Molecular Dynamics Simulations and Dynamic Network Analysis Reveal the Allosteric Unbinding of Monobody to H-Ras Triggered by R135K Mutation


Abstract
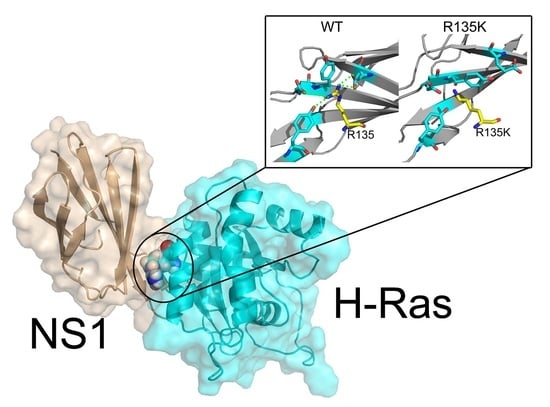
:
1. Introduction
2. Results and Discussion
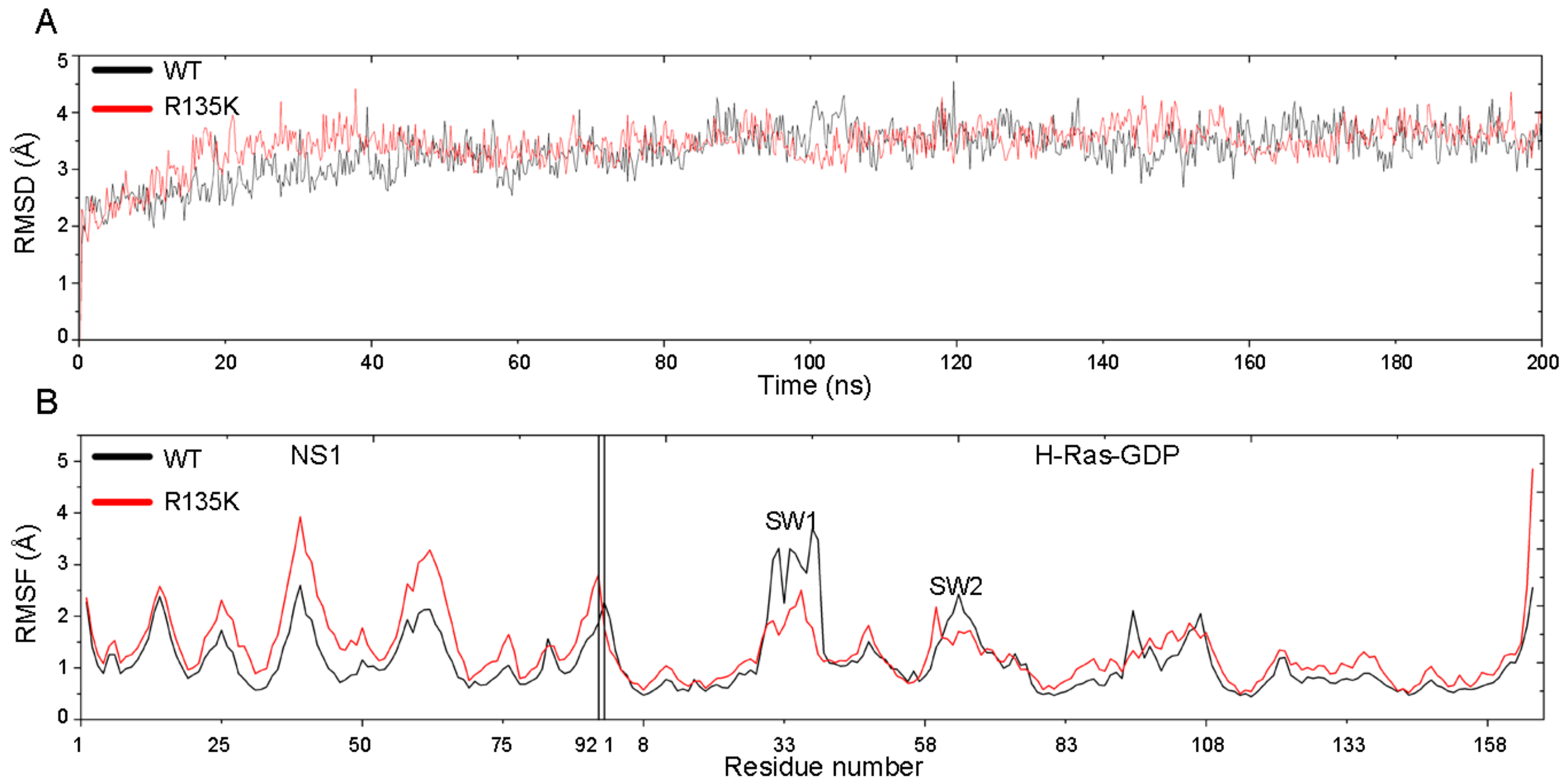
