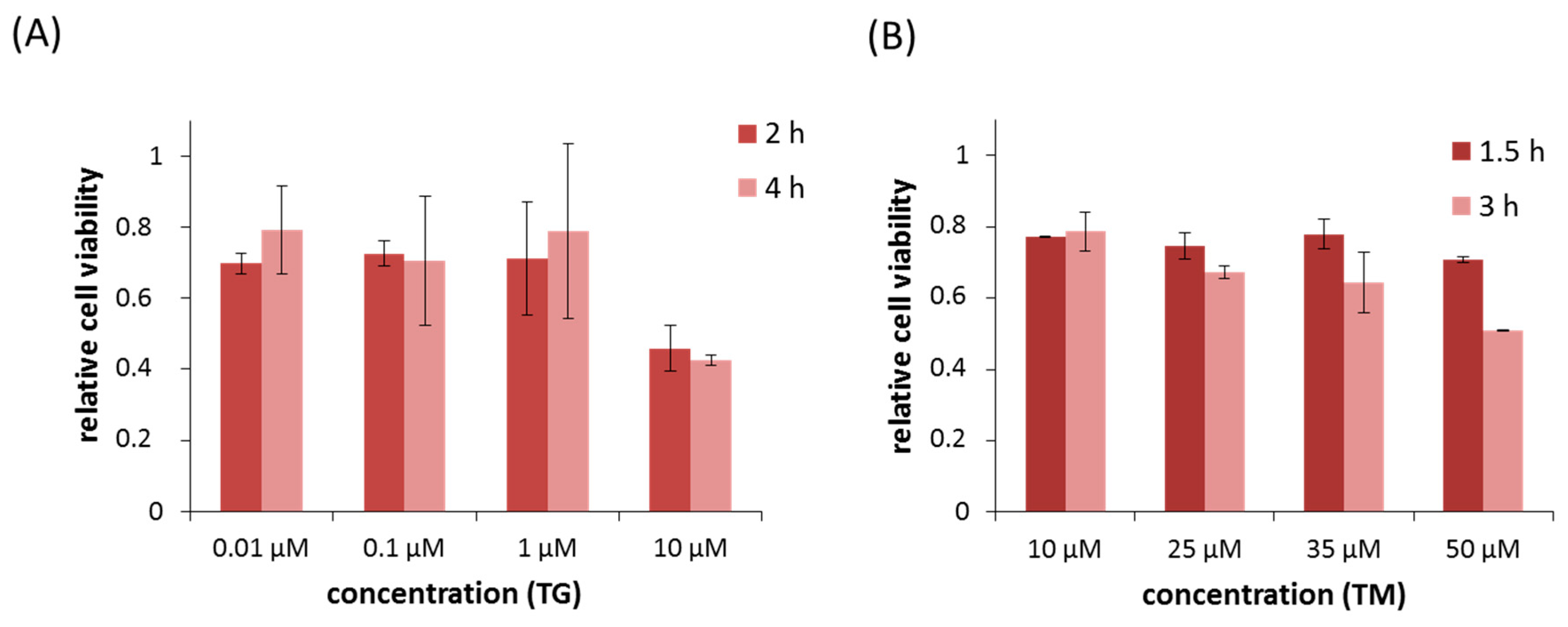
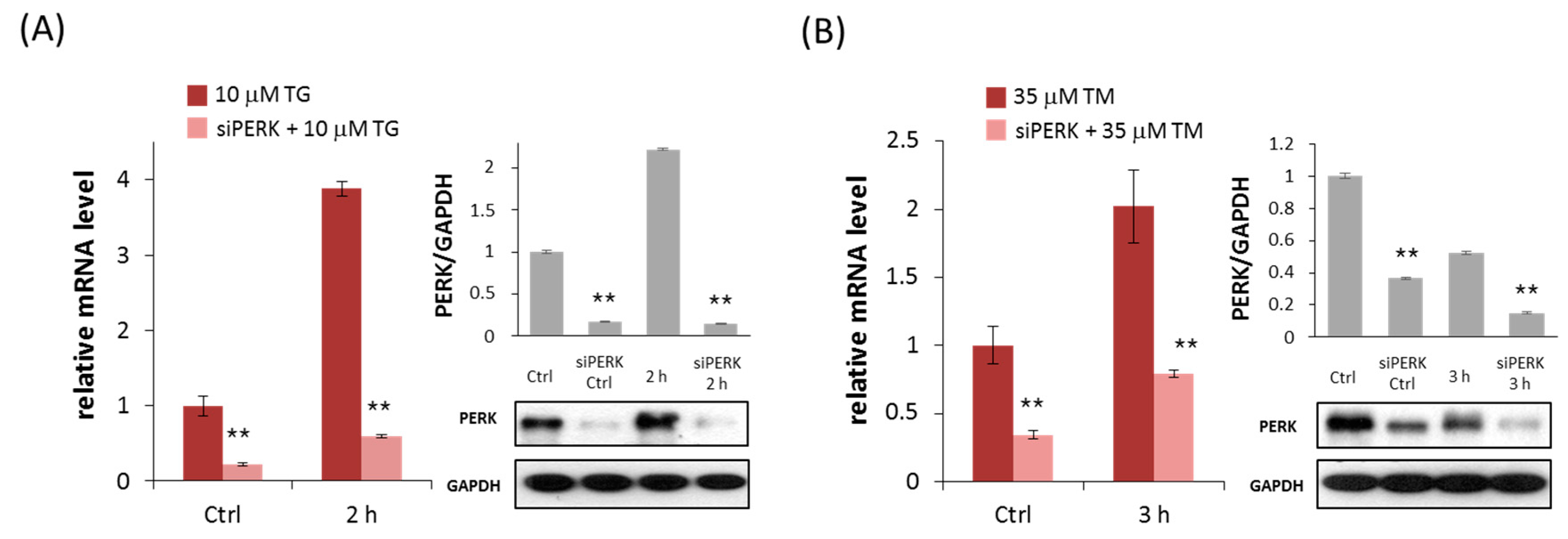
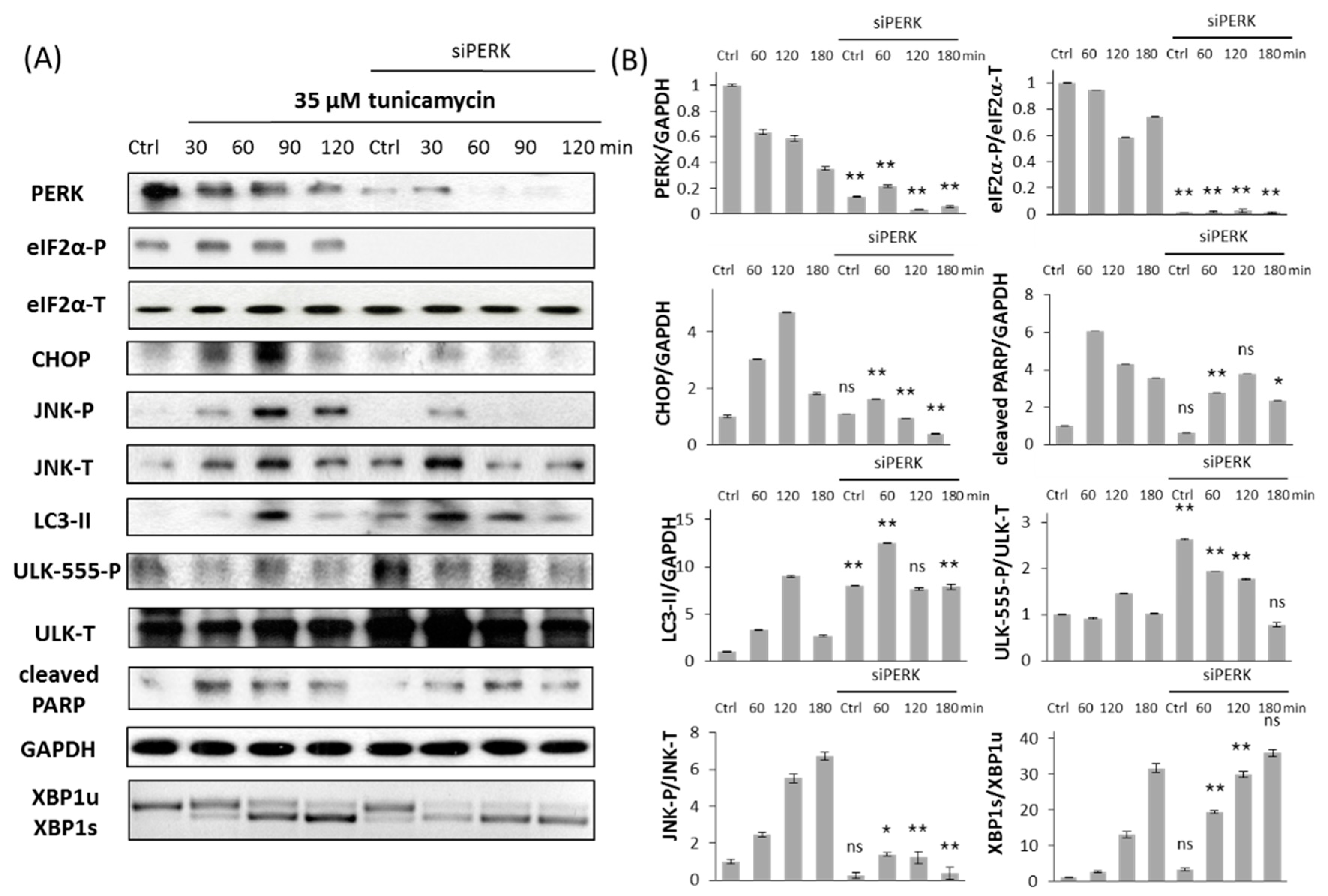
2.1. Silencing of PERK Increases Cell Viability with Respect to Endoplasmic Reticulum (ER) Stress
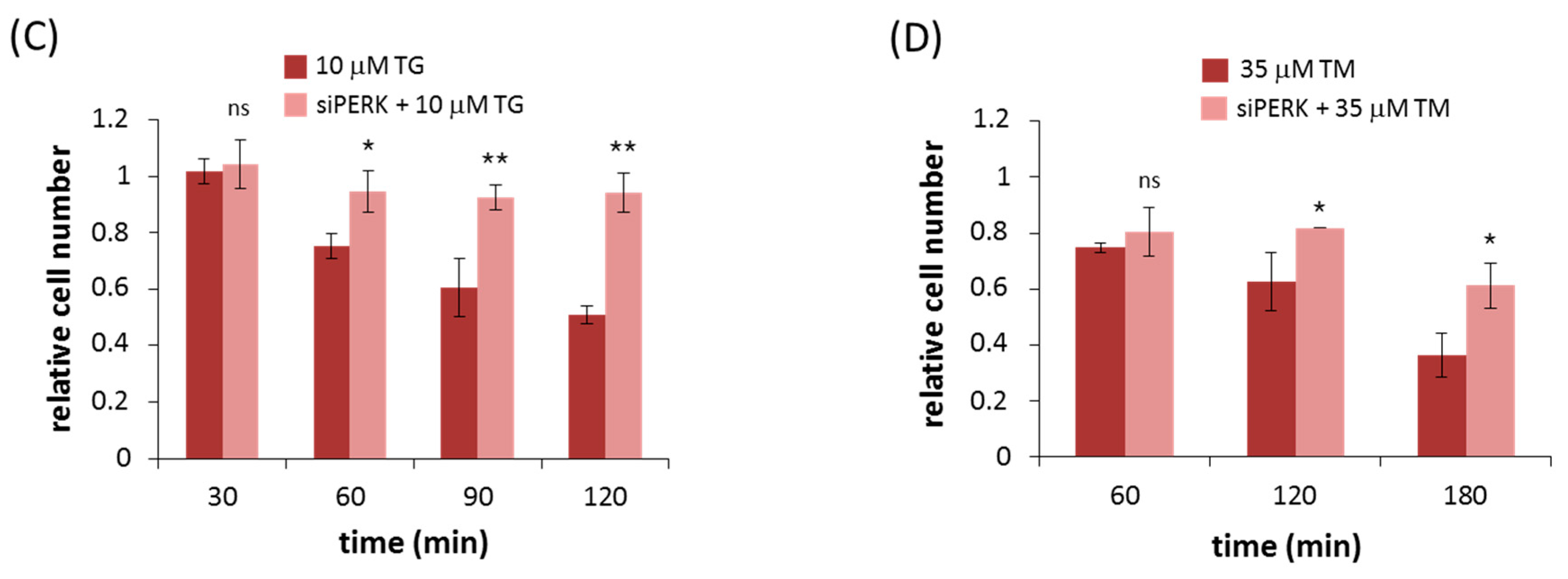
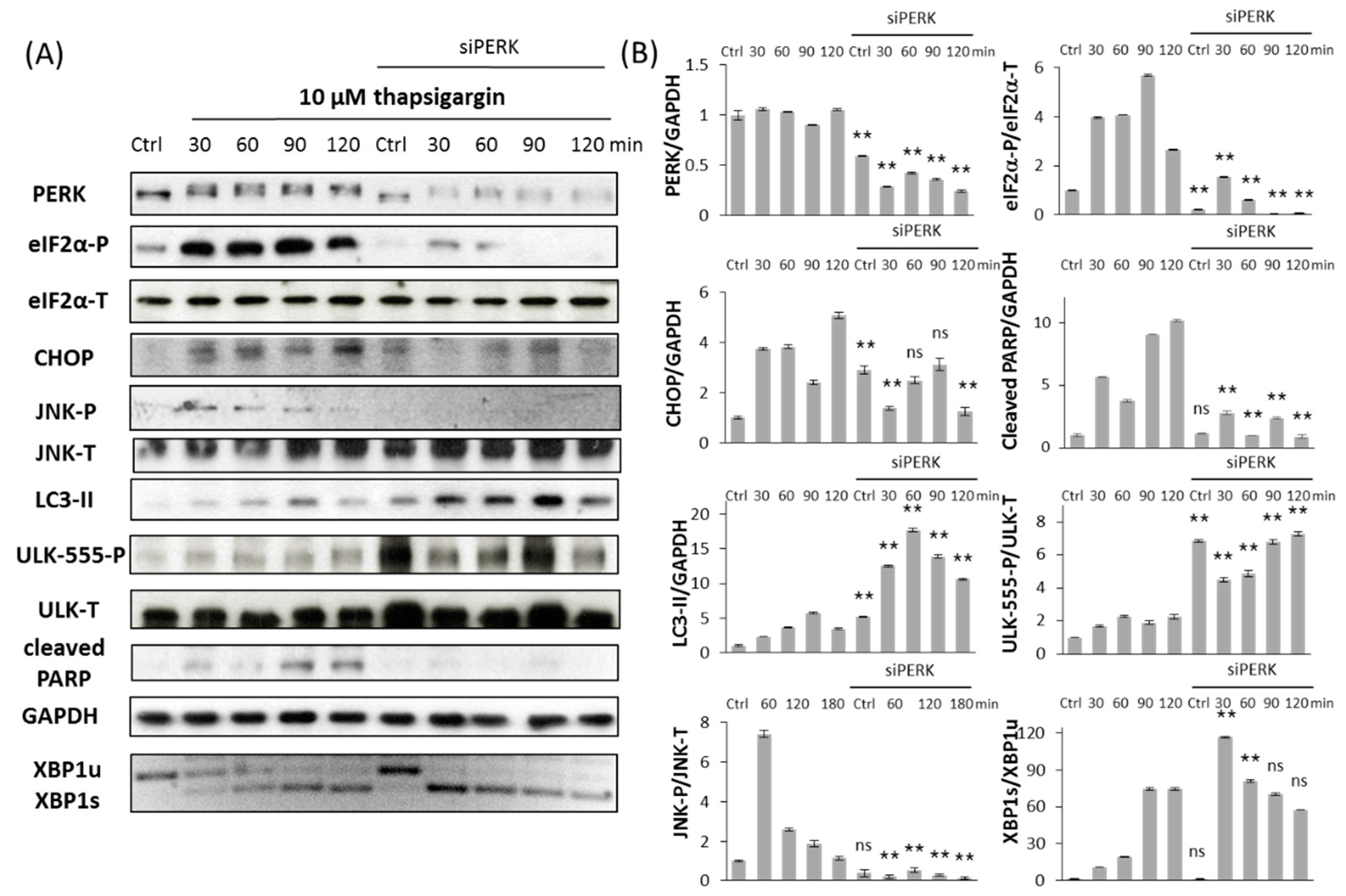
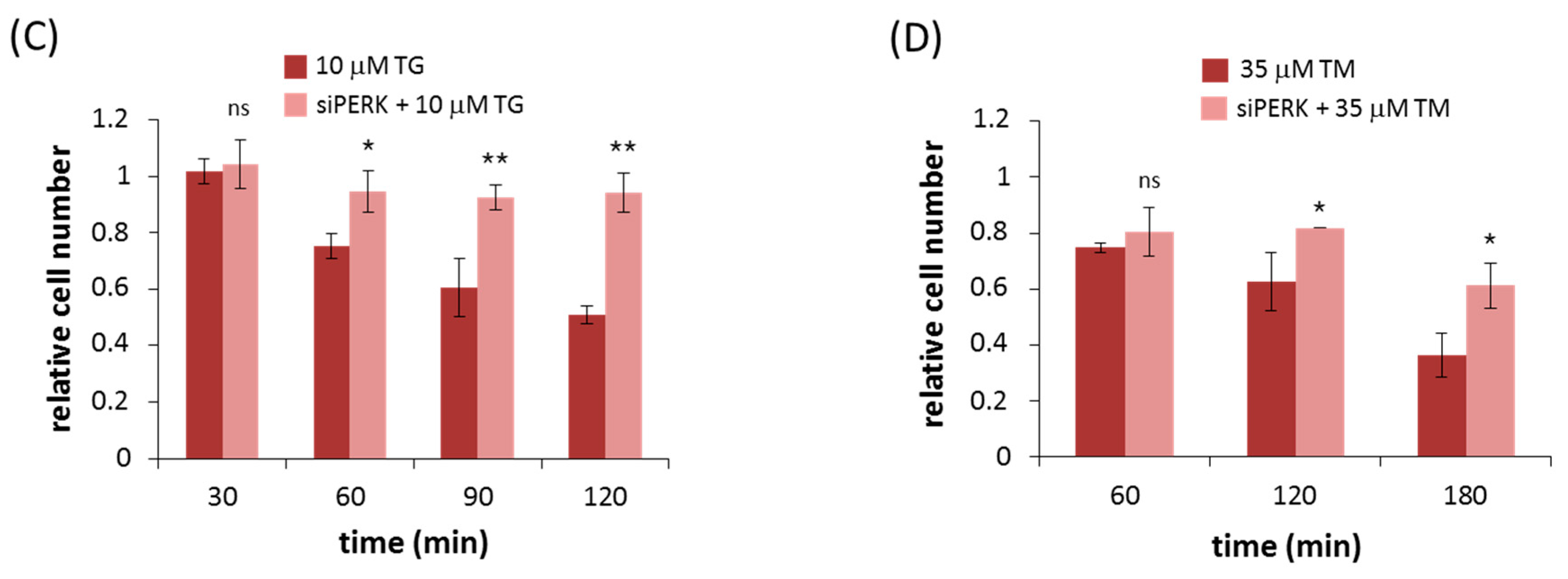
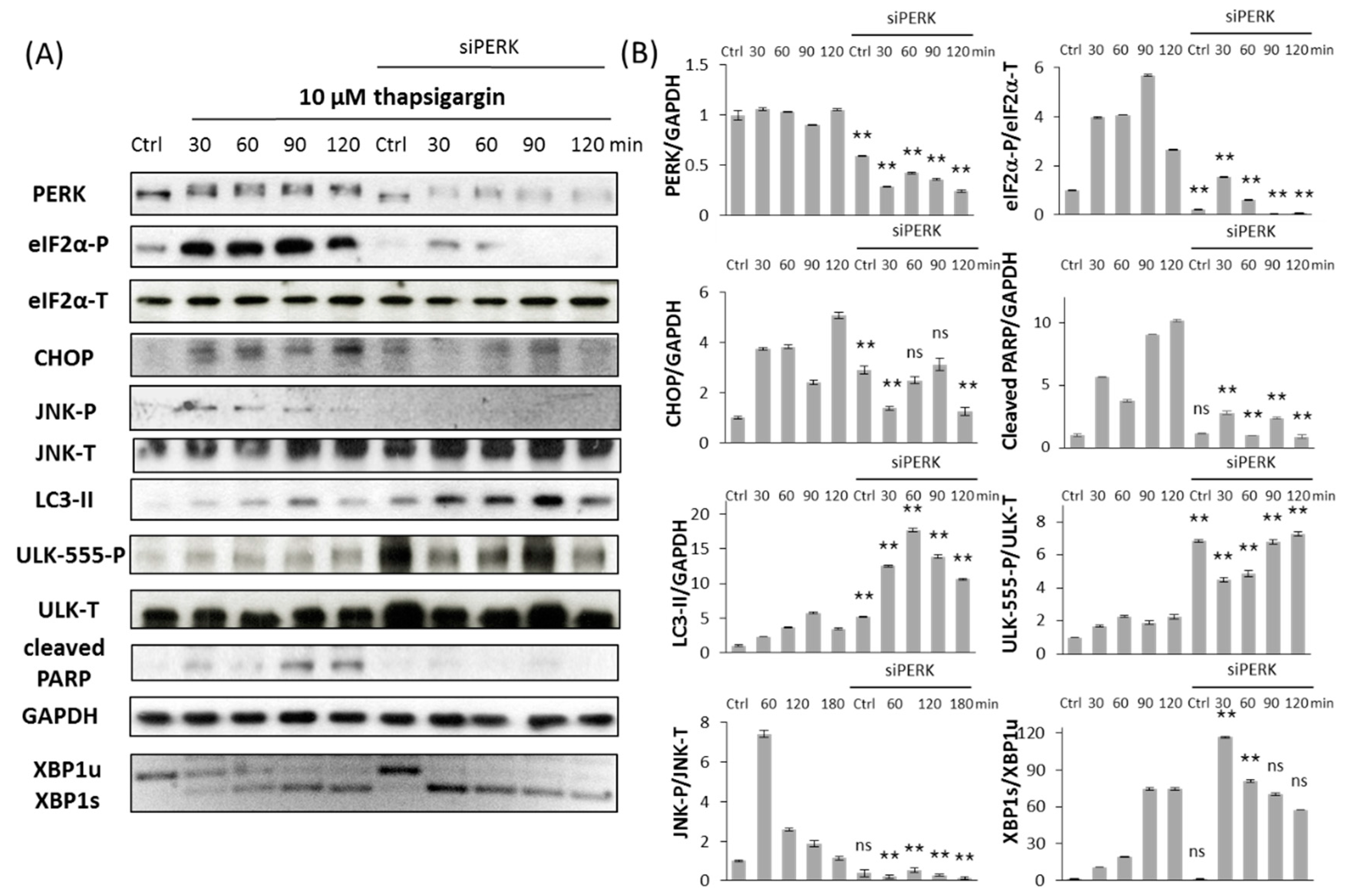
2.2. Silencing of PERK Extends Autophagy-Dependent Survival and Delays Apoptotic Cell Death during ER Stress
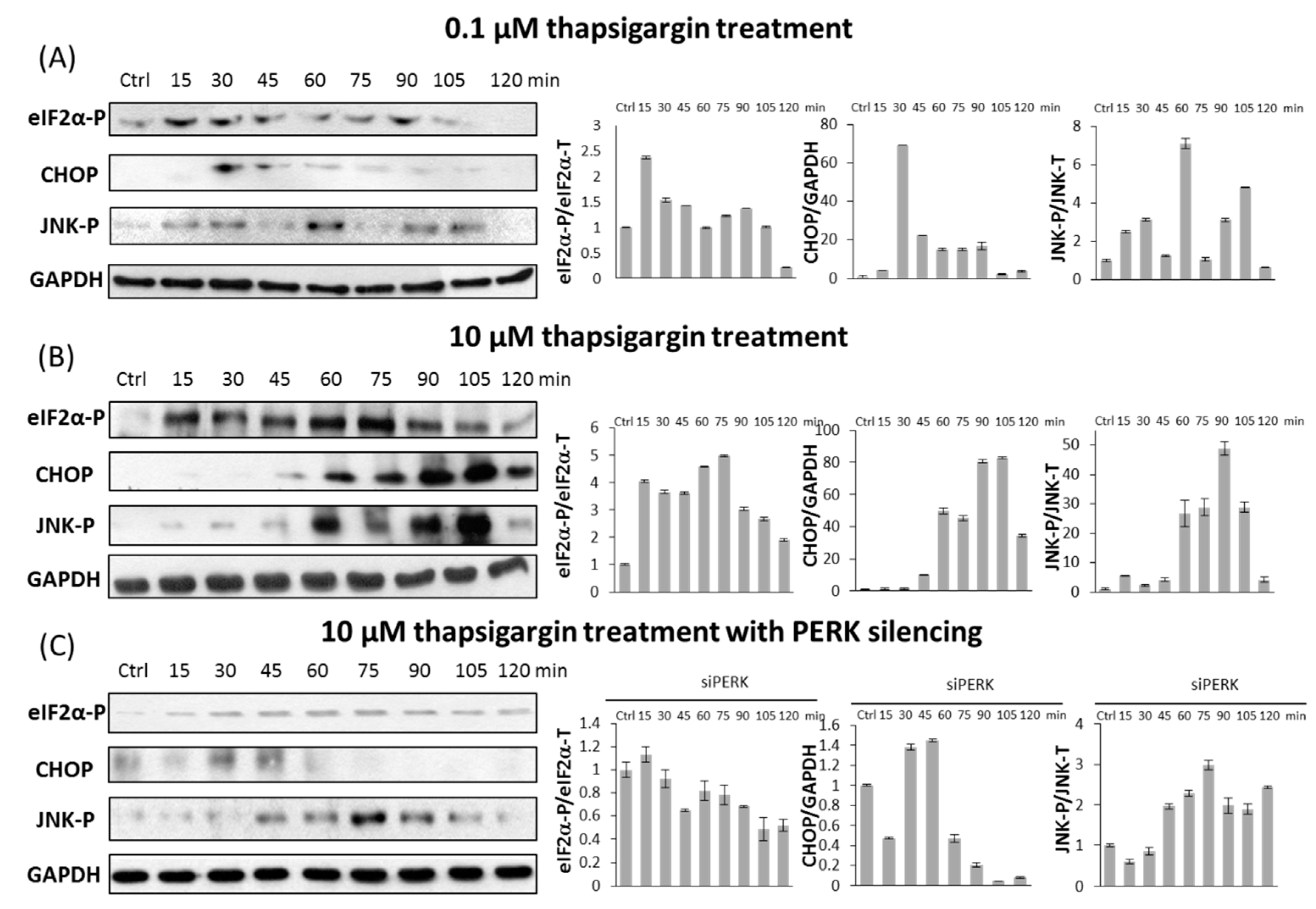
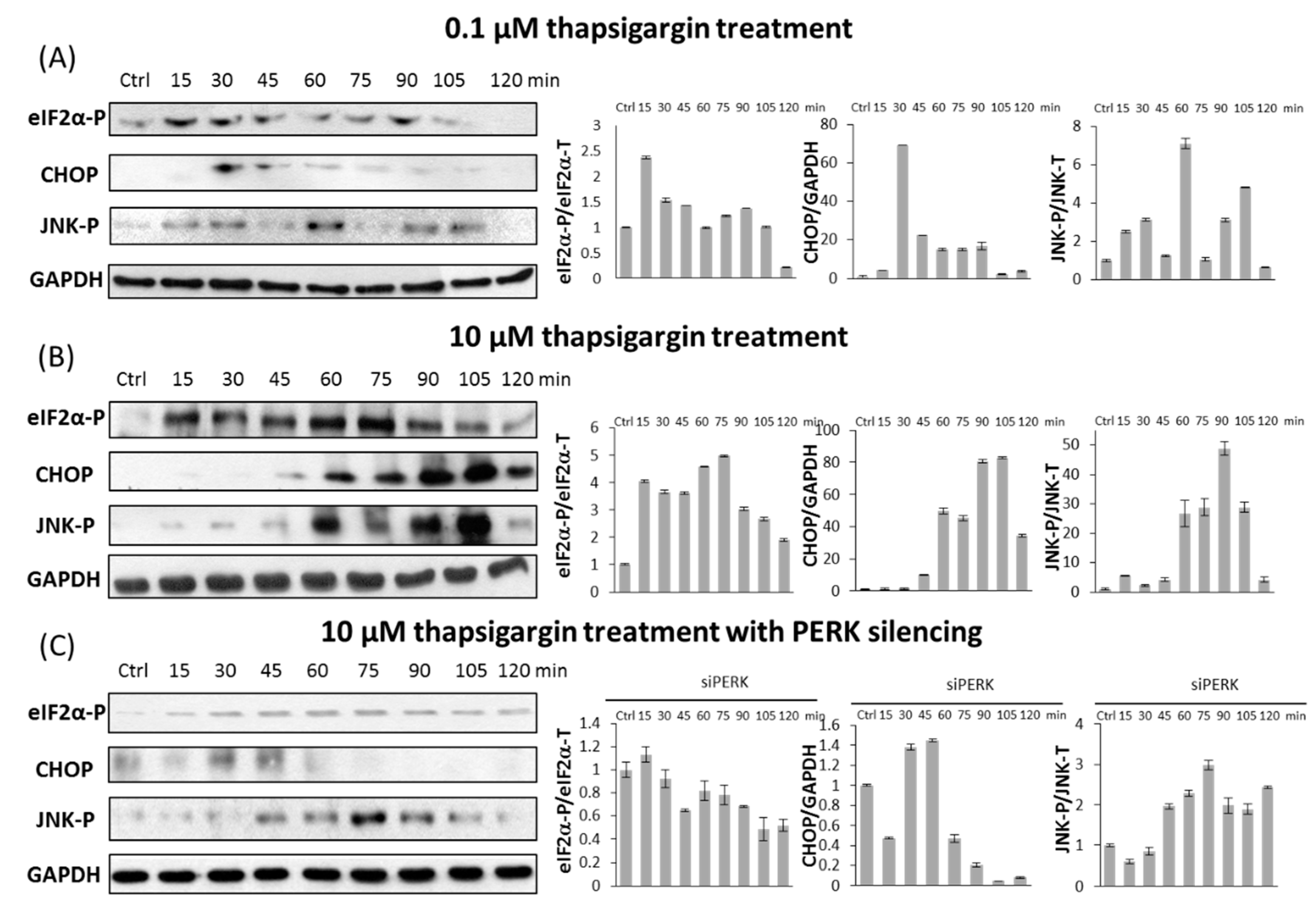
2.3. PERK Affects the Apoptosis Inducer of Inositol Requiring Kinase 1 (IRE-1) Arm Positively with Respect to ER Stress
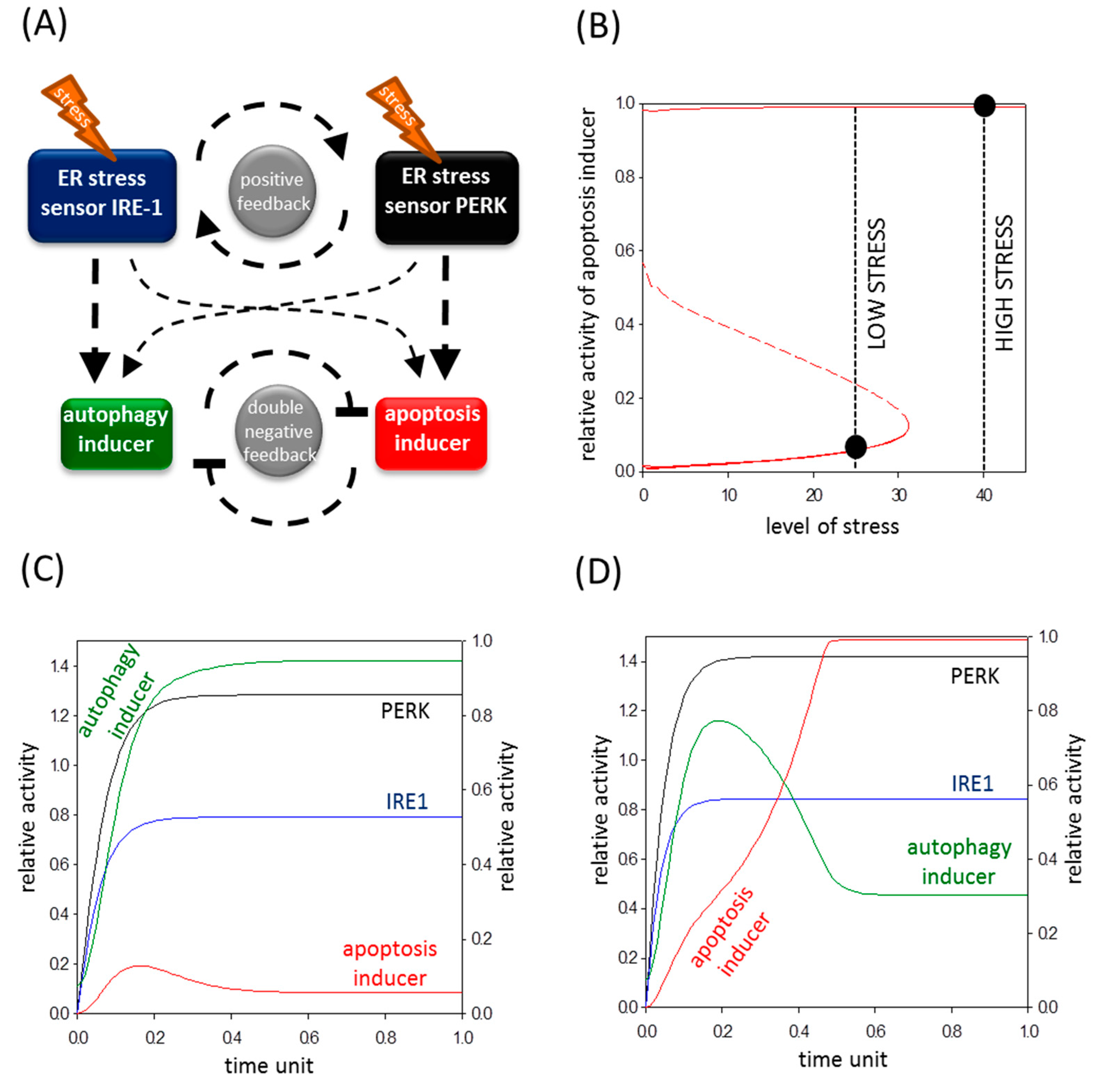
2.4. The Switch-Like Activation of Apoptosis Inducers Is Precisely Regulated during ER Stress
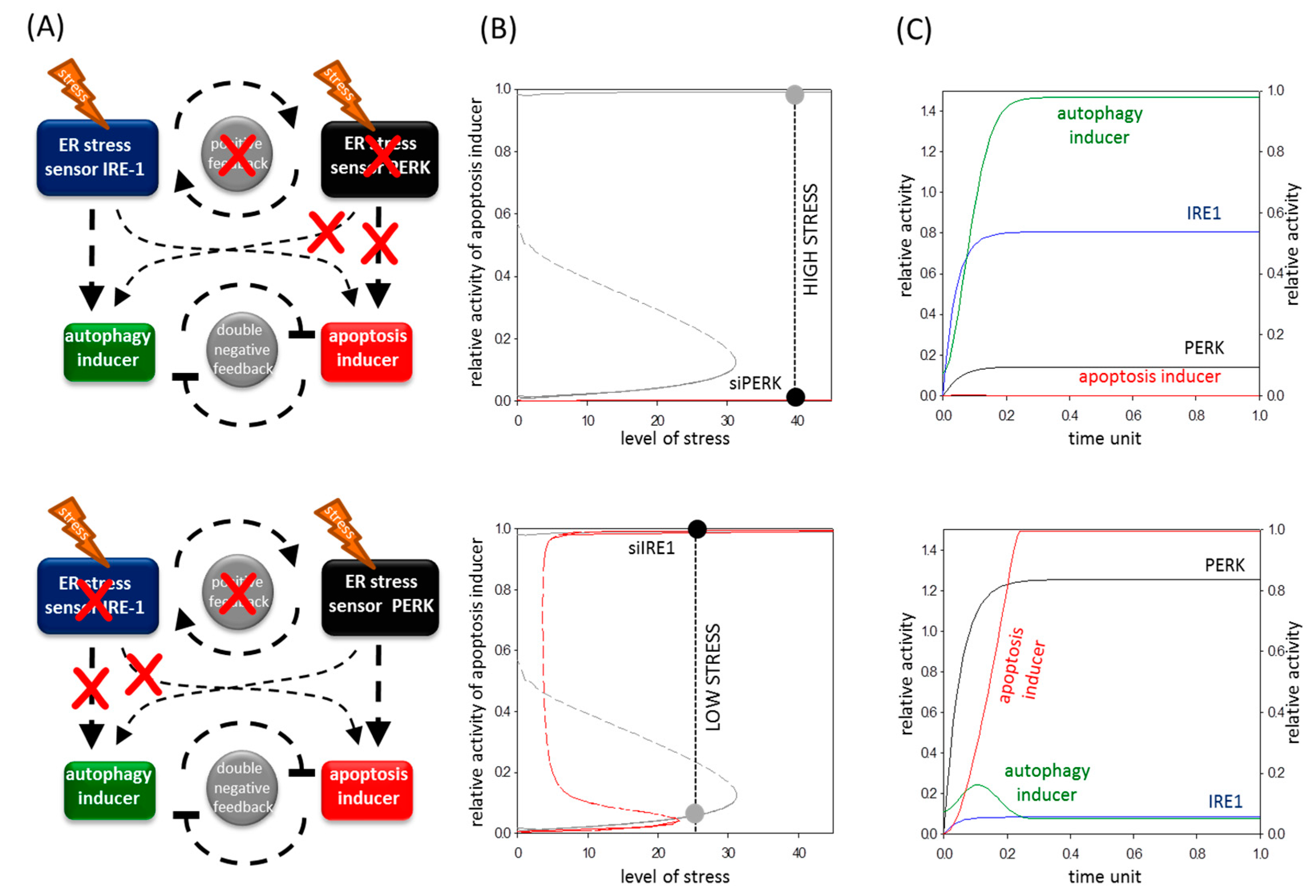
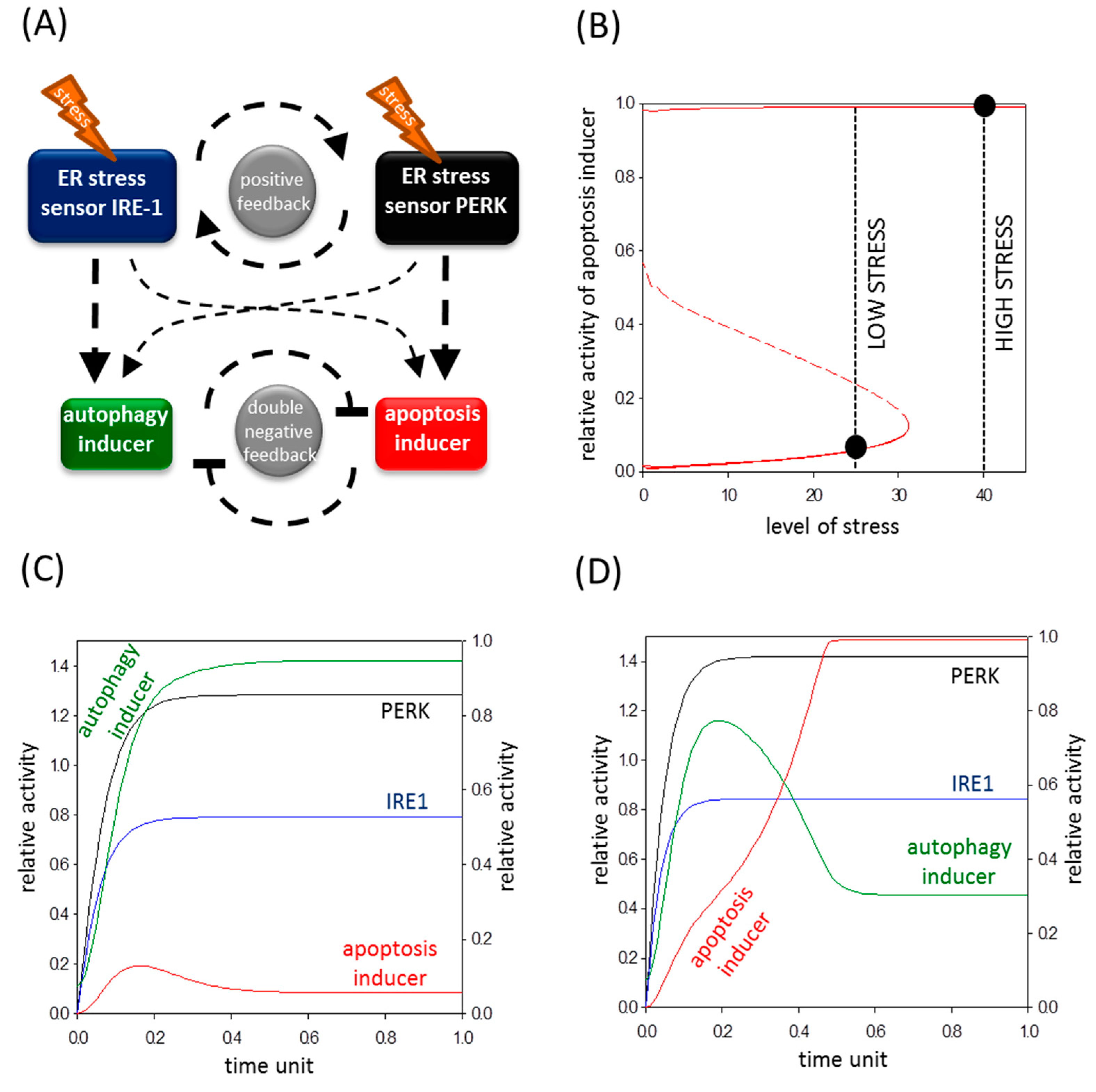
2.5. A Mathematical Model Suggests a Positive Feedback Loop between IRE-1 (Inositol Requiring Protein 1) and PERK (Protein Kinase (RNA)-Like Endoplasmic Reticulum Kinase)
2.6. The Altered Upregulation of Autophagy and Apoptosis Inducers by IRE-1 and PERK Determines the Life-and-Death Decision
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture and Maintenance
