Spatiotemporal Control of Doxorubicin Delivery from “Stealth-Like” Prodrug Micelles


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
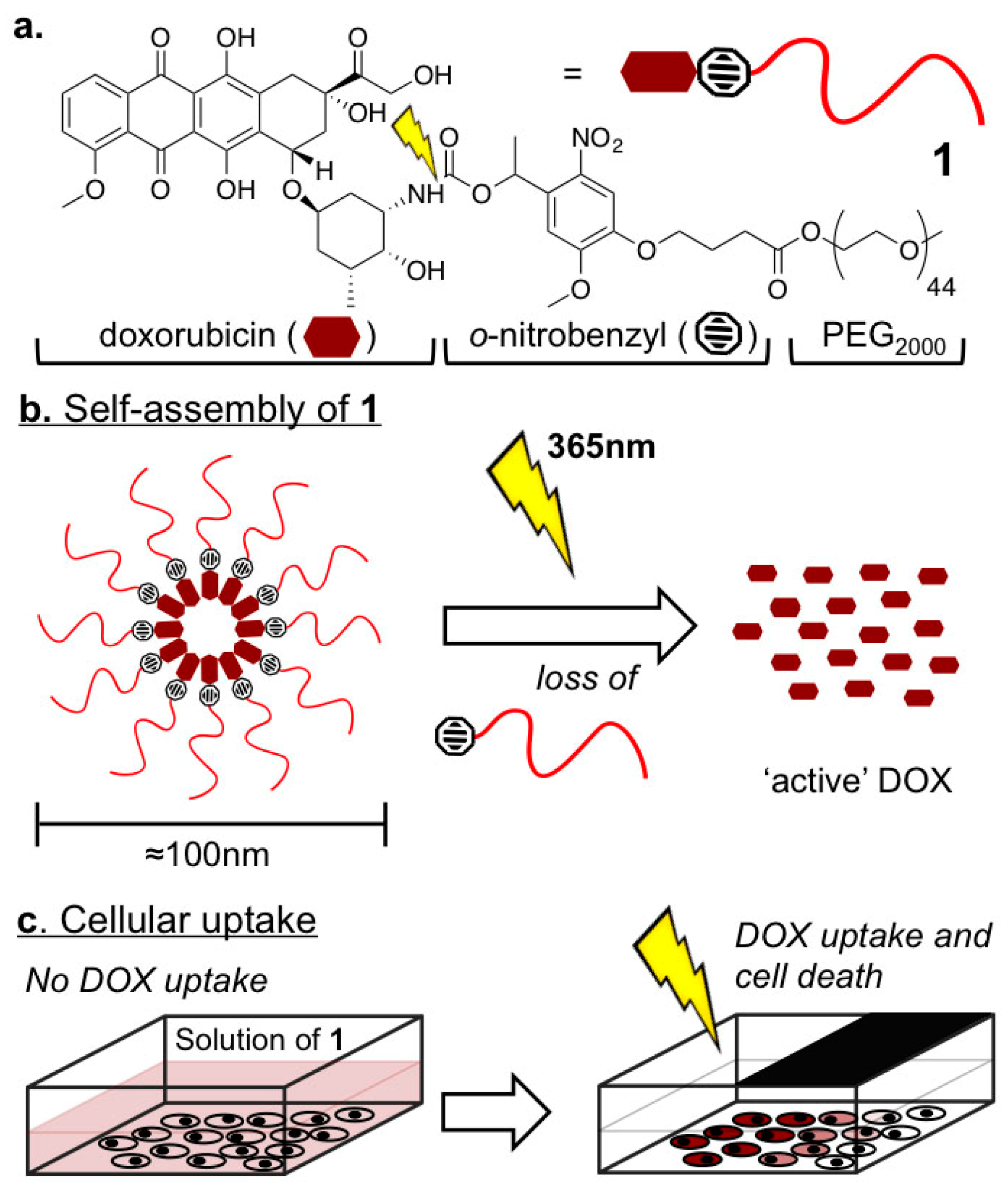
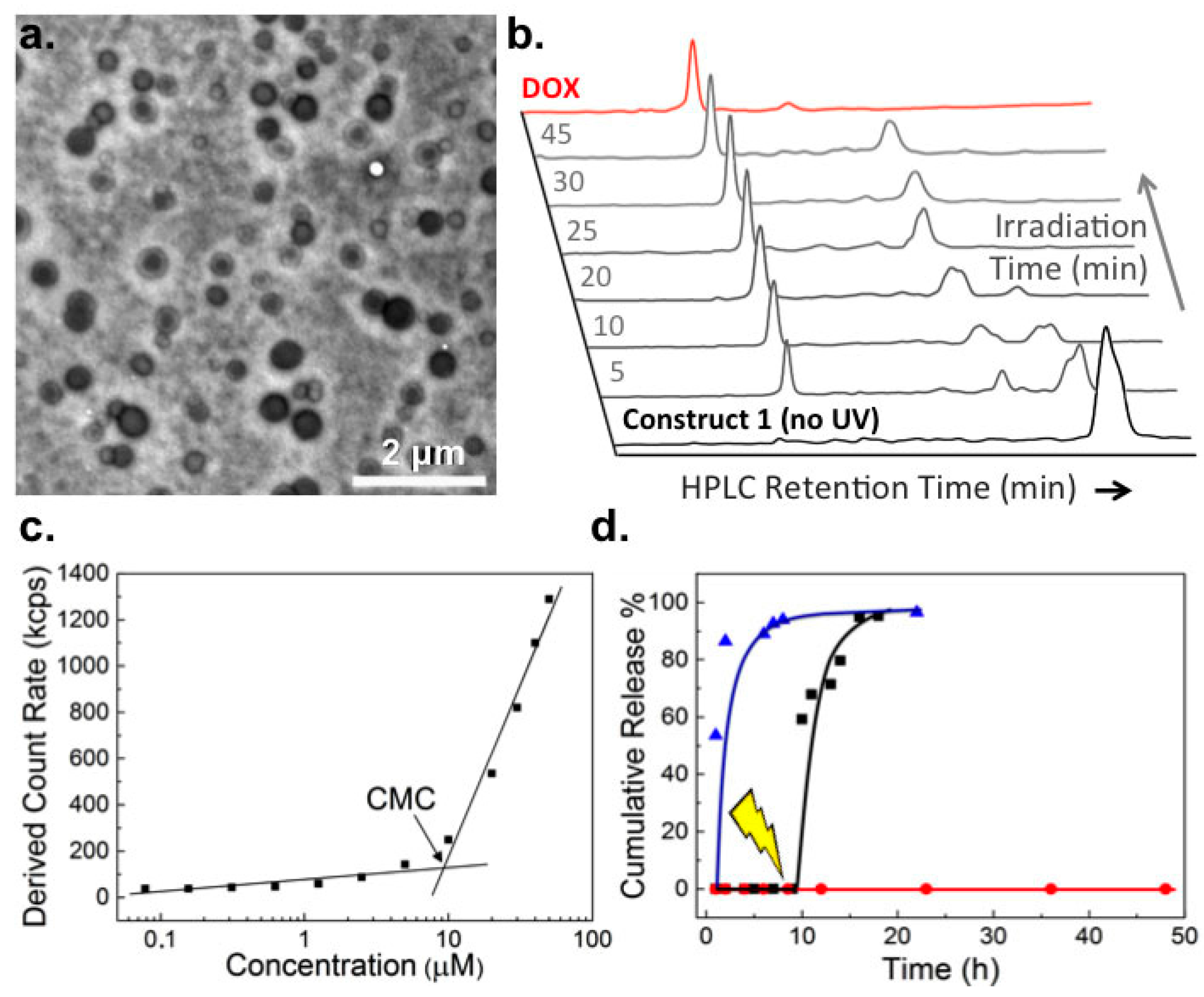
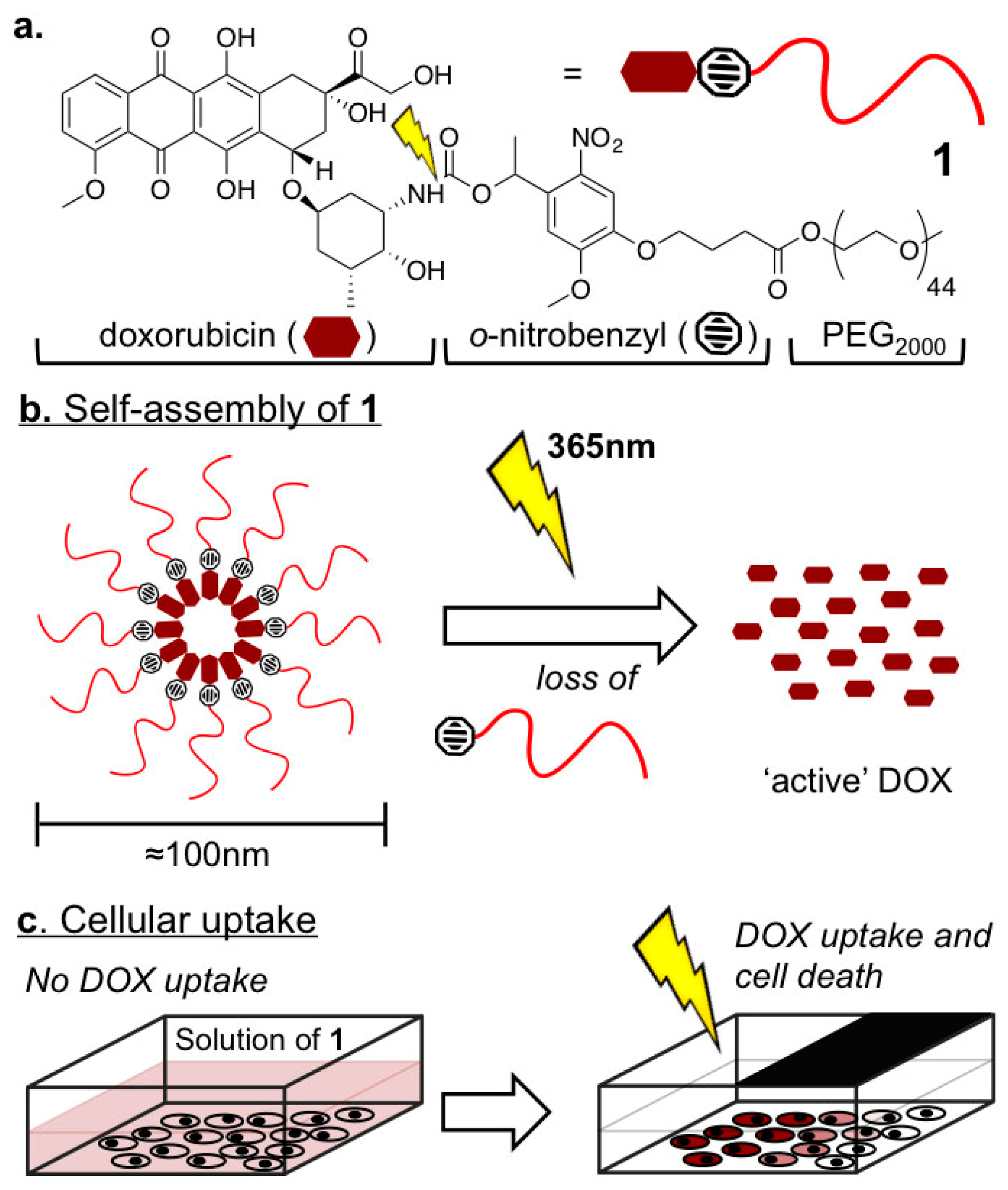
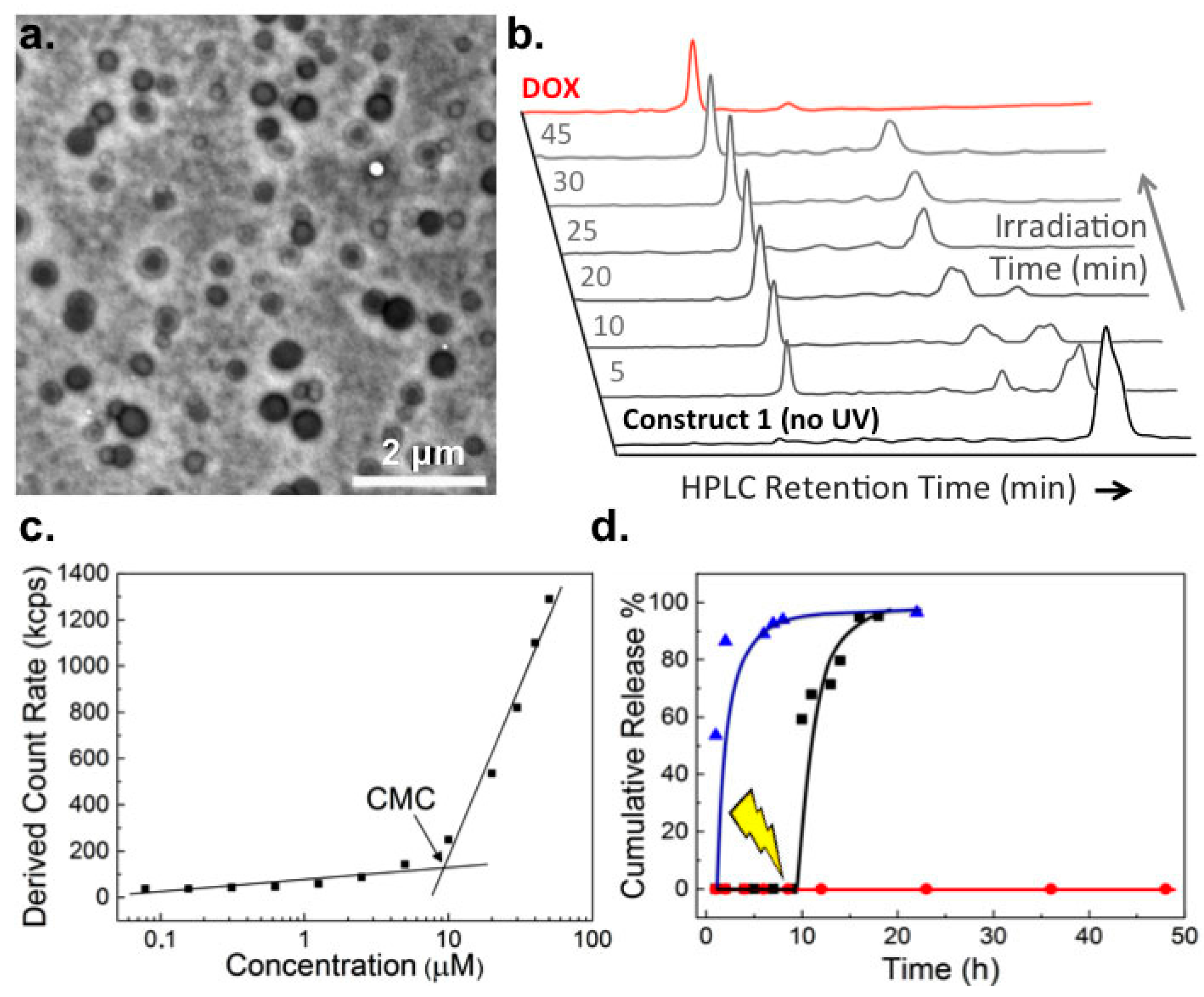
2.1. Biophysical Characterization of Light-Activated DOX-PEG Prodrug Micelles
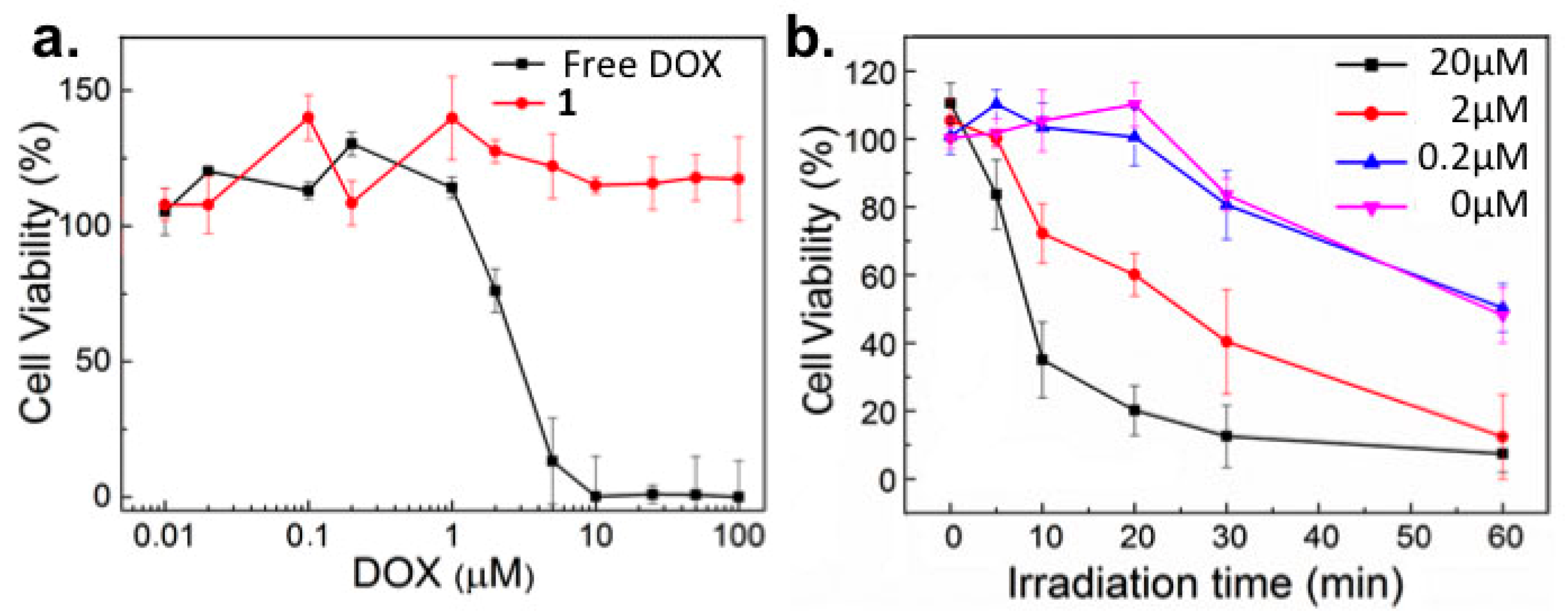
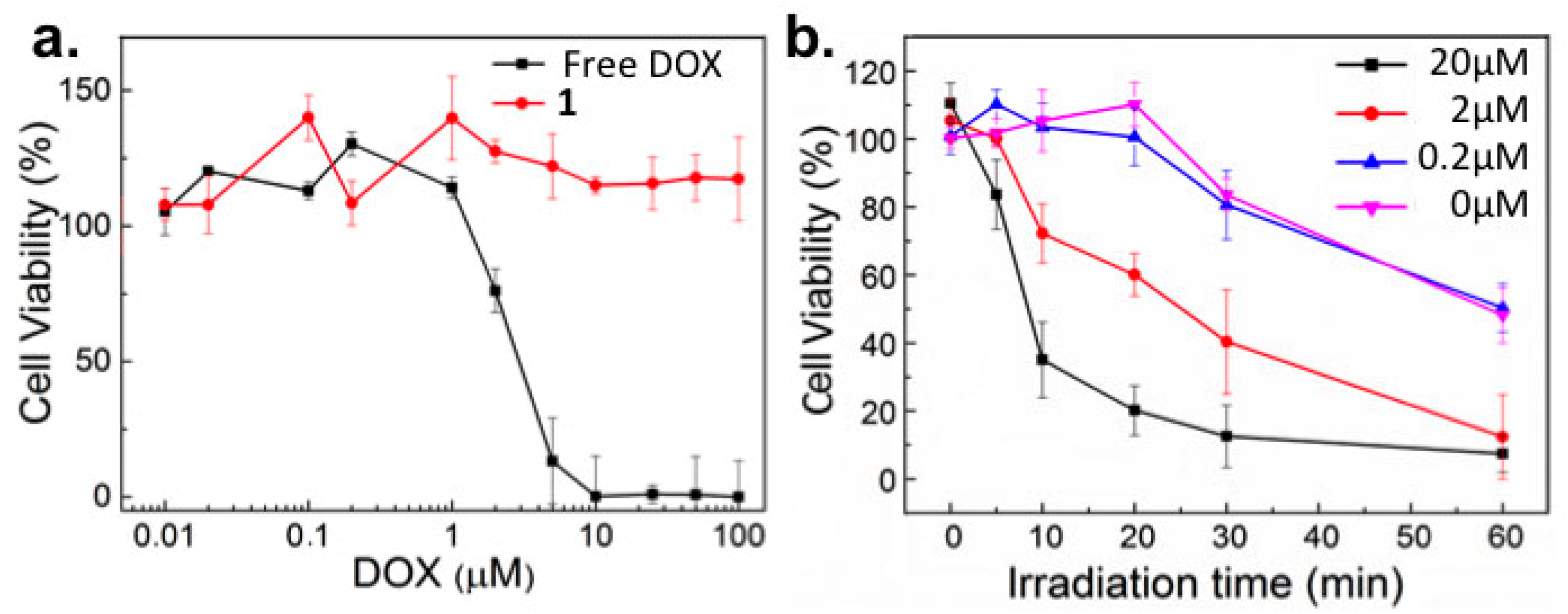
2.2. In Vitro Toxicity
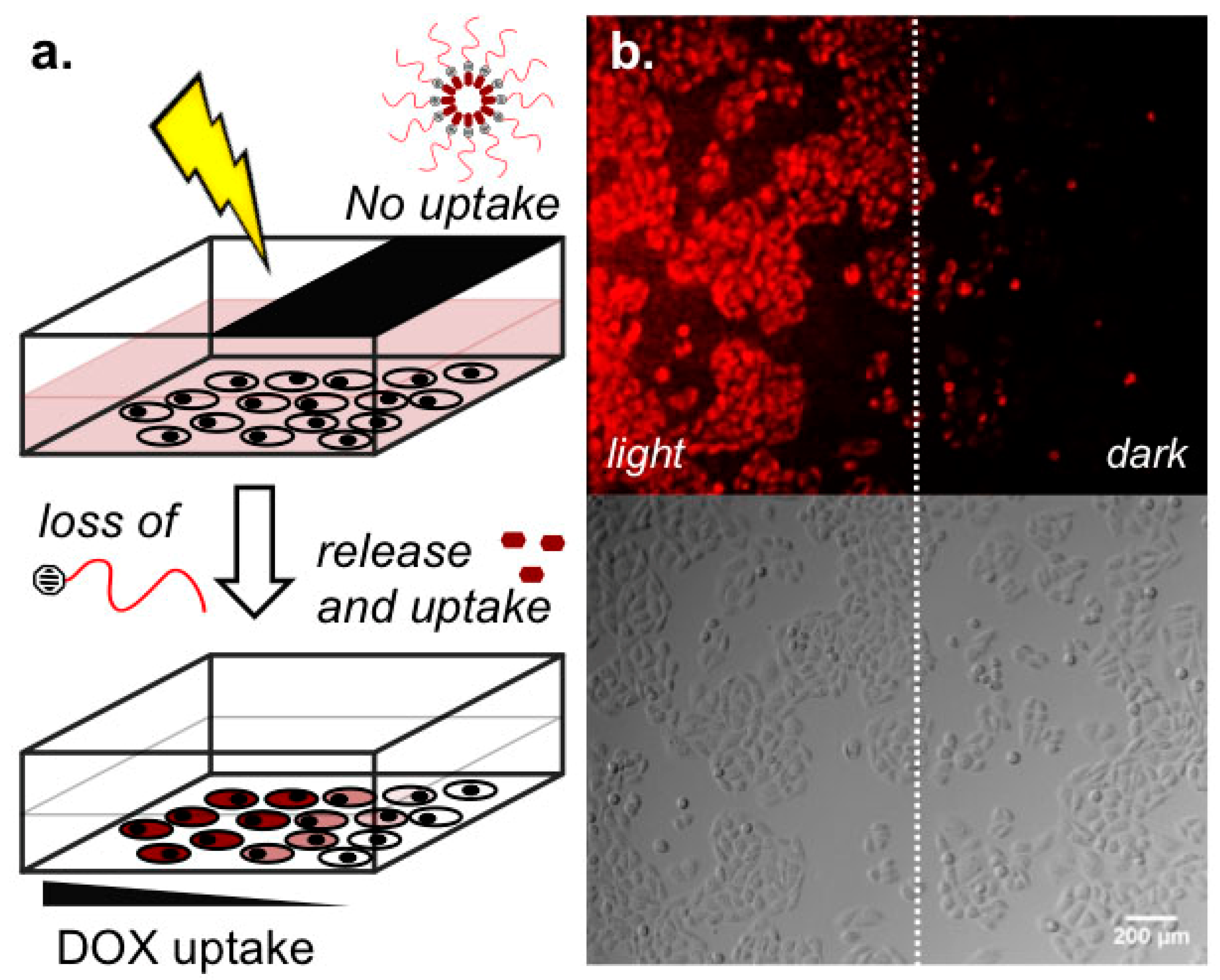
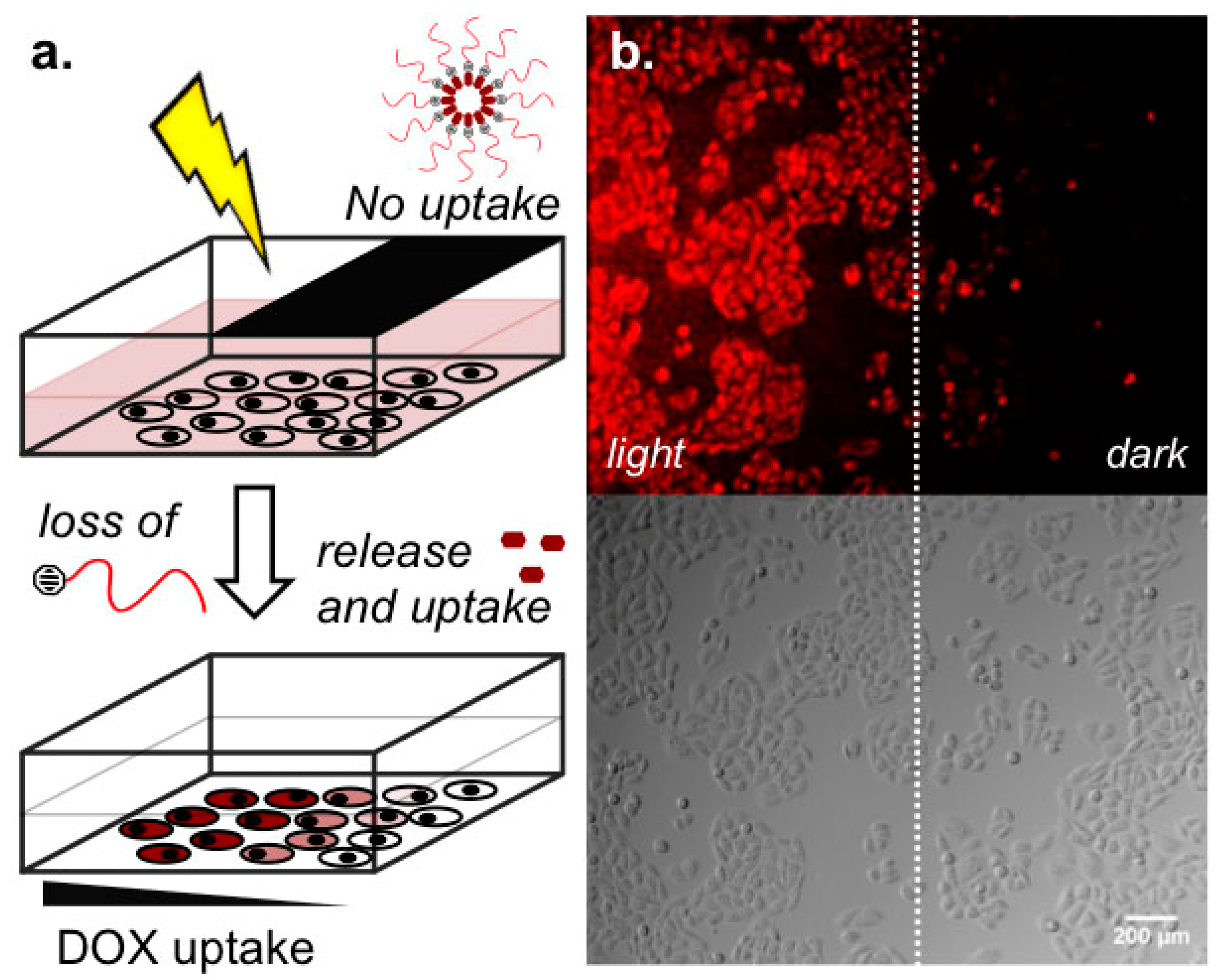
2.3. Light-Templated Cellular Uptake of DOX
3. Discussion
4. Materials and Methods
4.1. Materials and Instruments
4.2. Preperation and Characterization of Light-Activated DOX-PEG Prodrug Micelles
4.3. In Vitro Drug Release
4.4. WST Cell Proliferation Assay
4.5. FACS Analysis