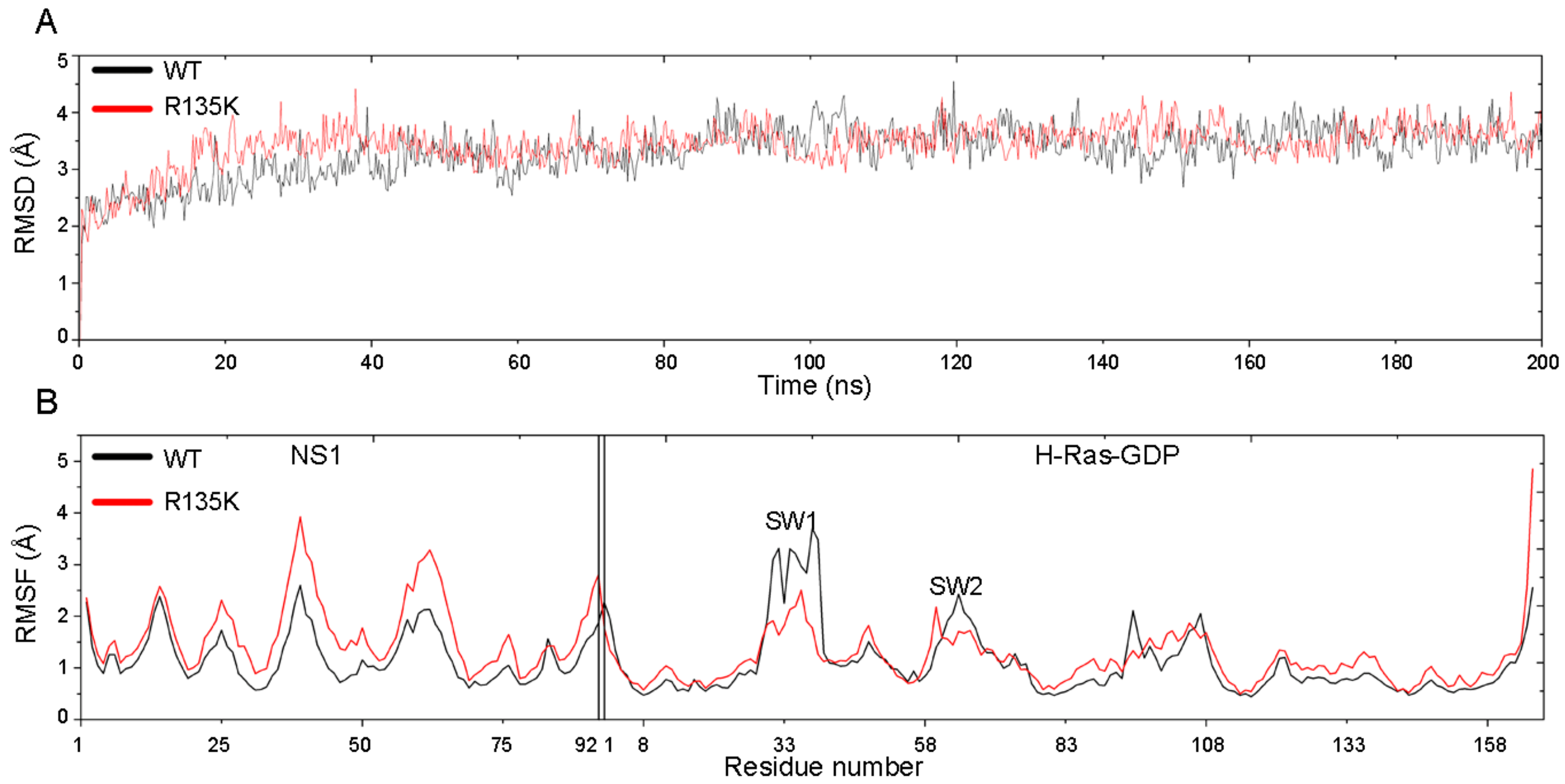
2.1. RMSD and RMSF Analysis
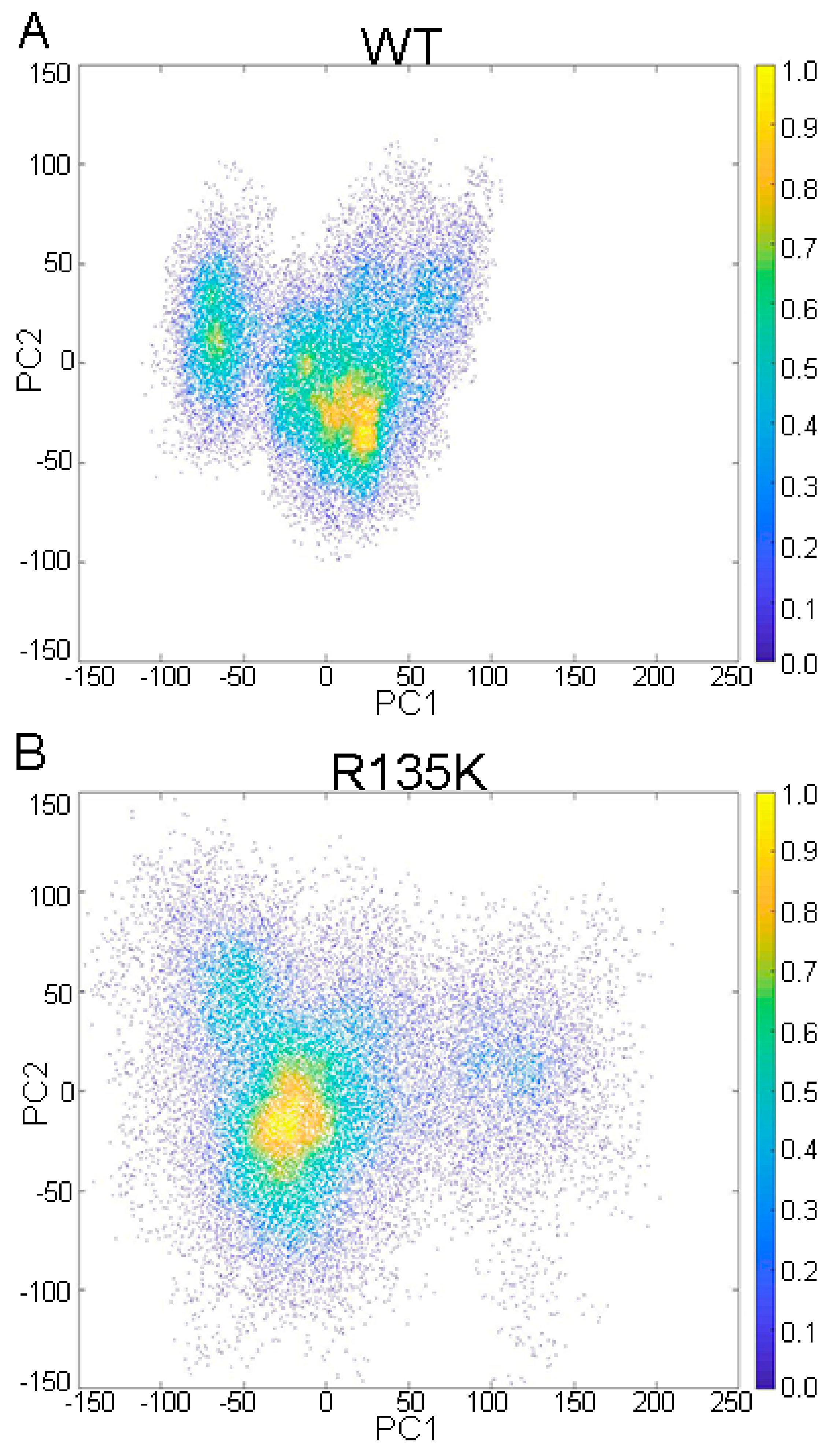
