Differential Functional Roles of ALDH1A1 and ALDH1A3 in Mediating Metastatic Behavior and Therapy Resistance of Human Breast Cancer Cells

 , and
, and
Abstract
:1. Introduction
2. Results
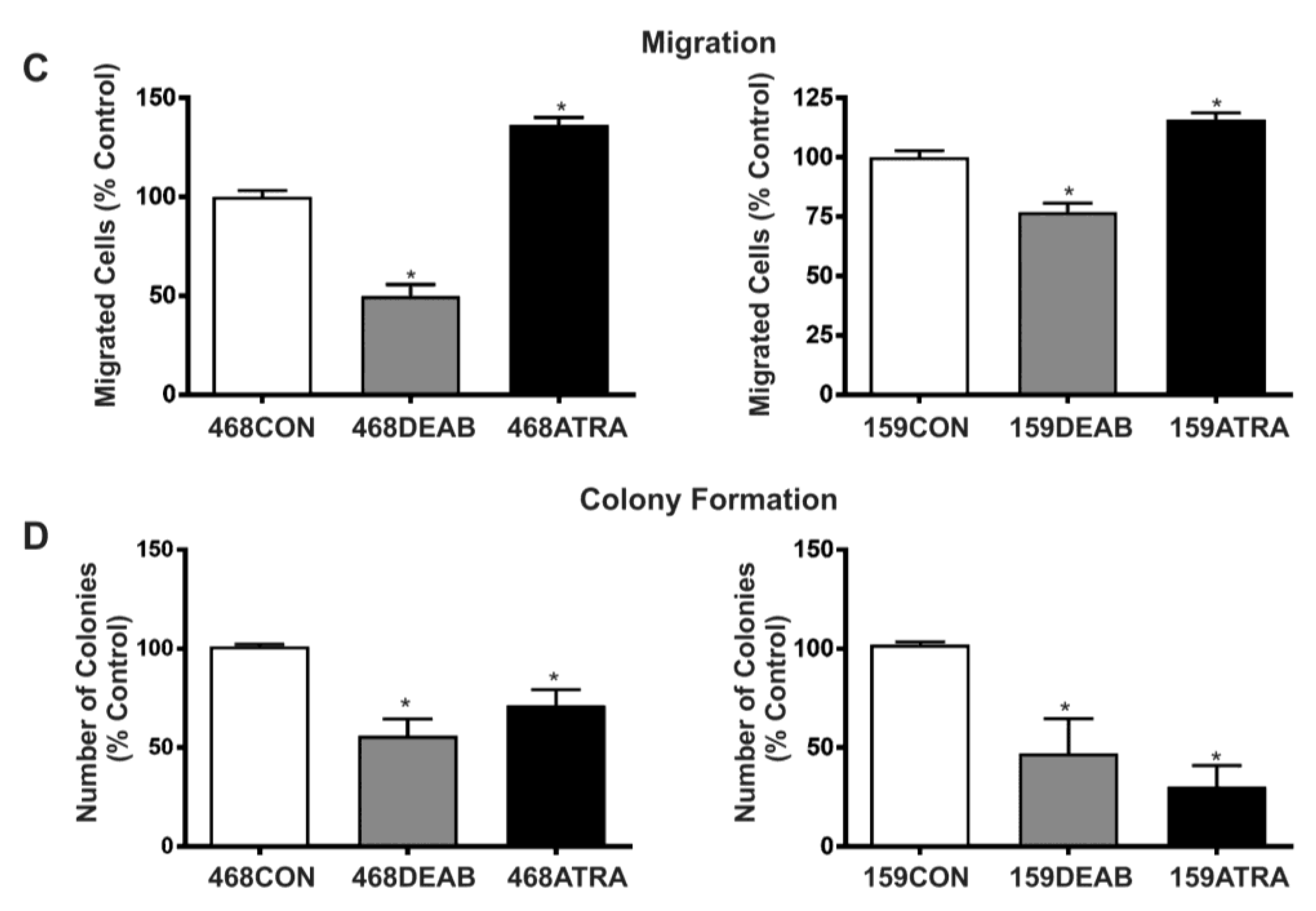
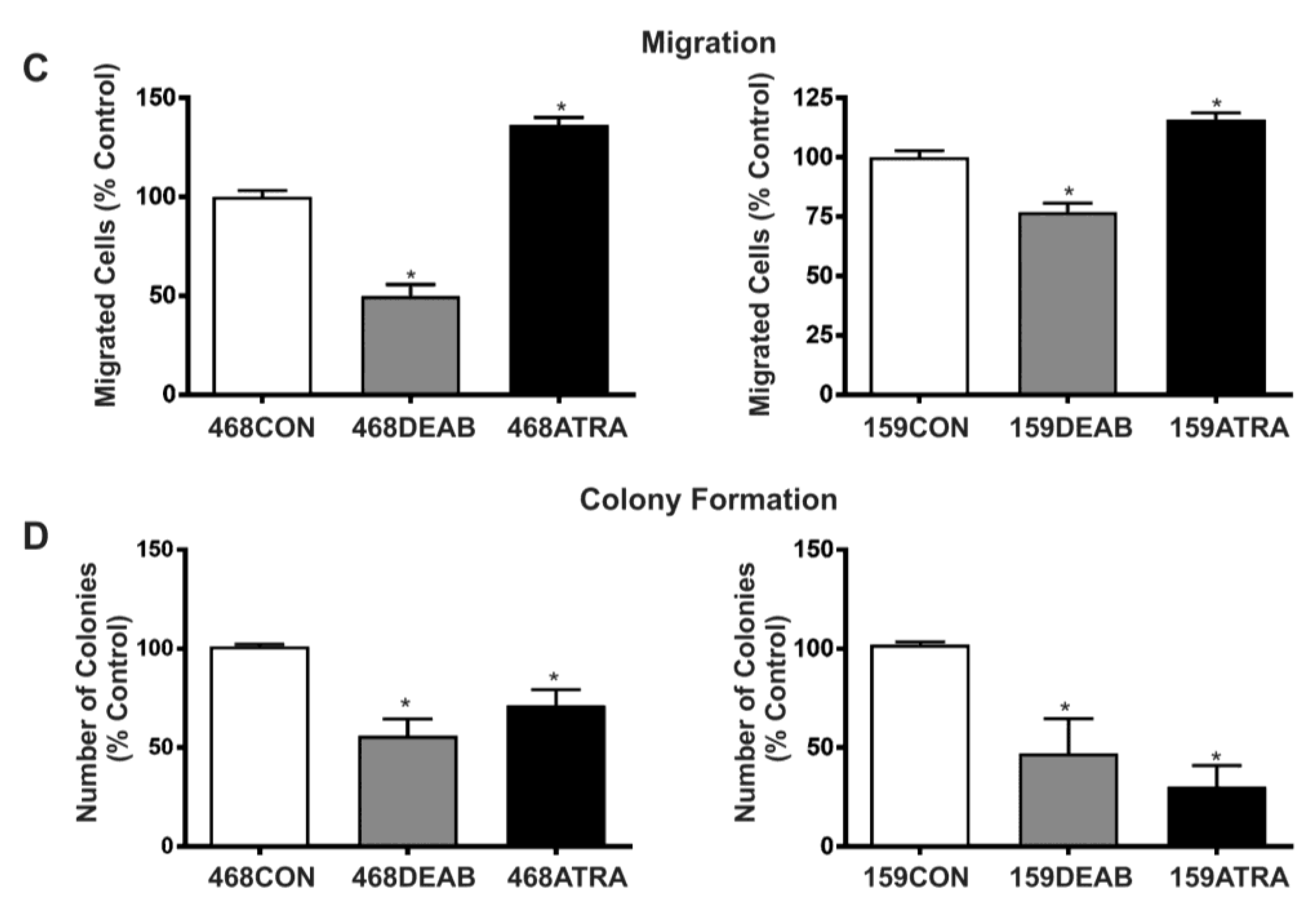
2.1. Treatment with DEAB (Diethylaminobenzaldehyde) Reduces Breast Cancer Cell Proliferation, Adhesion, Migration, and Colony Formation In Vitro
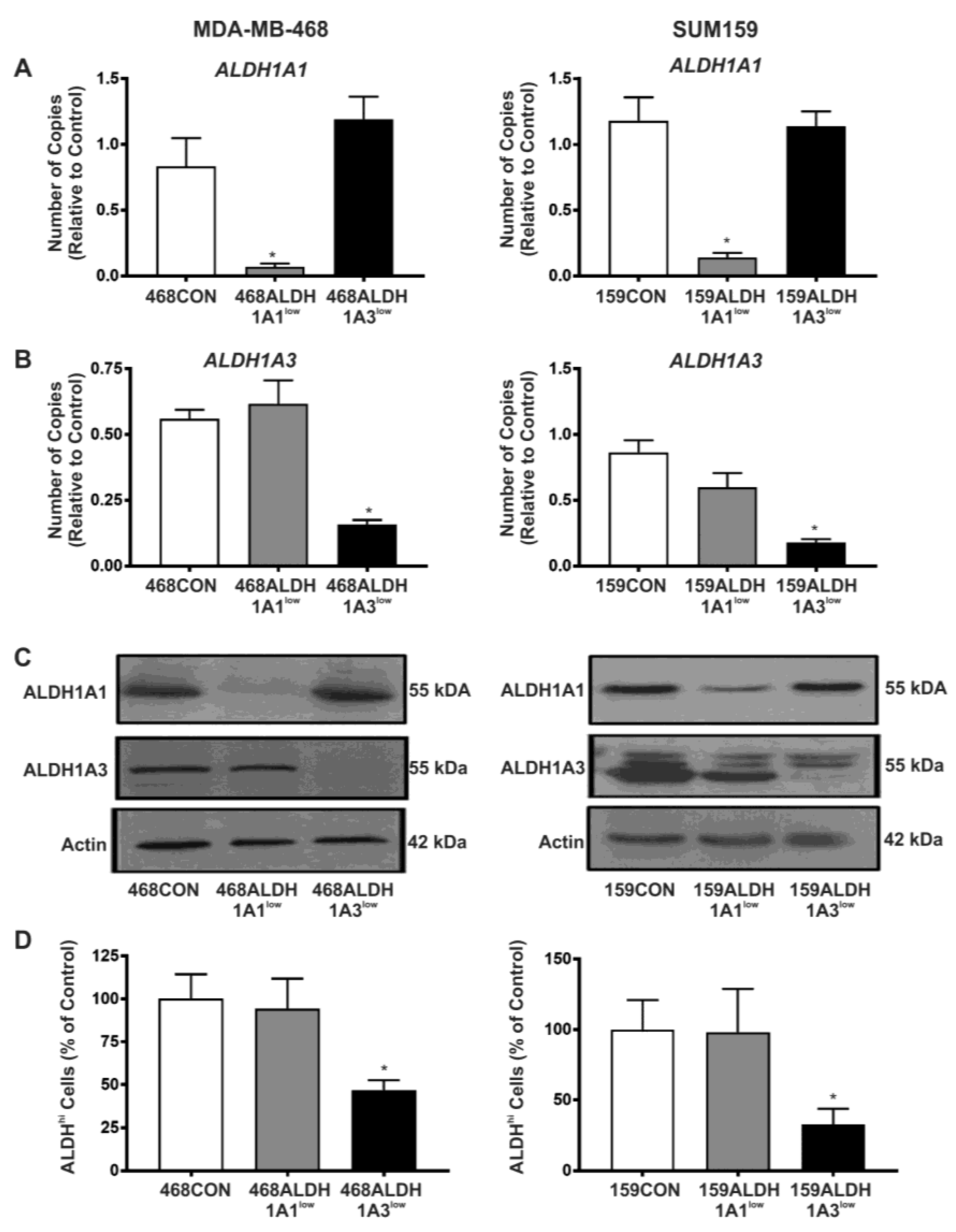
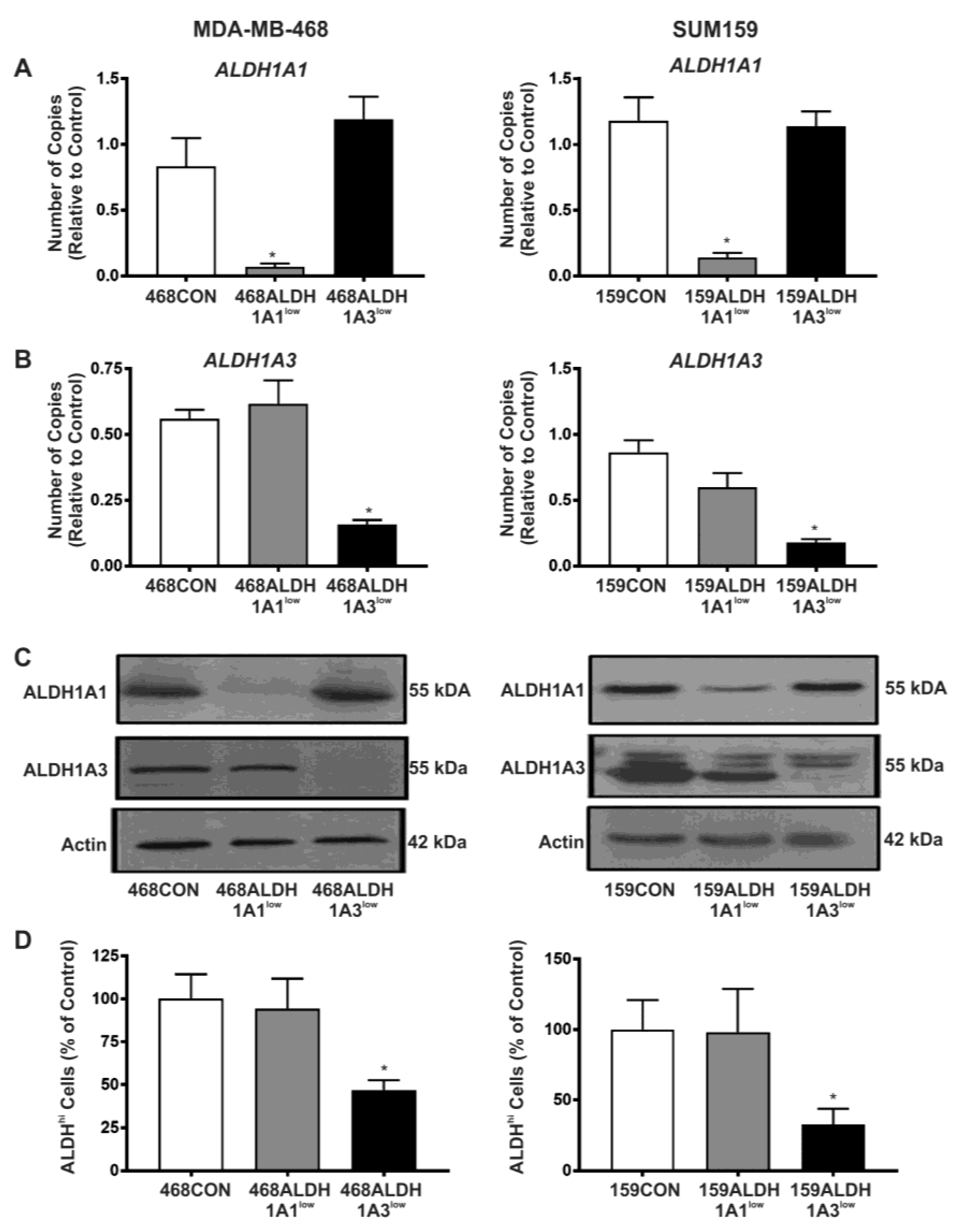
2.2. Decreased Expression of ALDH1A3 but Not ALDH1A1 Reduces ALDH Activity as Measured by the ALDEFLUOR® Assay
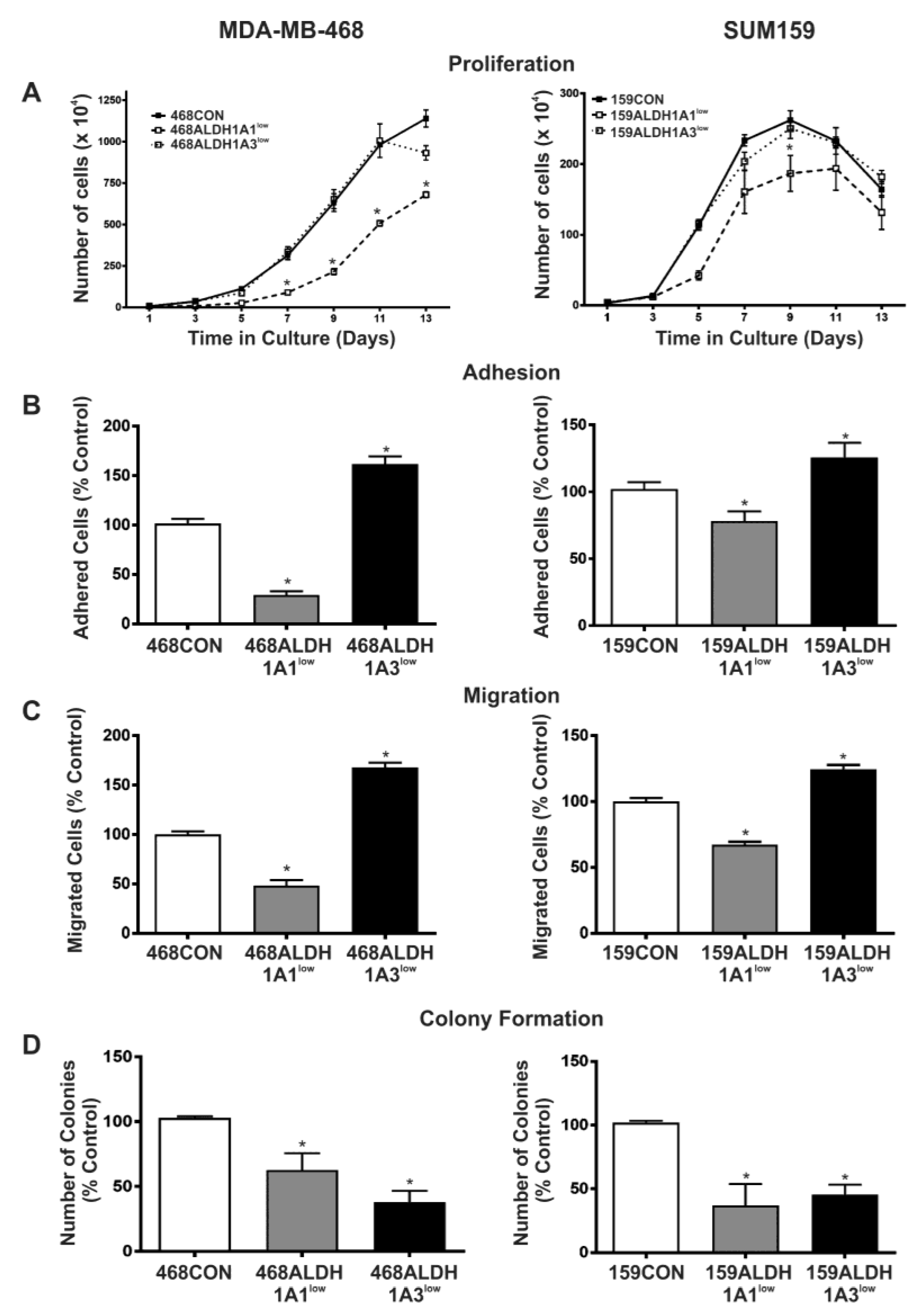
2.3. Decreased Expression of ALDH1A1 Reduces Breast Cancer Cell Proliferation, but Adhesion and Migration of Human Breast Cancer Cells Is Differentially Influenced by ALDH1A1 versus ALDH1A3 In Vitro
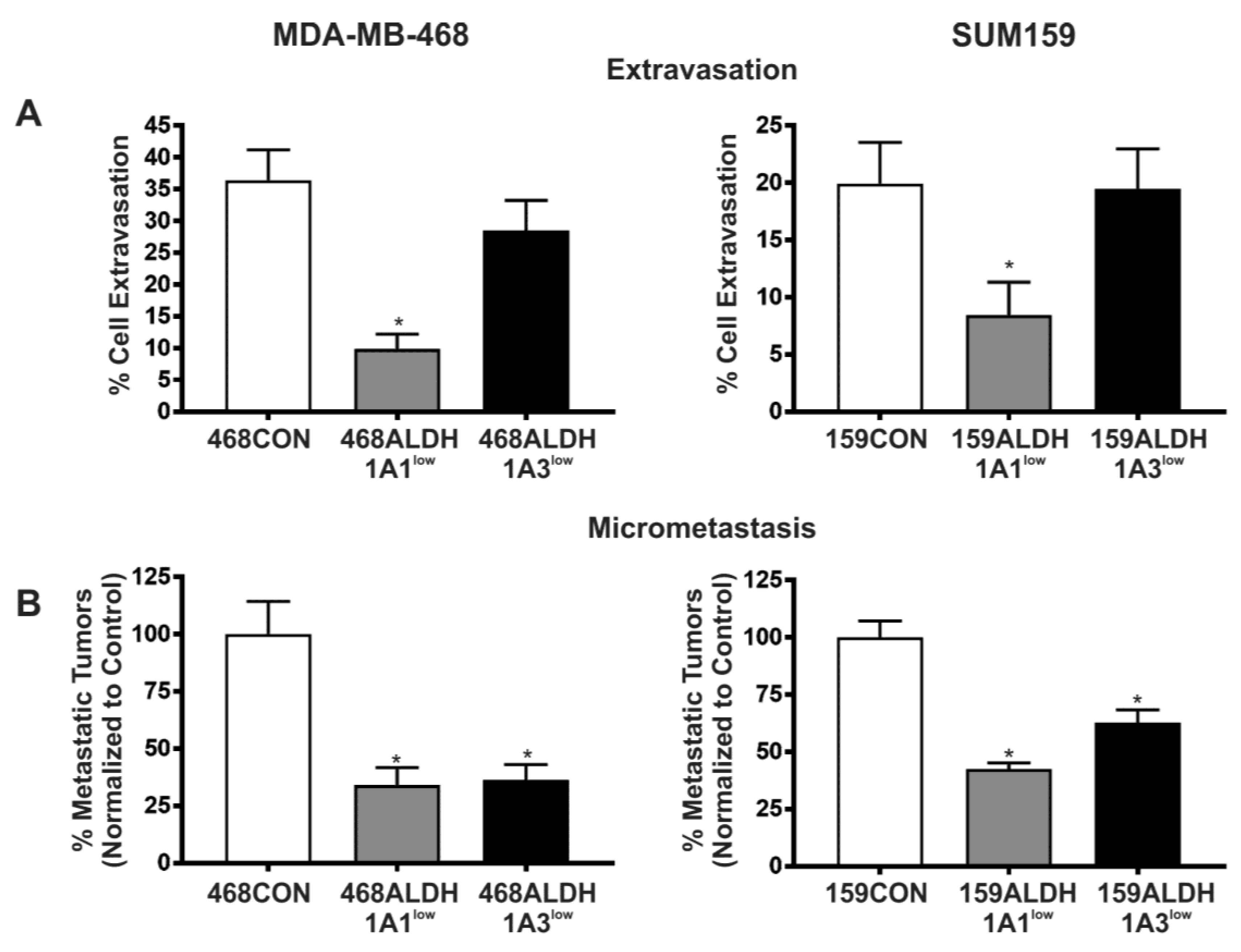
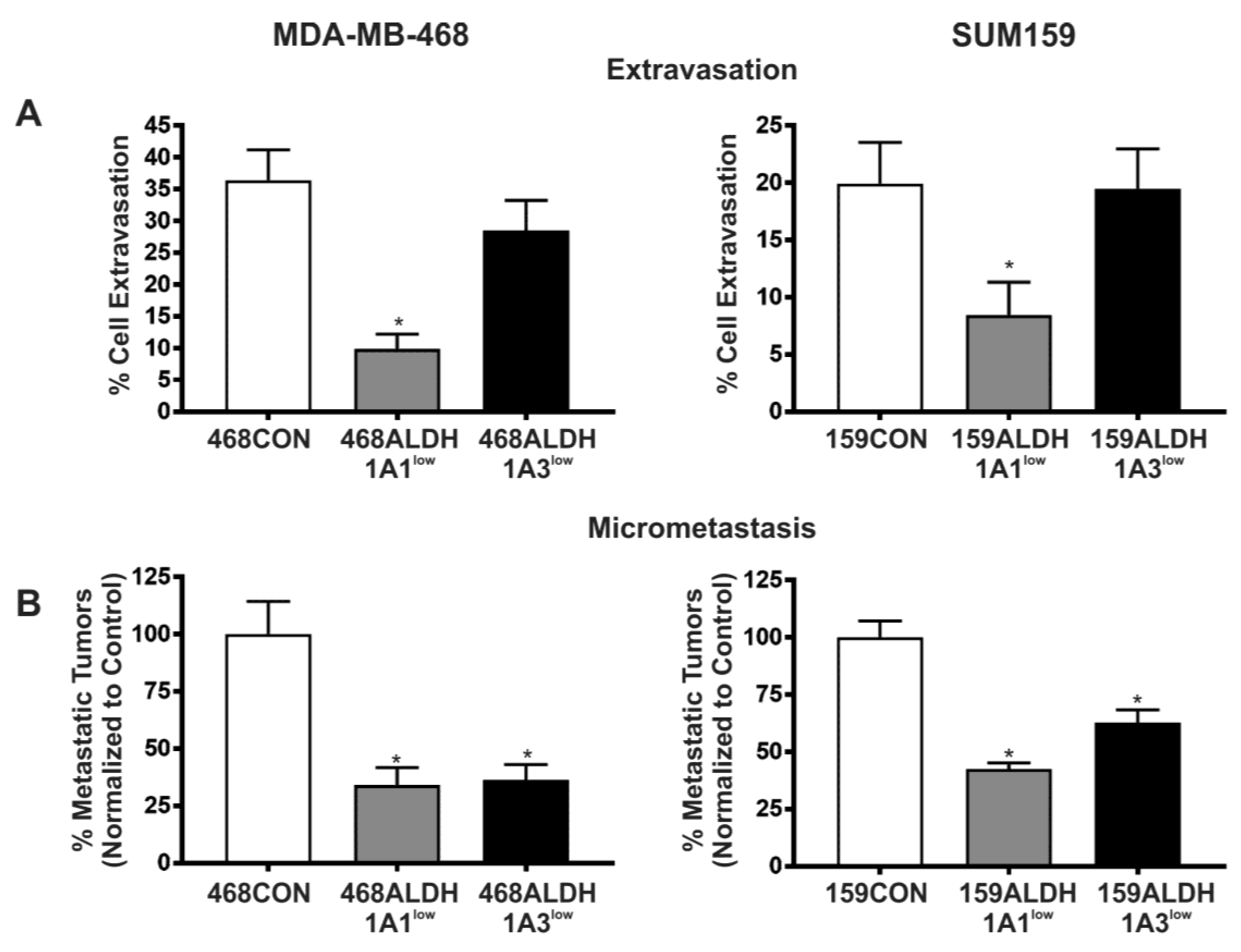
2.4. Decreased Expression of ALDH1A1 and ALDH1A3 Reduces In Vivo Metastatic Ability of Breast Cancer Cells in the Chick Chorioallantoic Membrane (CAM) Assay
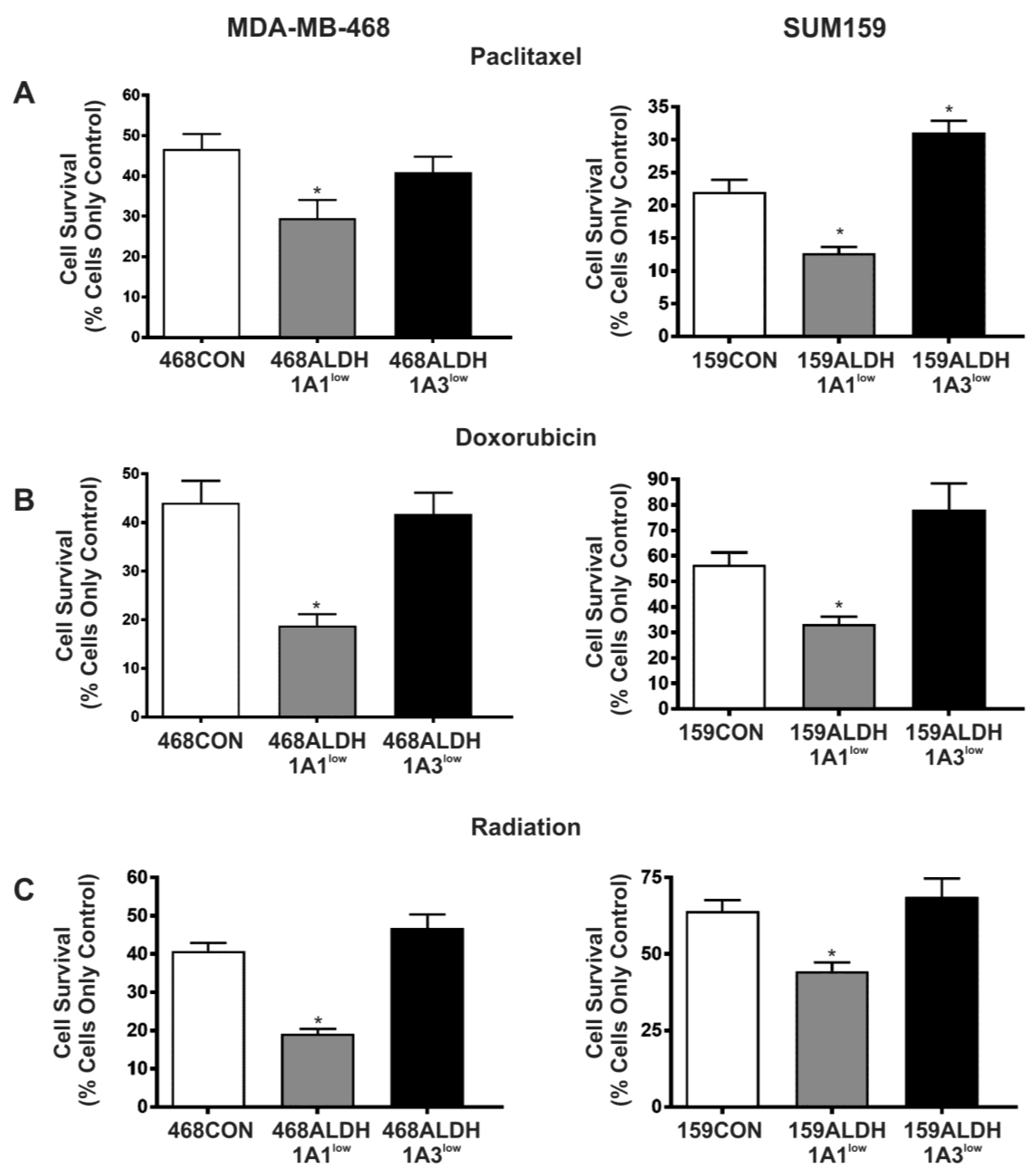
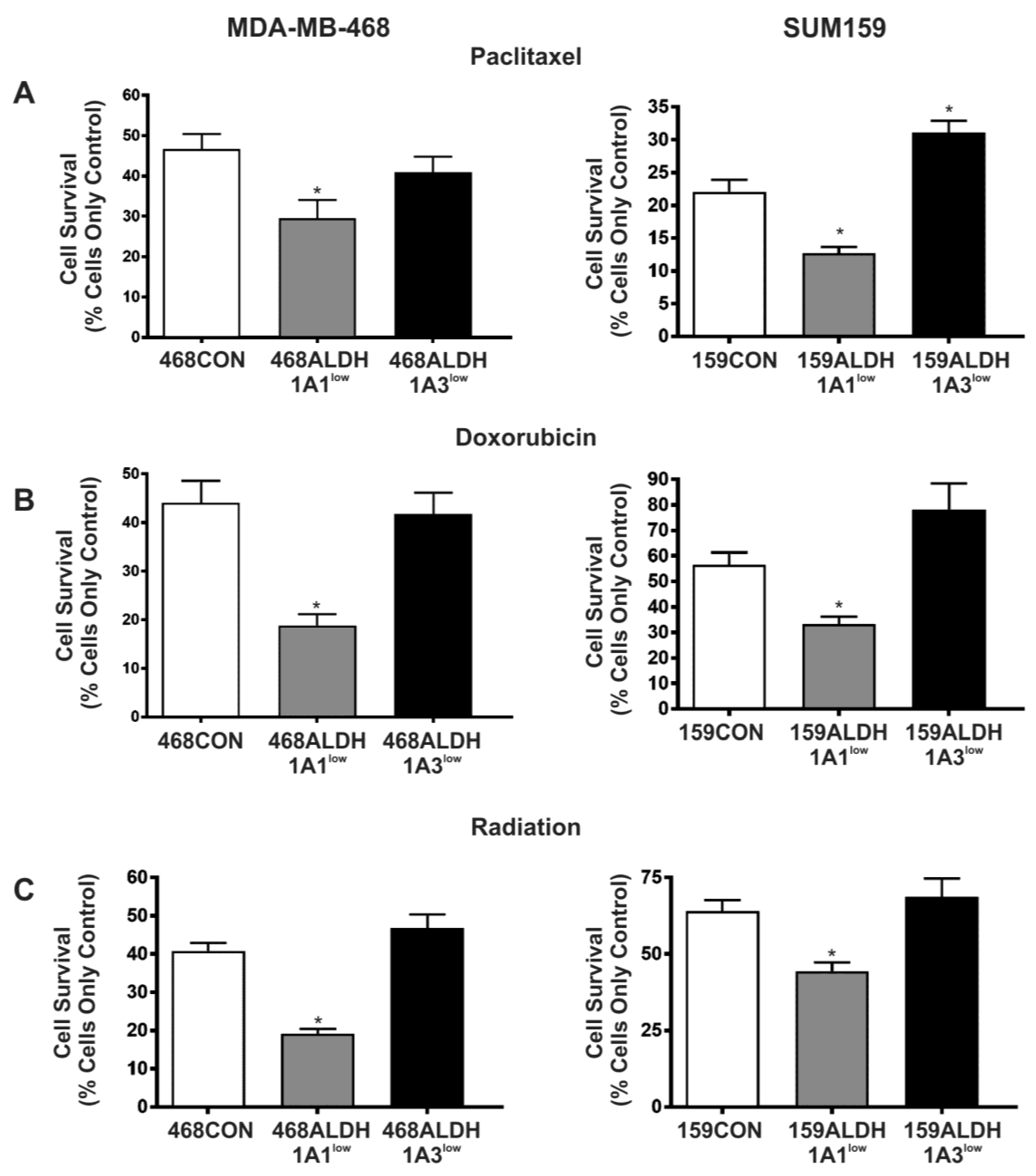
2.5. Decreased Expression of ALDH1A1 but Not ALDH1A3 Sensitizes Breast Cancer Cells to Chemotherapy and Radiation In Vitro
3. Discussion
4. Materials and Methods
4.1. Cell Culture, Reagents, and Therapy Conditions
4.2. Cell Proliferation Assays
4.3. Cell Adhesion Assays
4.4. Cell Migration Assays
4.5. Colony Forming Assays
4.6. siRNA Knockdown of ALDH1A1 and ALDH1A3
4.7. RNA Isolation and Quantitative RT-PCR
4.8. Immunoblotting
4.9. ALDEFLUOR® Assay
4.10. Chick Embryo Chorioallantoic Membrane (CAM) Assay
4.11. Chemotherapy and Radiation Treatment
4.12. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ALDH | Aldehyde dehydrogenase |
| ANOVA | Analysis of variance |
| APL | Acute promyelocytic leukemia |
| ATRA | All-trans retinoic acid |
| BSA | Bovine serum albumin |
| CAM | Choroiallantoic membrane assay |
| CD | Cluster of differentiation |
| CMFDA | 5-chloromethylfluorescein diacetate |
| CYP | Cytochrome P450 |
| DEAB | Diethylaminobenzaldehyde |
| ECL | Enhanced chemiluminescence |
| ER | Estrogen receptor |
| EtOH | Ethanol |
| FBS | Fetal bovine serum |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| GFP | Green fluorescent protein |
| Gy | Gray |
| HPF | High-powered field |
| LC50 | Lethal concentration (50%) |