4.3. Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE) and Western Blot Analysis
4.4. Statistics
4.5. RNA Interference
4.6. Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
4.7. Real-Time PCR
4.8. Cell Viability Assays
4.9. Mathematical Modeling
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A. The Minimal Model of ER Stress Response Mechanism
Appendix A.1. The Code for Simulating Time Series
Appendix A.2. The Code for Simulating Signal Response Curves
References
- Malhi, H.; Kaufman, R.J. Endoplasmic reticulum stress in liver disease. J. Hepatol. 2011, 54, 795–809. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Bernales, S.; Papa, F.R.; Walter, P. Intracellular signaling by the unfolded protein response. Annu. Rev. Cell Dev. Biol. 2006, 22, 487–508. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H. ER stress and diseases. FEBS J. 2007, 274, 630–658. [Google Scholar] [CrossRef] [PubMed]
- Lindholm, D.; Wootz, H.; Korhonen, L. ER stress and neurodegenerative diseases. Cell Death Differ. 2006, 13, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Morito, D.; Nagata, K. ER Stress Proteins in Autoimmune and Inflammatory Diseases. Front. Immunol. 2012, 3, 48. [Google Scholar] [CrossRef] [PubMed]
- Ogata, M.; Hino, S.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell Biol. 2006, 26, 9220–9231. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [PubMed]
- Holczer, M.; Marton, M.; Kurucz, A.; Banhegyi, G.; Kapuy, O. A comprehensive systems biological study of autophagy-apoptosis crosstalk during endoplasmic reticulum stress. BioMed Res. Int. 2015, 2015, 319589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapuy, O.; Vinod, P.K.; Mandl, J.; Banhegyi, G. A cellular stress-directed bistable switch controls the crosstalk between autophagy and apoptosis. Mol. Biosyst. 2013, 9, 296–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.S.; Shamu, C.E.; Walter, P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 1993, 73, 1197–1206. [Google Scholar] [CrossRef]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Brandizzi, F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 2013, 23, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Pehar, M.; Jonas, M.C.; Hare, T.M.; Puglielli, L. SLC33A1/AT-1 protein regulates the induction of autophagy downstream of IRE1/XBP1 pathway. J. Biol. Chem. 2012, 287, 29921–29930. [Google Scholar] [CrossRef] [PubMed]
