Viral-Cellular DNA Junctions as Molecular Markers for Assessing Intra-Tumor Heterogeneity in Cervical Cancer and for the Detection of Circulating Tumor DNA

 ,
,
Abstract
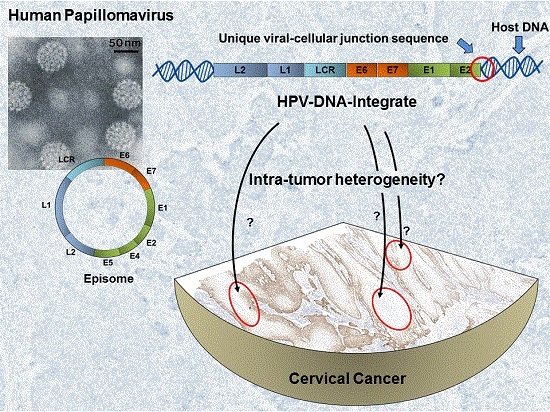
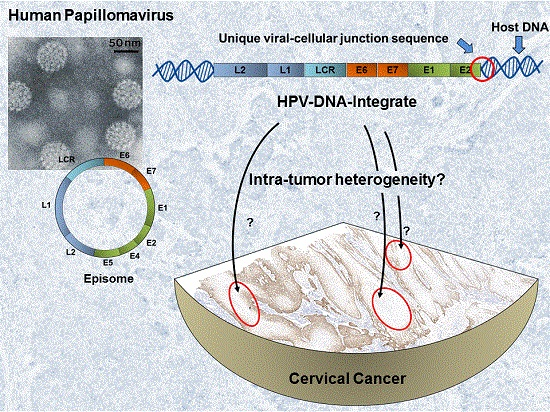
:
1. Introduction
2. Results
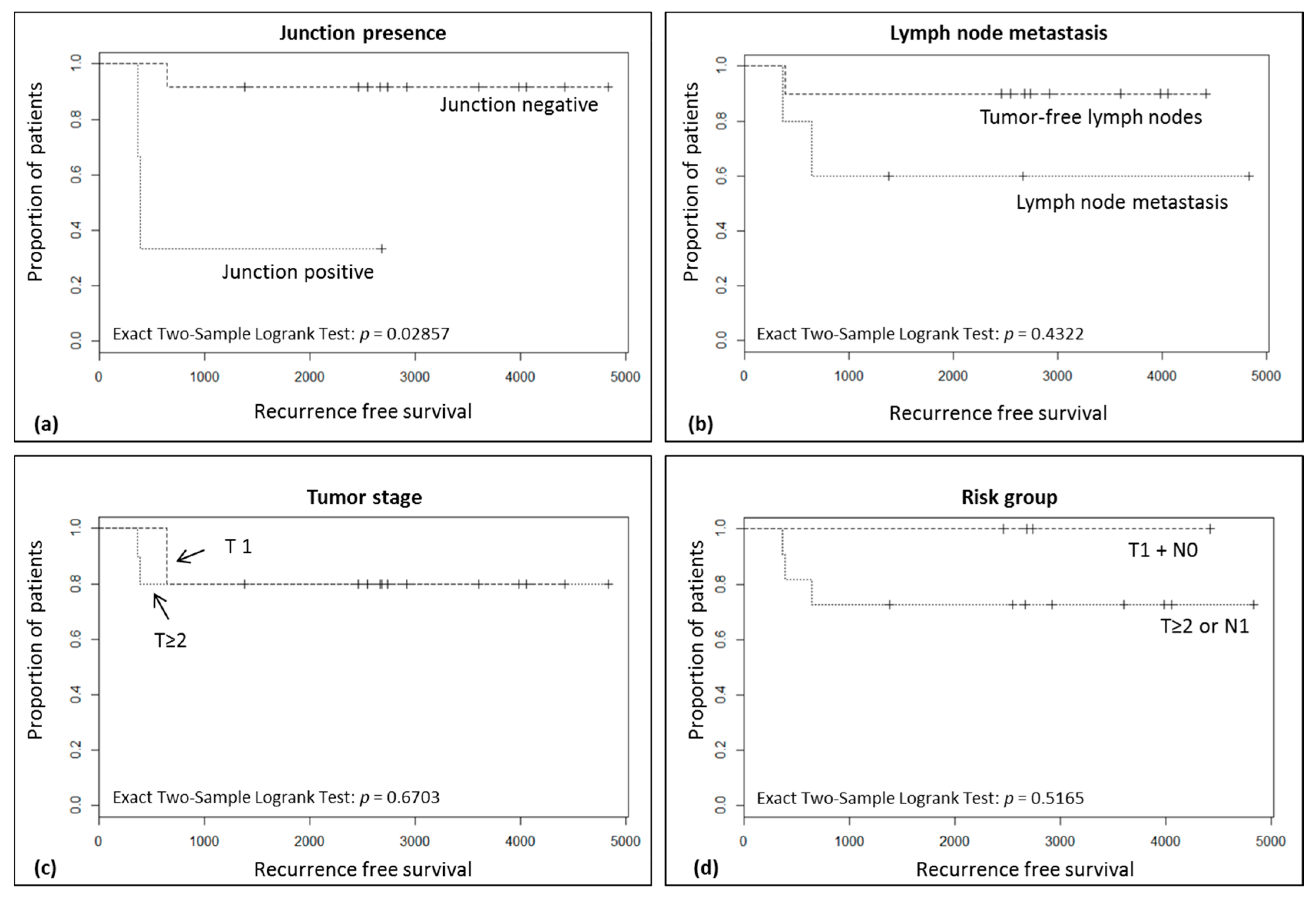
2.1. The Majority of Tumors Show Intra-Tumor Homogeneity with Respect to Junction Distribution
2.2. Detection of Viral-Cellular Junctions Fragments in Cell-Free Serum DNA
3. Discussion
4. Materials and Methods
4.1. Samples and Sample Processing
4.2. Intra-Tumor Heterogeneity Analyses: Assay Design and Optimization
4.3. Intra-Tumor Heterogeneity Analyses: Laser Micro-Dissection
4.4. ctDNA Analyses: Detection of Viral-Cellular Junctions in Cell-Free Tumor DNA from Sera
4.5. Statistical Analyses
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ADC | Adenocarcinoma |
| cfDNA | Circulating cell-free DNA |
| Ct | Cycle threshold |
| ctDNA | Circulating tumor DNA |
| FIGO | Fédération internationale de gynécologie et d’obstétrique |
| HE | Hematoxylin and eosin stain |
| HPV | Human papillomavirus |
| ITH | Intra-tumor heterogeneity |
| NSCLC | Non-small cell lung cancer |
| SCC | Squamous cell carcinoma |
| TM | Melting temperature |
| vcj-PCR | Viral-cellular junction PCR |
References
- Kovac, M.; Navas, C.; Horswell, S.; Salm, M.; Bardella, C.; Rowan, A.; Stares, M.; Castro-Giner, F.; Fisher, R.; de Bruin, E.C.; et al. Recurrent chromosomal gains and heterogeneous driver mutations characterise papillary renal cancer evolution. Nat. Commun. 2015, 6, 6336. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.S.; Eeles, R.; Wedge, D.C.; van Loo, P.; Gundem, G.; Alexandrov, L.B.; Kremeyer, B.; Butler, A.; Lynch, A.G.; Camacho, N.; et al. Analysis of the genetic phylogeny of multifocal prostate cancer identifies multiple independent clonal expansions in neoplastic and morphologically normal prostate tissue. Nat. Genet. 2015, 47, 367–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012, 486, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Navin, N.; Krasnitz, A.; Rodgers, L.; Cook, K.; Meth, J.; Kendall, J.; Riggs, M.; Eberling, Y.; Troge, J.; Grubor, V.; et al. Inferring tumor progression from genomic heterogeneity. Genome Res. 2010, 20, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Bachtiary, B.; Boutros, P.C.; Pintilie, M.; Shi, W.; Bastianutto, C.; Li, J.H.; Schwock, J.; Zhang, W.; Penn, L.Z.; Jurisica, I.; et al. Gene expression profiling in cervical cancer: An exploration of intratumor heterogeneity. Clin. Cancer Res. 2006, 12, 5632–5640. [Google Scholar] [CrossRef] [PubMed]