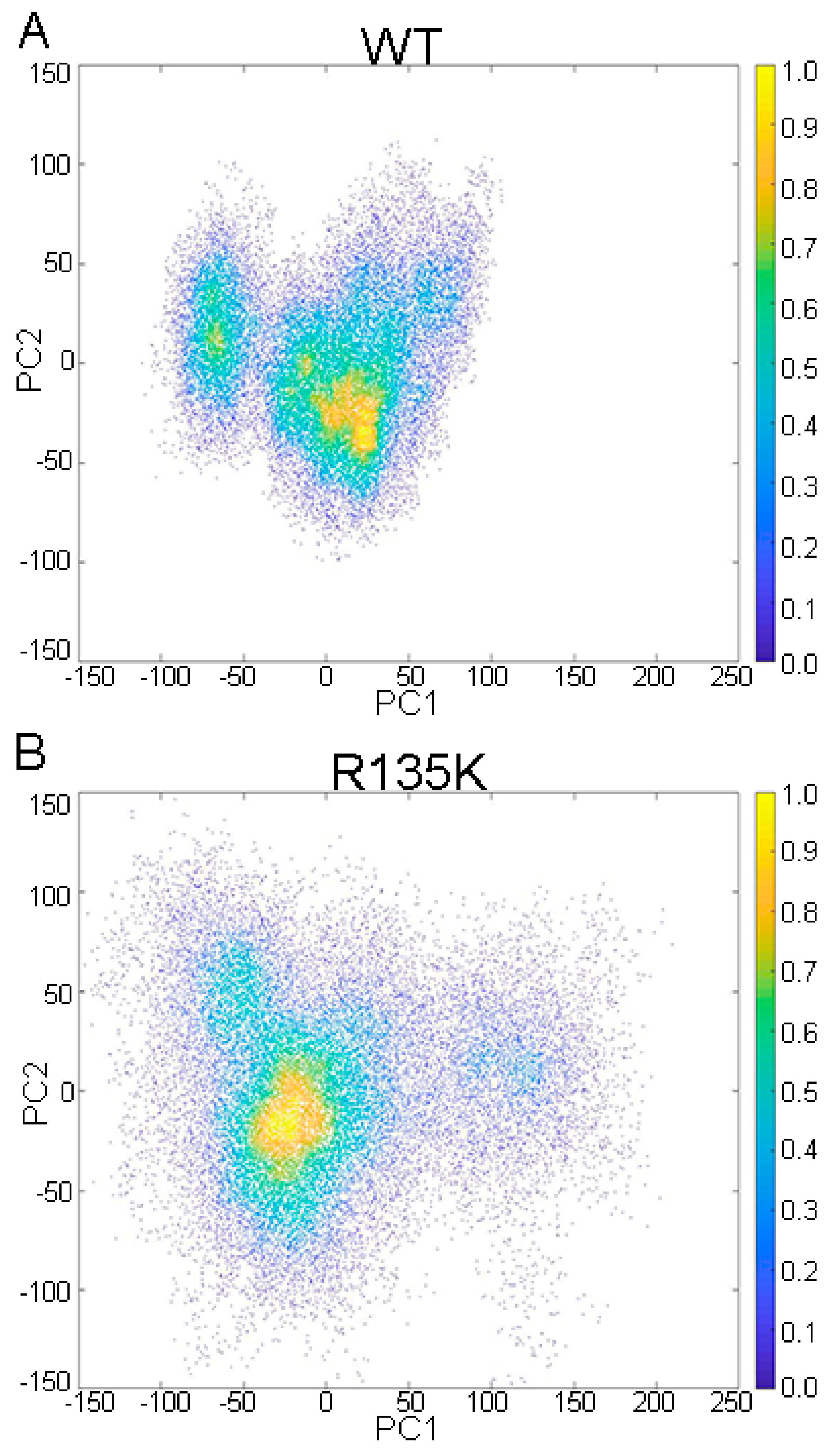
2.2. DCCM and PCA Analysis
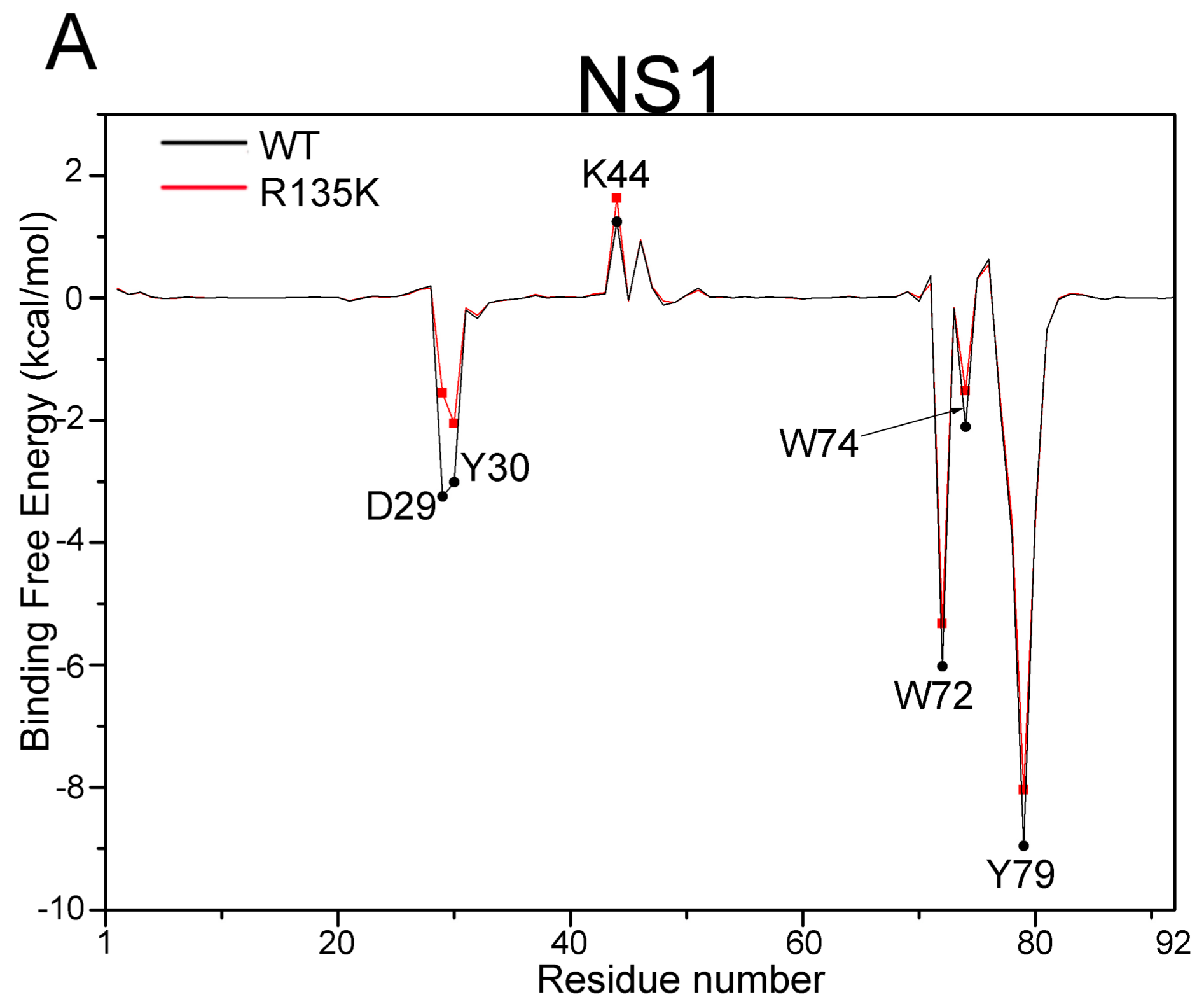
