1. Introduction
As a promising strategy for cancer treatment, recent studies have focused on the development of drugs for inhibiting receptor tyrosine kinases (RTKs), such as the epidermal growth factor receptor (EGFR), vascular endothelial growth factor receptors (VEGFRs), platelet-derived growth factor receptor (PDGFR), and the tropomyosin-related kinase (TRK) family [
1]. In particular, TRKA, a member of the RTK family, is emerging as a potential therapeutic target for the treatment of human cancers, owing to its oncogenic function associated with cell growth, proliferation, survival, and evasion of apoptosis [
2]. Moreover, it has been reported that synthetic azaindole derivatives, such as TRKA inhibitors, confer anti-cancer effects by suppressing v-Akt murine thymoma viral oncogene (AKT) -mediated growth signaling in vitro [
3]. Since VEGFR2 is closely linked to pathological angiogenesis, including cancer and inflammation, an efficient inhibitor targeting VEGFR2 would be useful for the treatment of human cancer [
4]. Indeed, a recent preclinical study has demonstrated that tubeimoside-1 can suppress cancer growth and angiogenesis by attenuating mTOR-AKT signaling in non-small cell lung cancer [
5].
Fascaplysin, originally identified as a natural compound from the marine sponge, and its synthetic derivatives have been reported as potential CDK4 inhibitors [
6,
7,
8]. However, the anti-cancer effects of fascaplysin and its derivatives via CDK4 have not been fully studied. Kumar et al. recently suggested that fascaplysin induces apoptosis and autophagy by suppressing the PI3K-AKT-mTOR signaling cascade in HL-60 cells [
9], indicating that the identification of unknown molecules, namely key targets for the development of fascaplysin-derived anti-cancer drugs, is necessary.
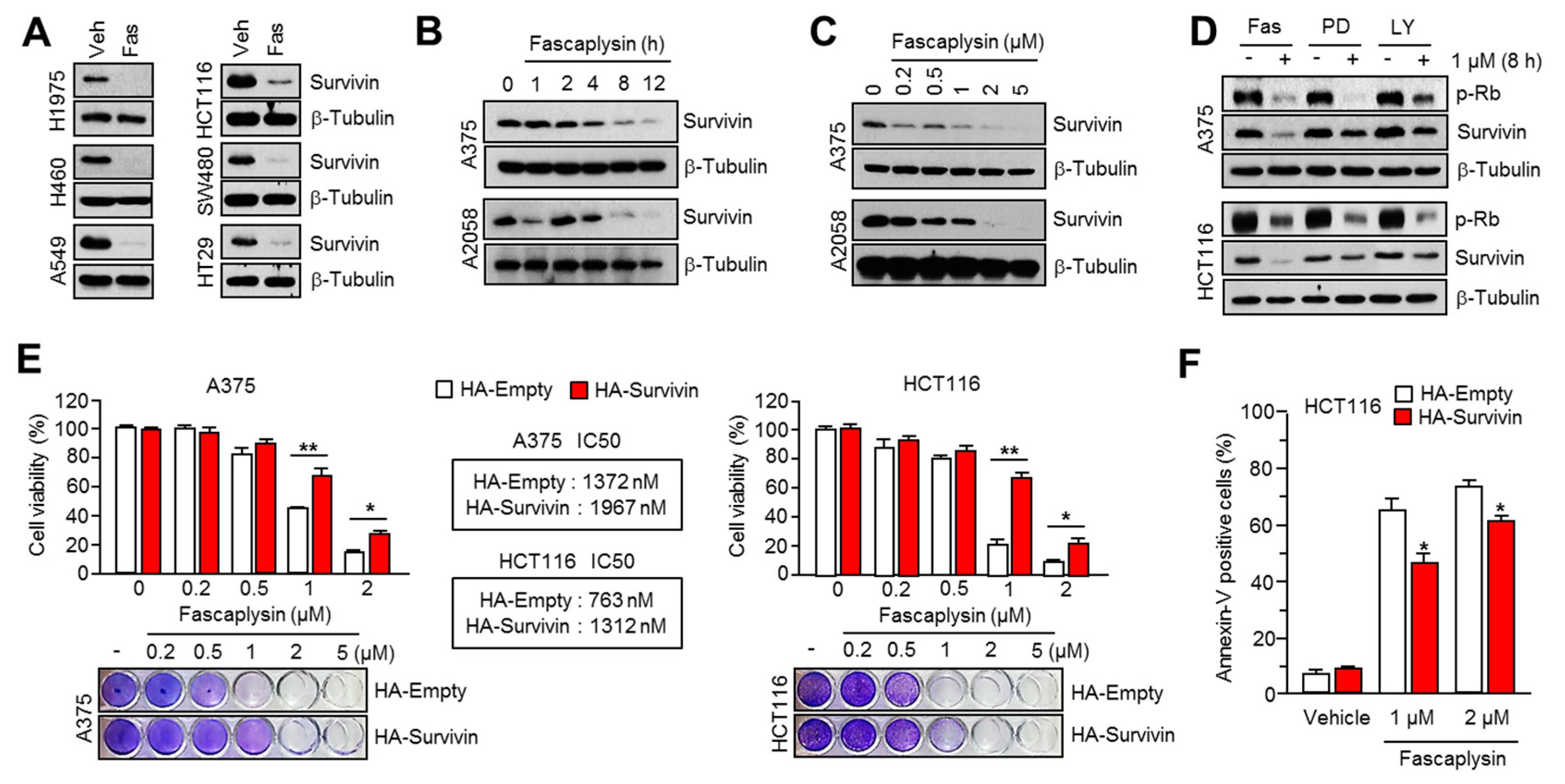
It is becoming clear that survivin, as an anti-apoptotic factor owing to its suppressive function in the pro-apoptotic signal-mediated caspase cascade, is an attractive therapeutic target because of its differential expression in cancer versus normal tissues, which allows for specificity in cancer therapies [
10,
11]. Indeed, targeting survivin with genetic silencing using antisense oligonucleotides or pharmacological inhibition using small molecules, such as YM155, induced apoptosis by triggering caspase activation in tumor cell lines whereas did not in normal cells [
12]. Moreover, many reports suggest that the suppression of survivin sensitizes tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-based cancer therapy that requires the incorporation of other anti-cancer agents because of its limited anti-cancer efficacy due to TRAIL-resistance [
13,
14,
15,
16,
17].
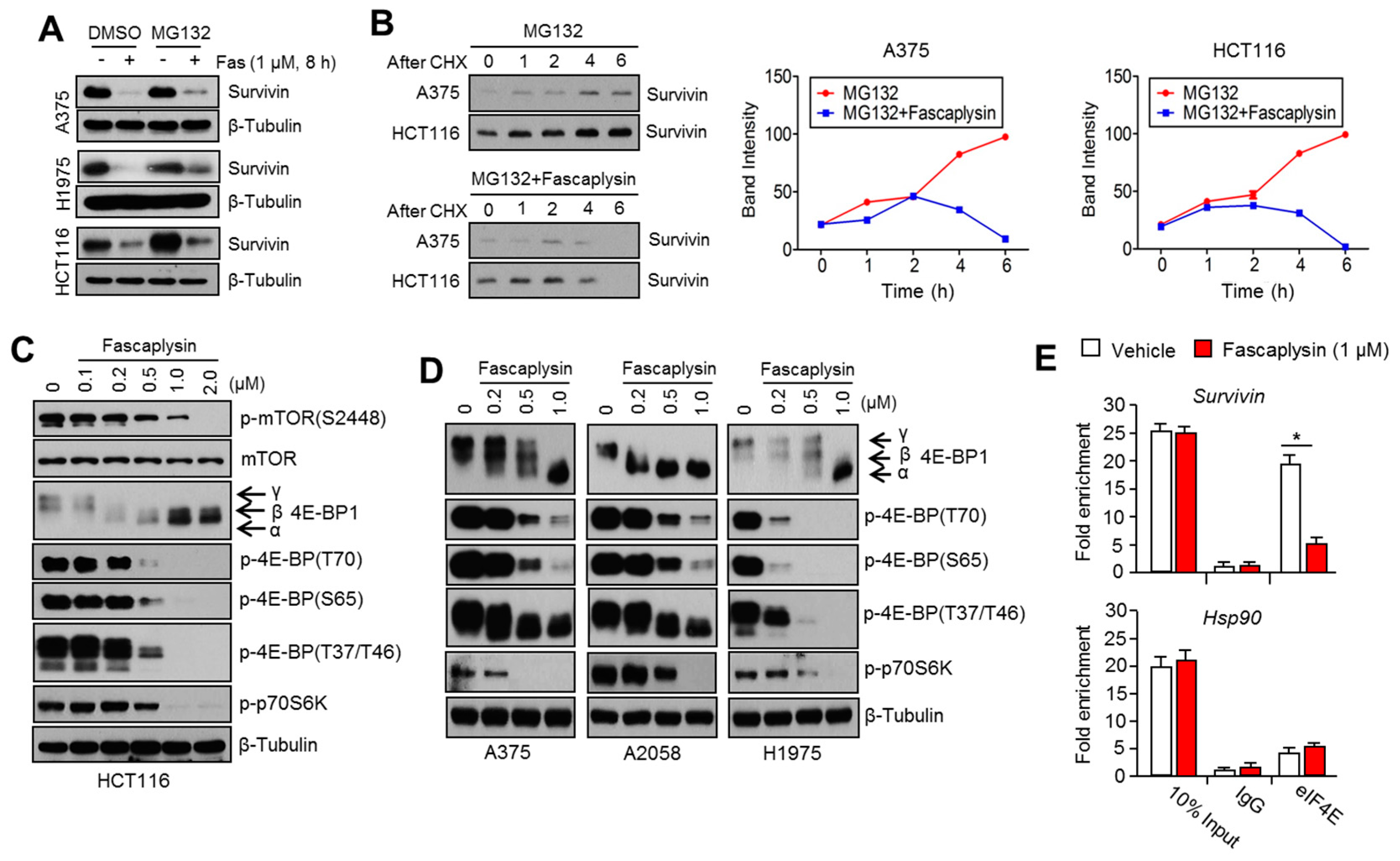
Furthermore, 4EBP1 and p70S6K1, which are regulated by growth factor-mediated mTOR signaling, control tumor development and progression through eIF4E-mediated cap-dependent translation of tumor promoting genes, such as survivin, HIF-1α, cyclin D1, and c-Myc [
18,
19]. Several preclinical studies have reported potential therapeutic tools for the treatment of human cancers, that target the translation machinery, such as mTOR inhibitors (e.g., Rapamycin, Torin 1), eIF4E inhibitors (e.g., 4EGI-1, 4E1RCat), and MNK inhibitors (e.g., Cercosporamide, CGP57380) [
20].