- Dawson, S.J.; Tsui, D.W.; Murtaza, M.; Biggs, H.; Rueda, O.M.; Chin, S.F.; Dunning, M.J.; Gale, D.; Forshew, T.; Mahler-Araujo, B.; et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 2013, 368, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Reinert, T.; Scholer, L.V.; Thomsen, R.; Tobiasen, H.; Vang, S.; Nordentoft, I.; Lamy, P.; Kannerup, A.S.; Mortensen, F.V.; Stribolt, K.; et al. Analysis of circulating tumour DNA to monitor disease burden following colorectal cancer surgery. Gut 2015, 65, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, R.; Wang, S.; Zhong, J.; Wu, M.; Zhao, J.; Duan, J.; Zhuo, M.; An, T.; Wang, Y.; et al. Quantification and dynamic monitoring of EGFR T790M in plasma cell-free DNA by digital PCR for prognosis of EGFR-TKI treatment in advanced NSCLC. PLoS ONE 2014, 9, e110780. [Google Scholar] [CrossRef] [PubMed]
- Wimberger, P.; Roth, C.; Pantel, K.; Kasimir-Bauer, S.; Kimmig, R.; Schwarzenbach, H. Impact of platinum-based chemotherapy on circulating nucleic acid levels, protease activities in blood and disseminated tumor cells in bone marrow of ovarian cancer patients. Int. J. Cancer 2011, 128, 2572–2580. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, T.; Fujita, M.; Inoue, M.; Tanizawa, O.; Nomura, T.; Shroyer, K.R. Analysis of clonality by amplification of short tandem repeats. Carcinomas of the female reproductive tract. Diagn. Mol. Pathol. 1994, 3, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Pang, T.; Asplund, A.; Ponten, J.; Nister, M. Clonality analysis of synchronous lesions of cervical carcinoma based on X chromosome inactivation polymorphism, human papillomavirus type 16 genome mutations, and loss of heterozygosity. J. Exp. Med. 2002, 195, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Wu, F.; Asplund, A.; Hu, X.; Mazurenko, N.; Kisseljov, F.; Ponten, J.; Wilander, E. Analysis of intratumoral heterogeneity of chromosome 3p deletions and genetic evidence of polyclonal origin of cervical squamous carcinoma. Mod. Pathol. 2001, 14, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Ueda, Y.; Miyatake, T.; Okazawa, M.; Kimura, T.; Miyake, T.; Fujiwara, K.; Yoshino, K.; Nakashima, R.; Fujita, M.; Enomoto, T. Clonality and HPV infection analysis of concurrent glandular and squamous lesions and adenosquamous carcinomas of the uterine cervix. Am. J. Clin. Pathol. 