Challenges for Super-Resolution Localization Microscopy and Biomolecular Fluorescent Nano-Probing in Cancer Research

 ,
, 

Abstract
:1. Introduction
1.1. Challenges of Cancer Research
1.2. Localization Microscopy
1.3. Nano-Probing for Localization Microscopy
2. Results and Discussion
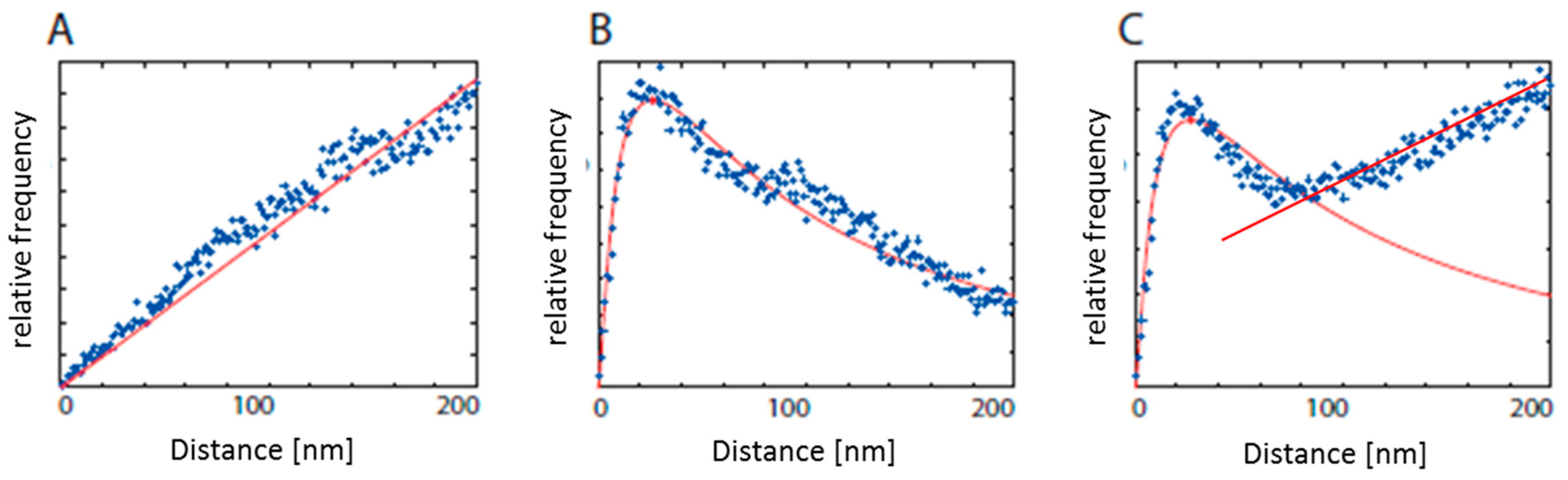
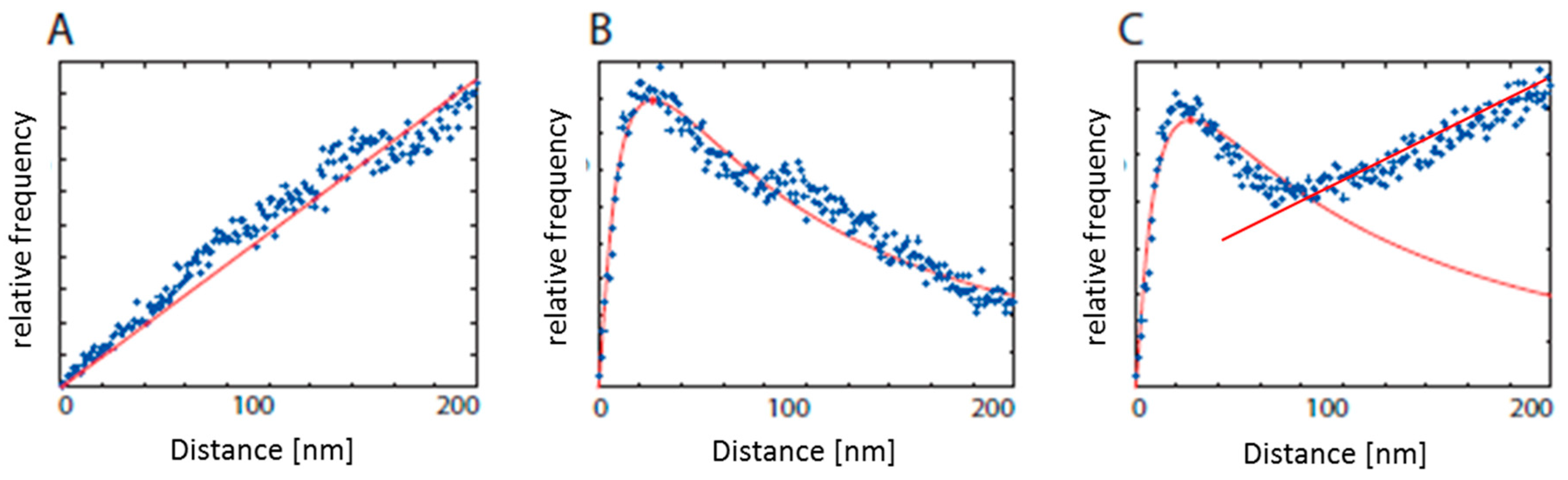
2.1. Data Interpretation: From Points to Structural Organization
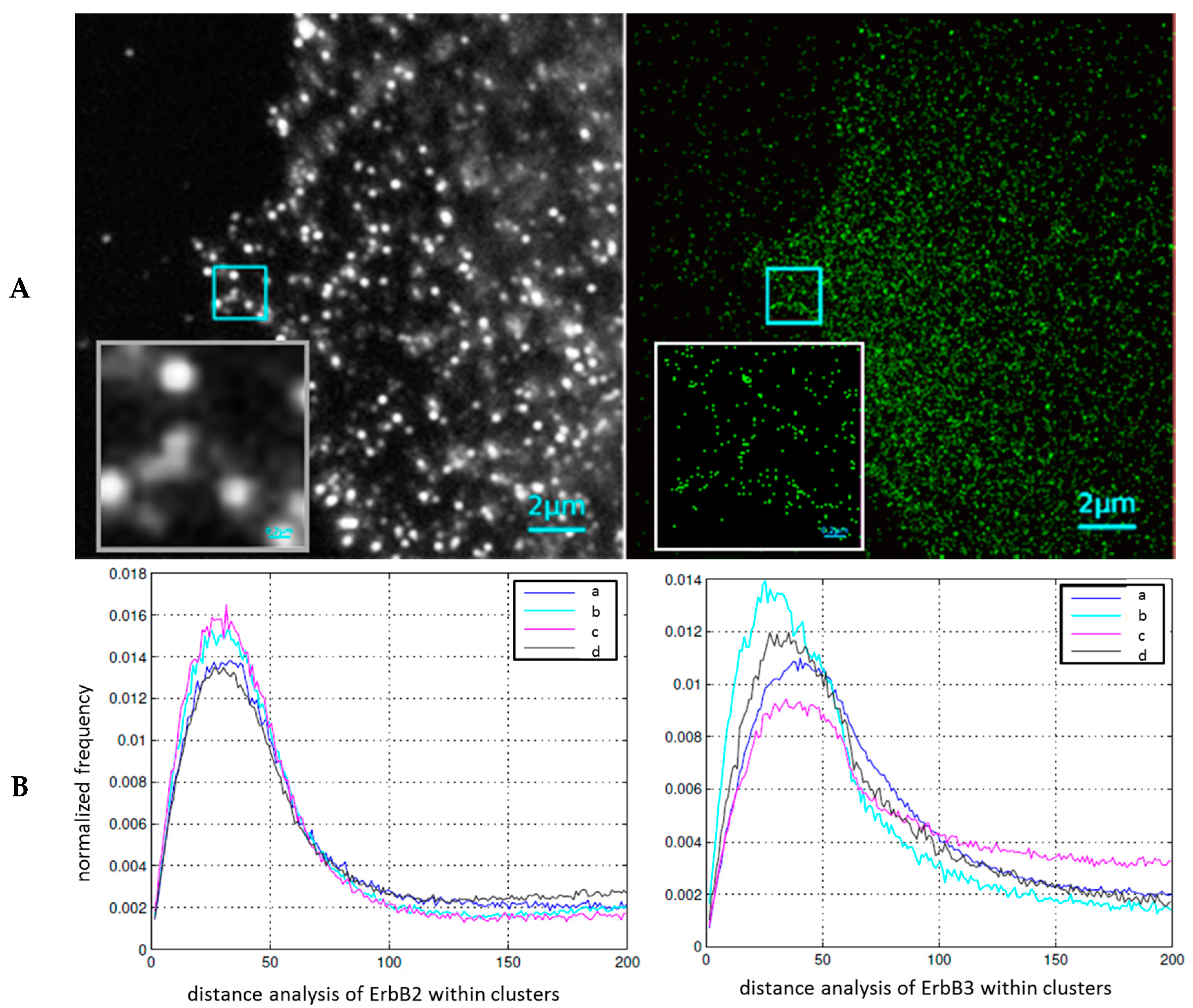
2.2. Cluster Formation of Epidermal Growth Factor Receptors (EGFR) and Their Response to Cancer Treatment
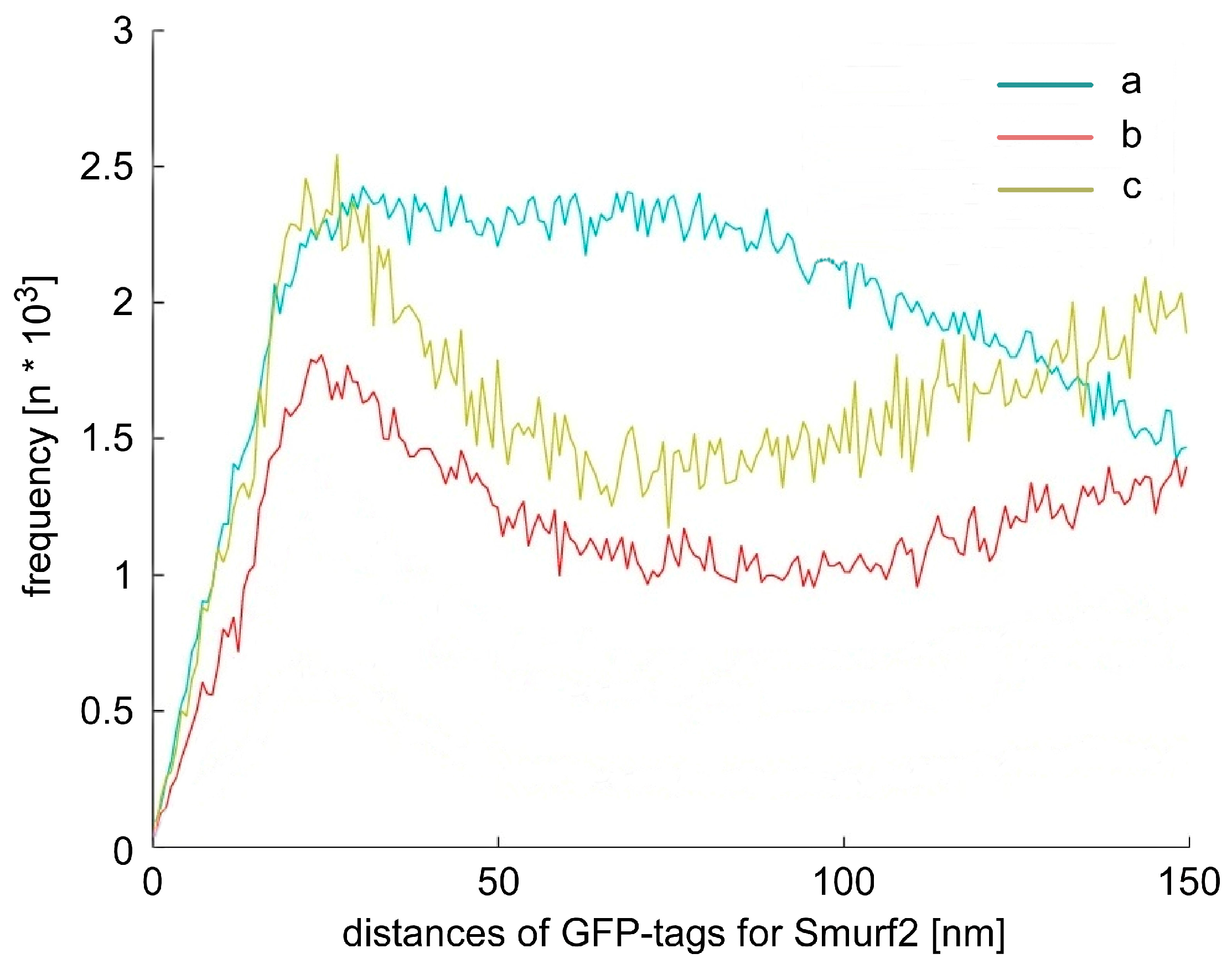
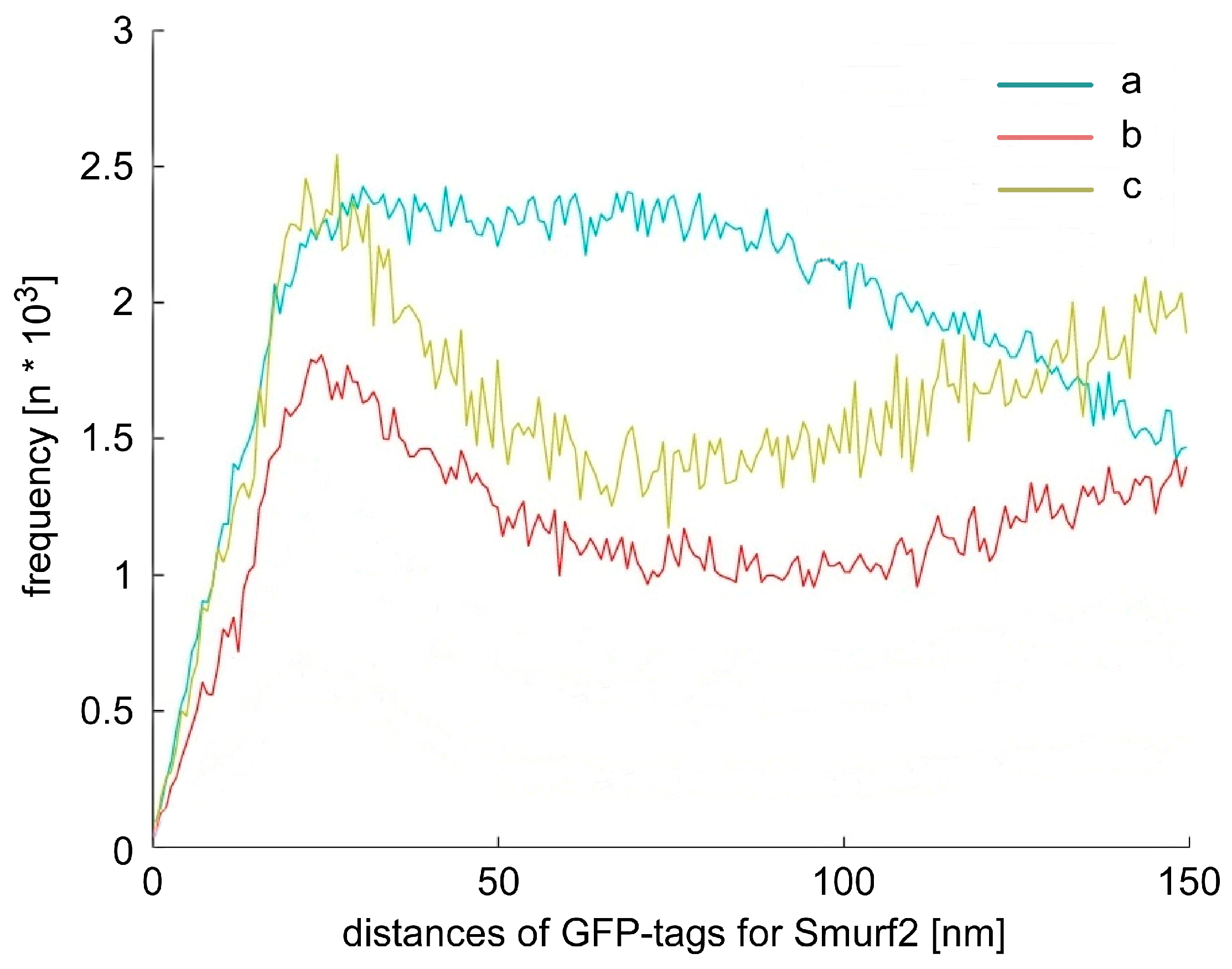
2.3. Chromatin Response after Smad Specific Uboquitin Protein Ligase 2 (Smurf2) Over-Expression and Cell Treatment with the Chemotherapeutic Drug Etoposide
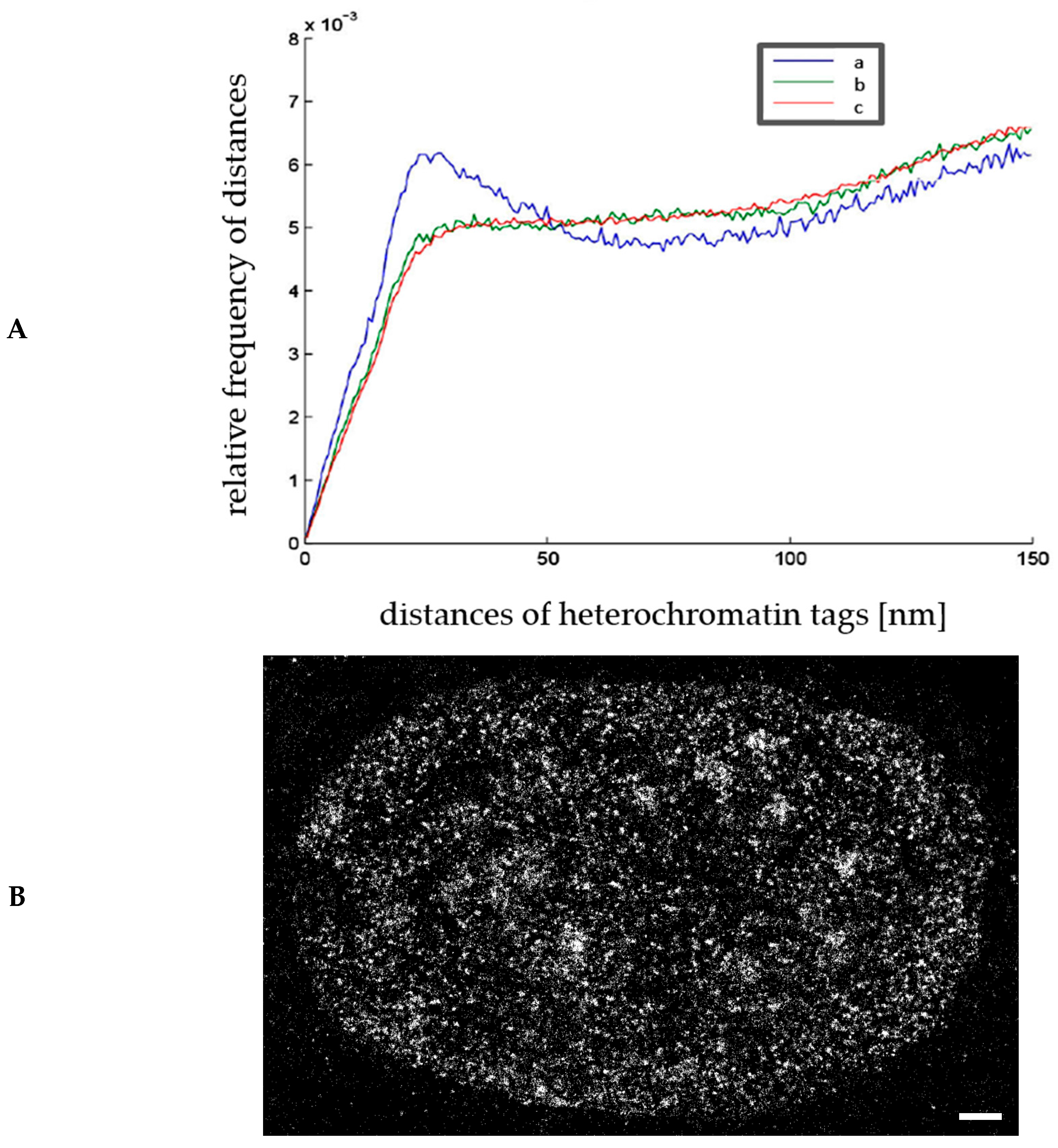
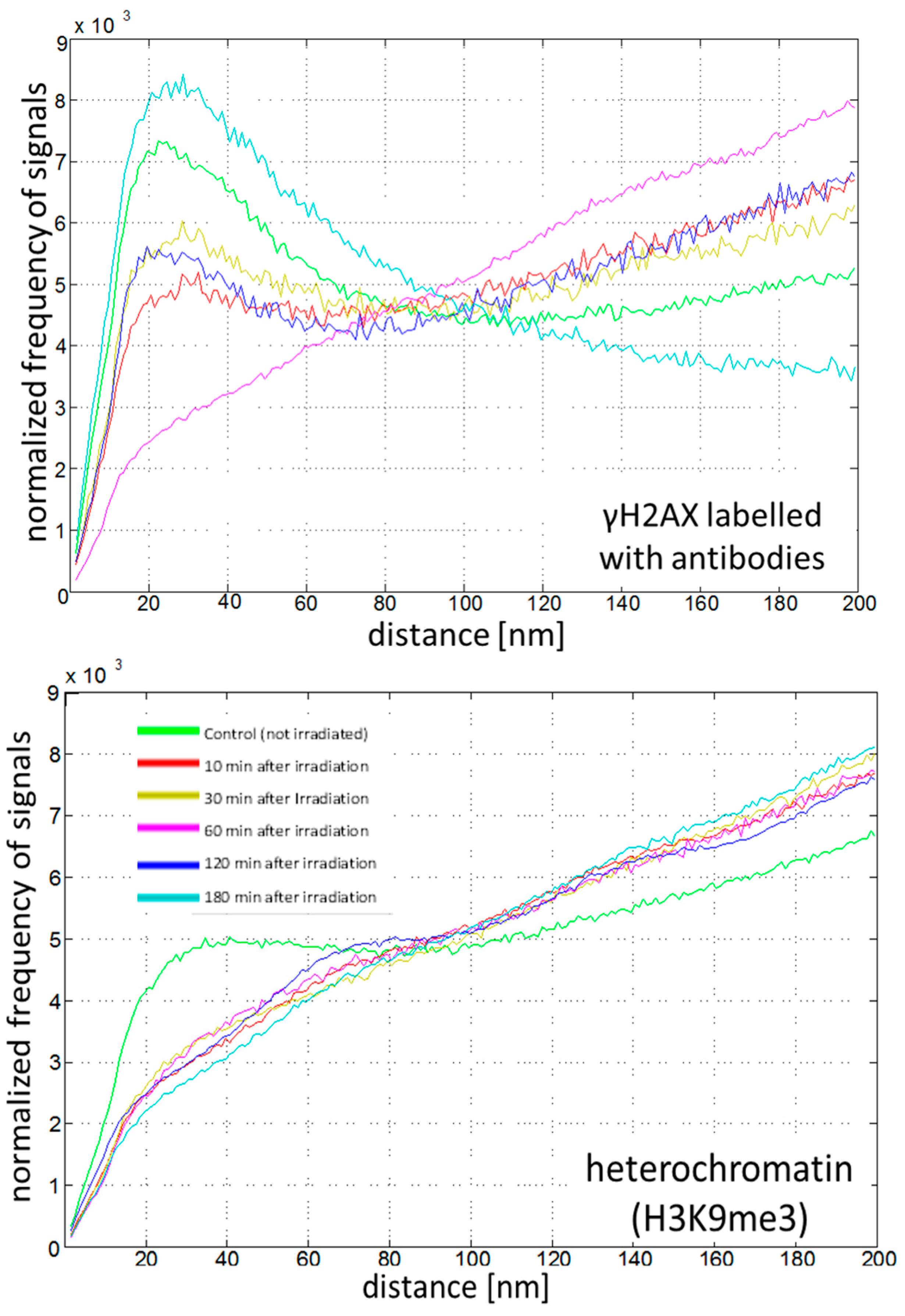
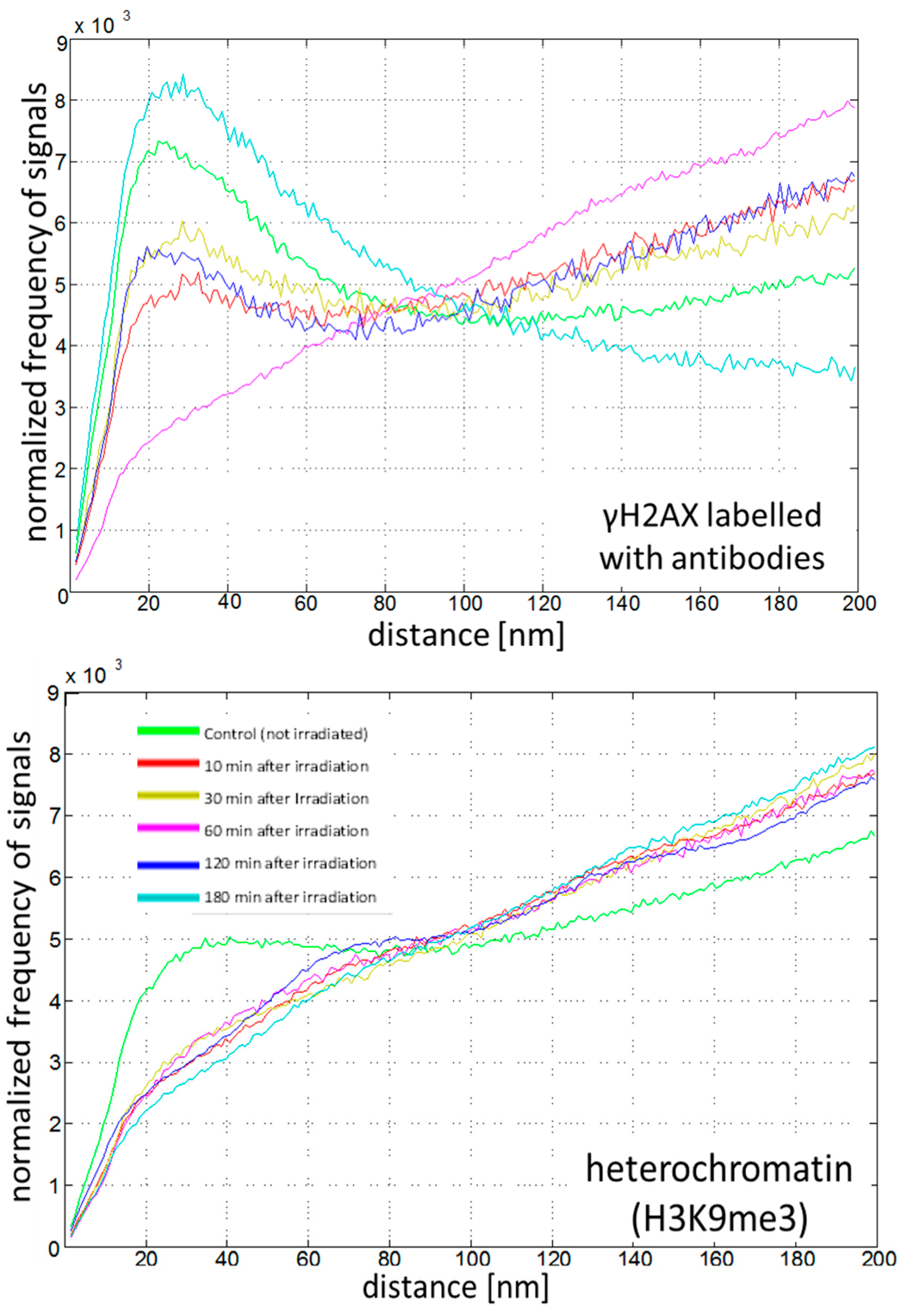
2.4. Chromatin Response to Radiation Treatment Detected by γH2AX Foci Formation and Heterochromatin (HC) Reorganisation
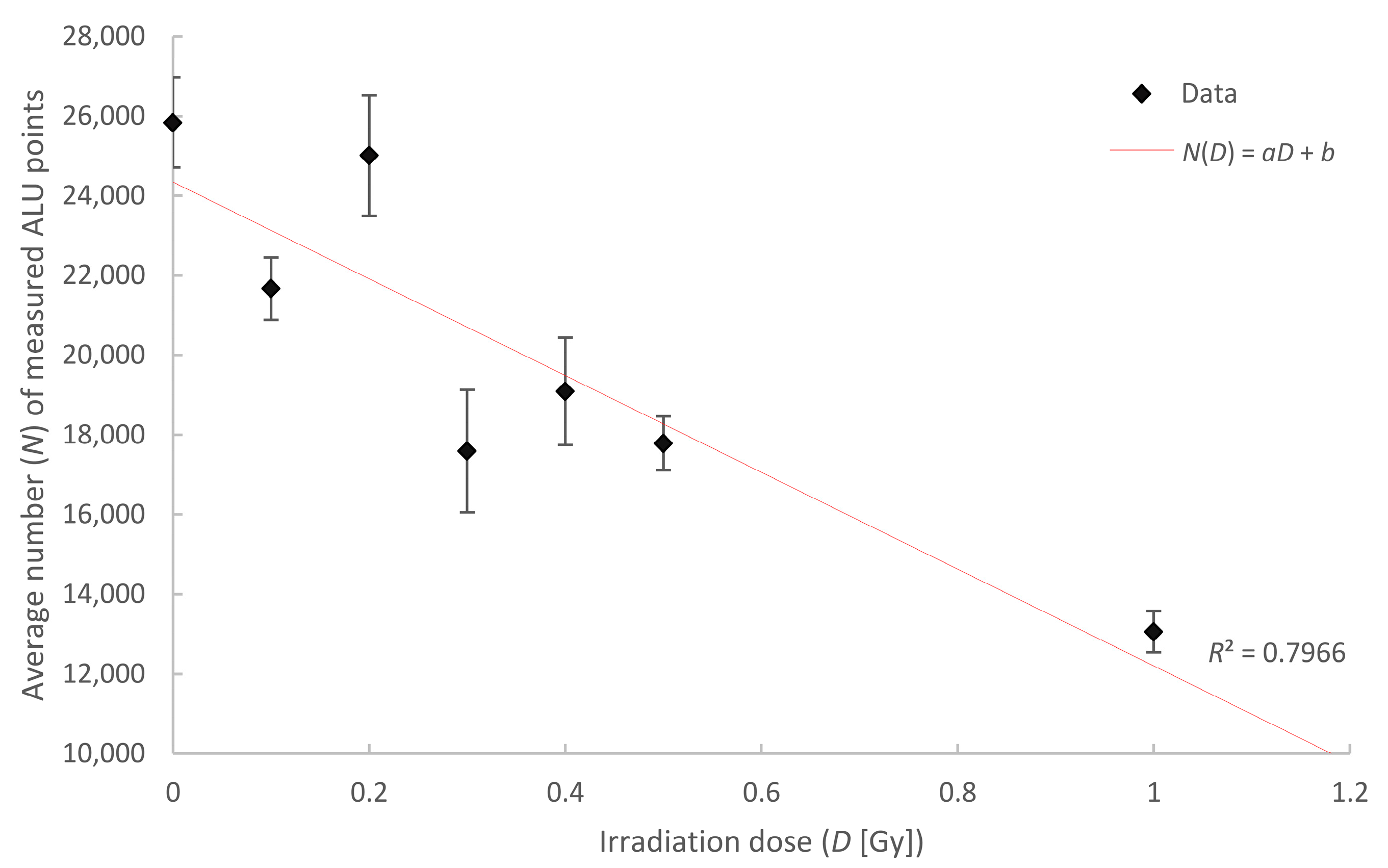
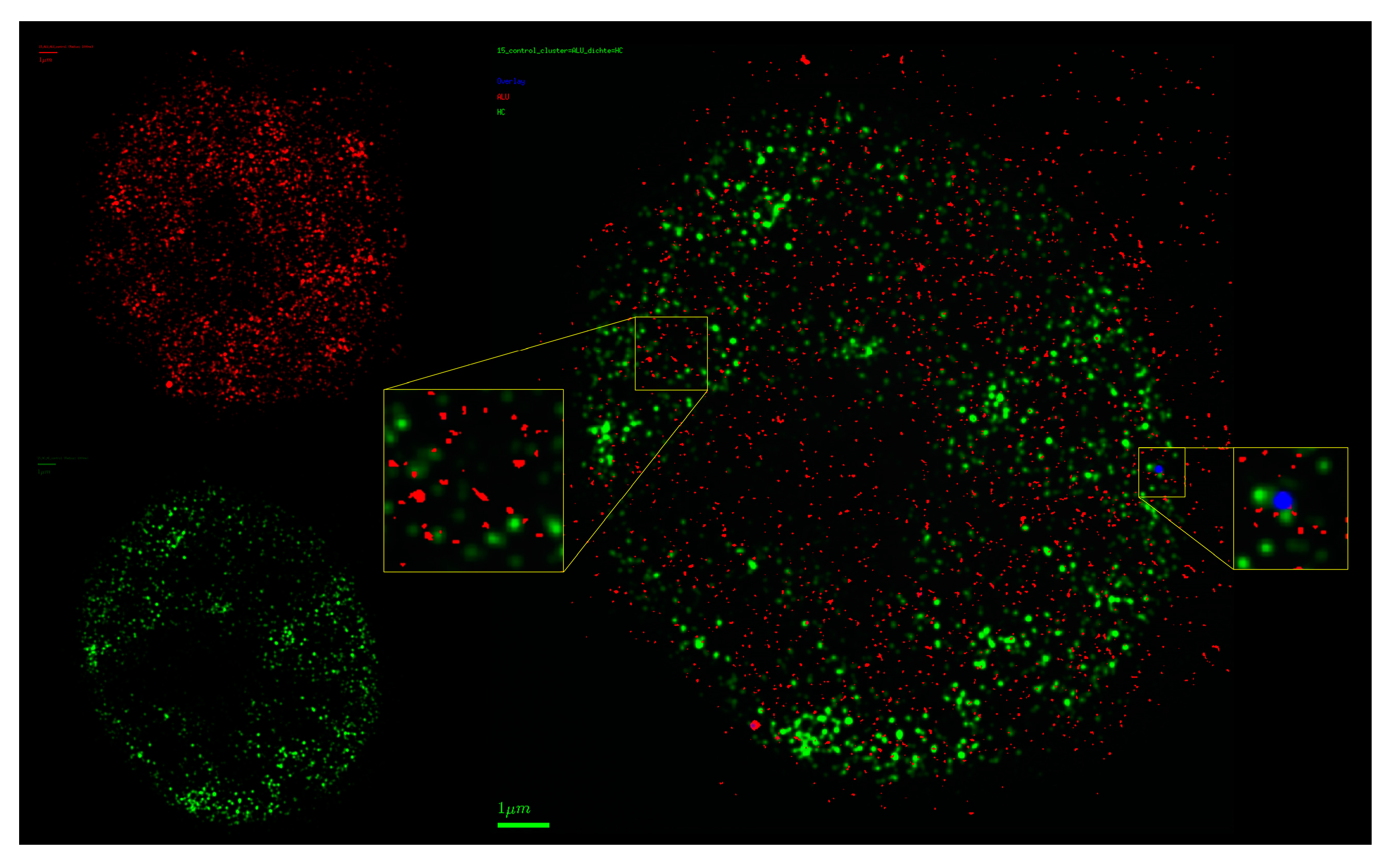
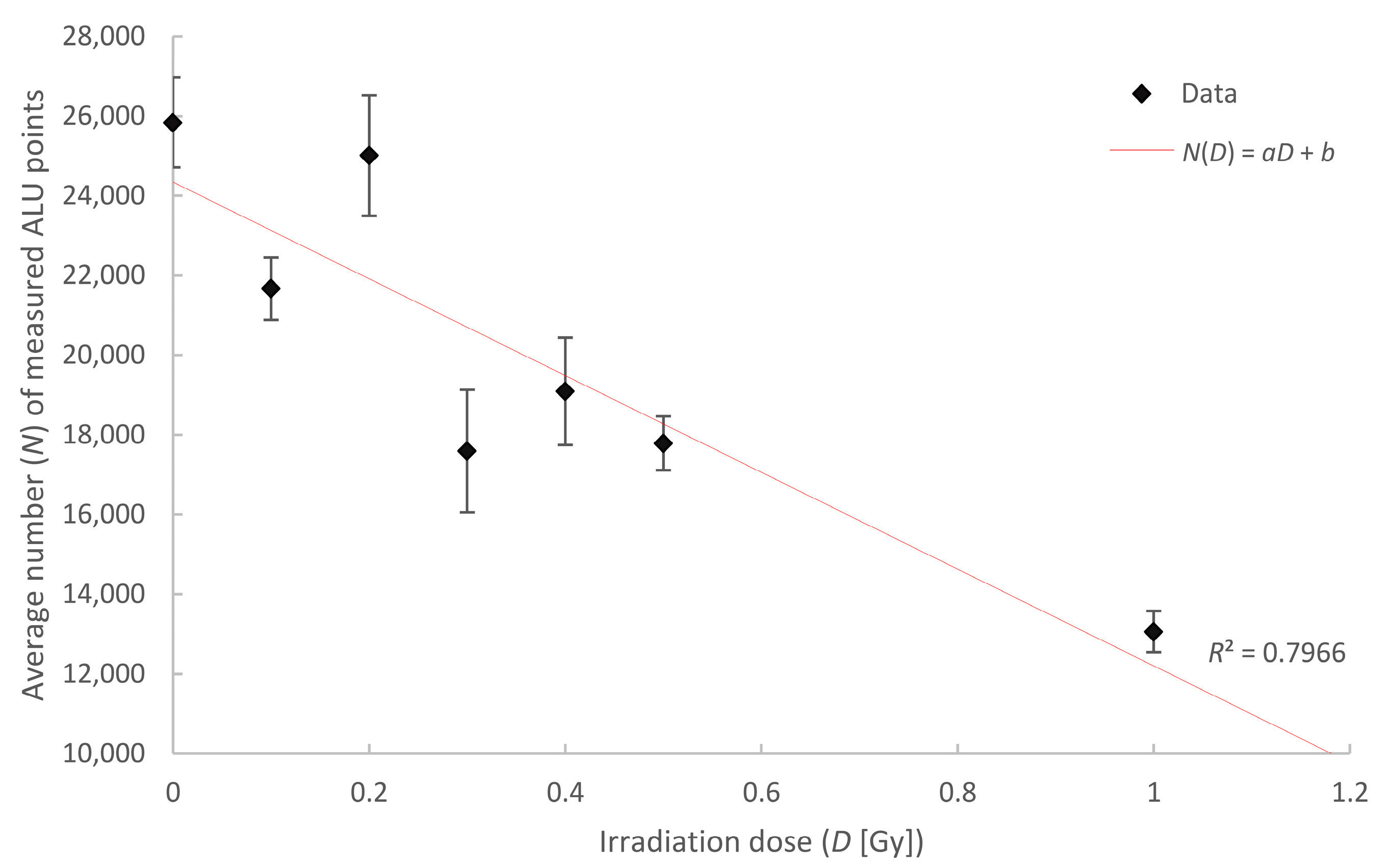
2.5. Chromatin Response to Radiation Treatment Detected by COMBO-FISH of the ALU Consensus Regions
3. Materials and Methods
3.1. Cell Culture and Treatment
3.2. Immunostaining and COMBOinatorial Oligonucleotide Fluorescence In Situ Hybridization (COMBO-FISH)
3.3. Localization Microscopy
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Broustas, C.G.; Lieberman, H.B. DNA damage response genes and the development of cancer metastasis. Radiat. Res. 2014, 181, 111–130. [Google Scholar] [CrossRef] [PubMed]
- Dietlein, F.; Thelen, L.; Reinhardt, H.C. Cancer-specific defects in DNA repair pathways as targets for personalized therapeutic approaches. Trends Genet. 2014, 30, 326–339. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Le Cao, K.A.; Ghenevix-Trench, G.; Simpson, P.T.; Ragan, M.A.; Khanna, K.K. A fine scale dissection of the DNA double-strand break repair machinery and its implications for breast cancer therapy. Nucleic Acids Res. 2014, 42, 6106–6127. [Google Scholar] [CrossRef] [PubMed]
- Pillai, R.N.; Owonikoko, T.K. Small cell lung cancer: Therapies and targets. Semin. Oncol. 2014, 41, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.C.; Kostyanovskaya, E.; Huang, R.S. Pharmacogenomics of cisplatin sensitivity in non-small cell lung cancer. Genom. Proteom. Bioinf. 2014, 12, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Villanueva, A.; Lachenmayer, A.; Finn, R.S. Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat. Rev. Clin. Oncol. 2015, 12, 408–424. [Google Scholar] [CrossRef] [PubMed]
