Molecular Evolution of MERS Coronavirus: Dromedaries as a Recent Intermediate Host or Long-Time Animal Reservoir?

 and
and
Abstract
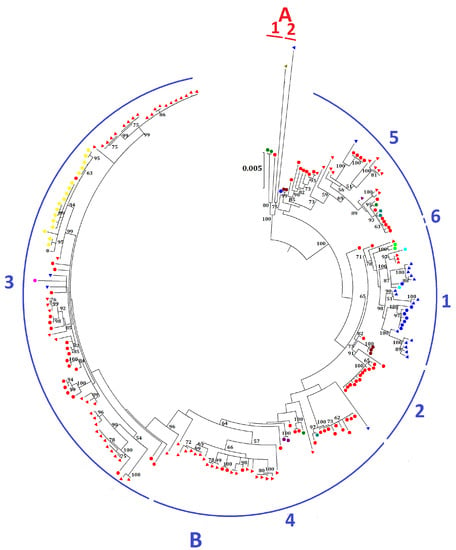
:
1. Introduction
2. Results
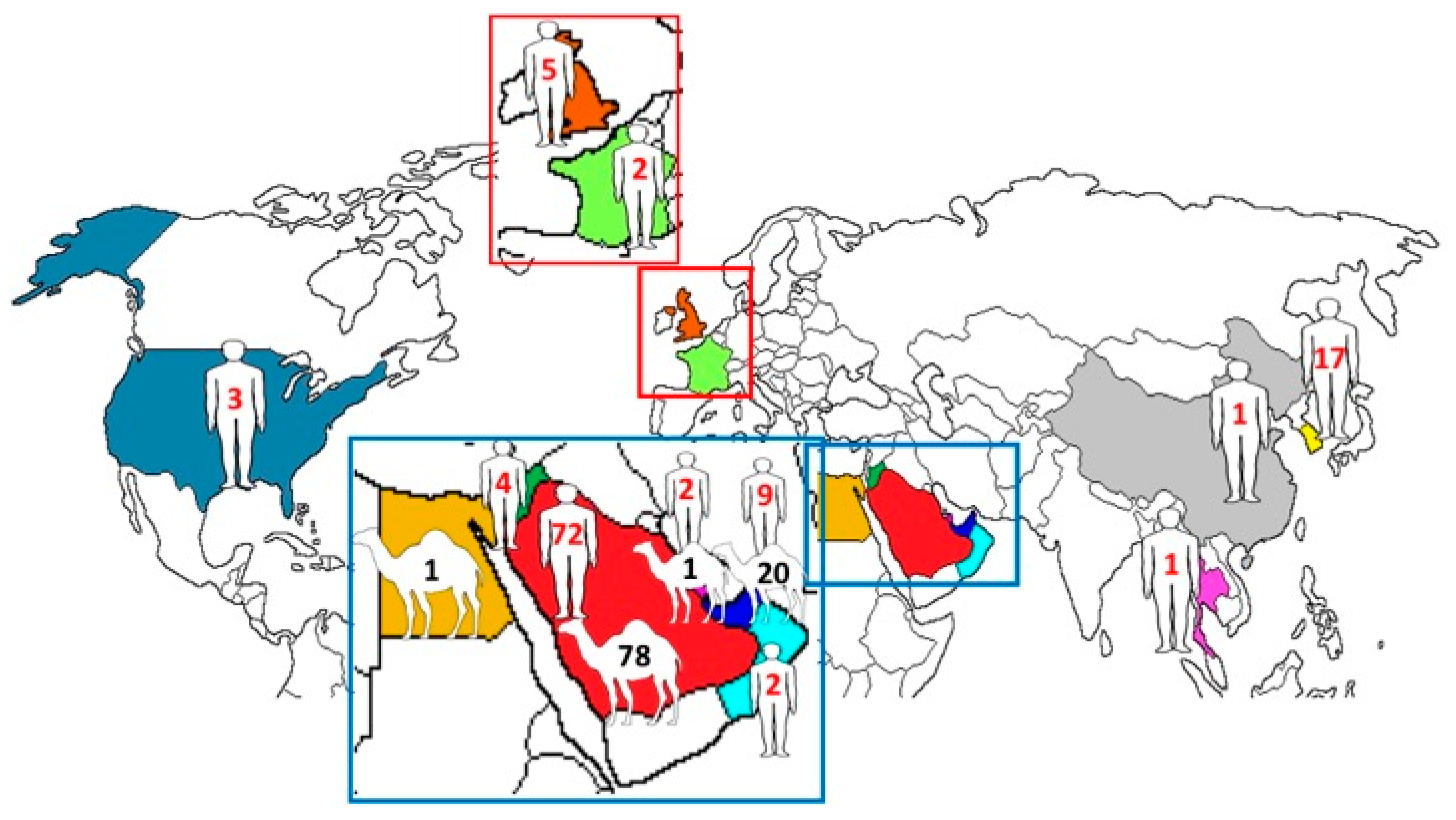
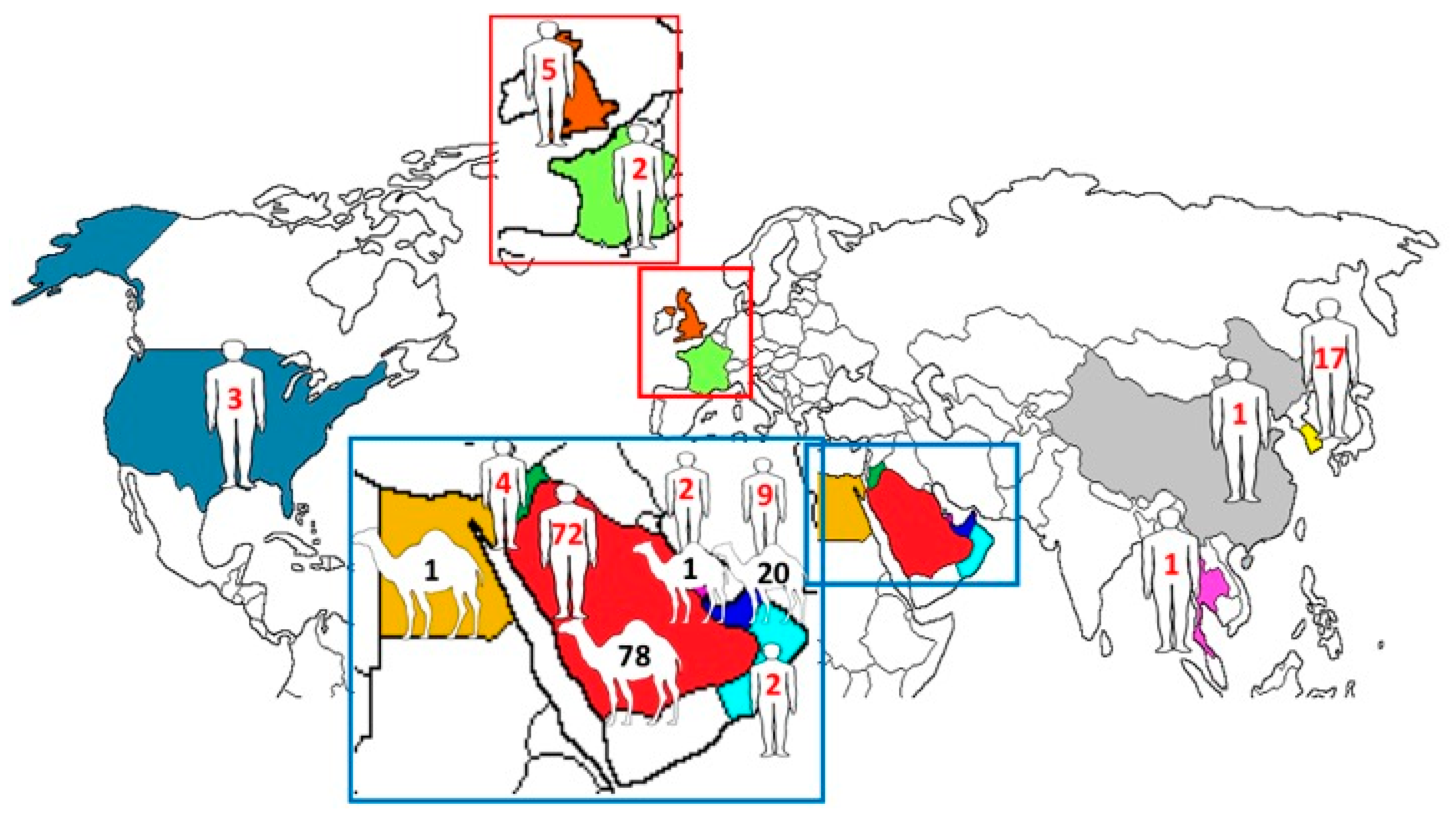
2.1. Human and Dromedary MERS-CoV Genomes
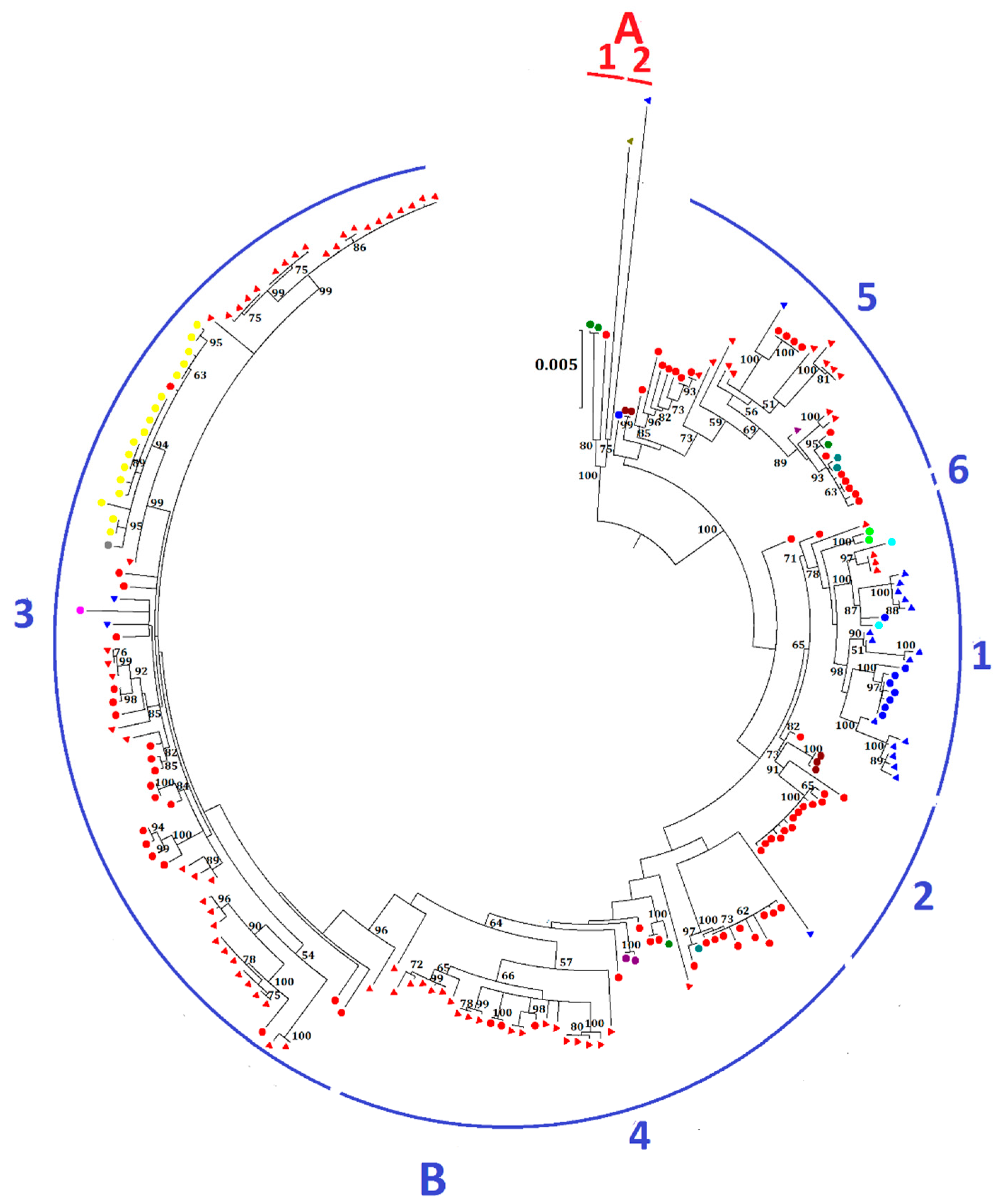
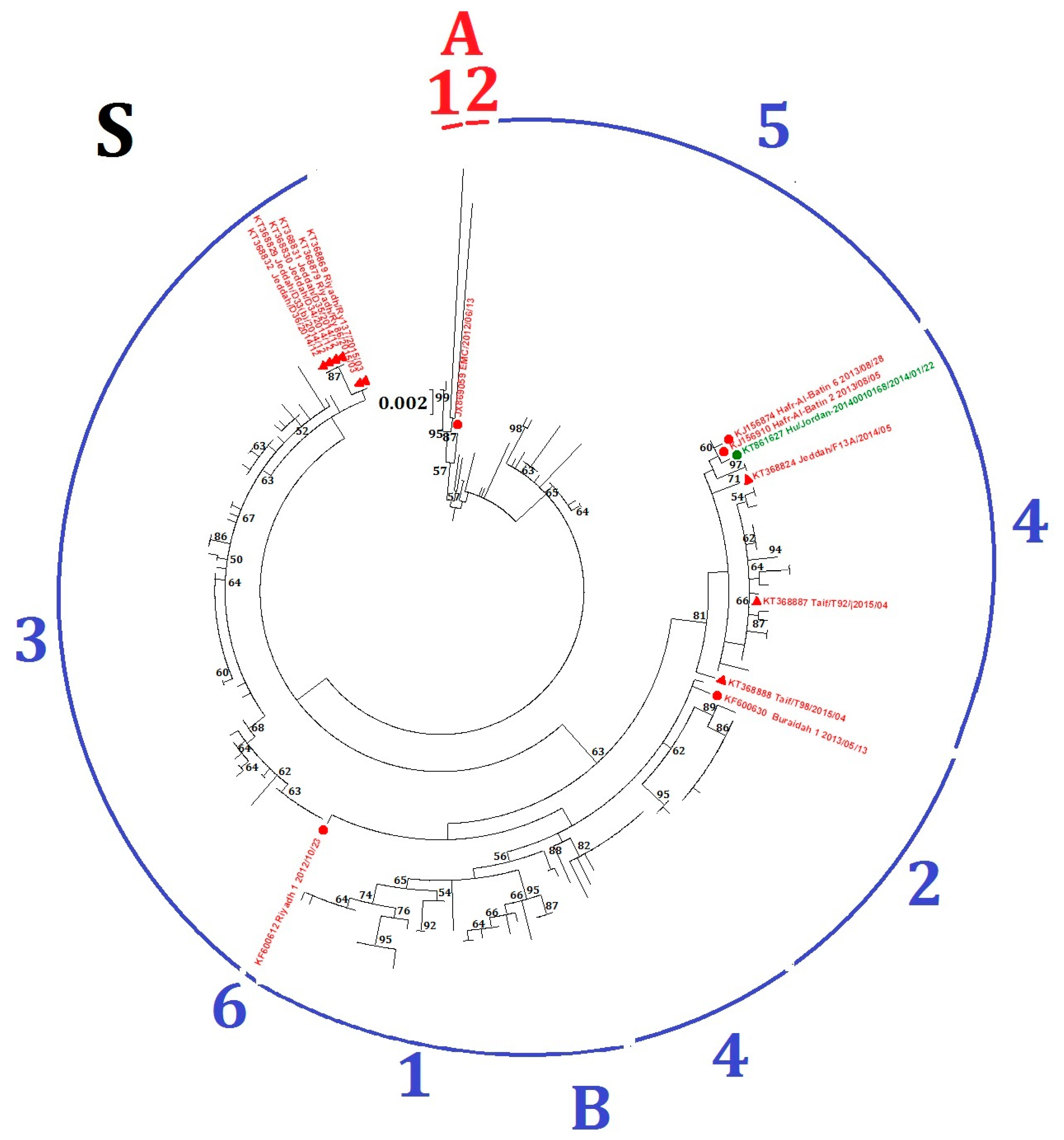
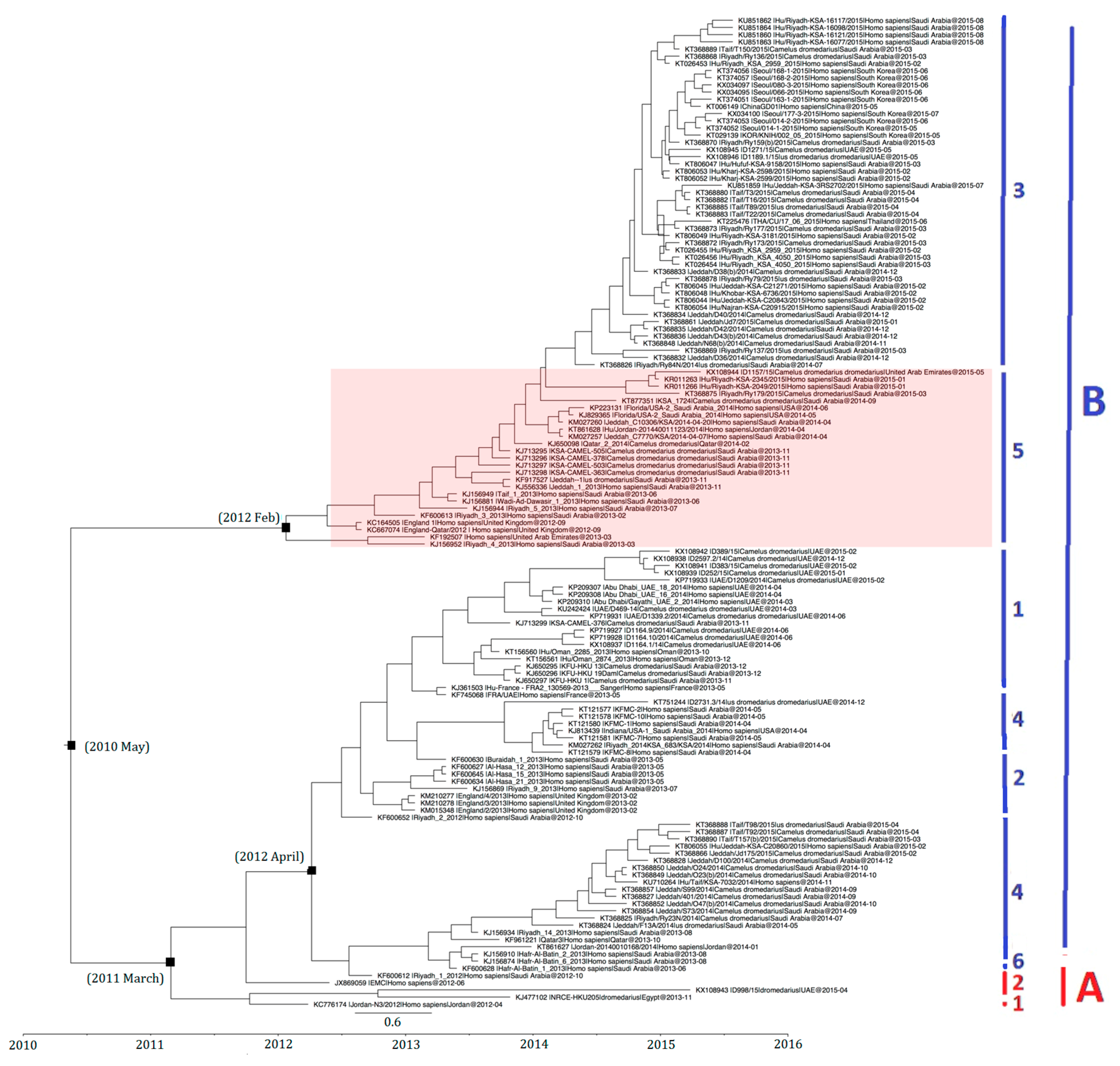
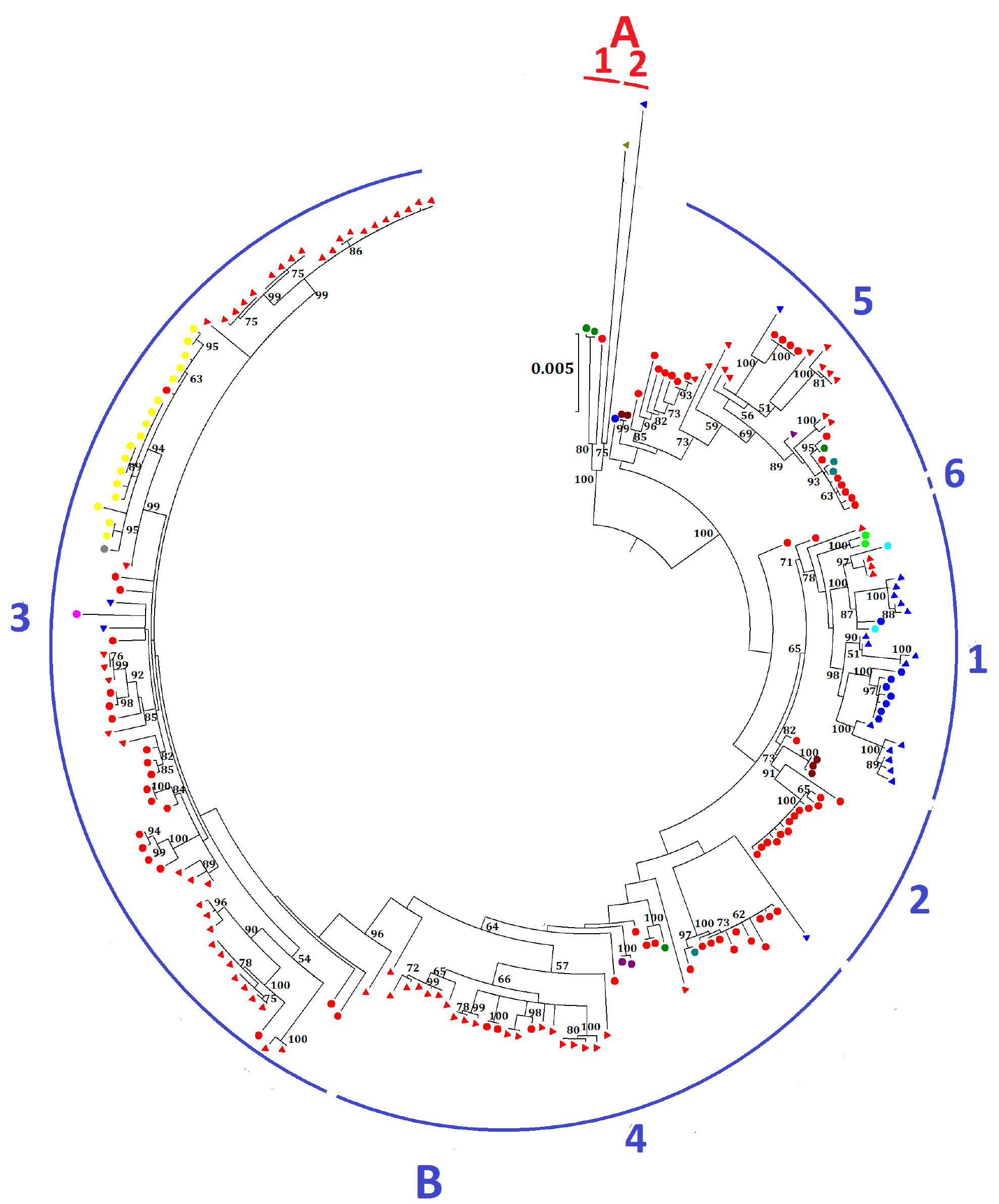
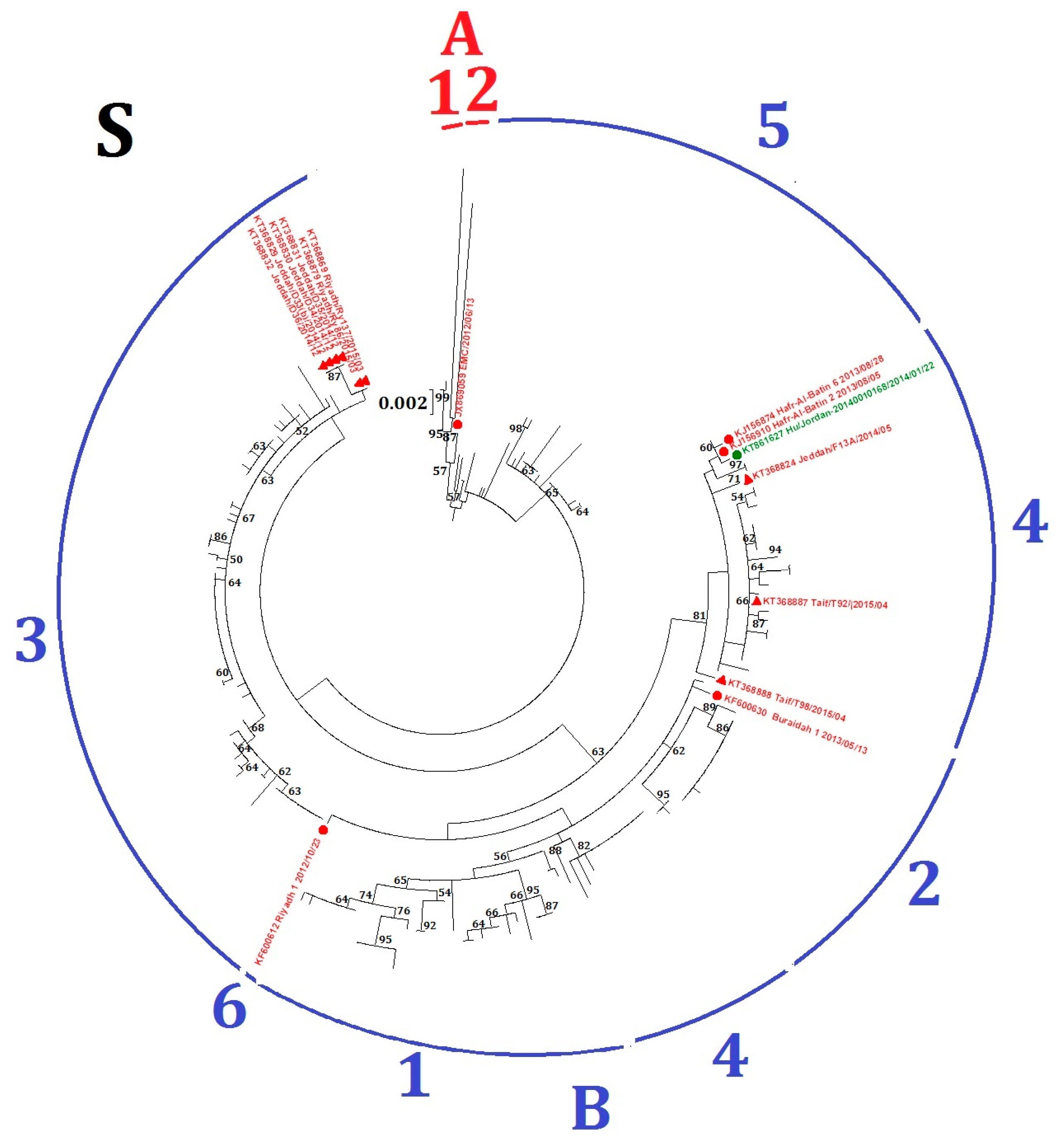
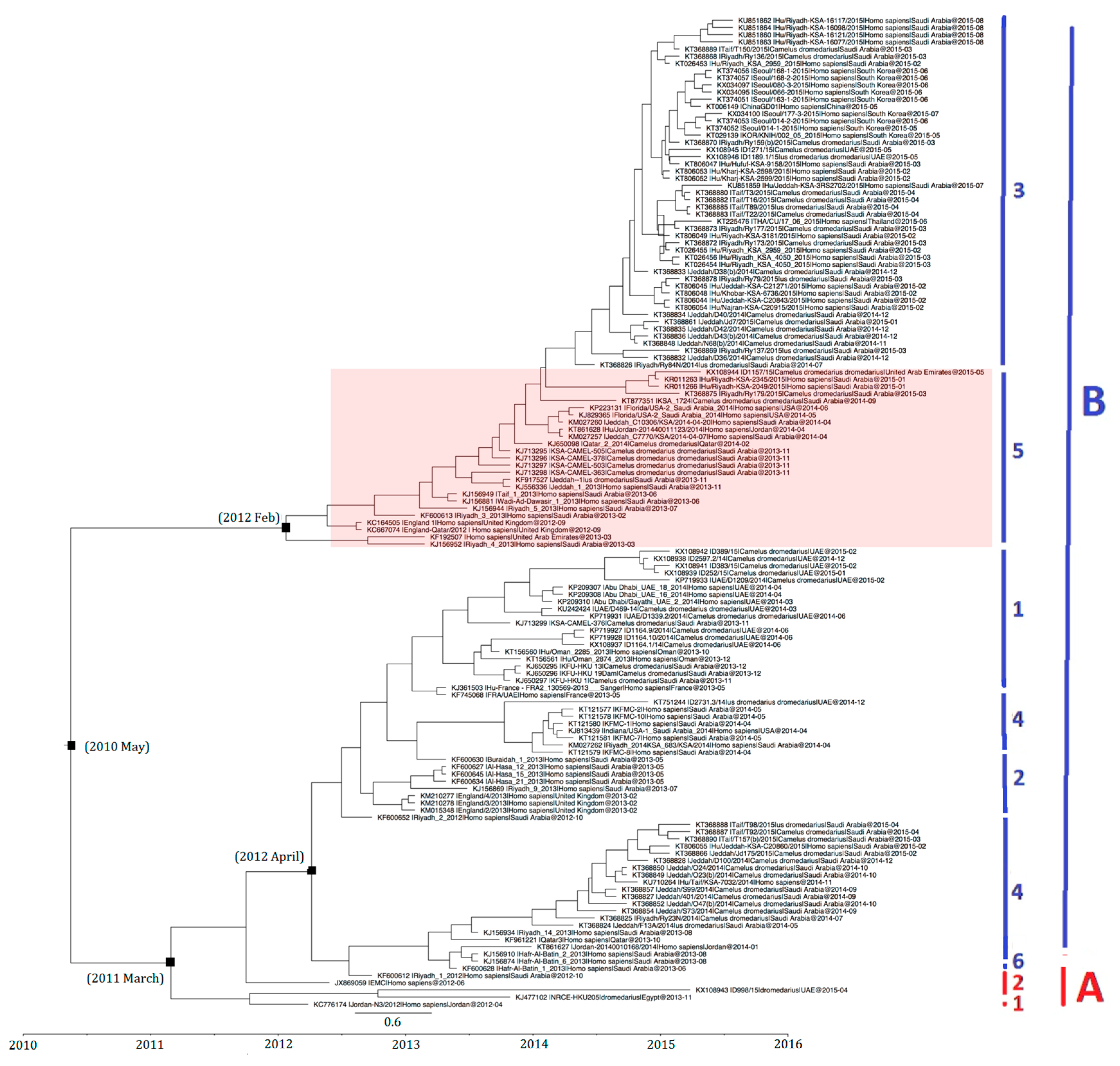
2.2. Phylogenetic Analysis and Genomic Sequence Comparison
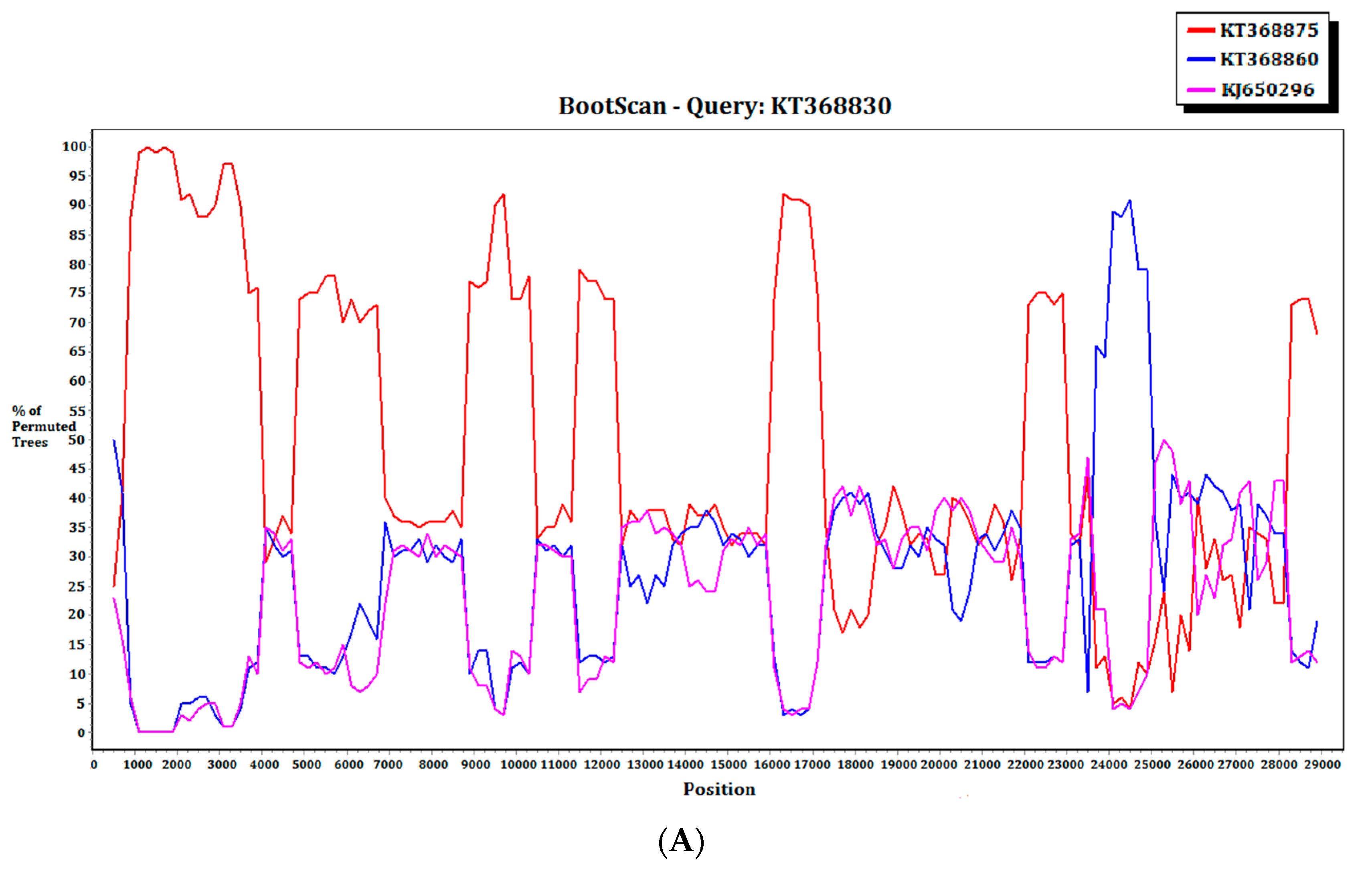
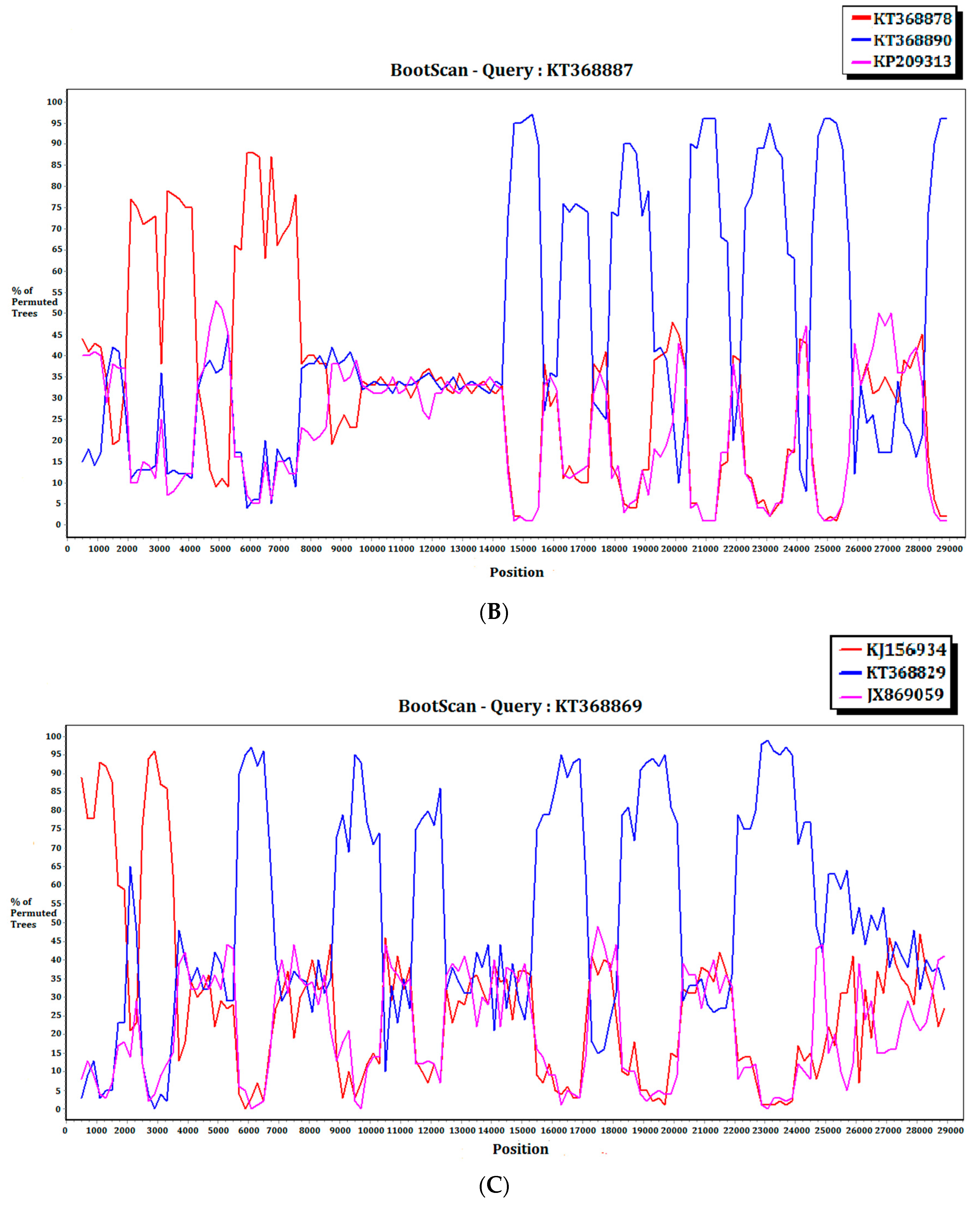
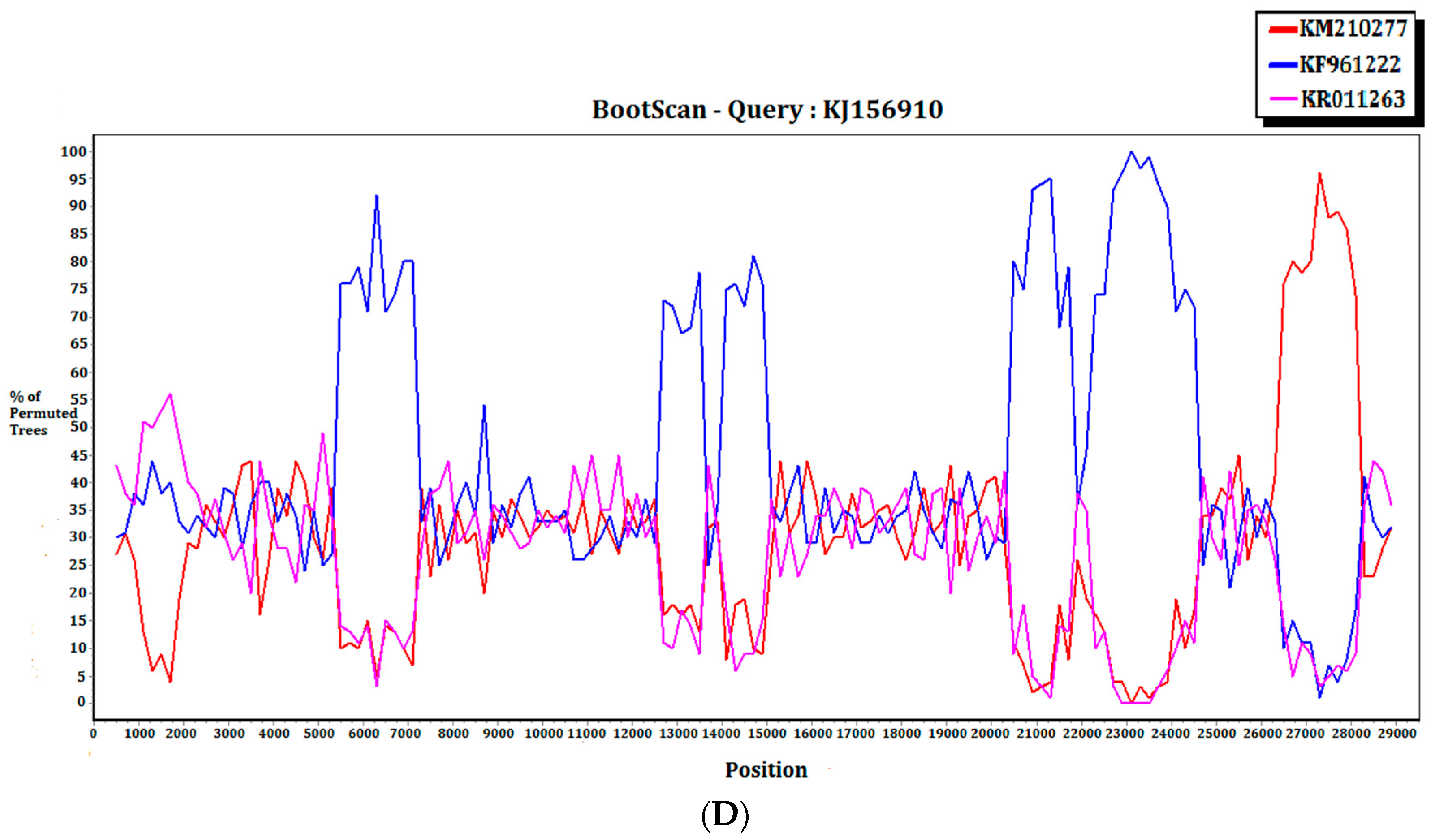
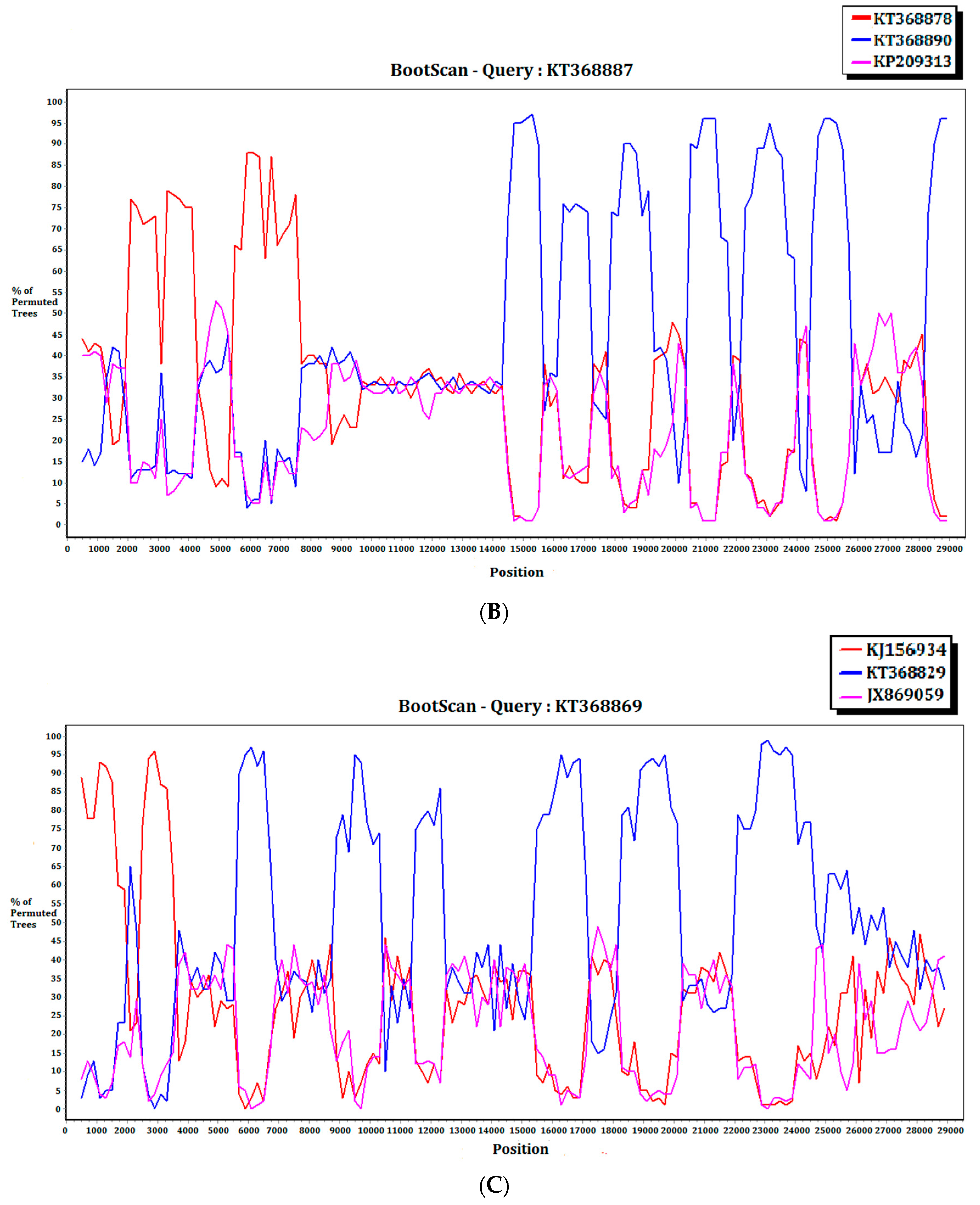
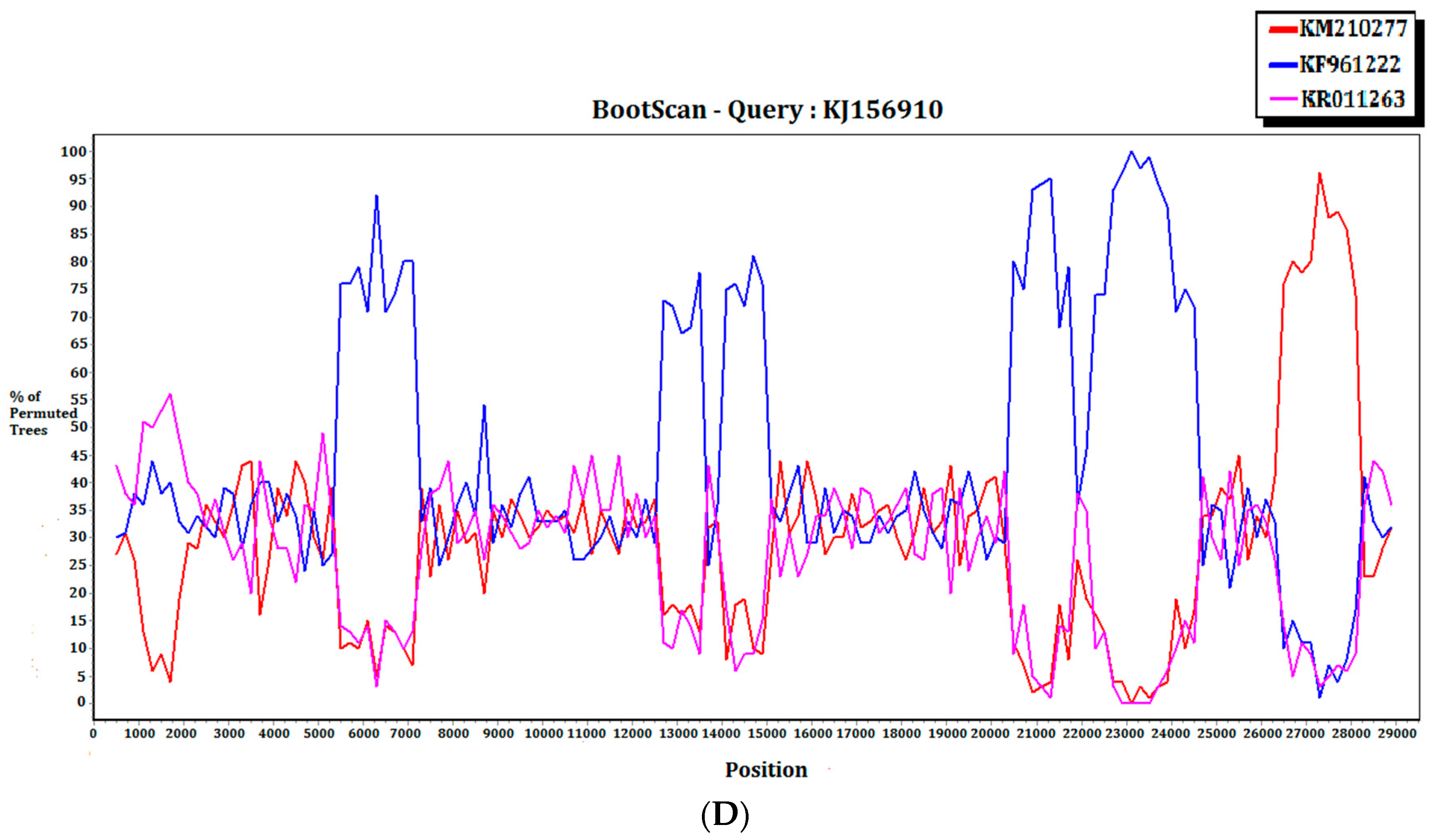
2.3. Recombination Analysis
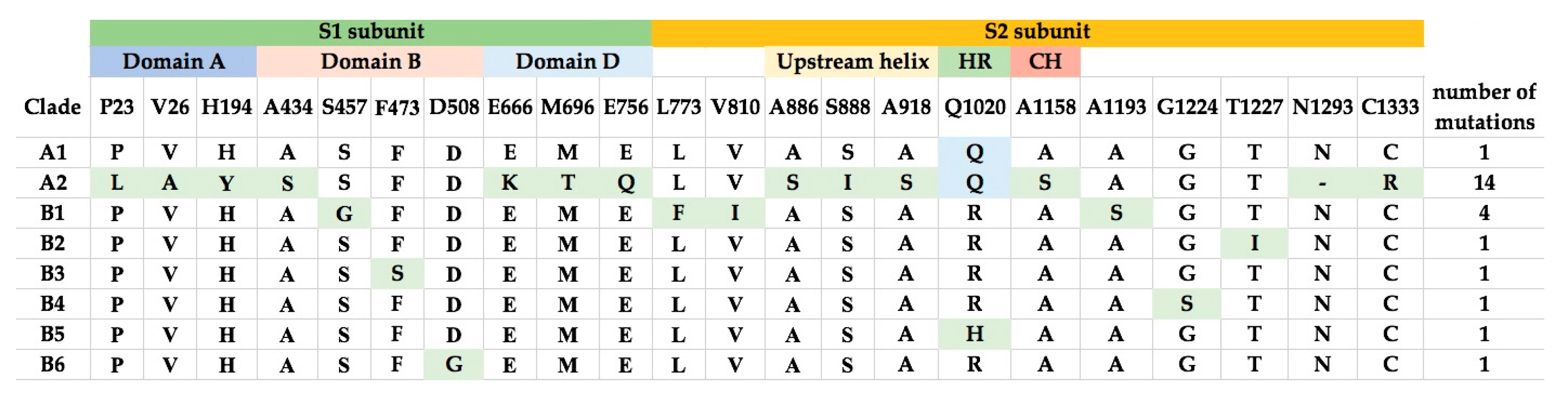
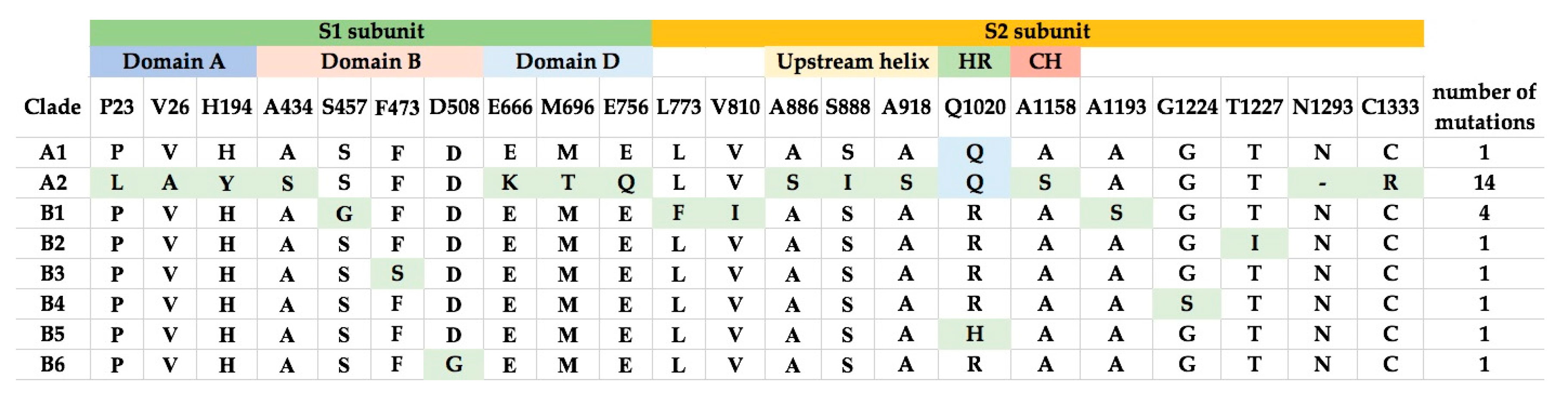
2.4. Multiple Alignment Analysis
2.5. Selective Pressure Analysis
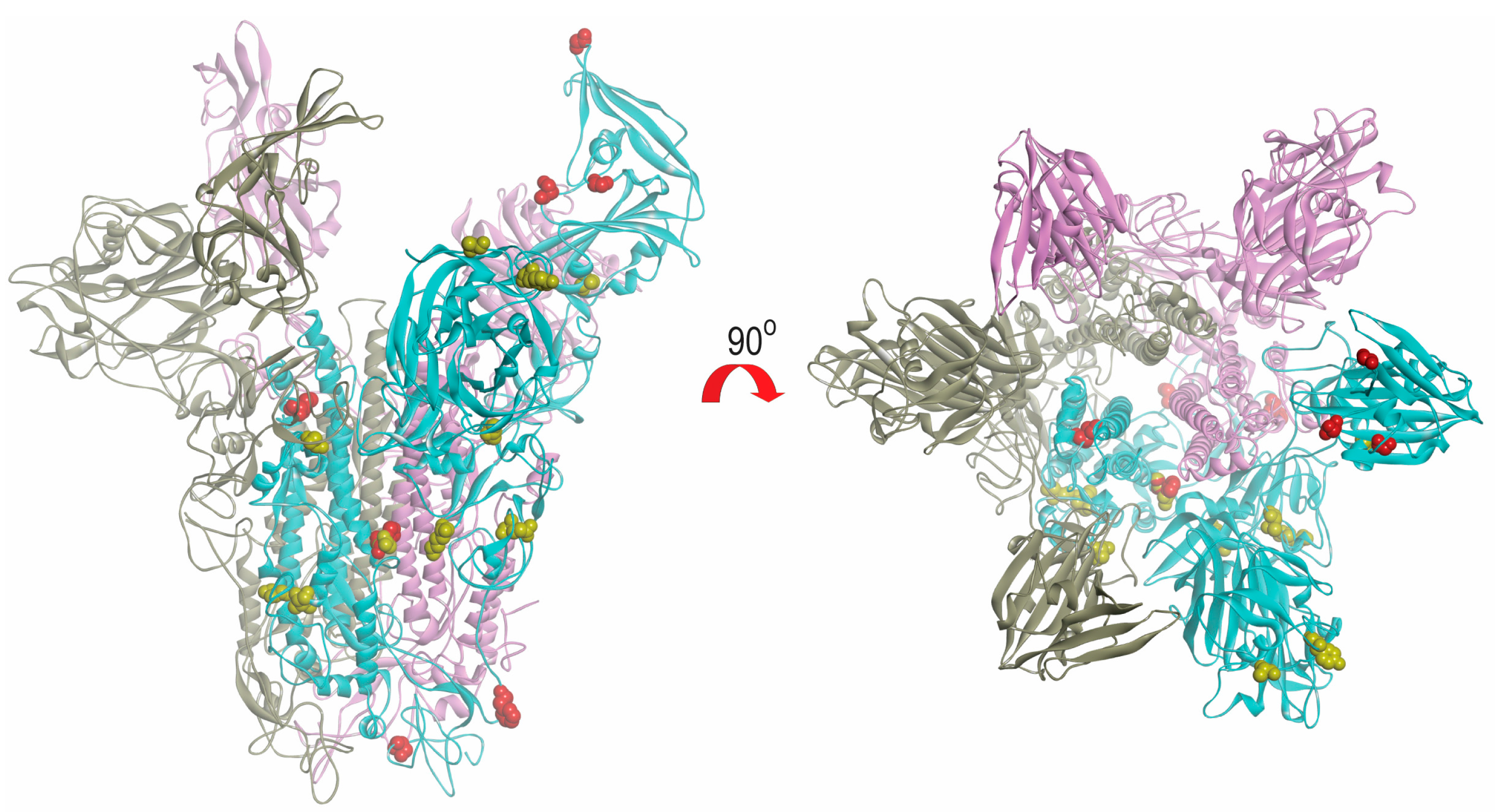
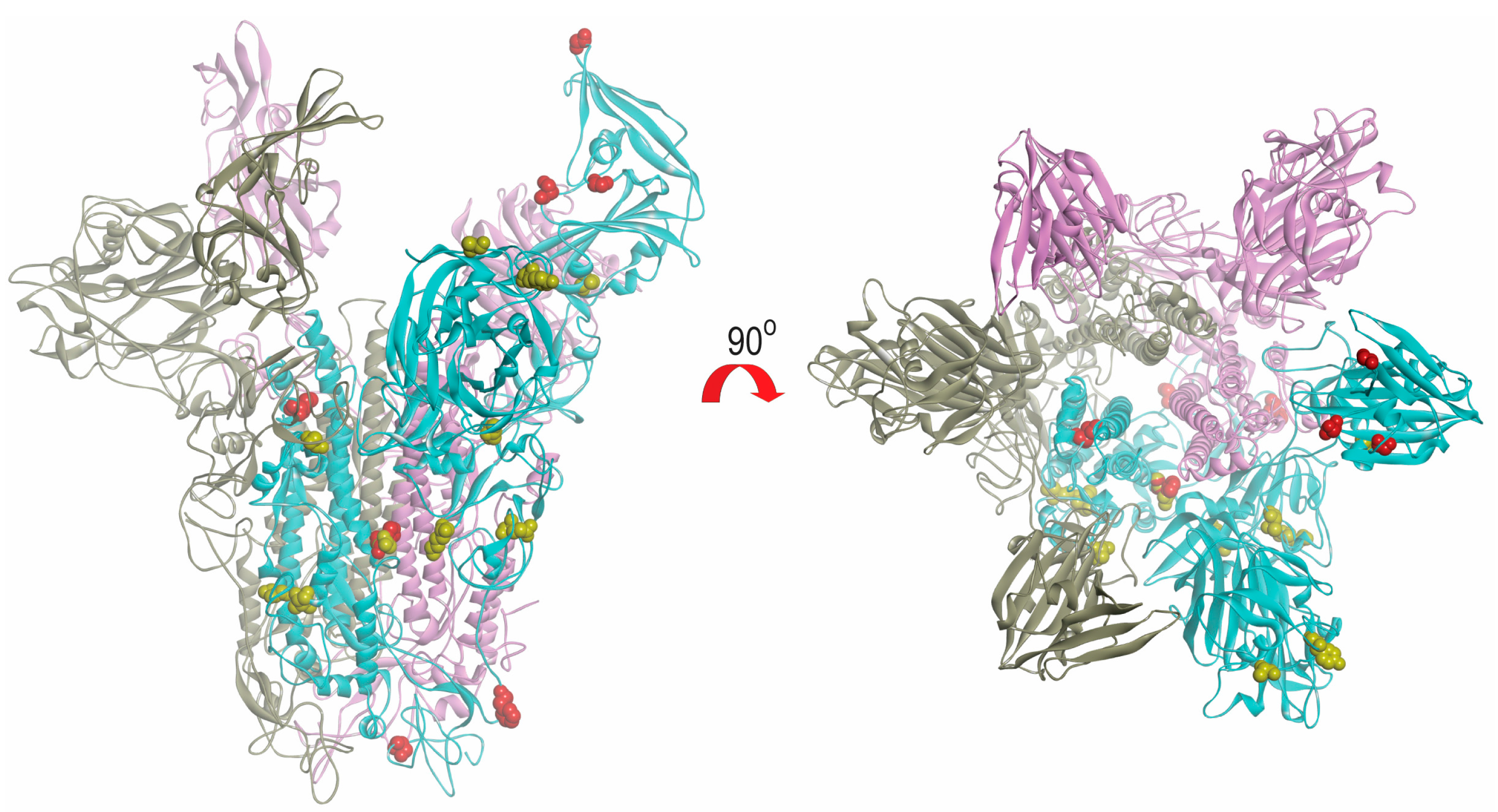
2.6. Modeling of Spike Protein
2.7. Estimation of Divergence Dates
3. Discussion
4. Materials and Methods
4.1. Human and Dromedary MERS-CoV Genomes
4.2. Phylogenetic Analysis and Genomic Sequence Comparison
4.3. Recombination Analysis
4.4. Multiple Alignment Analysis
4.5. Selective Pressure Analysis on Spike Protein
4.6. Modeling of Spike Protein