- Tian, P.G.; Jiang, Z.X.; Li, J.H.; Zhou, Z.; Zhang, Q.H. Spliced XBP1 promotes macrophage survival and autophagy by interacting with Beclin-1. Biochem. Biophys. Res. Commun. 2015, 463, 518–523. [Google Scholar] [CrossRef] [PubMed]
- Deegan, S.; Koryga, I.; Glynn, S.A.; Gupta, S.; Gorman, A.M.; Samali, A. A close connection between the PERK and IRE arms of the UPR and the transcriptional regulation of autophagy. Biochem. Biophys. Res. Commun. 2015, 456, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.Y.; Wek, R.C. Phosphorylation of the α-subunit of the eukaryotic initiation factor-2 (eIF2α) reduces protein synthesis and enhances apoptosis in response to proteasome inhibition. J. Biol. Chem. 2005, 280, 14189–14202. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.D.; Harding, H.P.; Ron, D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J. Cell. Biol. 2004, 167, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Hendershot, L.M. Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress. J. Biol. Chem. 2003, 278, 34864–34873. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Zinszner, H.; Kuroda, M.; Wang, X.; Batchvarova, N.; Lightfoot, R.T.; Remotti, H.; Stevens, J.L.; Ron, D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998, 12, 982–995. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Z.; Lawson, B.; Brewer, J.W.; Zinszner, H.; Sanjay, A.; Mi, L.J.; Boorstein, R.; Kreibich, G.; Hendershot, L.M.; Ron, D. Signals from the stressed endoplasmic reticulum induce C/EBP-homologous protein (CHOP/GADD153). Mol. Cell. Biol. 1996, 16, 4273–4280. [Google Scholar] [CrossRef] [PubMed]
- B’Chir, W.; Chaveroux, C.; Carraro, V.; Averous, J.; Maurin, A.C.; Jousse, C.; Muranishi, Y.; Parry, L.; Fafournoux, P.; Bruhat, A. Dual role for CHOP in the crosstalk between autophagy and apoptosis to determine cell fate in response to amino acid deprivation. Cell Signal. 2014, 26, 1385–1391. [Google Scholar] [CrossRef] [PubMed]
- Brush, M.H.; Weiser, D.C.; Shenolikar, S. Growth arrest and DNA damage-inducible protein GADD34 targets protein phosphatase 1α to the endoplasmic reticulum and promotes dephosphorylation of the α subunit of eukaryotic translation initiation factor 2. Mol. Cell. Biol. 2003, 23, 1292–1303. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [PubMed]
- Novoa, I.; Zhang, Y.; Zeng, H.; Jungreis, R.; Harding, H.P.; Ron, D. Stress-induced gene expression requires programmed recovery from translational repression. EMBO J. 2003, 22, 1180–1187. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 2000, 5, 897–904. [Google Scholar] [CrossRef]