- Cornell, L.; Munck, J.M.; Alsinet, C.; Villanueva, A.; Ogle, L.; Willoughby, C.E.; Televantou, D.; Thomas, H.D.; Jackson, J.; Burt, A.D.; et al. DNA-PK a candidate driver of hepatocarcinogenesis and tissue biomarker that predicts response to treatment and survival. Clin. Cancer Res. 2015, 21, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Ji, L.; Yu, Y.; Wang, J. Novel molecular targets for diagnosis and treatment of hepatocellular carcinoma. Discov. Med. 2015, 19, 7–14. [Google Scholar] [PubMed]
- Tapia-Laliena, M.A.; Korzeniewski, N.; Hohenfellner, M.; Duensing, S. High-risk prostate cancer: A disease of genomic instability. Urol. Oncol. 2014, 32, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Jarzen, J.; Diamanduros, A.; Scarpinato, K.D. Mismatch repair proteins in recurrent prostate cancer. Adv. Clin. Chem. 2014, 60, 65–84. [Google Scholar]
- Brand, T.M.; Iida, M.; Li, C.; Wheeler, D.L. The nuclear epidermal growth factor receptor signaling network and its role in cancer. Discov. Med. 2011, 12, 419–432. [Google Scholar] [PubMed]
- Bertelsen, V.; Stang, E. The mysterious ways of ErbB2/HER2 trafficking. Membranes 2014, 4, 424–446. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.K.; Hung, M.C. Proteolytic cleavage, trafficking, and functions of nuclear receptor tyrosine kinases. FEBS J. 2015, 282, 3693–3721. [Google Scholar] [CrossRef] [PubMed]
- Cremer, C.; Masters, B. Resolution enhancement techniques in microscopy. Eur. Phys. J. H 2013. [Google Scholar] [CrossRef]
- Cremer, C.; Kaufmann, R.; Gunkel, M.; Pres, S.; Weiland, Y.; Müller, P.; Ruckelshausen, T.; Lemmer, P.; Geiger, F.; Degenhard, M.; et al. Superresolution imaging of biological nanostructures by Spectral Precision Distance Microscopy (SPDM). Review. Biotechnol. J. 2011, 6, 1037–1051. [Google Scholar] [CrossRef] [PubMed]
- Hell, S.W. Far-field optical nanoscopy. Science 2007, 316, 1153–1158. [Google Scholar] [CrossRef] [PubMed]
- Schermelleh, L.; Heintzmann, R.; Leonhardt, H. A guide to super-resolution fluorescence microscopy. J. Cell Biol. 2010, 190, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Esa, A.; Edelmann, P.; Kreth, G.; Trakhtenbrot, L.; Amariglio, N.; Rechavi, G.; Hausmann, M.; Cremer, C. Three-dimensional spectral precision distance microscopy of chromatin nano-structures after triple-colour DNA labelling: A study of the BCR region on chromosome 22 and the Philadelphia chromosome. J. Microsc. 2000, 199, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef] [PubMed]
- Hess, S.T.; Girirajan, T.P.; Mason, M.D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 2006, 91, 4258–4272. [Google Scholar] [CrossRef] [PubMed]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Meth. 2006, 3, 793–795. [Google Scholar] [CrossRef] [PubMed]
- Heilemann, M.; Herten, D.P.; Heintzmann, R.; Cremer, C.; Mueller, C.; Tinnefeld, P.; Weston, K.D.; Wolfrum, J.; Sauer, M. High-resolution colocalization of single dye molecules by fluorescence lifetime imaging microscopy. Anal. Chem. 2002, 74, 3511–3517. [Google Scholar] [CrossRef] [PubMed]