4.7. Estimation of Divergence Dates
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AIC | Akaike’s information criterion |
| BEAST | bayesian evolutionary analysis by sampling trees |
| dN | the number of non-synonymous substitutions per non-synonymous site |
| dS | The number of synonymous substitutions per synonymous site |
| FEL | fixed effects likelihood |
| HPD | highest posterior density |
| MCMC | markov chain Monte Carlo |
| MEME | mixed-effects model of evolution |
| MERS-CoV | middle east respiratory syndrome coronavirus |
| ORF | open reading frame |
| S | spike |
| SARS-CoV | severe acute respiratory syndrome coronavirus |
| SLAC | single-likelihood ancestor counting |
| tMRCA | the most recent common ancestor |
References
- Chan, J.F.; Lau, S.K.; To, K.K.; Cheng, V.C.; Woo, P.C.; Yuen, K.Y. Middle east respiratory syndrome coronavirus: Another zoonotic betacoronavirus causing sars-like disease. Clin. Microbiol. Rev. 2015, 28, 465–522. [Google Scholar] [CrossRef] [PubMed]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- Van Boheemen, S.; de Graaf, M.; Lauber, C.; Bestebroer, T.M.; Raj, V.S.; Zaki, A.M.; Osterhaus, A.D.; Haagmans, B.L.; Gorbalenya, A.E.; Snijder, E.J.; et al. Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. MBio 2012, 3, e00473-12. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.A.; Wang, P.; Gomaa, M.R.; El-Shesheny, R.; Kandeil, A.; Bagato, O.; Siu, L.Y.; Shehata, M.M.; Kayed, A.S.; Moatasim, Y.; et al. Seroepidemiology for MERS coronavirus using microneutralisation and pseudoparticle virus neutralisation assays reveal a high prevalence of antibody in dromedary camels in Egypt, June 2013. Eurosurveillance 2013, 18, 20574. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.; Lau, S.K.; Wernery, U.; Wong, E.Y.; Tsang, A.K.; Johnson, B.; Yip, C.C.; Lau, C.C.; Sivakumar, S.; Cai, J.P.; et al. Novel betacoronavirus in dromedaries of the middle east, 2013. Emerg. Infect. Dis. 2014, 20, 560–572. [Google Scholar] [CrossRef] [PubMed]
- Reusken, C.B.; Haagmans, B.L.; Muller, M.A.; Gutierrez, C.; Godeke, G.J.; Meyer, B.; Muth, D.; Raj, V.S.; Smits-De Vries, L.; Corman, V.M.; et al. Middle east respiratory syndrome coronavirus neutralising serum antibodies in dromedary camels: A comparative serological study. Lancet Infect. Dis. 2013, 13, 859–866. [Google Scholar] [CrossRef]
- Corman, V.M.; Kallies, R.; Philipps, H.; Gopner, G.; Muller, M.A.; Eckerle, I.; Brunink, S.; Drosten, C.; Drexler, J.F. Characterization of a novel betacoronavirus related to middle east respiratory syndrome coronavirus in european hedgehogs. J. Virol. 2014, 88, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.; Wang, M.; Lau, S.K.; Xu, H.; Poon, R.W.; Guo, R.; Wong, B.H.; Gao, K.; Tsoi, H.W.; Huang, Y.; et al. Comparative analysis of twelve genomes of three novel group 2c and group 2d coronaviruses reveals unique group and subgroup features. J. Virol. 2007, 81, 1574–1585. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.; Li, K.S.; Tsang, A.K.; Lam, C.S.; Ahmed, S.; Chen, H.; Chan, K.H.; Woo, P.C.; Yuen, K.Y. Genetic characterization of betacoronavirus lineage C viruses in bats reveals marked sequence divergence in the spike protein of pipistrellus bat coronavirus HKU5 in Japanese pipistrelle: Implications for the origin of the novel middle east respiratory syndrome coronavirus. J. Virol. 2013, 87, 8638–8650. [Google Scholar] [PubMed]
- Memish, Z.A.; Mishra, N.; Olival, K.J.; Fagbo, S.F.; Kapoor, V.; Epstein, J.H.; Alhakeem, R.; Durosinloun, A.; Al Asmari, M.; Islam, A.; et al. Middle east respiratory syndrome coronavirus in bats, Saudi Arabia. Emerg. Infect. Dis. 2013, 19, 1819–1823. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Du, L.; Liu, C.; Wang, L.; Ma, C.; Tang, J.; Baric, R.S.; Jiang, S.; Li, F. Receptor usage and cell entry of bat coronavirus HKU4 provide insight into bat-to-human transmission of MERS coronavirus. Proc. Natl. Acad. Sci. USA 2014, 111, 12516–12521. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Qi, J.; Yuan, Y.; Xuan, Y.; Han, P.; Wan, Y.; Ji, W.; Li, Y.; Wu, Y.; Wang, J.; et al. Bat origins of MERS-CoV supported by bat coronavirus HKU4 usage of human receptor CD26. Cell Host Microbe 2014, 16, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.; Wernery, R.; Wong, E.Y.; Joseph, S.; Tsang, A.K.; Patteril, N.A.; Elizabeth, S.K.; Chan, K.H.; Muhammed, R.; Kinne, J.; et al. Polyphyletic origin of MERS coronaviruses and isolation of a novel clade a strain from dromedary camels in the United Arab Emirates. Emerg. Microbes Infect. 2016, 5, e128. [Google Scholar] [CrossRef] [PubMed]