| NAD(P) | Nicotinamide adenine dinucleotide phosphate |
| PBS | Phosphate buffered saline |
| PCR | Polymerase chain reaction |
| PR | Progesterone receptor |
| PVDF | Polyvinylidene fluoride |
| RA | Retinoic acid |
| RT | Reverse transcriptase |
| SDS-PAGE | Sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| SEM | Standard error of the mean |
| siRNA | Small interfering RNA |
| STR | Short tandem repeat |
| TBST | Tris-buffered saline + Tween-20 |
References
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Luzzi, K.J.; MacDonald, I.C.; Schmidt, E.E.; Kerkvliet, N.; Morris, V.L.; Chambers, A.F.; Groom, A.C. Multistep nature of metastatic inefficiency: Dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am. J. Pathol. 1998, 153, 865–873. [Google Scholar] [CrossRef]
- Weiss, L. Metastatic inefficiency. Adv. Cancer Res. 1990, 54, 159–211. [Google Scholar] [PubMed]
- Goss, P.; Allan, A.L.; Rodenhiser, D.I.; Foster, P.J.; Chambers, A.F. New clinical and experimental approaches for studying tumor dormancy: Does tumor dormancy offer a therapeutic target? APMIS 2008, 116, 552–568. [Google Scholar] [CrossRef] [PubMed]
- Croker, A.K.; Goodale, D.; Chu, J.; Postenka, C.; Hedley, B.D.; Hess, D.A.; Allan, A.L. High aldehyde dehydrogenase and expression of cancer stem cell markers selects for breast cancer cells with enhanced malignant and metastatic ability. J. Cell. Mol. Med. 2009, 13, 2236–2252. [Google Scholar] [CrossRef] [PubMed]
- Charafe-Jauffret, E.; Ginestier, C.; Iovino, F.; Tarpin, C.; Diebel, M.; Esterni, B.; Houvenaeghel, G.; Extra, J.M.; Bertucci, F.; Jacquemier, J.; et al. Aldehyde dehydrogenase 1-positive cancer stem cells mediate metastasis and poor clinical outcome in inflammatory breast cancer. Clin. Cancer Res. 2010, 16, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Charafe-Jauffret, E.; Ginestier, C.; Iovino, F.; Wicinski, J.; Cervera, N.; Finetti, P.; Hur, M.H.; Diebel, M.E.; Monville, F.; Dutcher, J.; et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. 2009, 69, 1302–1313. [Google Scholar] [CrossRef] [PubMed]