- Lemmer, P.; Gunkel, M.; Baddeley, D.; Kaufmann, R.; Urich, A.; Weiland, Y.; Reymann, J.; Müller, P.; Hausmann, M.; Cremer, C. SPDM: Light microscopy with single molecule resolution at the nanoscale. Appl. Phys. B 2008, 93, 1–12. [Google Scholar] [CrossRef]
- Fölling, J.; Bossi, M.; Bock, H.; Medda, R.; Wurm, C.; Hein, B.; Jakobs, S.; Eggeling, C.; Hell, S.W. Fluorescence nanoscopy by ground-state depletion and single-molecule return. Nat. Meth. 2008, 5, 943–945. [Google Scholar] [CrossRef] [PubMed]
- Lemmer, P.; Gunkel, M.; Weiland, Y.; Müller, P.; Baddeley, D.; Kaufmann, R.; Urich, A.; Eipel, H.; Amberger, R.; Hausmann, M.; et al. Using conventional fluorescent markers for far-field fluorescence localization nanoscopy allows resolution in the 10 nm range. J. Microsc. 2009, 235, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, R.; Müller, P.; Hildenbrand, G.; Hausmann, M.; Cremer, C. Analysis of Her2/neu membrane protein clusters in different types of breast cancer cells using localization microscopy. J. Microsc. 2011, 242, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Boyd, P.S.; Struve, N.; Bach, M.; Eberle, J.P.; Gote, M.; Schock, F.; Cremer, C.; Kriegs, M.; Hausmann, M. Clustered localization of EGFRvIII in glioblastoma cells as detected by high precision localization microscopy. Nanoscale 2016, 8, 20037–20047. [Google Scholar] [CrossRef] [PubMed]
- Bohn, M.; Diesinger, P.; Kaufmann, R.; Weiland, Y.; Müller, P.; Gunkel, M.; von Ketteler, A.; Lemmer, P.; Hausmann, M.; Heermann, D.W.; et al. Localization microscopy reveals expression-dependent parameters of chromatin nanostructure. Biophys. J. 2010, 99, 1358–1367. [Google Scholar] [CrossRef] [PubMed]
- Müller, P.; Weiland, Y.; Kaufmann, R.; Gunkel, M.; Hillebrandt, S.; Cremer, C.; Hausmann, M. Analysis of fluorescent nanostructures in biological systems by means of Spectral Position Determination Microscopy (SPDM). In Current Microscopy Contributions to Advances in Science and Technology; Méndez-Vilas, A., Ed.; Formatex Research Center: Badajoz, Spain, 2012; Volume 1, pp. 3–12. [Google Scholar]
- Zhang, Y.; Máté, G.; Müller, P.; Hillebrandt, S.; Krufczik, M.; Bach, M.; Kaufmann, R.; Hausmann, M.; Heermann, D.W. Radiation induced chromatin conformation changes analysed by fluorescent localization microscopy, statistical physics, and graph theory. PLoS ONE 2015, 10, e0128555. [Google Scholar] [CrossRef] [PubMed]