2008, 130, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Lyng, H.; Beigi, M.; Svendsrud, D.H.; Brustugun, O.T.; Stokke, T.; Kristensen, G.B.; Sundfor, K.; Skjonsberg, A.; de Angelis, P.M. Intratumor chromosomal heterogeneity in advanced carcinomas of the uterine cervix. Int. J. Cancer 2004, 111, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Walboomers, J.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.; Peto, J.; Meijer, C.J.; Munoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999, 189, 12–19. [Google Scholar] [CrossRef]
- Zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.; Allen-Hoffmann, B.L.; Lambert, P.F. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J. Virol. 1995, 69, 2989–2997. [Google Scholar] [PubMed]
- Hudelist, G.; Manavi, M.; Pischinger, K.I.; Watkins-Riedel, T.; Singer, C.F.; Kubista, E.; Czerwenka, K.F. Physical state and expression of HPV DNA in benign and dysplastic cervical tissue: Different levels of viral integration are correlated with lesion grade. Gynecol. Oncol. 2004, 92, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Badaracco, G.; Venuti, A.; Sedati, A.; Marcante, M.L. HPV16 and HPV18 in genital tumors: Significantly different levels of viral integration and correlation to tumor invasiveness. J. Med. Virol. 2002, 67, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhu, D.; Wang, W.; Li, W.; Jia, W.; Zeng, X.; Ding, W.; Yu, L.; Wang, X.; Wang, L.; et al. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat. Genet. 2015, 47, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Badaracco, G.; Venuti, A. Physical status of HPV types 16 and 18 in topographically different areas of genital tumours and in paired tumour-free mucosa. Int. J. Oncol. 2005, 27, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Galehouse, D.; Jenison, E.; DeLucia, A. Differences in the integration pattern and episomal forms of human papillomavirus type 16 DNA found within an invasive cervical neoplasm and its metastasis. Virology 1992, 186, 339–341. [Google Scholar] [CrossRef]
- Vinokurova, S.; Wentzensen, N.; Einenkel, J.; Klaes, R.; Ziegert, C.; Melsheimer, P.; Sartor, H.; Horn, L.C.; Hockel, M.; von Knebel Doeberitz, M. Clonal history of papillomavirus-induced dysplasia in the female lower genital tract. J. Natl. Cancer Inst. 2005, 97, 1816–1821. [Google Scholar] [CrossRef] [PubMed]
- Campitelli, M.; Jeannot, E.; Peter, M.; Lappartient, E.; Saada, S.; de la Rochefordiere, A.; Fourchotte, V.; Alran, S.; Petrow, P.; Cottu, P.; et al. Human papillomavirus mutational insertion: Specific marker of circulating tumor DNA in cervical cancer patients. PLoS ONE 2012, 7, e43393. [Google Scholar] [CrossRef] [PubMed]