- Croker, A.K.; Allan, A.L. Inhibition of aldehyde dehydrogenase (ALDH) activity reduces chemotherapy and radiation resistance of stem-like ALDHhiCD44(+) human breast cancer cells. Breast Cancer Res. Treat. 2012, 133, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Torres, M.; Allan, A.L. Aldehyde dehydrogenase as a marker and functional mediator of metastasis in solid tumors. Clin. Exp. Metastasis 2016, 33, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Pors, K.; Moreb, J.S. Aldehyde dehydrogenases in cancer: An opportunity for biomarker and drug development? Drug Discov. Today 2014, 19, 1953–1963. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.A.; Watt, F.M. Dynamic regulation of retinoic acid-binding proteins in developing, adult and neoplastic skin reveals roles for β-catenin and notch signalling. Dev. Biol. 2008, 324, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Sladek, N.E. Human aldehyde dehydrogenases: Potential pathological, pharmacological, and toxicological impact. J. Biochem. Mol. Toxicol. 2003, 17, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, P.; Castaigne, S.; Dombret, H.; Archimbaud, E.; Duarte, M.; Morel, P.; Lamy, T.; Tilly, H.; Guerci, A.; Maloisel, F.; et al. All-transretinoic acid followed by intensive chemotherapy gives a high complete remission rate and may prolong remissions in newly diagnosed acute promyelocytic leukemia: A pilot study on 26 cases. Blood 1992, 80, 2176–2181. [Google Scholar] [PubMed]
- Sanz, M.A.; Lo-Coco, F. Modern approaches to treating acute promyelocytic leukemia. J. Clin. Oncol. 2011, 29, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Elizondo, G.; Corchero, J.; Sterneck, E.; Gonzalez, F.J. Feedback inhibition of the retinaldehyde dehydrogenase gene ALDH1 by retinoic acid through retinoic acid receptor alpha and ccaat/enhancer-binding protein β. J. Biol. Chem. 2000, 275, 39747–39753. [Google Scholar] [CrossRef] [PubMed]
- Ozeki, M.; Shively, J.E. Differential cell fates induced by all-trans retinoic acid-treated HL-60 human leukemia cells. J. Leukoc. Biol. 2008, 84, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Ginestier, C.; Wicinski, J.; Cervera, N.; Monville, F.; Finetti, P.; Bertucci, F.; Wicha, M.S.; Birnbaum, D.; Charafe-Jauffret, E. Retinoid signaling regulates breast cancer stem cell differentiation. Cell Cycle 2009, 8, 3297–3302. [Google Scholar] [CrossRef] [PubMed]
- Tanei, T.; Morimoto, K.; Shimazu, K.; Kim, S.J.; Tanji, Y.; Taguchi, T.; Tamaki, Y.; Noguchi, S. Association of breast cancer stem cells identified by aldehyde dehydrogenase 1 expression with resistance to sequential paclitaxel and epirubicin-based chemotherapy for breast cancers. Clin. Cancer Res. 2009, 15, 4234–4241. [Google Scholar] [CrossRef] [PubMed]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Lin, Y.; Shen, S.; Zhou, Y.; Mao, F.; Guan, J.; Sun, Q. Expression of ALDH1 in breast invasive ductal carcinoma: An independent predictor of early tumor relapse. Cancer Cell Int. 2013, 13, 60. [Google Scholar] [CrossRef] [PubMed]