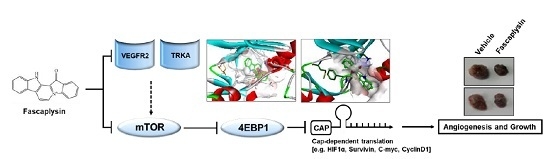
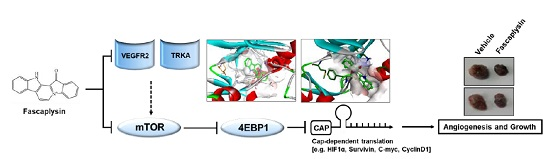
In the present study, we determined the dependency of the CDK4-retinoblastoma (CDK4-RB) axis in the suppression of cancer growth by fascaplysin, and identified TRKA and VEGFR2 as novel target molecules, which are directly targeted by fascaplysin. Our results showed that fascaplysin downregulates survivin and HIF-1α, resulting in increased apoptosis and decreased angiogenesis due to the suppression of 4EBP1-mediated cap-dependent translation.
3. Discussion
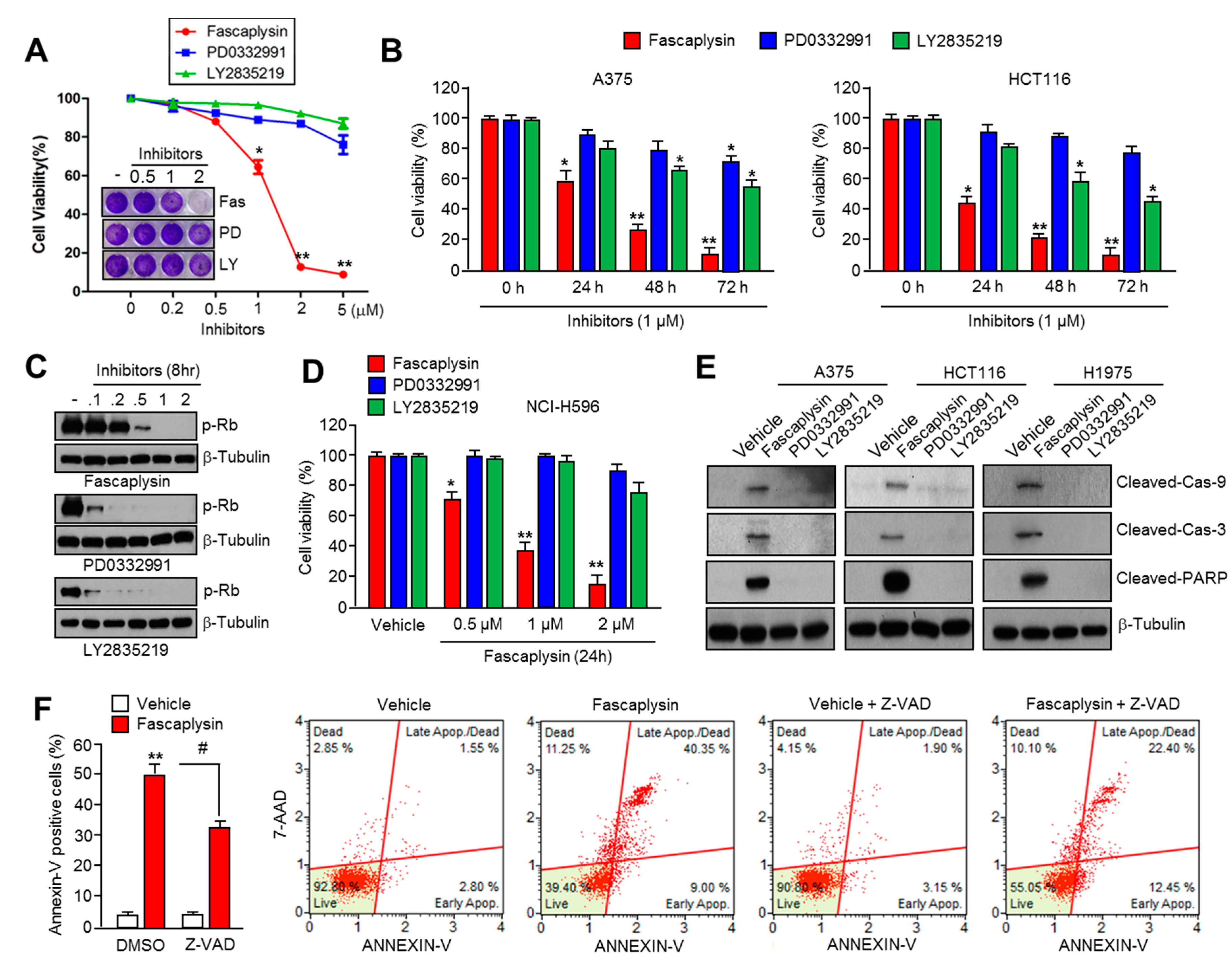
Previous studies have shown the anti-cancer effect of fascaplysin through the suppression of CDK4 and cell cycle progression in various cancer cells [
6,
7,
8,
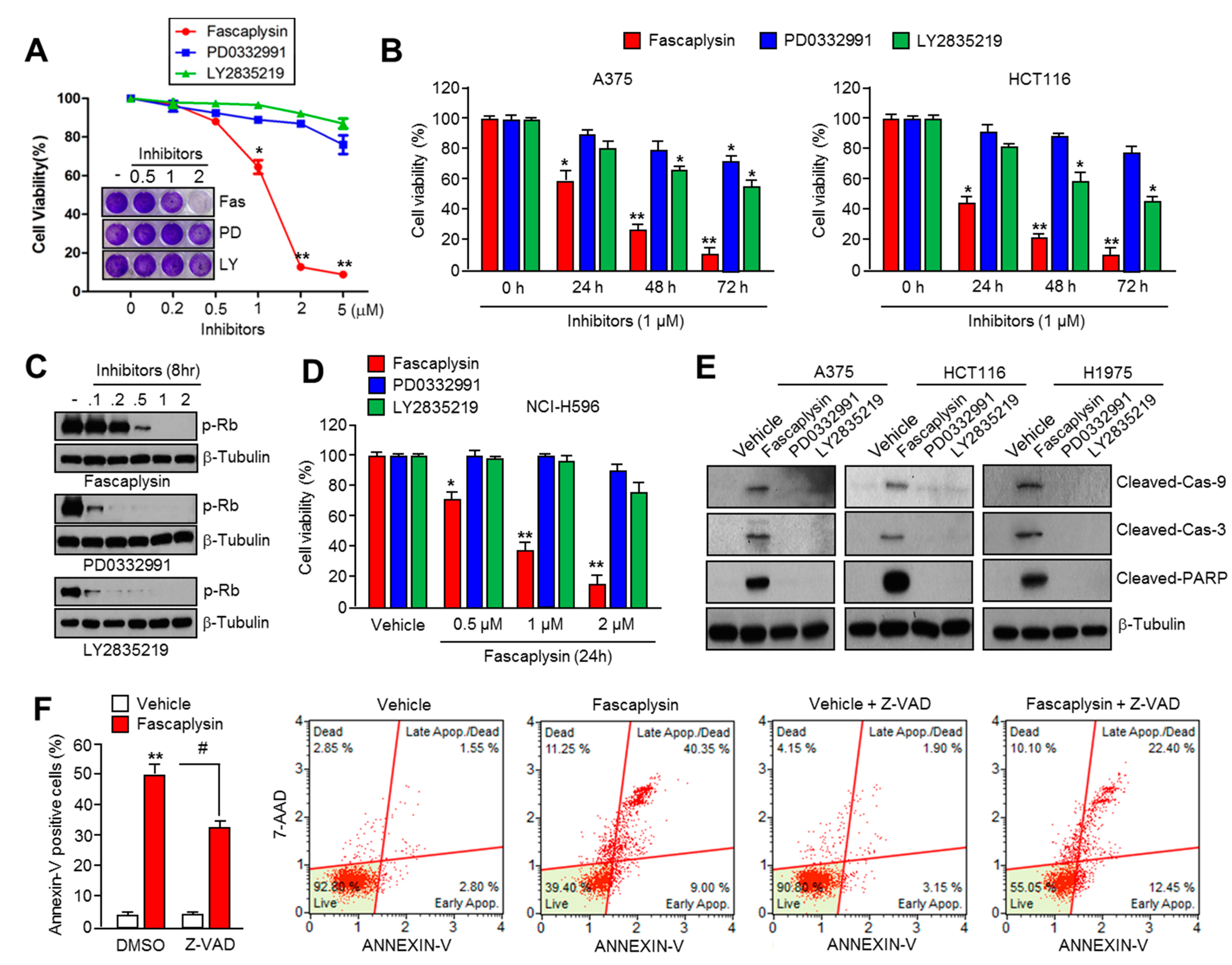
22], but the precise molecular mechanism by which fascaplysin attenuates cancer cell growth remained unexplored. Here, for the first time, we examined the dependence of the CDK4-RB axis in the anti-cancer effect of fascaplysin. Interestingly, our results revealed that fascaplysin rapidly decreased cell viability and increased apoptosis when compared to other CDK4 inhibitors, such as PD0332991 and LY2835219, in various cancer