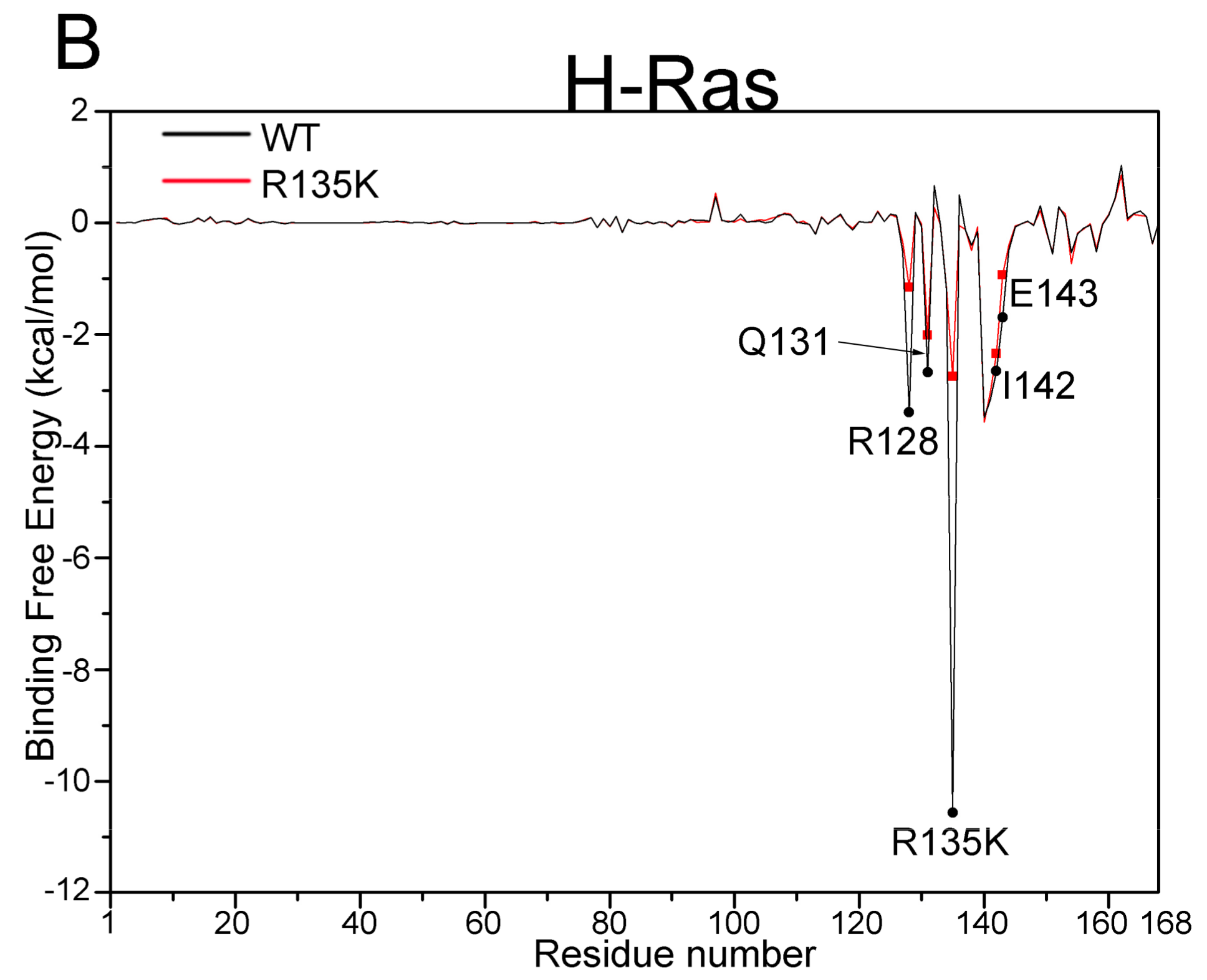
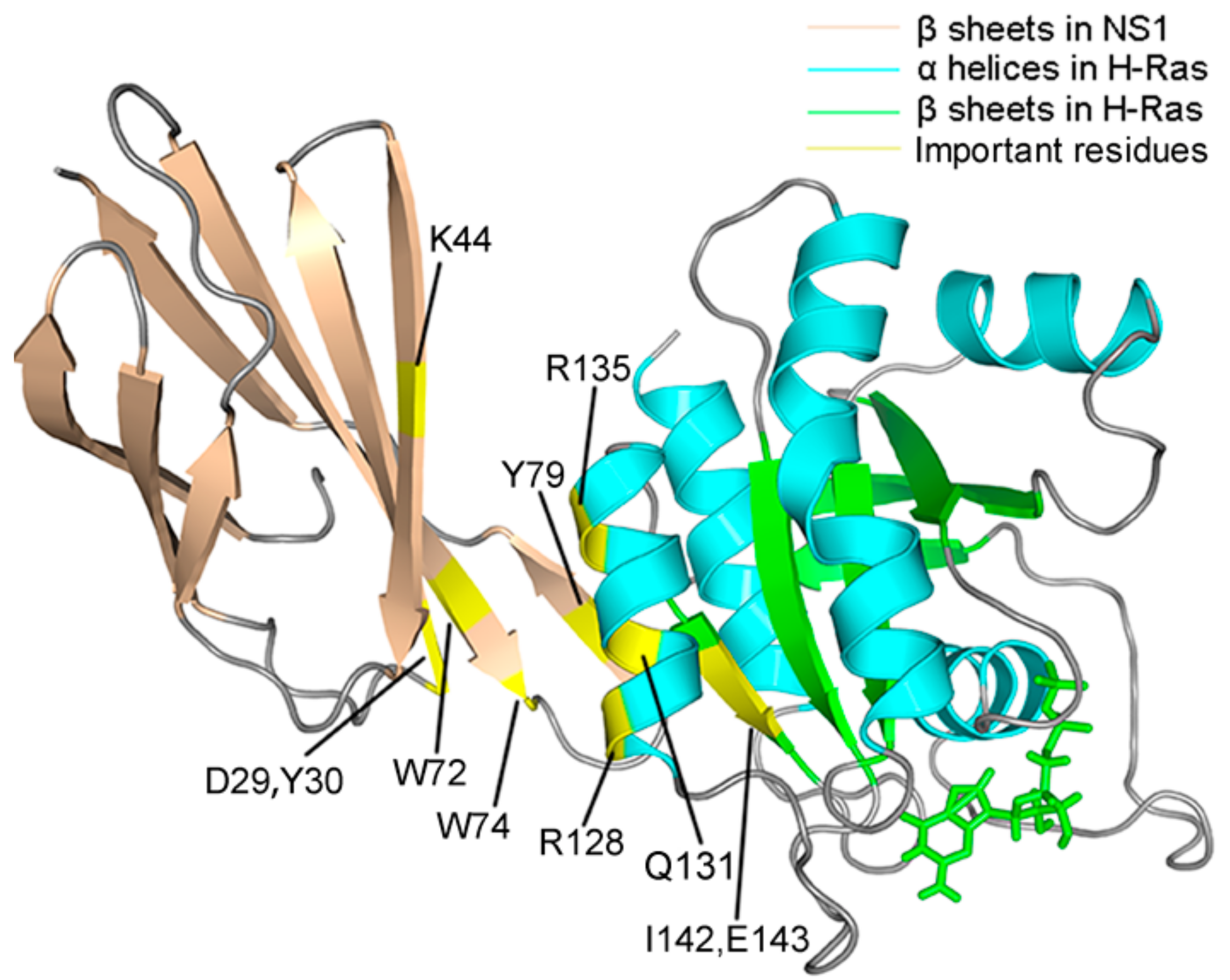