- Kida, K.; Ishikawa, T.; Yamada, A.; Shimada, K.; Narui, K.; Sugae, S.; Shimizu, D.; Tanabe, M.; Sasaki, T.; Ichikawa, Y.; et al. Effect of aldh1 on prognosis and chemoresistance by breast cancer subtype. Breast Cancer Res. Treat. 2016, 156, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, Y.; Shien, T.; Ogiya, A.; Ishida, N.; Yamazaki, K.; Horii, R.; Horimoto, Y.; Masuda, N.; Yasojima, H.; Inao, T.; et al. Differences in expression of the cancer stem cell marker aldehyde dehydrogenase 1 among estrogen receptor-positive/human epidermal growth factor receptor type 2-negative breast cancer cases with early, late, and no recurrence. Breast Cancer Res. BCR 2016, 18, 73. [Google Scholar] [CrossRef] [PubMed]
- Khoury, T.; Ademuyiwa, F.O.; Chandrasekhar, R.; Jabbour, M.; Deleo, A.; Ferrone, S.; Wang, Y.; Wang, X. Aldehyde dehydrogenase 1A1 expression in breast cancer is associated with stage, triple negativity, and outcome to neoadjuvant chemotherapy. Mod. Pathol. 2012, 25, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Jiang, Y.; Yan, T.; Di, G.; Shen, Z.; Shao, Z.; Lu, J. The prognostic role of cancer stem cells in breast cancer: A meta-analysis of published literatures. Breast Cancer Res. Treat. 2010, 122, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Visus, C.; Wang, Y.; Lozano-Leon, A.; Ferris, R.L.; Silver, S.; Szczepanski, M.J.; Brand, R.E.; Ferrone, C.R.; Whiteside, T.L.; Ferrone, S.; et al. Targeting ALDH(bright) human carcinoma-initiating cells with ALDH1A1-specific CD8(+) T cells. Clin. Cancer Res. 2011, 17, 6174–6184. [Google Scholar] [CrossRef] [PubMed]
- Marcato, P.; Dean, C.A.; Pan, D.; Araslanova, R.; Gillis, M.; Joshi, M.; Helyer, L.; Pan, L.; Leidal, A.; Gujar, S.; et al. Aldehyde dehydrogenase activity of breast cancer stem cells is primarily due to isoform ALDH1A3 and its expression is predictive of metastasis. Stem Cells 2011, 29, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Koppaka, V.; Thompson, D.C.; Chen, Y.; Ellermann, M.; Nicolaou, K.C.; Juvonen, R.O.; Petersen, D.; Deitrich, R.A.; Hurley, T.D.; Vasiliou, V. Aldehyde dehydrogenase inhibitors: A comprehensive review of the pharmacology, mechanism of action, substrate specificity, and clinical application. Pharmacol. Rev. 2012, 64, 520–539. [Google Scholar] [CrossRef] [PubMed]