cells. Moreover, the anti-cancer effect of fascaplysin was also observed in RB-null NCI-H596 lung cancer cells. These results indicated that key endogenous targets of fascaplysin, which might be critical factors for tumor growth, have not yet been explored.
Recently, Kumar et al. have reported that fascaplysin attenuated PI3K-AKT-mTOR signaling in HL-60 cells [
9]; however, the molecular mechanism by which fascaplysin inhibits the PI3K-AKT-mTOR pathway has not been explored. In the present study, we performed a high-throughput kinase binding assay and identified that multiple types of cancer-related kinases, such as VEGFR3, VEGFR2, and TRKA, were inhibited by fascaplysin at concentrations lower than 3 µM. This finding suggests that the PI3K-AKT-mTOR pathway might be indirectly inhibited by fascaplysin via the suppression of the VEGFR and TRKA pathways. Further studies whether fascaplysin attenuates autophosphorylation or activation of VEGFR2 or TRKA that stimulate signal cascades related to cancer cell growth and proliferation would strengthen our finding as well. Similarly, the high-throughput kinase binding assay revealed that PI3K, AKT, and mTOR might not be directly affected by fascaplysin. Given the fact that VEGFRs and TRKs are emerging potential targets for cancer-targeted therapies, these findings provide a possibility that fascaplysin can be a valuable candidate for the development of cancer-targeting drugs.
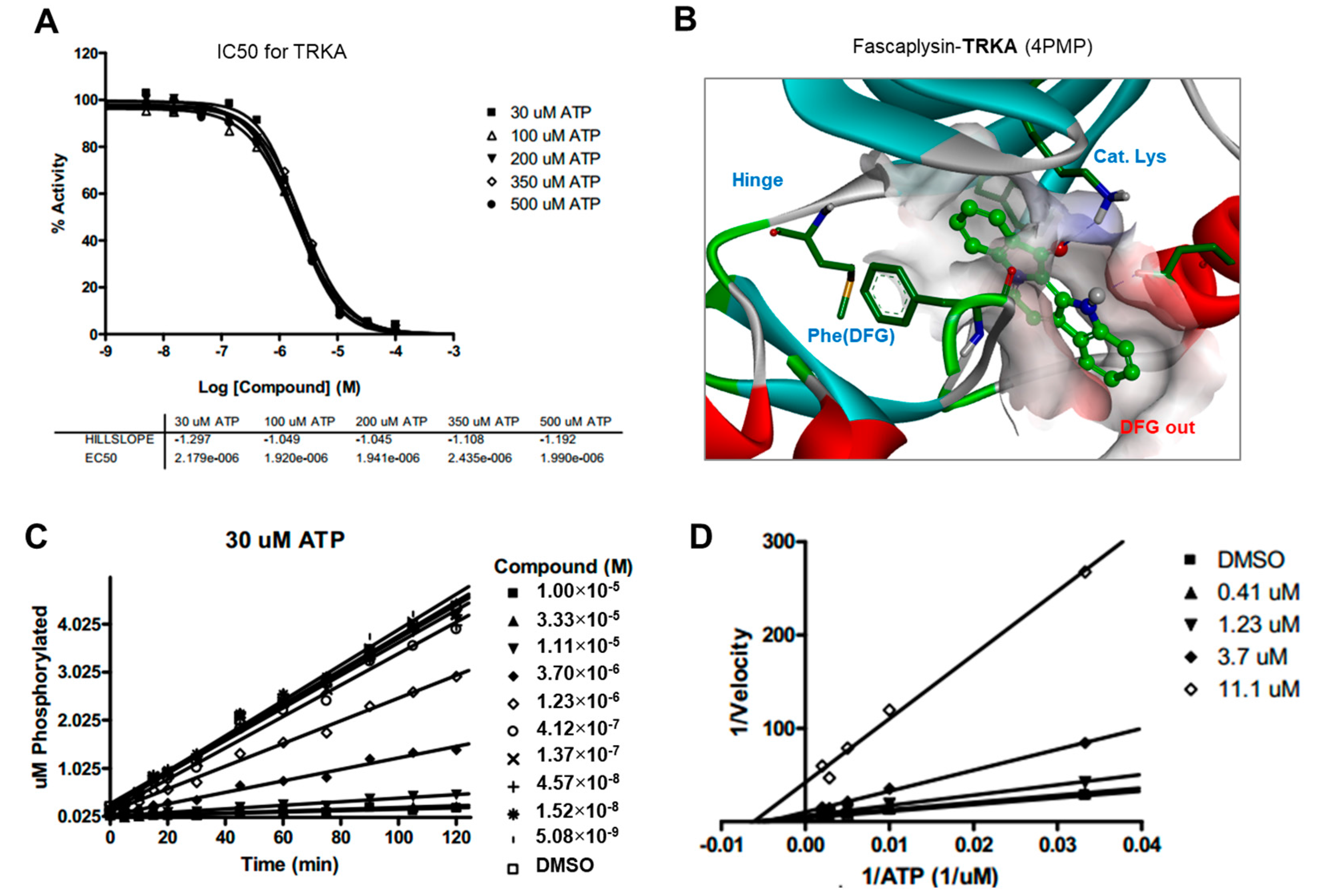
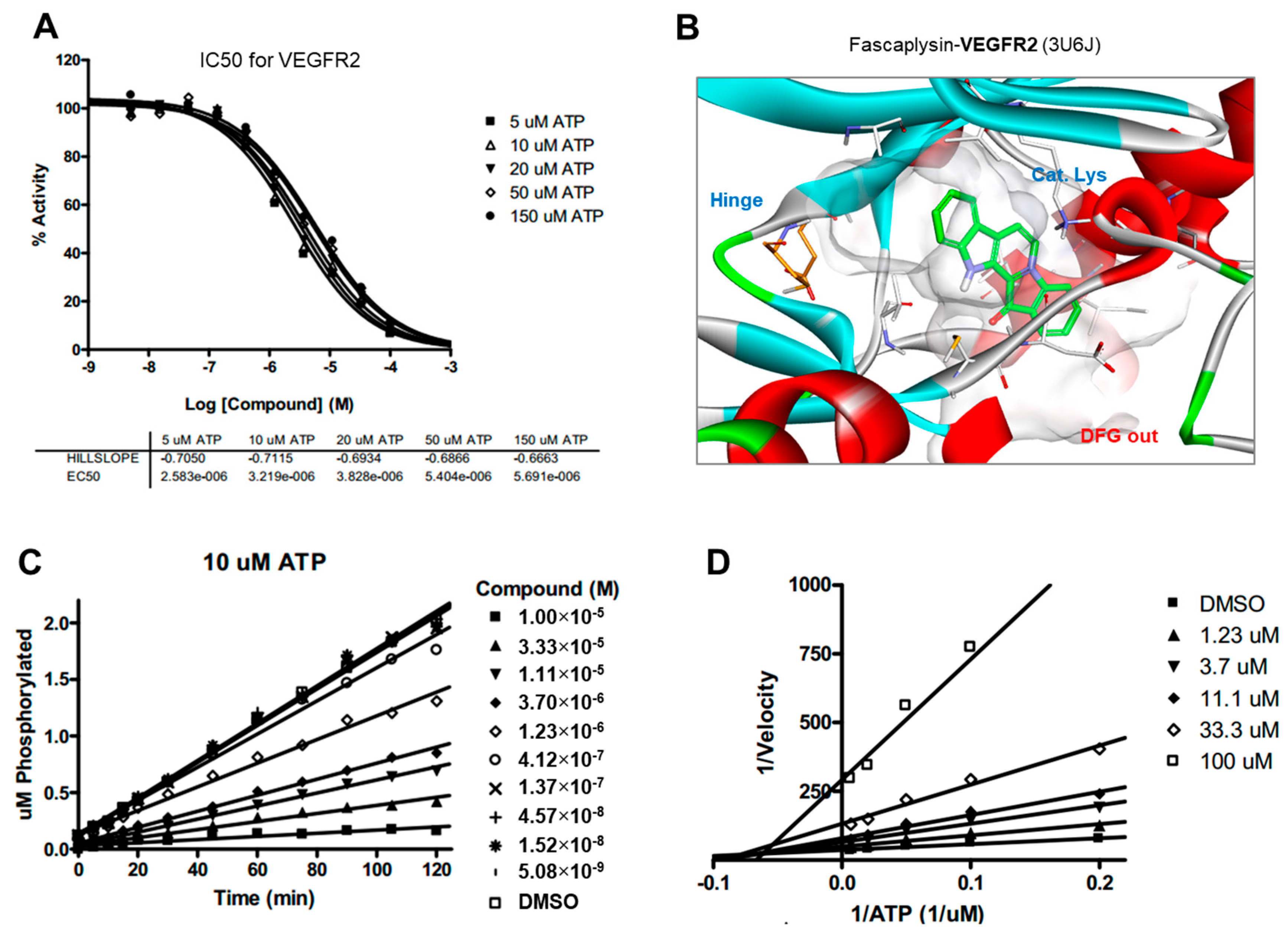
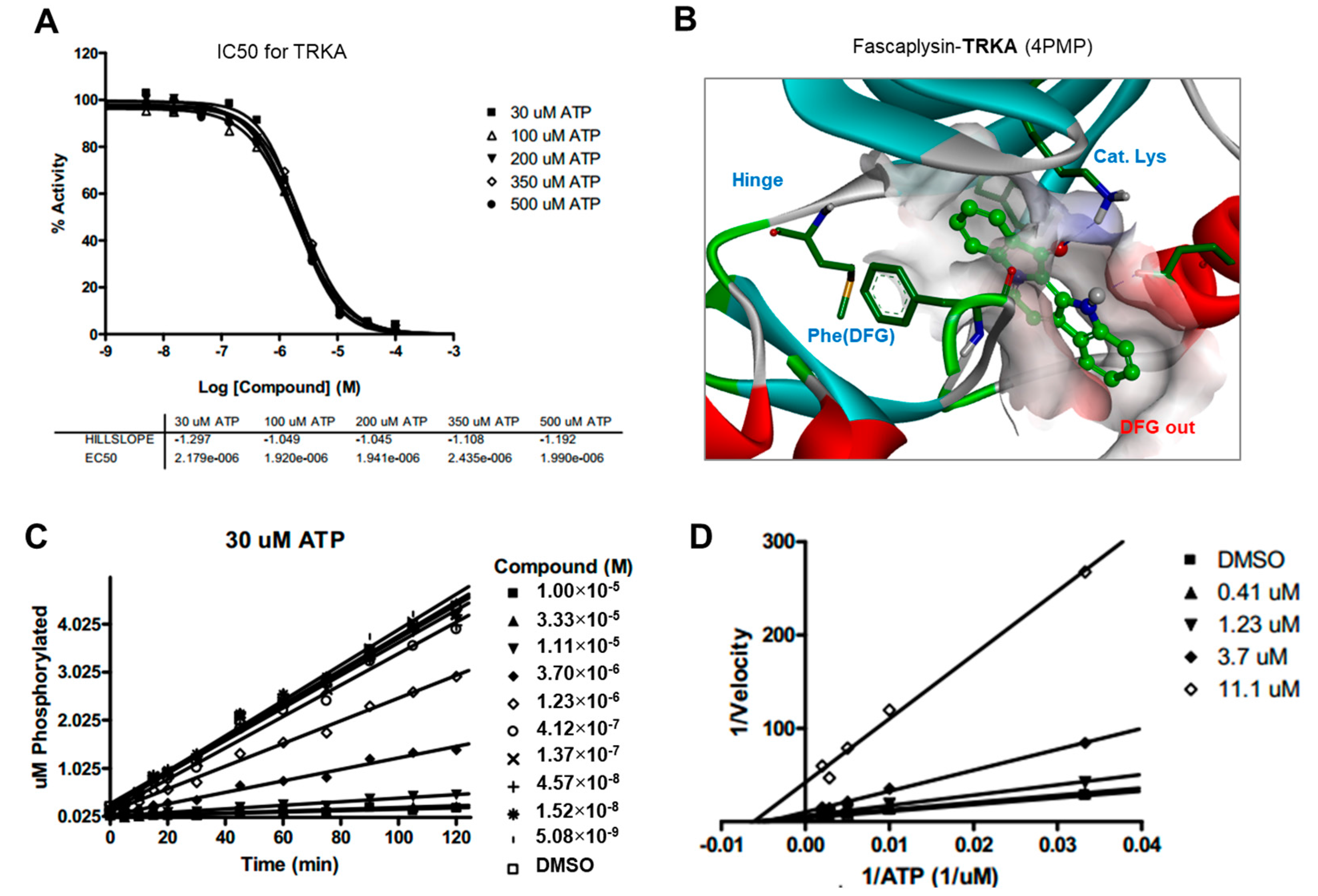
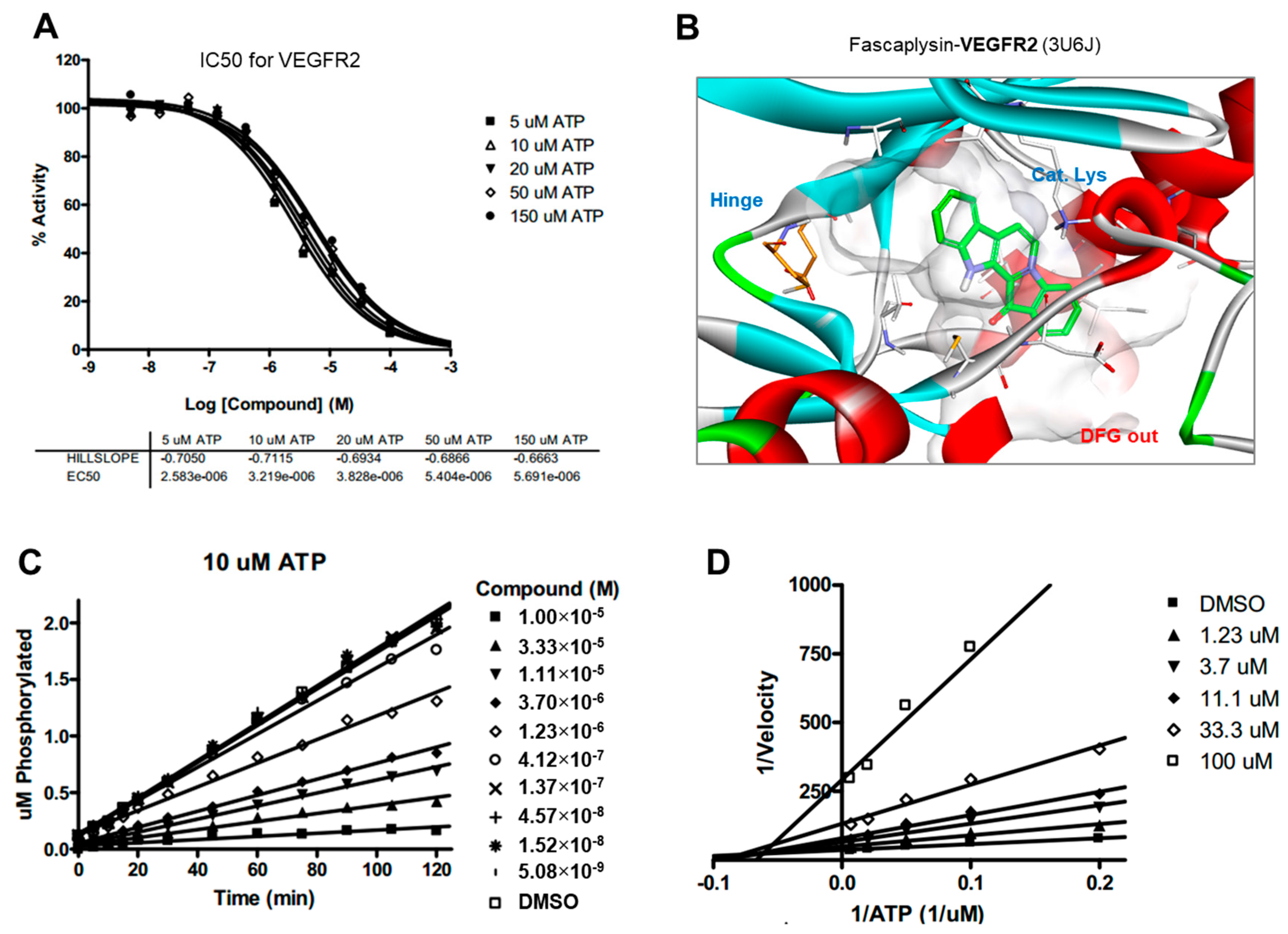
Drug and protein docking simulations were analyzed to obtain energetic and structural insight into the binding modes between fascaplysin and TRKA or fascaplysin and VEGFR2. In the calculated TRKA–fascaplysin complex shown in
Figure 5B, fascaplysin appears to be in close contact with residues Val573, Leu657, Ile572, Phe646, Leu563, and Phe669, all of which are close to the DFG motif. It is noted that the C=O and NH bond of fascaplysin form two hydrogen bonds with a catalytic lysine (Lys544) and Glu560 of the α-C helix, respectively. Fascaplysin may be further stabilized via an attractive charge interaction between the endocyclic ammonium cation with the side chain of Asp668 and via a π-stacking interaction with the benzyl side chain of the gatekeeper Phe589. Fascaplysin also displayed high affinity towards VEGFR2 and VEGFR3. This binding