- Dall, K.L.; Scarpini, C.G.; Roberts, I.; Winder, D.M.; Stanley, M.A.; Muralidhar, B.; Herdman, M.T.; Pett, M.R.; Coleman, N. Characterization of naturally occurring HPV16 integration sites isolated from cervical keratinocytes under noncompetitive conditions. Cancer Res. 2008, 68, 8249–8259. [Google Scholar] [CrossRef] [PubMed]
- Hanna, W.; Nofech-Mozes, S.; Kahn, H.J. Intratumoral heterogeneity of HER2/neu in breast cancer—A rare event. Breast J. 2007, 13, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.K.; Martelotto, L.G.; Gauthier, A.; Wen, H.C.; Piscuoglio, S.; Lim, R.S.; Cowell, C.F.; Wilkerson, P.M.; Wai, P.; Rodrigues, D.N.; et al. Intra-tumor genetic heterogeneity and alternative driver genetic alterations in breast cancers with heterogeneous HER2 gene amplification. Genome Biol. 2015, 16, 107. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Horswell, S.; Larkin, J.; Rowan, A.J.; Salm, M.P.; Varela, I.; Fisher, R.; McGranahan, N.; Matthews, N.; Santos, C.R.; et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat. Genet. 2014, 46, 225–233. [Google Scholar] [CrossRef] [PubMed]
- De Bruin, E.C.; McGranahan, N.; Mitter, R.; Salm, M.; Wedge, D.C.; Yates, L.; Jamal-Hanjani, M.; Shafi, S.; Murugaesu, N.; Rowan, A.J.; et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 2014, 346, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.L.; Kan, M.; Zhang, M.M.; Yu, S.S.; Xie, H.J.; Gu, Z.H.; Wang, H.N.; Zhao, S.X.; Zhou, G.B.; Song, H.D.; et al. Multiregion sequencing reveals the intratumor heterogeneity of driver mutations in TP53-driven non-small cell lung cancer. Int. J. Cancer 2017, 140, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Kinzler, K.W. Digital PCR. Proc. Natl. Acad. Sci. USA 1999, 96, 9236–9241. [Google Scholar] [CrossRef] [PubMed]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [PubMed]
- Lecomte, T.; Berger, A.; Zinzindohoue, F.; Micard, S.; Landi, B.; Blons, H.; Beaune, P.; Cugnenc, P.H.; Laurent-Puig, P. Detection of free-circulating tumor-associated DNA in plasma of colorectal cancer patients and its association with prognosis. Int. J. Cancer 2002, 100, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Bidard, F.C.; Madic, J.; Mariani, P.; Piperno-Neumann, S.; Rampanou, A.; Servois, V.; Cassoux, N.; Desjardins, L.; Milder, M.; Vaucher, I.; et al. Detection rate and prognostic value of circulating tumor cells and circulating tumor DNA in metastatic uveal melanoma. Int. J. Cancer 2014, 134, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Chotewutmontri, S.; Wolf, S.; Klos, U.; Schmitz, M.; Durst, M.; Schwarz, E. Multiplex Identification of Human Papillomavirus 16 DNA Integration Sites in Cervical Carcinomas. PLoS ONE 2013, 8, e66693. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.; Driesch, C.; Beer-Grondke, K.; Jansen, L.; Runnebaum, I.B.; Durst, M. Loss of gene function as a consequence of human papillomavirus DNA integration. Int. J. Cancer 2012, 131, E593–E602. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient-ID | Age at Diagnosis | Histologic Subtype | Tumor Type | HPV Type | TNM-Classification | HPV-Integrates | 3′ Integration Site | Tumor Blocks for ITH Analyses | Serum Samples for ctDNA Analyses | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| T | N | M | |||||||||
| 841 | 58 | SCC | P | 16 | 2a | 0 | 1 | 1 | 2q33.3 | 6 | 3 |
| 2 | 13q22.2 | ||||||||||
| 3 | 13q22.1 | ||||||||||
| 4 | Xp22.11 | ||||||||||
| 5 | Xp22.11 | ||||||||||
| 1509 | 70 | SCC | P | 16 | 2b | 1 | 0 | 1 | 9p13.3 | 6 | 1 |
| 2 | 8q23.3 | ||||||||||
| 1907 | 33 | SCC | P | 16 | 4 | 1 | 14q23.2 | - | 3 | ||
| 2349 | 58 | SCC | P | 16 | 1b1 | 0 | 0 | 1 | Xp22.31 | - | 4 |
| 2555 | 48 | SCC | P | 16 | 2a | 1 | 0 | 1 | 1p36.22 | 3 | 2 |
| 2723 | 52 | SCC | P | 16 | 2b | 0 | x | 1 | 17q23.1 | - | 3 |
| 3817 | 42 | SCC | P | 16 | 1b1 | 1 | 0 | 1 | 16q23.1 | - | 1 |
| 3986/4112 * | 47 | SCC | P | 18 | 3b | 1 | 1 | 17p13.1 | 10 | 2 | |
| 4154 | 46 | SCC | P | 18 | 1b | 0 | x | 1 | 2q24.2 | - | 1 |
| 4338 | 60 | SCC | P | 16 | 2 | 0 | x | 1 | 9p24.1 | 11 | 1 |
| 4497 | 49 | ADC | P | 18 | 2b | 0 | x | 1 | 3p24.2 | - | 2 |
| 4502 | 37 | ADC | P | 18 | 3b | 1 | x | 1 | 7q34 | - | 1 |
| 4749 | 40 | SCC | P | 16 | 1b1 | 0 | 0 | 1 | 18p11.32 | - | 1 |
| 2 | 18p11.32 | ||||||||||
| 3 | 18q12.2 | ||||||||||
| 4977 | 67 | SCC | P | 16 | 2b | 0 | 0 | 1 | 6q22.32 | 4 | 1 |
| 2 | 7p15.1 | ||||||||||
| 5234 | 33 | SCC | P | 16 | 1b2 | 0 | 1 | 3p21.31 | 5 | 2 | |
| 5254 | 29 | SCC | P | 18 | 2b2 | 0 | 1 | 9p21.3 | - | 1 | |
| 5613 | 37 | ADC | P | 18 | 1b1 | 1 | 8p12 | - | 1 | ||
| 3719 | 38 | SCC | R | 16 | 1b | 1 | 0 | 1 | 2p22.3 | - | 1 |
| 2 | 4q21.1 | ||||||||||
| 4601 | 63 | SCC | R | 16 | 4 | 0 | x | 1 | 9p23 | - | 1 |
| 4995 | 52 | SCC | R | 18 | 2b | 0 | 0 | 1 | 19p13.3 | - | 1 |
| 5189 | 48 | SCC | R | 16 | 1b2 | 0 | 0 | 1 | 8q24.21 | 3 | 1 |
| 2 | 8q23.2 | ||||||||||
| 3 | 5p14.1 | ||||||||||