- Vandewynckel, Y.P.; Laukens, D.; Bogaerts, E.; Paridaens, A.; van den Bussche, A.; Verhelst, X.; van Steenkiste, C.; Descamps, B.; Vanhove, C.; Libbrecht, L.; et al. Modulation of the unfolded protein response impedes tumor cell adaptation to proteotoxic stress: A PERK for hepatocellular carcinoma therapy. Hepatol. Int. 2015, 9, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Dai, D.L.; Yao, L.; Yu, H.H.; Ning, B.; Zhang, Q.; Chen, J.; Cheng, W.H.; Shen, W.; Yang, Z.X. Saturated fatty acid induction of endoplasmic reticulum stress and apoptosis in human liver cells via the PERK/ATF4/CHOP signaling pathway. Mol. Cell. Biochem. 2012, 364, 115–129. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Piao, M.J.; Kim, K.C.; Kim, A.D.; Choi, J.Y.; Choi, J.; Hyun, J.W. Endoplasmic reticulum stress signaling is involved in silver nanoparticles-induced apoptosis. Int. J. Biochem. Cell Biol. 2012, 44, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Walter, F.; Schmid, J.; Dussmann, H.; Concannon, C.G.; Prehn, J.H. Imaging of single cell responses to ER stress indicates that the relative dynamics of IRE1/XBP1 and PERK/ATF4 signalling rather than a switch between signalling branches determine cell survival. Cell Death Differ. 2015, 22, 1502–1516. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Li, H.; Yasumura, D.; Cohen, H.R.; Zhang, C.; Panning, B.; Shokat, K.M.; Lavail, M.M.; Walter, P. IRE1 signaling affects cell fate during the unfolded protein response. Science 2007, 318, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Upton, J.P.; Wang, L.; Han, D.; Wang, E.S.; Huskey, N.E.; Lim, L.; Truitt, M.; McManus, M.T.; Ruggero, D.; Goga, A.; et al. IRE1α cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science 2012, 338, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Martinon, F.; Rodriguez, D.; Glimcher, L.H. The unfolded protein response: Integrating stress signals through the stress sensor IRE1α. Physiol. Rev. 2011, 91, 1219–1243. [Google Scholar] [CrossRef] [PubMed]
- Schonthal, A.H. Endoplasmic reticulum stress: Its role in disease and novel prospects for therapy. Scientifica (Cairo) 2012, 2012, 857516. [Google Scholar] [CrossRef] [PubMed]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Chomczynski, P.; Sacchi, N. The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: Twenty-something years on. Nat. Protoc. 2006, 1, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, D.; Glass, L. Understanding Nonlinear Dynamics; Springer: New York, NY, USA, 1995. [Google Scholar]
- Tyson, J.J.; Chen, K.C.; Novak, B. Sniffers, buzzers, toggles and blinkers: Dynamics of regulatory and signaling pathways in the cell. Curr. Opin. Cell Biol. 2003, 15, 221–231. [Google Scholar] [CrossRef]
- Strogatz, S.H. Nonlinear Dynamics and Chaos; Addison-Wesley Co.: Reading, MA, USA, 1994. [Google Scholar]

© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Márton, M.; Kurucz, A.; Lizák, B.; Margittai, É.; Bánhegyi, G.; Kapuy, O. A Systems Biological View of Life-and-Death Decision with Respect to Endoplasmic Reticulum Stress—The Role of PERK Pathway. Int. J. Mol. Sci. 2017, 18, 58. https://doi.org/10.3390/ijms18010058
Márton M, Kurucz A, Lizák B, Margittai É, Bánhegyi G, Kapuy O. A Systems Biological View of Life-and-Death Decision with Respect to Endoplasmic Reticulum Stress—The Role of PERK Pathway. International Journal of Molecular Sciences. 2017; 18(1):58. https://doi.org/10.3390/ijms18010058
Chicago/Turabian StyleMárton, Margita, Anita Kurucz, Beáta Lizák, Éva Margittai, Gábor Bánhegyi, and Orsolya Kapuy. 2017. "A Systems Biological View of Life-and-Death Decision with Respect to Endoplasmic Reticulum Stress—The Role of PERK Pathway" International Journal of Molecular Sciences 18, no. 1: 58. https://doi.org/10.3390/ijms18010058