- Chute, J.P.; Muramoto, G.G.; Whitesides, J.; Colvin, M.; Safi, R.; Chao, N.J.; McDonnell, D.P. Inhibition of aldehyde dehydrogenase and retinoid signaling induces the expansion of human hematopoietic stem cells. Proc. Natl. Acad. Sci. USA 2006, 103, 11707–11712. [Google Scholar] [CrossRef] [PubMed]
- Vasiliou, V.; Nebert, D.W. Analysis and update of the human aldehyde dehydrogenase (ALDH) gene family. Hum. Genom. 2005, 2, 138–143. [Google Scholar]
- Moreb, J.S.; Baker, H.V.; Chang, L.J.; Amaya, M.; Lopez, M.C.; Ostmark, B.; Chou, W. Aldh isozymes downregulation affects cell growth, cell motility and gene expression in lung cancer cells. Mol. Cancer 2008, 7, 87. [Google Scholar] [CrossRef] [PubMed]
- Moreb, J.S.; Zucali, J.R.; Ostmark, B.; Benson, N.A. Heterogeneity of aldehyde dehydrogenase expression in lung cancer cell lines is revealed by aldefluor flow cytometry-based assay. Cytom. Part B Clin. Cytom. 2007, 72, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Hoogen, C.; Horst, G.; Cheung, H.; Buijs, J.T.; Pelger, R.C.M.; Pluijm, G. The aldehyde dehydrogenase enzyme 7A1 is functionally involved in prostate cancer bone metastasis. Clin. Exp. Metastasis 2011, 28, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Moreb, J.S.; Ucar, D.; Han, S.; Amory, J.K.; Goldstein, A.S.; Ostmark, B.; Chang, L.J. The enzymatic activity of human aldehyde dehydrogenases 1A2 and 2 (ALDH1A2 and ALDH2) is detected by aldefluor, inhibited by diethylaminobenzaldehyde and has significant effects on cell proliferation and drug resistance. Chem. Biol. Interact. 2012, 195, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Canuto, R.A.; Muzio, G.; Salvo, R.A.; Maggiora, M.; Trombetta, A.; Chantepie, J.; Fournet, G.; Reichert, U.; Quash, G. The effect of a novel irreversible inhibitor of aldehyde dehydrogenases 1 and 3 on tumour cell growth and death. Chem. Biol. Interact. 2001, 130–132, 209–218. [Google Scholar] [CrossRef]
- Muzio, G.; Maggiora, M.; Paiuzzi, E.; Oraldi, M.; Canuto, R.A. Aldehyde dehydrogenases and cell proliferation. Free Radic. Biol. Med. 2012, 52, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Muzio, G.; Trombetta, A.; Martinasso, G.; Canuto, R.A.; Maggiora, M. Antisense oligonucleotides against aldehyde dehydrogenase 3 inhibit hepatoma cell proliferation by affecting map kinases. Chem. Biol. Interact. 2003, 143–144, 37–43. [Google Scholar] [CrossRef]