| Tissue | Patient | Follow-Up (Days) | Status at End of Follow-Up | Junctions | Junction-Detection in Primary Serum | Junction-Detection in Sequential Sera |
|---|---|---|---|---|---|---|
| Primary Tumor | 841 | 2549 | deceased | J1 | No | No |
| J2 | No | No | ||||
| J3 | No | No | ||||
| J4 | No | No | ||||
| J5 | No | No | ||||
| 1509 | 2663 | deceased | J1 | No | x | |
| J2 | No | x | ||||
| 1907 | 257 | Deceased * | J1 | No | No | |
| 2349 | 2462 | tumor-free | J1 | No | No | |
| 2555 | 4831 | tumor-free | J1 | No | No | |
| 2723 | 3605 | tumor-free | J1 | No | No | |
| 3817 | 643 | Deceased * | J1 | No | x | |
| 3986 | 360 | Deceased * | J1 | Yes | Yes | |
| 4154 | 4420 | tumor-free | J1 | No | x | |
| 4338 | 1350 | Deceased * | J1 | Yes | Yes | |
| 4497 | 4055 | tumor-free | J1 | No | No | |
| 4502 | 1376 | deceased | J1 | No | x | |
| 4749 | 0 | tumor-free | J1 | No | x | |
| J2 | No | x | ||||
| J3 | No | x | ||||
| 4977 | 3980 | tumor-free | J1 | No | x | |
| J2 | No | x | ||||
| 5234 | 2681 | tumor-free | J1 | Yes | Yes | |
| 5254 | 2923 | tumor-free | J1 | No | x | |
| 5613 | 2740 | tumor-free | J1 | No | x | |
| Relapse | 3719 | - | Deceased * | J1 | No | x |
| J2 | No | x | ||||
| 4601 | - | Deceased * | J1 | Yes | x | |
| 4995 | - | Deceased * | J1 | No | x | |
| 5189 | - | Deceased * | J1 | Yes | x | |
| J2 | Yes | x | ||||
| J3 | Yes | x |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carow, K.; Gölitz, M.; Wolf, M.; Häfner, N.; Jansen, L.; Hoyer, H.; Schwarz, E.; Runnebaum, I.B.; Dürst, M. Viral-Cellular DNA Junctions as Molecular Markers for Assessing Intra-Tumor Heterogeneity in Cervical Cancer and for the Detection of Circulating Tumor DNA. Int. J. Mol. Sci. 2017, 18, 2032. https://doi.org/10.3390/ijms18102032
Carow K, Gölitz M, Wolf M, Häfner N, Jansen L, Hoyer H, Schwarz E, Runnebaum IB, Dürst M. Viral-Cellular DNA Junctions as Molecular Markers for Assessing Intra-Tumor Heterogeneity in Cervical Cancer and for the Detection of Circulating Tumor DNA. International Journal of Molecular Sciences. 2017; 18(10):2032. https://doi.org/10.3390/ijms18102032
Chicago/Turabian StyleCarow, Katrin, Mandy Gölitz, Maria Wolf, Norman Häfner, Lars Jansen, Heike Hoyer, Elisabeth Schwarz, Ingo B. Runnebaum, and Matthias Dürst. 2017. "Viral-Cellular DNA Junctions as Molecular Markers for Assessing Intra-Tumor Heterogeneity in Cervical Cancer and for the Detection of Circulating Tumor DNA" International Journal of Molecular Sciences 18, no. 10: 2032. https://doi.org/10.3390/ijms18102032