affinity was found to be similar to that of TRKA, in that fascaplysin is stabilized by Van der Waals interactions with Ile892, Leu1035, Thr916, and Val914 in the DFG-out conformation. The carbonyl group of fascaplysin forms a hydrogen bond with Asp1046 and an attractive charge interaction of the endocyclic ammonium cation with the side chain of Glu885 in the α-C helix, respectively. These findings may be helpful for understanding the design of fascaplysin-based derivatives as potential anticancer drugs.
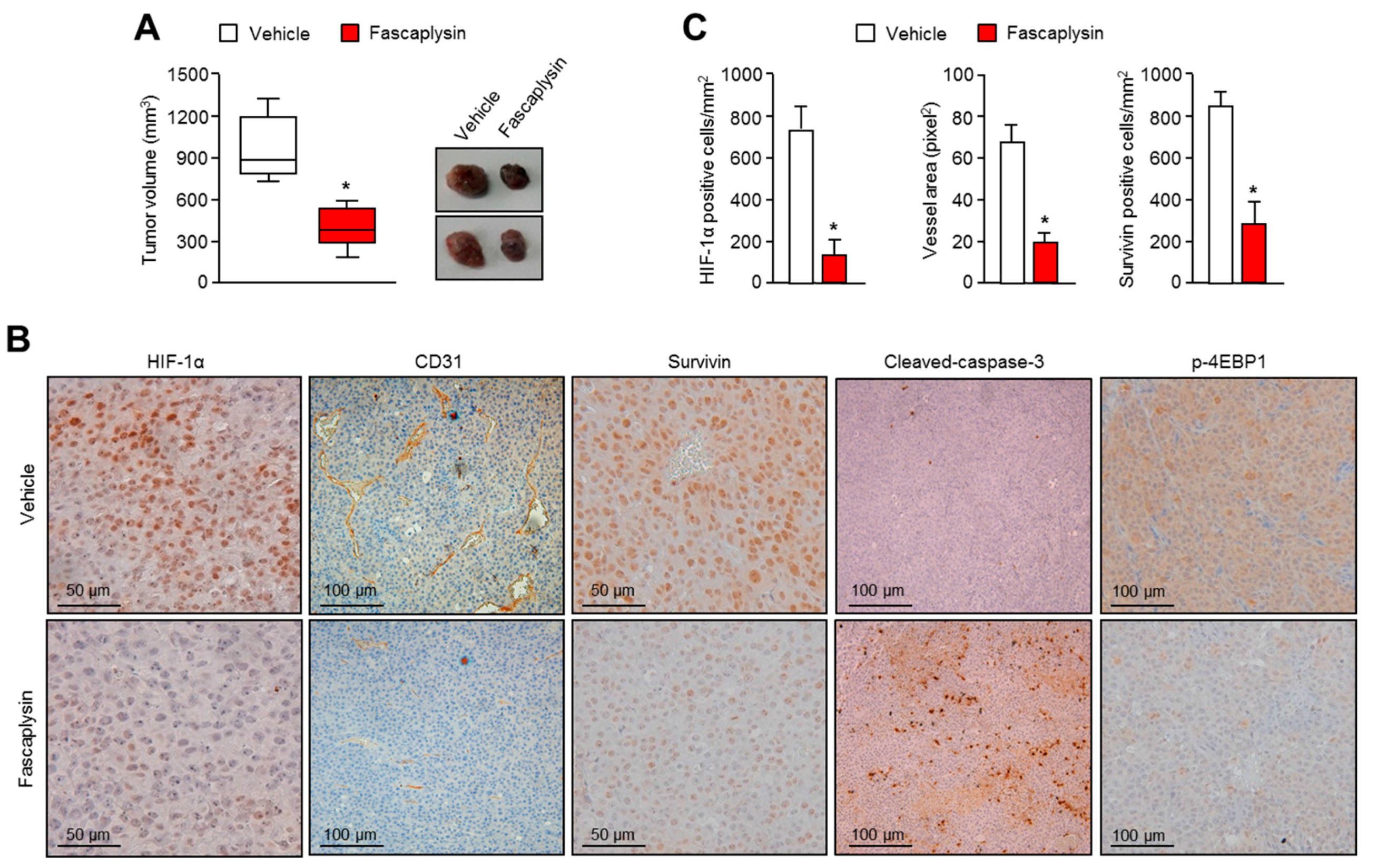
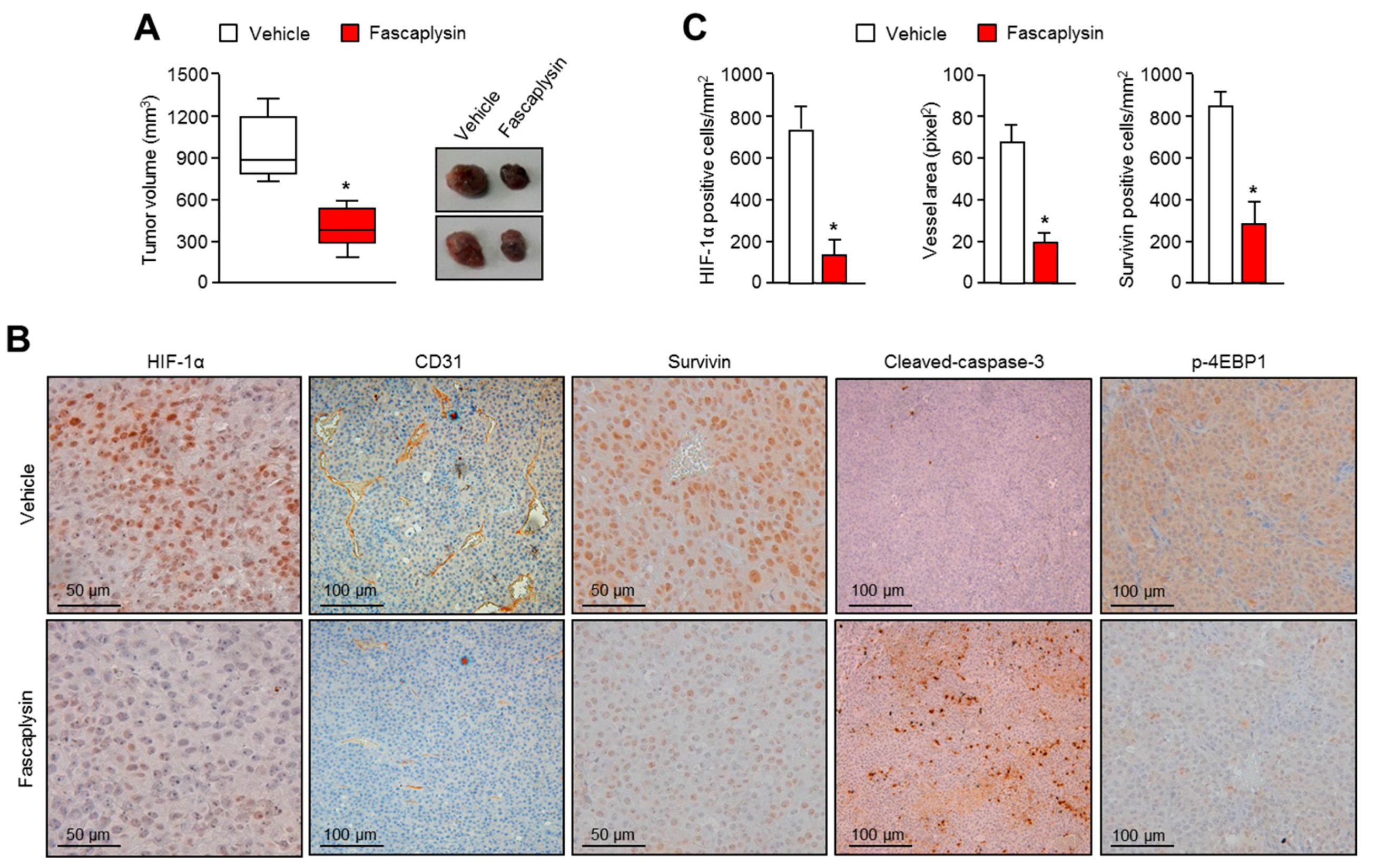
Although the precise molecular mechanism is not clearly understood, previous studies have revealed that fascaplysin attenuates tumor angiogenesis in vitro and in vivo [
21,
23]. In this study, a drug–protein docking simulation and several biochemical assays suggest that fascaplysin inhibits VEGFR2, which is a potential target for cancer treatment. Moreover, the administration of fascaplysin strongly reduced the accumulation of HIF-1α, which is a key transcriptional factor for regulating tumor angiogenesis in hypoxic tumor in vitro and in vivo. Consistently, histological analysis revealed that fascaplysin decreased blood vessels in xenografted tumor tissues. These findings suggest that fascaplysin attenuates tumor angiogenesis via the suppression of HIF-1α and VEGFR2.