- Müller, P.; Lemmermann, N.A.; Kaufmann, R.; Gunkel, M.; Paech, D.; Hildenbrand, G.; Holtappels, R.; Cremer, C.; Hausmann, M. Spatial distribution and structural arrangement of a murine cytomegalovirus glycoprotein detected by SPDM localization microscopy. Histochem. Cell Biol. 2014, 142, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Stuhlmüller, M.; Schwarz-Finsterle, J.; Fey, E.; Lux, J.; Bach, M.; Cremer, C.; Hinderhofer, K.; Hausmann, M.; Hildenbrand, G. In situ optical sequencing and nano-structure analysis of a trinucleotide expansion region by localization microscopy after specific COMBO-FISH labelling. Nanoscale 2015, 7, 17938–17946. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.; Hausmann, M.; Lukášová, E.; Biswas, A.; Hildenbrand, G.; Davídková, M.; Krasavin, E.; Kleibl, Z.; Falková, I.; Ježková, L.; et al. Giving OMICS spatiotemporal dimensions by challenging microscopy: From functional networks to structural organization of cell nuclei elucidating mechanisms of complex radiation damage response and chromatin repair—PART B (Structuromics). Crit. Rev. Eukaryot. Gene Express 2014, 24, 225–247. [Google Scholar] [CrossRef]
- Hausmann, M.; Winkler, R.; Hildenbrand, G.; Finsterle, J.; Weisel, A.; Rapp, A.; Schmitt, E.; Janz, S.; Cremer, C. COMBO-FISH: Specific labelling of nondenatured chromatin targets by computer-selected DNA oligonucleotide probe combinations. Biotechniques 2003, 35, 564–577. [Google Scholar] [PubMed]
- Stuhlmüller, M.; Hausmann, M. Selection of COMBO-FISH probes for multi-purpose applications. J. Theor. Comput. Sci. 2015, 2, 131–132. [Google Scholar] [CrossRef]
- Schmitt, E.; Wagner, J.; Hausmann, M. Combinatorial selection of short triplex forming oligonucleotides for fluorescence in situ hybridisation COMBO-FISH. J. Comput. Sci. 2012, 3, 328–334. [Google Scholar] [CrossRef]
- Krufczik, M.; Sievers, A.; Hausmann, A.; Lee, J.-H.; Hildenbrand, G.; Schaufler, W.; Hausmann, M. Combining low temperature fluorescence DNA-hybridization, immunostaining, and super-resolution localization microscopy for nano-structure analysis of ALU elements and their influence on chromatin structure. Int. J. Mol. Sci. 2017, 18, 1005. [Google Scholar] [CrossRef] [PubMed]
- Schwarz-Finsterle, J.; Stein, S.; Großmann, C.; Schmitt, E.; Schneider, H.; Trakhtenbrot, L.; Rechavi, G.; Amariglio, N.; Cremer, C.; Hausmann, M. COMBO-FISH for focussed fluorescence labelling of gene domains: 3D-analysis of the genome architecture of abl and bcr in human blood cells. Cell Biol. Int. 2005, 29, 1038–1046. [Google Scholar] [CrossRef] [PubMed]
- Schwarz-Finsterle, J.; Stein, S.; Großmann, C.; Schmitt, E.; Trakhtenbrot, L.; Rechavi, G.; Amariglio, N.; Cremer, C.; Hausmann, M. Comparison of triplehelical COMBO-FISH and standard FISH by means of quantitative microscopic image analysis of abl/bcr genome organisation. J. Biophys. Biochem. Meth. 2007, 70, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Zeller, D.; Kepper, N.; Hausmann, M.; Schmitt, E. Sequential and structural biophysical aspects of combinatorial oligo-FISH in Her2/neu breast cancer diagnostics. IFMBE Proc. 2013, 38, 82–85. [Google Scholar]