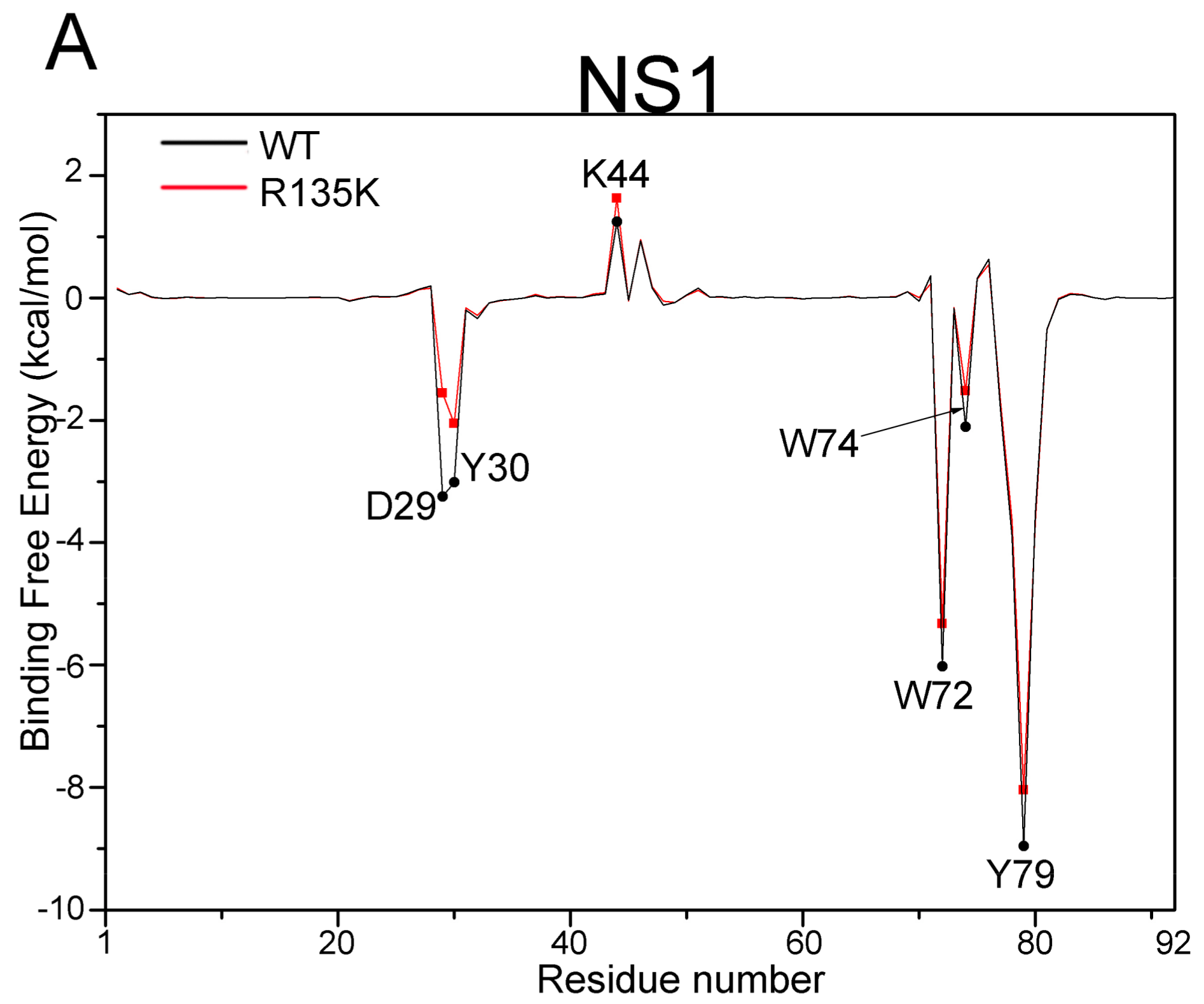
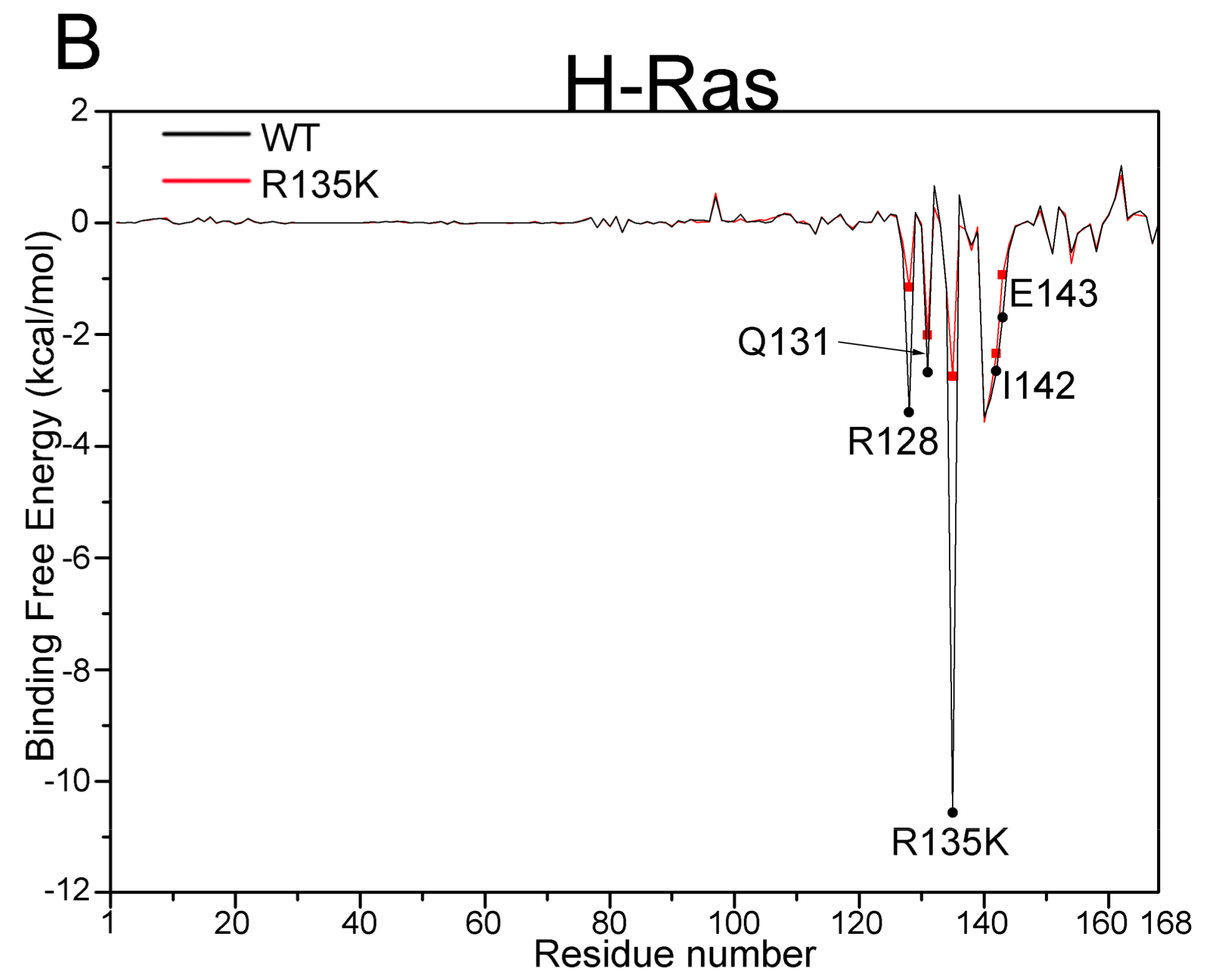