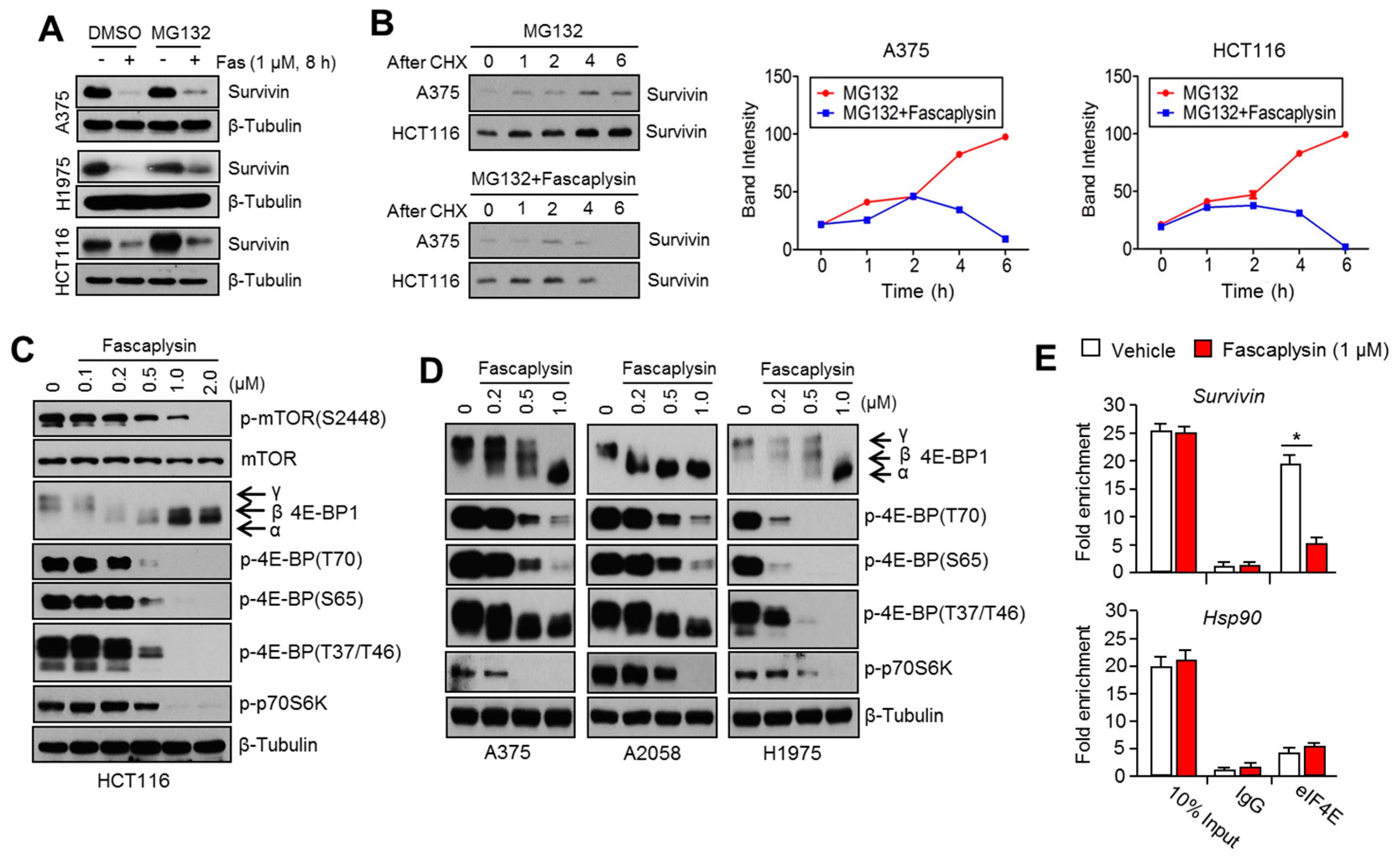
Given the fact that cap-dependent translation regulated by the mTOR-4EBP1-eIF4E axis fine-tunes the expression of several oncoproteins, such as survivin, c-myc, cyclin D1, VEGF, and HIF-1α, all of which are called eIF4E-sensitive mRNAs, at the post-transcriptional level [
18,
19], therapeutic suppression of cap-dependent translation has been considered as a valuable strategy for cancer-targeted therapy. Indeed, several reports have shown that small molecules targeting the mTOR-4EBP1-eIF4E axis reduces cancer growth in many experimental models [
18,
19,
24]. Herein, we found that fascaplysin suppresses the phosphorylation of mTOR, 4EBP1, and p70S6K1, all of which are pivotal signaling molecules for the cap-dependent translation machinery. Additionally, we showed that fascaplysin attenuated survivin protein synthesis by dissociating eIF4E from survivin mRNA. These findings reveal that fascaplysin could be considered as a candidate for the development of anti-cancer drugs targeting cap-dependent translation machinery.