4.6. Light Templated DOX Devlivery to Cells
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chatterjee, K.; Zhang, J.; Honbo, N.; Karliner, J.S. Doxorubicin cardiomyopathy. Cardiology 2010, 115, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Whaley, F.S.; Ewer, M.S. Congestive heart failure in patients treated with doxorubicin: A retrospective analysis of three trials. Cancer 2003, 97, 2869–2879. [Google Scholar] [CrossRef] [PubMed]
- Byrne, J.D.; Betancourt, T.; Brannon-Peppas, L. Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv. Drug Deliv. Rev. 2008, 60, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- Immordino, M.L.; Dosio, F.; Cattel, L. Stealth liposomes: Review of the basic science, rationale, and clinical applications, existing and potential. Int. J. Nanomed. 2006, 1, 297–315. [Google Scholar]
- Du, J.; Lane, L.A.; Nie, S. Stimuli-responsive nanoparticles for targeting the tumor microenvironment. J. Control. Release 2015, 219, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V.P. Multifunctional, stimuli-sensitive nanoparticulate systems for drug delivery. Nat. Rev. Drug Discov. 2014, 13, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Nakamura, H.; Fang, J. The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv. Drug Deliv. Rev. 2013, 65, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Gou, P.; Liu, W.; Mao, W.; Tang, J.; Shen, Y.; Sui, M. Self-assembling doxorubicin prodrug forming nanoparticles for cancer chemotherapy: Synthesis and anticancer study in vitro and in vivo. J. Mater. Chem. B Mater. Biol. Med. 2013, 1, 284–292. [Google Scholar] [CrossRef]