- Müller, P.; Rößler, J.; Schwarz-Finsterle, J.; Pedersen, E.B.; Géci, I.; Schmitt, E.; Hausmann, M. PNA-COMBO-FISH: From combinatorial probe design in silico to vitality compatible, specific labelling of gene targets in cell nuclei. Exp. Cell Res. 2016, 345, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Müller, P.; Schmitt, E.; Jacob, A.; Hoheisel, J.; Kaufmann, R.; Cremer, C.; Hausmann, M. COMBO-FISH enables high precision localization microscopy as a prerequisite for nanostructure analysis of genome loci. Int. J. Mol. Sci. 2010, 11, 4094–4105. [Google Scholar] [CrossRef] [PubMed]
- Winkler, R.; Perner, B.; Rapp, A.; Durm, M.; Cremer, C.; Greulich, K.O.; Hausmann, M. Labelling quality and chromosome morphology after low temperature FISH analysed by scanning far-field and scanning near-field optical microscopy. J. Microsc. 2003, 209, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Lanzano, L.; Coto Hernández, I.; Castello, M.; Gratton, E.; Diaspro, A.; Vicidomini, G. Encoding and decoding spatio-temporal information for super-resolution microscopy. Nat. Commun. 2015, 6, 6701. [Google Scholar] [CrossRef] [PubMed]
- Aurenhammer, F. Voronoi diagrams—A survey of a fundamental geometric data structure. Am. Comput. Surveys 1991, 23, 345–405. [Google Scholar] [CrossRef]
- Magenau, A.; Owen, D.M.; Yamamoto, Y.; Tran, J.; Kwiatek, J.M.; Parton, R.G.; Gaus, K. Discreet and distinct clustering of five model membrane proteins revealed by single molecule localization microscopy. Mol. Membrane Biol. 2015, 32, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Morozova, D.; Guigas, G.; Weiss, M. Dynamic Structure Formation of Peripheral Membrane Proteins. PLoS Comput. Biol. 2011, 7, e1002067. [Google Scholar] [CrossRef] [PubMed]
- Guigas, G.; Weiss, M. Membrane protein mobility depends on the length of extra-membrane domains and on protein concentration. Soft Matter 2015, 11, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Guigas, G.; Weiss, M. Influence of hydrophobic mismatching on membrane protein diffusion. Biophys. J. 2008, 95, L25–L27. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, U.; Guigas, G.; Weiss, M. Cluster formation of transmembrane proteins due to hydrophobic mismatching. Phys. Rev. Lett. 2008, 101, 128104. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, Y.; Cai, M.; Pan, Y.; Xu, H.; Jiang, J.; Ji, H.; Wang, H. Mechanistic insights into EGFR membrane clustering revealed by super-resolution imaging. Nanoscale 2015, 7, 2511–2519. [Google Scholar] [CrossRef] [PubMed]
- Pilarczyk, G.; Nesnidal, I.; Gunkel, M.; Bach, M.; Bestvater, F.; Hausmann, M. Localisation microscopy of breast epithelial ErbB-2 receptors and gap junctions: Trafficking after gamma-irradiation, Neuregulin-1b and Herceptin application. Int. J. Mol. Sci. 2017, 18, 362. [Google Scholar] [CrossRef] [PubMed]