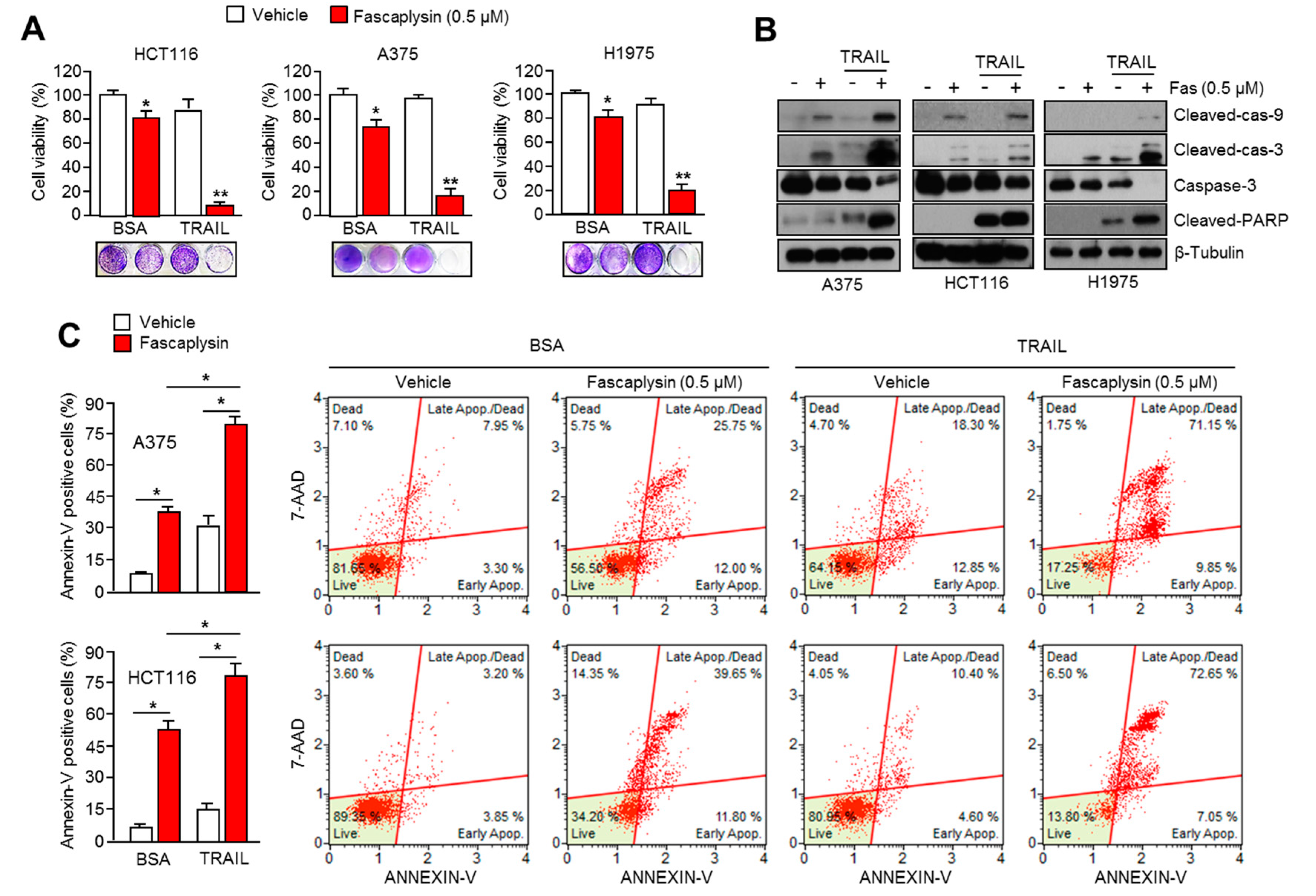
Although it is clear that TRAIL is a promising agent for cancer-targeted therapy because of its tumor selectivity, its low efficacy has been observed in many tumors due to the resistance to TRAIL-based therapies [
17,
25]. Many reports have shown that several chemopreventive agents and anti-cancer drugs, including PPARγ ligands, resveratrol, aspirin, HDAC inhibitors, and proteasome inhibitors could improve the anti-cancer efficacy of TRAIL-based therapies [
13,
14,
15,
16]. The importance of survivin suppression in cancer treatment has also been demonstrated by its ability to overcome resistance to TRAIL-based cancer therapy. Indeed, a previous report has shown that the pharmacological inhibition or genetic silencing of survivin sensitizes tumors to TRAIL-based cancer therapy [
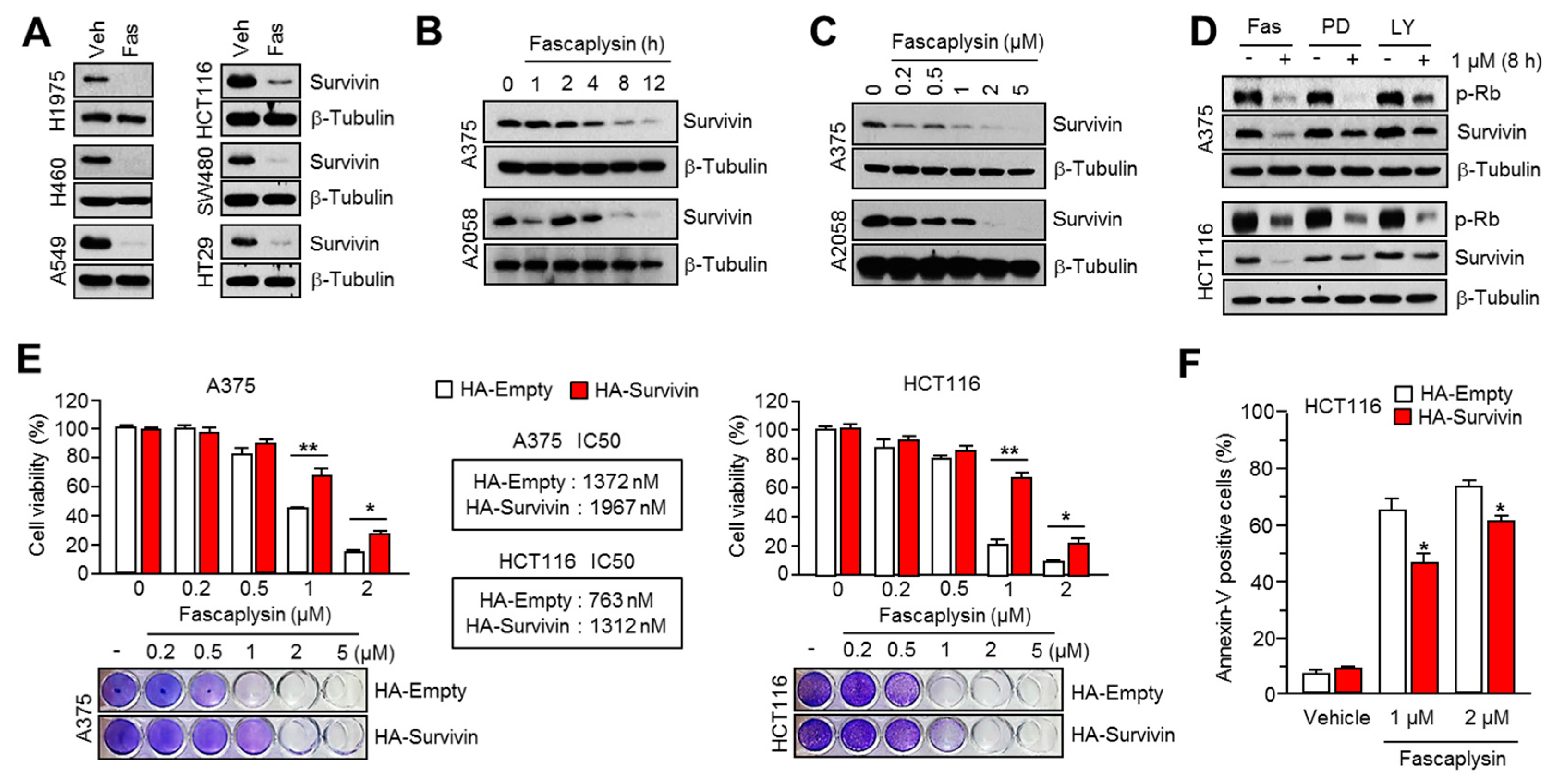
16]. Consistently, fascaplysin suppresses survivin expression, and here we found that TRAIL-induced cancer cell death was significantly increased by fascaplysin in malignant melanoma, colorectal, and lung cancer cells. These results suggest that fascaplysin might be a potential agent for overcoming resistance to TRAIL-based cancer therapies.
4. Materials and Methods
4.1. Reagents and Antibodies
Fascaplysin, PD0332991, and LY2835219 were purchased from Selleck Chemicals (Houston, TX, USA). Z-VAD-FMK, MG132, MAZ51, and cycloheximide were purchased from Sigma Aldrich (St. Louis, MO, USA). Recombinant TRAIL was purchased from PeproTech (Rocky Hill, NJ, USA). For Western blotting, antibodies recognizing phospho-mTOR (CST-5536), mTOR (CST-2983), phospho-4EBP1T70(sc-18092), phospho-4EBP1S65(sc-18091), phospho-4EBP1T37/T46(CST-2855), 4EBP1 (CST-9452), phospho-p70S6K1 (CST-9234), phospho-RB (CST-8516), cleaved-caspase-9 (CST-9505), cleaved-caspase-3 (CST-9664), cleaved-PARP (CST-5625), c-myc (sc-40), cyclin D1 (sc-8396), and β-tubulin (sc-9104) were purchased from Cell Signaling Technology (Danvers, MA, USA) and Santa Cruz Biotechnology (Dallas, TX, USA). Antibodies recognizing survivin (AF886), and HIF-1α (ab1) were purchased from R&D systems, Inc. (Minneapolis, MN, USA) and Abcam (Cambridge, MA, USA).