- Cowling, B.J.; Park, M.; Fang, V.J.; Wu, P.; Leung, G.M.; Wu, J.T. Preliminary epidemiological assessment of MERS-CoV outbreak in South Korea, May to June 2015. Eurosurveillance 2015, 20, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, D.; Shi, W.; Lu, R.; Wang, W.; Zhao, Y.; Deng, Y.; Zhou, W.; Ren, H.; Wu, J.; et al. Origin and possible genetic recombination of the middle east respiratory syndrome coronavirus from the first imported case in China: Phylogenetics and coalescence analysis. Mbio 2015, 6, e01280-15. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.; Lau, S.K.; Yip, C.C.; Huang, Y.; Tsoi, H.W.; Chan, K.H.; Yuen, K.Y. Comparative analysis of 22 coronavirus HKU1 genomes reveals a novel genotype and evidence of natural recombination in coronavirus HKU1. J. Virol. 2006, 80, 7136–7145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, S.K.; Lee, P.; Tsang, A.K.; Yip, C.C.; Tse, H.; Lee, R.A.; So, L.Y.; Lau, Y.L.; Chan, K.H.; Woo, P.C.; et al. Molecular epidemiology of human coronavirus OC43 reveals evolution of different genotypes over time and recent emergence of a novel genotype due to natural recombination. J. Virol. 2011, 85, 11325–11337. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Wang, Y.; Chen, Y.; Wu, B.; Qin, K.; Zhao, J.; Lou, Y.; Tan, W. Discovery of a novel canine respiratory coronavirus support genetic recombination among betacoronavirus 1. Virus Res. 2017, 237, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.A.; Corman, V.M.; Jores, J.; Meyer, B.; Younan, M.; Liljander, A.; Bosch, B.J.; Lattwein, E.; Hilali, M.; Musa, B.E.; et al. MERS coronavirus neutralizing antibodies in camels, eastern Africa, 1983–1997. Emerg. Infect. Dis. 2014, 20, 2093–2095. [Google Scholar] [CrossRef] [PubMed]
- Corman, V.M.; Jores, J.; Meyer, B.; Younan, M.; Liljander, A.; Said, M.Y.; Gluecks, I.; Lattwein, E.; Bosch, B.J.; Drexler, J.F.; et al. Antibodies against MERS coronavirus in dromedary camels, Kenya, 1992–2013. Emerg. Infect. Dis. 2014, 20, 1319–1322. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.; Yip, C.C.; Zhao, P.S.; Chow, W.N.; To, K.K.; Wu, A.K.; Yuen, K.Y.; Woo, P.C. Enterovirus D68 infections associated with severe respiratory illness in elderly patients and emergence of a novel clade in Hong Kong. Sci. Rep. 2016, 6, 25147. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Frost, S.D. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [PubMed]
- Pond, S.L.; Frost, S.D. Datamonkey: Rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 2005, 21, 2531–2533. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Rambaut, A. Beast: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, S.K.; Woo, P.C.; Li, K.S.; Tsang, A.K.; Fan, R.Y.; Luk, H.K.; Cai, J.P.; Chan, K.H.; Zheng, B.J.; Wang, M.; et al. Discovery of a novel coronavirus, china rattus coronavirus HKU24, from Norway rats supports the murine origin of betacoronavirus 1 and has implications for the ancestor of betacoronavirus lineage A. J. Virol. 2015, 89, 3076–3092. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GenBank Accession No. | Country of Isolation | Human/Dromedary | Lineage Whole Genome | ORF1ab | S |
|---|---|---|---|---|---|
| JX869059 | Saudi Arabia | Human | A1 | A1 | A2 |
| KF600630 | Saudi Arabia | Human | B1 | B1 | B2 |
| KT368869 | Saudi Arabia | Dromedary | B3 | B4 | B3 |
| KT368879 | Saudi Arabia | Dromedary | B3 | B4 | B3 |
| KT368831 | Saudi Arabia | Dromedary | B5 | B5 | B3 |
| KT368830 | Saudi Arabia | Dromedary | B5 | B5 | B3 |
| KT368829 | Saudi Arabia | Dromedary | B5 | B5 | B3 |
| KT368832 | Saudi Arabia | Dromedary | B5 | B5 | B3 |
| KT861627 | Jordan | Human | B4 | B2 | B4 |
| KJ156910 | Saudi Arabia | Human | B4 | B2 | B4 |
| KJ156874 | Saudi Arabia | Human | B4 | B2 | B4 |
| KT368888 | Saudi Arabia | Dromedary | B3 | B3 | B4 |
| KT368887 | Saudi Arabia | Dromedary | B3 | B3 | B4 |
| KT368824 | Saudi Arabia | Dromedary | B4 | B5 | B4 |
| Clade/Lineage | dN/dS | Positively Selected Sites by SLAC Method | Positively Selected Sites by FEL Method | Positively Selected Sites by MEME Method | Best Fit Model |
|---|---|---|---|---|---|