- Ritter, C.A.; Perez-Torres, M.; Rinehart, C.; Guix, M.; Drzgger, T.; Engelman, J.A.; Arteaga, C.L. Human breast cancer cells selected for resistance to trastuzumab in vivo overexpress epidermal growth factor receptor and ErbB ligands and remain dependent on the ErbB receptor network. Clin. Cancer Res. 2007, 13, 4909–4919. [Google Scholar] [CrossRef] [PubMed]
- Schoeberl, B.; Pace, E.A.; Fitzgerals, J.B.; Harms, B.D.; Xu, L.; Nie, L.; Linggi, B.; Kalra, A.; Paragas, V.; Bukhalid, R.; et al. Therapeutically targeting ErbB3: A key role in lignad-induced activation of the ErbB receptor-PI3K axis. Sci. Signal. 2009, 2, ra31. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Levy-Cohen, G.; Blank, M. Molecular functions of NEDD4 E3 ubiquitin ligases in cancer. Biochim. Biophys. Acta 2015, 1856, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Blank, M.; Tang, Y.; Yamashita, M.; Burkett, S.S.; Cheng, S.Y.; Zhang, Y.E. A tumor suppressor function of Smurf2 associated with controlling chromatin landscape and genome stability through RNF20. Nat. Med. 2012, 18, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Fierz, B.; Chatterjee, C.; McGinty, R.K.; Bar-Dagan, M.; Raleigh, D.P.; Muir, T.W. Histone H2B ubiquitylation disrupts local and higher-order chromatin compaction. Nat. Chem. Biol. 2011, 7, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Minsky, N.; Shema, E.; Field, Y.; Schuster, M.; Segal, E.; Oren, M. Monoubiquitinated H2B is associated with the transcribed region of highly expressed genes in human cells. Nat. Cell Biol. 2008, 10, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Moyal, L.; Lerenthal, Y.; Gana-Weisz, M.; Mass, G.; So, S.; Wang, S.Y.; Eppink, B.; Chung, Y.M.; Shalev, G.; Shema, E.; et al. Requirement of ATM-Dependent Monoubiquitylation of Histone H2B for Timely Repair of DNA Double-Strand Breaks. Mol. Cell 2011, 41, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Ramkumar, C.; Kong, Y.; Cui, H.; Hao, S.; Jones, S.N.; Gerstein, R.M.; Zhang, H. Smurf2 regulates the senescence response and suppresses tumorigenesis in mice. Cancer Res. 2012, 72, 2714–2719. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Gu, X.; Sun, L.; Flowers, A.B.; Rademaker, A.W.; Zhou, Y.; Kiyokawa, H. Downregulation of Smurf2, a tumor-suppressive ubiquitin ligase, in triple-negative breast cancers: Involvement of the RB-microRNA axis. BMC Cancer 2014, 14, 57. [Google Scholar] [CrossRef] [PubMed]
- David, D.; Nair, S.A.; Pillai, M.R. Smurf E3 ubiquitin ligases at the cross roads of oncogenesis and tumor suppression. Biochim. Biophys. Acta 2013, 1835, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Emanuelli, A.; Borroni, A.P.; Apel-Sarid, L.; Shah, P.A.; Ayyathan, D.M.; Koganti, P.; Levy-Cohen, G.; Blank, M. Smurf2-mediated stabilization of DNA topoisomerase IIα controls genomic integrity. Cancer Res. 2017, 77, 4217–4227. [Google Scholar] [CrossRef] [PubMed]