- Moreb, J.S.; Mohuczy, D.; Ostmark, B.; Zucali, J.R. Rnai-mediated knockdown of aldehyde dehydrogenase class-1A1 and class-3A1 is specific and reveals that each contributes equally to the resistance against 4-hydroperoxycyclophosphamide. Cancer Chemother. Pharmacol. 2007, 59, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Marcato, P.; Dean, C.A.; Liu, R.Z.; Coyle, K.M.; Bydoun, M.; Wallace, M.; Clements, D.; Turner, C.; Mathenge, E.G.; Gujar, S.A.; et al. Aldehyde dehydrogenase 1A3 influences breast cancer progression via differential retinoic acid signaling. Mol. Oncol. 2015, 9, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Su, Y.; Mei, Y.; Leng, Q.; Leng, B.; Liu, Z.; Stass, S.A.; Jiang, F. ALDH1A1 is a marker for malignant prostate stem cells and predictor of prostate cancer patients’ outcome. Lab. Investig. 2010, 90, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wan, L.; Geng, J.; Wu, C.L.; Bai, X. Aldehyde dehydrogenase 1A1 possesses stem-like properties and predicts lung cancer patient outcome. J. Thorac. Oncol. 2012, 7, 1235–1245. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, K.; Kim, S.J.; Tanei, T.; Shimazu, K.; Tanji, Y.; Taguchi, T.; Tamaki, Y.; Terada, N.; Noguchi, S. Stem cell marker aldehyde dehydrogenase 1-positive breast cancers are characterized by negative estrogen receptor, positive human epidermal growth factor receptor type 2, and high ki67 expression. Cancer Sci. 2009, 100, 1062–1068. [Google Scholar] [CrossRef] [PubMed]
- Neumeister, V.; Agarwal, S.; Bordeaux, J.; Camp, R.L.; Rimm, D.L. In situ identification of putative cancer stem cells by multiplexing ALDH1, CD44, and cytokeratin identifies breast cancer patients with poor prognosis. Am. J. Pathol. 2010, 176, 2131–2138. [Google Scholar] [CrossRef] [PubMed]
- Cristofanilli, M. Advancements in the treatment of metastatic breast cancer (MBC): The role of ixabepilone. J. Oncol. 2012, 2012, 703858. [Google Scholar] [CrossRef] [PubMed]
- Price, J.E.; Polyzos, A.; Zhang, R.D.; Daniels, L.M. Tumorigenicity and metastasis of human breast carcinoma cell lines in nude mice. Cancer Res. 1990, 50, 717–721. [Google Scholar] [PubMed]