| A and B | 0.424737 | Nil | Codon 26 *, 312 *, 1224 * | Codon 1224 *, 1250 * | 010121 |
| A | 0.392404 | Nil | Nil | Codon 879 * | 010121 |
| B | 0.439492 | Nil | Codon 312 *, 1224 * | Codon 1020 *, 1224 *, 1250 * | 010121 |
| A1 | 0.234930 | Nil | Nil | Nil | F81 |
| A2 | Insufficient available strains for analysis | ||||
| B1 | 0.382699 | Nil | Nil | Nil | 010011 |
| B2 | 0.405071 | Nil | Nil | Nil | 010121 |
| B3 | 0.400104 | Nil | Nil | Codon 1250 * | 001020 |
| B4 | 0.641872 | Nil | Nil | Nil | 000010 |
| B5 | 0.343586 | Nil | Nil | Codon 1020 * | 010121 |
| B6 | Insufficient available strains for analysis | ||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lau, S.K.P.; Wong, A.C.P.; Lau, T.C.K.; Woo, P.C.Y. Molecular Evolution of MERS Coronavirus: Dromedaries as a Recent Intermediate Host or Long-Time Animal Reservoir? Int. J. Mol. Sci. 2017, 18, 2138. https://doi.org/10.3390/ijms18102138
Lau SKP, Wong ACP, Lau TCK, Woo PCY. Molecular Evolution of MERS Coronavirus: Dromedaries as a Recent Intermediate Host or Long-Time Animal Reservoir? International Journal of Molecular Sciences. 2017; 18(10):2138. https://doi.org/10.3390/ijms18102138
Chicago/Turabian StyleLau, Susanna K. P., Antonio C. P. Wong, Terrence C. K. Lau, and Patrick C. Y. Woo. 2017. "Molecular Evolution of MERS Coronavirus: Dromedaries as a Recent Intermediate Host or Long-Time Animal Reservoir?" International Journal of Molecular Sciences 18, no. 10: 2138. https://doi.org/10.3390/ijms18102138