- Wei, T.; Chen, C.; Liu, J.; Liu, C.; Posocco, P.; Liu, X.; Cheng, Q.; Huo, S.; Liang, Z.; Fermeglia, M.; Pricl, S.; Liang, X.-J.; Rocchi, P.; Peng, L. Anticancer drug nanomicelles formed by self-assembling amphiphilic dendrimer to combat cancer drug resistance. Proc. Natl. Acad. Sci. USA 2015, 112, 2978–2983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; He, J.; Cao, D.; Zhang, M.; Li, F.; Tam, K.C.; Ni, P. Synthesis of an acid-labile polymeric prodrug DOX-acetal-PEG-acetal-DOX with high drug loading content for pH-triggered intracellular drug release. Polym. Chem. 2015, 6, 4809–4818. [Google Scholar] [CrossRef]
- Xu, Z.; Zhang, K.; Hou, C.; Wang, D.; Liu, X.; Guan, X.; Zhang, X.; Zhang, H. A novel nanoassembled doxorubicin prodrug with a high drug loading for anticancer drug delivery. J. Mater. Chem. B Mater. Biol. Med. 2014, 2, 3433–3437. [Google Scholar] [CrossRef]
- Tu, Y.; Zhu, L. Enhancing cancer targeting and anticancer activity by a stimulus-sensitive multifunctional polymer-drug conjugate. J. Control. Release 2015, 212, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Ding, J.; Xiao, C.; Chen, J.; Zhuang, X.; Chen, X. Preclinical evaluation of antitumor activity of acid-sensitive PEGylated doxorubicin. ACS Appl. Mater. Interfaces 2014, 6, 21202–21214. [Google Scholar] [CrossRef] [PubMed]
- Ibsen, S.; Zahavy, E.; Wrasidlo, W.; Hayashi, T.; Norton, J.; Su, Y.; Adams, S.; Esener, S. Localized in vivo activation of a photoactivatable doxorubicin prodrug in deep tumor tissue. Photochem. Photobiol. 2013, 89, 698–708. [Google Scholar] [CrossRef] [PubMed]
- Dcona, M.M.; Mitra, D.; Goehe, R.W.; Gewirtz, D.A.; Lebman, D.A.; Hartman, M.C.T. Photocaged permeability: A new strategy for controlled drug release. Chem. Commun. 2012, 48, 4755–4757. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Lu, Y.Y.; Burts, A.O.; Lim, Y.H.; Finn, M.G.; Koberstein, J.T.; Turro, N.J.; Tirrell, D.A.; Grubbs, R.H. Core-clickable PEG-branch-azide bivalent-bottle-brush polymers by ROMP: Grafting-through and clicking-to. J. Am. Chem. Soc. 2011, 133, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.K.; Thomas, T.; Li, M.H.; Kotlyar, A.; Desai, A.; Baker, J. Light-controlled release of caged doxorubicin from folate receptor-targeting PAMAM dendrimer nanoconjugate. Chem. Commun. 2010, 46, 2632–2634. [Google Scholar] [CrossRef] [PubMed]
- Ibsen, S.; Zahavy, E.; Wrasdilo, W.; Berns, M.; Chan, M.; Esener, S. A novel doxorubicin prodrug with controllable photolysis activation for cancer chemotherapy. Pharm. Res. 2010, 27, 1848–1860. [Google Scholar] [CrossRef] [PubMed]
- McMillan, T.J.; Leatherman, E.; Ridley, A.; Shorrocks, J.; Tobi, S.E.; Whiteside, J.R. Cellular effects of long wavelength UV light (UVA) in mammalian cells. J. Pharm. Pharmacol. 2008, 60, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z. A review of progress in clinical photodynamic therapy. Technol. Cancer Res. Treat. 2005, 4, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Tomatsu, I.; Korobko, A.V.; Kros, A. Cyclodextrin–dextran based in situ hydrogel formation: A carrier for hydrophobic drugs. Soft Matter 2010, 6, 85–87. [Google Scholar] [CrossRef]
- Shanmugam, V.; Selvakumar, S.; Yeh, C.-S. Near-infrared light-responsive nanomaterials in cancer therapeutics. Chem. Soc. Rev. 2014, 43, 6254–6287. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, R.; Kompella, U.B. Nanomicellar formulations for sustained drug delivery: Strategies and underlying principles. Nanomedicine 2010, 5, 485–505. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kong, L.; Poulcharidis, D.; Schneider, G.F.; Campbell, F.; Kros, A. Spatiotemporal Control of Doxorubicin Delivery from “Stealth-Like” Prodrug Micelles. Int. J. Mol. Sci. 2017, 18, 2033. https://doi.org/10.3390/ijms18102033
Kong L, Poulcharidis D, Schneider GF, Campbell F, Kros A. Spatiotemporal Control of Doxorubicin Delivery from “Stealth-Like” Prodrug Micelles. International Journal of Molecular Sciences. 2017; 18(10):2033. https://doi.org/10.3390/ijms18102033
Chicago/Turabian StyleKong, Li, Dimitrios Poulcharidis, Gregory F. Schneider, Frederick Campbell, and Alexander Kros. 2017. "Spatiotemporal Control of Doxorubicin Delivery from “Stealth-Like” Prodrug Micelles" International Journal of Molecular Sciences 18, no. 10: 2033. https://doi.org/10.3390/ijms18102033