- Delaney, G.; Jacob, S.; Featherstone, C.; Barton, M. The role of radiotherapy in cancer treatment: Estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer 2005, 104, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.S.; Duke, S.; Jena, R.; Williams, M.V.; Burnet, N.G. Advances in radiotherapy. BMJ 2012, 345, e7765. [Google Scholar] [CrossRef] [PubMed]
- Maier, P.; Hartmann, L.; Wenz, F.; Herskind, C. Cellular pathways in response to ionizing radiation and their targetability for tumor radiosensitization. Int. J. Mol. Sci. 2016, 17, 102. [Google Scholar] [CrossRef] [PubMed]
- Natale, F.; Rapp, A.; Yu, W.; Maiser, A.; Harz, H.; Schall, A.; Grulich, S.; Anton, T.; Hörl, D.; Chen, W.; et al. Identification of the elementary structural units of the DNA damage response. Nat. Commun. 2017, 8, 15760. [Google Scholar] [CrossRef] [PubMed]
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.W. Cancer and radiation therapy: Current advances and future directions. Int. J. Med. Sci. 2012, 9, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Purdy, J.A. Dose to normal tissues outside the radiation therapy patient’s treated volume: A review of different radiation therapy techniques. Health Phys. 2008, 95, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Wenz, F.; Tiefenbacher, U.; Willeke, F.; Weber, K.J. The search for therapeutic gain in radiation oncology. Onkologie 2001, 24 (Suppl. S5), 51–55. [Google Scholar] [PubMed]
- Eryilmaz, M.; Schmitt, E.; Krufczik, M.; Theda, F.; Schaufler, W.; Bestvater, F.; Hildenbrand, G.; Hausmann, M. Super-resolution microscopy meets radiation research: A localization microscopy study of MRE11 foci in 3D-conserved cell nuclei. Cancers 2017. in preparation. [Google Scholar]
- Hofmann, A.; Krufczik, M.; Hausmann, M.; Heermann, D.W. Topological data analysis of super- resolution microscopy. Nanoscale 2017. in preparation. [Google Scholar]
- Bach, M.; Savini, C.; Krufczik, M.; Cremer, C.; Rösl, F.; Hausmann, M. Super-resolution localization microscopy of γ-H2AX and heterochromatin after folate deficiency. Int. J. Mol. Sci. 2017, 18, 1726. [Google Scholar] [CrossRef] [PubMed]
- Deininger, P. ALU elements: Know the SINEs. Genome Biol. 2011, 12, 236. [Google Scholar] [CrossRef] [PubMed]
- Batzer, M.A.; Deininger, P.L. ALU repeats and human genomic diversity. Nat. Rev. Genet. 2002, 3, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ara, T.; Gautheret, D. Using ALU elements as polyadenylation sites: A case of retroposon exaptation. Mol. Biol. Evol. 2009, 26, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Morales, M.E.; White, T.B.; Streva, V.A.; DeFreece, C.B.; Hedges, D.J.; Deininger, P.L. The contribution of ALU elements to mutagenic DNA double-strand break repair. PLoS Genet. 2015, 11, e1005016. [Google Scholar] [CrossRef] [PubMed]