- Vantyghem, S.A.; Allan, A.L.; Postenka, C.O.; Al-Katib, W.; Keeney, M.; Tuck, A.B.; Chambers, A.F. A new model for lymphatic metastasis: Development of a variant of the MDA-MB-468 human breast cancer cell line that aggressively metastasizes to lymph nodes. Clin. Exp. Metastasis 2005, 22, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, L.; Van Weelden, K.; Ammerman, C.; Ethier, S.P.; Welsh, J. SUM-159PT cells: A novel estrogen independent human breast cancer model system. Breast Cancer Res. Treat. 1999, 58, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Allan, A.L.; George, R.; Vantyghem, S.A.; Lee, M.W.; Hodgson, N.C.; Engel, C.J.; Holliday, R.L.; Girvan, D.P.; Scott, L.A.; Postenka, C.O.; et al. Role of the integrin-binding protein osteopontin in lymphatic metastasis of breast cancer. Am. J. Pathol. 2006, 169, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Croker, A.K.; Allan, A.L. London Regional Cancer Program. London, ON, Canada, Unpublished work. 2011. [Google Scholar]
- Schulze, E.B.; Hedley, B.D.; Goodale, D.; Postenka, C.O.; Al-Katib, W.; Tuck, A.B.; Chambers, A.F.; Allan, A.L. The thrombin inhibitor argatroban reduces breast cancer malignancy and metastasis via osteopontin-dependent and osteopontin-independent mechanisms. Breast Cancer Res. Treat. 2008, 112, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Furger, K.A.; Allan, A.L.; Wilson, S.M.; Hota, C.; Vantyghem, S.A.; Postenka, C.O.; Al-Katib, W.; Chambers, A.F.; Tuck, A.B. β(3) integrin expression increases breast carcinoma cell responsiveness to the malignancy-enhancing effects of osteopontin. Mol. Cancer Res. 2003, 1, 810–819. [Google Scholar] [PubMed]
- Hess, D.A.; Craft, T.P.; Wirthlin, L.; Hohm, S.; Zhou, P.; Eades, W.C.; Creer, M.H.; Sands, M.S.; Nolta, J.A. Widespread non-hematopoietic tissue distribution by transplanted human progenitor cells with high aldehyde dehydrogenase activity. Stem Cells 2008, 26, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Hess, D.A.; Meyerrose, T.E.; Wirthlin, L.; Craft, T.P.; Herrbrich, P.E.; Creer, M.H.; Nolta, J.A. Functional characterization of highly purified human hematopoietic repopulating cells isolated according to aldehyde dehydrogenase activity. Blood 2004, 104, 1648–1655. [Google Scholar] [CrossRef] [PubMed]
- Hess, D.A.; Wirthlin, L.; Craft, T.P.; Herrbrich, P.E.; Hohm, S.A.; Lahey, R.; Eades, W.C.; Creer, M.H.; Nolta, J.A. Selection based on CD133 and high aldehyde dehydrogenase activity isolates long-term reconstituting human hematopoietic stem cells. Blood 2006, 107, 2162–2169. [Google Scholar] [CrossRef] [PubMed]