4.2. Cell Culture and Generation of Stable Cell Lines
All cancer cell lines were maintained in Dulbecco’s Modified Eagle’s medium (DMEM) containing 10% fetal bovine serum and 25 mM glucose. The human survivin–expressed lentiviral vector, pLenti-GIII-CMV-Survivin (LV089651), was purchased from Applied Biological Materials Inc. (Richmond, BC, Canada). The lentiviral and packaging vectors were co-transfected into HEK293T cells, the lentiviral particles were collected after 72 h of transfection. The lentiviral particles were infected into A375 or HCT116 cells, and survivin-overexpressing cells were selected by 2 µg/mL of puromycin (Invitrogen, Carlsbad, CA, USA) for six days.
4.3. Western Blotting
The cells were lysed with RIPA buffer containing 50 mM Tris-HCl (pH 7.9), 150 mM NaCl, 1% IGEPAL, 10 mM NaF, 0.1 mM EDTA, and a protease inhibitor cocktail. Protein samples were loaded onto sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred onto a PVDF membrane (Millipore, Billerica, MA, USA). Afterwards, primary antibodies were added to the membrane (1:1000–1:5000) at 4 °C for overnight. The second day, the membrane was reacted with secondary antibodies for 1 h at room temperature. The protein band appeared by adding substrate (Enhanced Chemiluminescence (ECL) Prime kit, GE healthcare, Pittsburgh, PA, USA).
4.4. RNA-Binding Protein Immunoprecipitation (RIP) Assay
As previously described [
26], cytoplasmic extracts were prepared using a lysis buffer containing IGEPAL (0.5%), KCl (120 mM), Tris-HCl (pH 7.9, 50 mM), EDTA (2 mM), RNase inhibitor (100 U/mL), and a protease inhibitor cocktail. Cell lysates were incubated with 1 µg/mL of antibody against eIF4E or normal IgG at 4 °C overnight, and then eIF4E-bound RNAs were measured by quantitative real time (RT)-PCR.
4.5. Quantitative Real Time-PCR
Total RNA was isolated from cells using TRIzol (Invitrogen), and RNA (2 µg) was reverse-transcribed—by a high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster city, CA, USA). Quantitative real time PCR was performed by equal amounts of cDNA mixed with SYBR Green PCR Master Mix (Applied Biosystems), and the following primers (5′–3′). CTTTCTCCGCAGTTTCCTCA and TTGGTGAATTTTTGAAACTGGA for survivin; GGCATTGATGACTCCAGTGTT and ATGGAGCCCAGCAGCAA for GLUT1; AGCTGCGCTGATAGACATCC and CTACCTCCACCATGCCAAGT for VEGF; GGAGATCCATCATCTCTCCC and GGCCTGTGCCATCAGTATCT for LDHA; ATTTTCCTCAAAGGAACGCC and CAACAGAGGTGTTTACCCCC for PDK1.
4.6. Tumor Xenograft Assay and Immunohistochemistry
As previously described [
27], A375 malignant melanoma cells (1 × 10
6) were subcutaneously injected into the flank of Balb/c-nude mice in 100 µL of FBS-free media. Three weeks after cell injection, mice were injected daily with fascaplysin (1 mg/kg) or DMSO by intraperitoneal injection in the control group for 10 days. Tumor volumes were measured with calipers and calculated using the following equation: volume = ab
2/2, where a is the maximal width and b is the maximal orthogonal width. The animal experiments were conducted and managed in accordance with the guidelines of the Konkuk University Institutional Animal Care and Use Committee (KU15072-1; approved at 21 September 2015). Immunohistochemical staining was performed as previously described [
28]. Tumor tissues were fixed with 4% paraformaldehyde, embedded in paraffin, and cut into 6-µM sections. They were deparaffinized and rehydrated, and then autoclaved for 10 min in 10 mM sodium citrate (pH 6.0) to retrieve the target antigens. These sections were then incubated with anti-HIF-1α (1:50; provided by Jong-Wan Park in Seoul National University), anti-CD31 (1:50; ab28364), anti-survivin (1:100; sc-17779), anti-cleaved-caspase-3 (1:50; CST-9991), or anti-phospho-4EBP1 (1:50; CST-2855) overnight at 4 °C. After the primary antibody reaction, sections were further incubated with biotinylated secondary antibodies. Finally, immunocomplexes were visualized using diaminobenzidine (DAB) staining.
4.7. Docking Simulation Studies
Docking simulation studies of TRKA and VEGFR2 in complex with fascaplysin were conducted with the Discovery Studio 4.5 (San Diego, CA, USA, version 4.5) using the all-atom model prepared by adding the missing atoms to the original X-ray crystal structure. The three-dimensional atomic coordinates required for the docking study were prepared from the X-ray crystal structure of TRKA (PDB entry: 4PMP) and VEGFR2 (PDB entry: 3U5J) in complex with fascaplysin. These uniquely defined potential grids for TRKA and VEGFR2 were then used in common for docking simulations. To construct the all-atom model for TRKA and VEGFR2, hydrogen atoms were added to the individual protein atoms after removing the crystallographic water molecules. The ionization states of the titratable residues (Asp, Glu, His, and Lys) were determined prior to building the model according to the hydrogen bond formation patterns revealed in the original X-ray structures.
4.8. Kinase Inhibition Assay
The inhibitory activities of fascaplysin with respect to TRKA and VEGFR2 were measured by means of radiometric kinase assays ([γ-33P]ATP) (Reaction Biology Corp. Malvern, PA, USA). The enzymatic activity of TRKA and VEGFR2 was monitored using 20 µM of peptide substrate, dissolved in the freshly-prepared reaction buffer (20 mM HEPES (pH 7.5), 10 mM MgCl2, 1 mM EGTA, 0.02% BRIJ-35, 0.02 mg/mL BSA, 0.1 mM Na3VO4, 2 mM DTT, 1% DMSO). Each putative fascaplysin was dissolved in 100% DMSO at the specific concentration and diluted in a serial manner with epMotion 5070 in DMSO. Either TRKA (recombinant human cytoplasmic domain (aa 441–796, accession number NP_002520), C-terminal His-tagged, expressed in insect cells. Molecular weight (Mw) = 42.8 kDa) or VEGFR2 (recombinant human catalytic domain (aa 789–1356; accession number: NM_002244), C-terminal His-tagged, expressed in insect cells. Activated in vitro via autophosphorylation; Mw = 68.4 kDa) were added into the reaction buffer including 20 µM of peptide substrate. After delivering the candidate inhibitor dissolved in DMSO into the kinase reaction mixture by acoustic technology (Echo 550; nanoliter range), and the reaction mixture was incubated for 20 min at room temperature. To initiate the enzymatic reaction, 33P-ATP with specific activity of 10 µCi/µL was delivered into the reaction mixture to reach the final ATP concentration of 10 µM. Radioactivity was then monitored by the filter binding method after the incubation of reaction mixture for 2 h at room temperature. At given concentrations of fascaplysin, biochemical potency was measured by the percent of kinase activity remaining with respect to the vehicle (dimethyl sulfoxide) reaction. Curve fits and IC50 values were then obtained using the PRISM program (GraphPad Software, La Jolla, CA, USA, version 5.01). The kinase assays were performed at room temperature.
4.9. Measurement of Cell Viability
To measure cell viability, cells were seeded onto 12- or 24-well tissue culture dishes and incubated for one, two, or three days in the absence or presence of fascaplysin. After incubation, cultured cells were washed with phosphate-buffered saline (PBS) and then stained with 0.5% crystal violet. After 20 min, the stained cells were dissolved with 1% SDS solution for three times at room temperature, and then the optical density was read at 570 nm using microplate reader (BioTek, Winooski, VT, USA).
4.10. Apoptosis Assays
For the apoptosis assay, cells were seeded at a density 1 × 105 cells/well onto a six-well cell culture plate and treated with fascaplysin (0.5, 1 µM), TRAIL (20 ng/mL), and Z-VAD-FMK (20 µM) at various concentrations. The adherent cells were collected and washed with cold PBS, and then centrifuged at 2000 rpm for 2 min at room temperature. The pellet was resuspended in 1 mL of medium, and then 100 µL of the suspension was transferred into a fresh tube. Then, 100 µL of Muse™ Annexin V and Dead Cell kit reagents (EMD Millipore, Billerica, MA, USA) were added to each tube and incubated for 20 min at room temperature in the dark. The stained samples were analyzed using a Mini Flow Cytometry Muse™ Cell Analyzer (EMD Millipore).
4.11. Statistical Analysis
For analyze between two experimental data, the unpaired Student’s t-test was used. For multiple comparisons, one-way ANOVA with Tukey post-test was performed. Data are shown as means ± standard deviations (SD). If the p-value was under 0.05, the data was regarded as statistically significant.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}