- White, T.B.; Morales, M.E.; Deininger, P.L. Alu elements and DNA double-strand break repair. Mob. Genet. Elements 2015, 5, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Welsch, P.L.; King, M.C. BRCA1 and BRCA2 and the genetics of breast and ovarian cancer. Hum. Mol. Genet. 2001, 10, 705–713. [Google Scholar]
- Chénais, B.; Caruso, A.; Hiard, S.; Casse, N. The impact of transposable elements on eukaryotic genomes: From genome size increase to genetic adaptation to stressful environments. Gene 2012, 509, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Huang, S. Nuclear function of ALUs. Nucleus 2014, 5, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Tsirigos, A.; Rigoutsos, I. ALU and b1 repeats have been selectively retained in the upstream and intronic regions of genes of specific functional classes. PLoS Comput. Biol. 2009, 5, e1000610. [Google Scholar] [CrossRef] [PubMed]
- Caudron-Herger, M.; Pankert, T.; Seiler, J.; Németh, A.; Voit, R.; Grummt, I.; Rippe, K. Alu element- containing RNAs maintain nucleolar structure and function. EMBO J. 2015, 34, 2758–2774. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Jin, K.; Crabbe, M.J.C.; Zhang, Y.; Liu, X.; Huang, Y.; Hua, M.; Nan, P.; Zhang, Z.; Zhong, Y. Enrichment analysis of ALU elements with different spatial chromatin proximity in the human genome. Protein Cell 2016, 7, 250–266. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Irradiation Dose (Gy) | Number of Cells Analyzed | Mean Number of Points | Error of the Mean (±) |
|---|---|---|---|
| control | 80 | 25,839 | 1129 |
| 0.1 | 83 | 21,669 | 783 |
| 0.2 | 40 | 25,011 | 1513 |
| 0.3 | 40 | 17,594 | 1538 |
| 0.4 | 40 | 19,091 | 1347 |
| 0.5 | 80 | 17,789 | 676 |
| 1 | 38 | 13,061 | 518 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hausmann, M.; Ilić, N.; Pilarczyk, G.; Lee, J.-H.; Logeswaran, A.; Borroni, A.P.; Krufczik, M.; Theda, F.; Waltrich, N.; Bestvater, F.; et al. Challenges for Super-Resolution Localization Microscopy and Biomolecular Fluorescent Nano-Probing in Cancer Research. Int. J. Mol. Sci. 2017, 18, 2066. https://doi.org/10.3390/ijms18102066
Hausmann M, Ilić N, Pilarczyk G, Lee J-H, Logeswaran A, Borroni AP, Krufczik M, Theda F, Waltrich N, Bestvater F, et al. Challenges for Super-Resolution Localization Microscopy and Biomolecular Fluorescent Nano-Probing in Cancer Research. International Journal of Molecular Sciences. 2017; 18(10):2066. https://doi.org/10.3390/ijms18102066
Chicago/Turabian StyleHausmann, Michael, Nataša Ilić, Götz Pilarczyk, Jin-Ho Lee, Abiramy Logeswaran, Aurora Paola Borroni, Matthias Krufczik, Franziska Theda, Nadine Waltrich, Felix Bestvater, and et al. 2017. "Challenges for Super-Resolution Localization Microscopy and Biomolecular Fluorescent Nano-Probing in Cancer Research" International Journal of Molecular Sciences 18, no. 10: 2066. https://doi.org/10.3390/ijms18102066