- Leong, H.S.; Chambers, A.F.; Lewis, J.D. Assessing cancer cell migration and metastatic growth in vivo in the chick embryo using fluorescence intravital imaging. Methods Mol. Biol. 2012, 872, 1–14. [Google Scholar] [PubMed]
- Seandel, M.; Noack-Kunnmann, K.; Zhu, D.; Aimes, R.T.; Quigley, J.P. Growth factor-induced angiogenesis in vivo requires specific cleavage of fibrillar type I collagen. Blood 2001, 97, 2323–2332. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Functional Behavior/Activity | ALDH1A1 Knockdown | ALDH1A3 Knockdown |
|---|---|---|
| ALDH Activity (Aldeflour) | No effect | |
| Proliferation | No effect | |
| Adhesion | ||
| Migration | ||
| Colony Formation | ||
| Extravasation | No effect | |
| Metastasis | ||
| Therapy Resistance | No effect |
| Gene | Primer Sequence | qPCR Cycling Conditions | Number of Cycles | Product Size (bp) |
|---|---|---|---|---|
| ALDH1A1 | Fwd: 5′-CGT TGG TTA TGC TCA TTT GGA A-3′ Rev: 5′-TGA TCA ACT TGC CAA CCT CTG T-3′ | 60 s 55 °C 60 s 72 °C 60 s 95 °C | 45 | 22 bp |
| ALDH1A3 | Fwd: 5′-ATG TGG GAA AAC CCC CTG TG-3′ Rev: 5′-GAA TGG TCC CAC CTT CAC CT-3′ | 60 s 57 °C 60 s 72 °C 60 s 95 °C | 45 | 20 bp |
| GAPDH | Fwd: 5′-CAT GTT CGT CAT GGG TGT GAA CCA-3′ Rev: 5′-ATG GCA TGG ACT GTG GTC ATG AGT -3′ | 45 s 60 °C 45 s 72 °C 60 s 95 °C | 40 | 24 bp |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Croker, A.K.; Rodriguez-Torres, M.; Xia, Y.; Pardhan, S.; Leong, H.S.; Lewis, J.D.; Allan, A.L. Differential Functional Roles of ALDH1A1 and ALDH1A3 in Mediating Metastatic Behavior and Therapy Resistance of Human Breast Cancer Cells. Int. J. Mol. Sci. 2017, 18, 2039. https://doi.org/10.3390/ijms18102039
Croker AK, Rodriguez-Torres M, Xia Y, Pardhan S, Leong HS, Lewis JD, Allan AL. Differential Functional Roles of ALDH1A1 and ALDH1A3 in Mediating Metastatic Behavior and Therapy Resistance of Human Breast Cancer Cells. International Journal of Molecular Sciences. 2017; 18(10):2039. https://doi.org/10.3390/ijms18102039
Chicago/Turabian StyleCroker, Alysha K., Mauricio Rodriguez-Torres, Ying Xia, Siddika Pardhan, Hon Sing Leong, John D. Lewis, and Alison L. Allan. 2017. "Differential Functional Roles of ALDH1A1 and ALDH1A3 in Mediating Metastatic Behavior and Therapy Resistance of Human Breast Cancer Cells" International Journal of Molecular Sciences 18, no. 10: 2039. https://doi.org/10.3390/ijms18102039