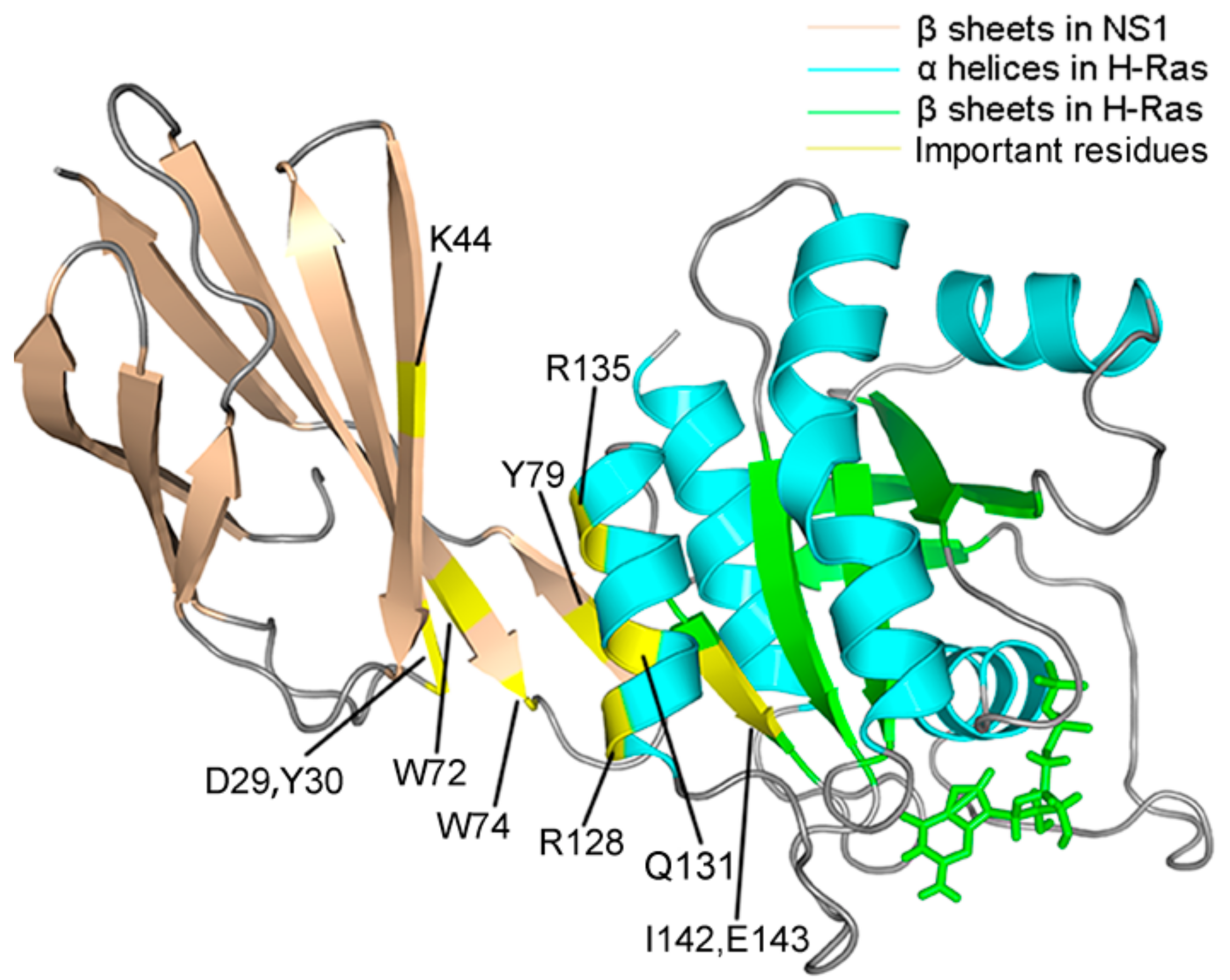
2.3. MM/GBSA Free Energy Analysis
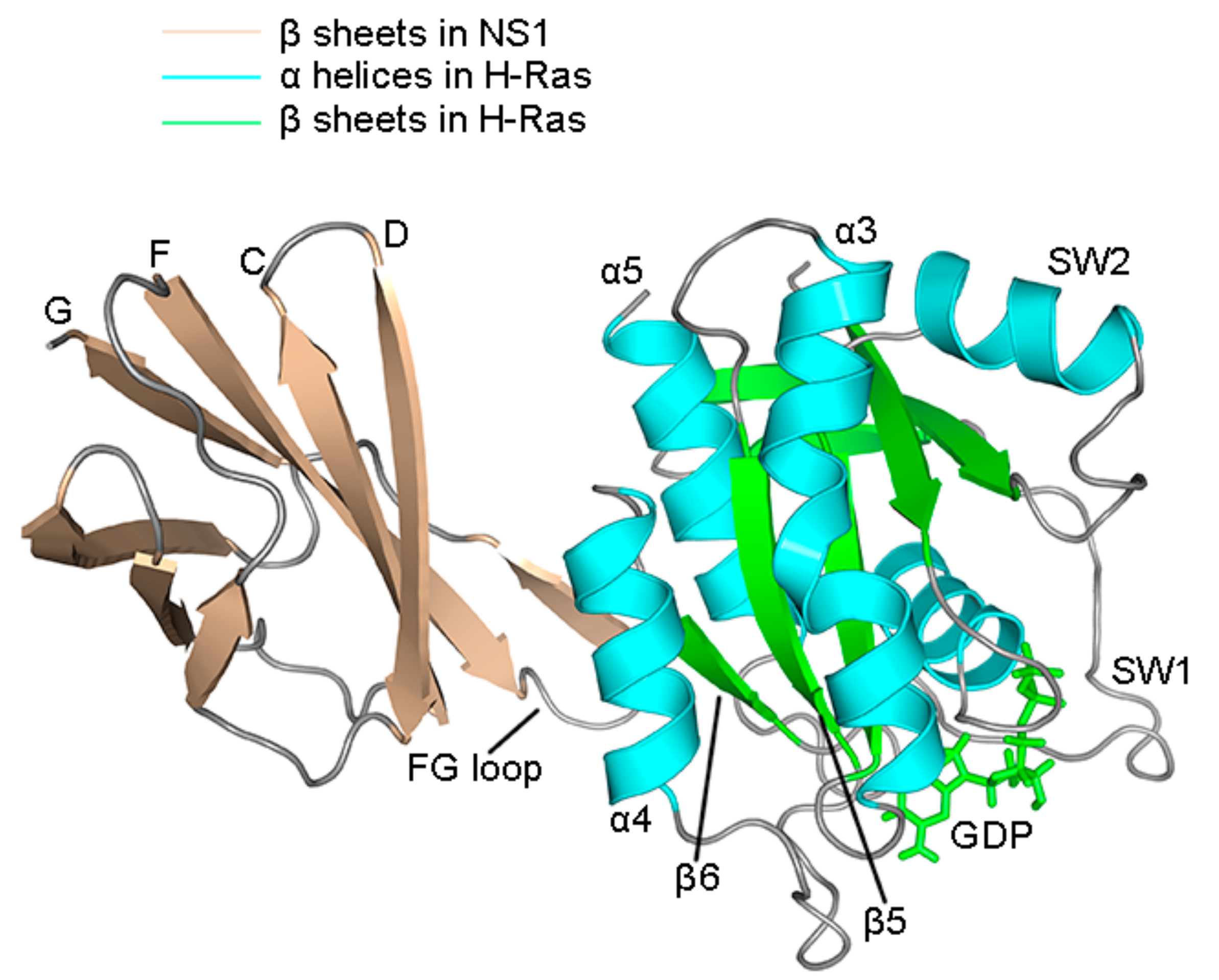
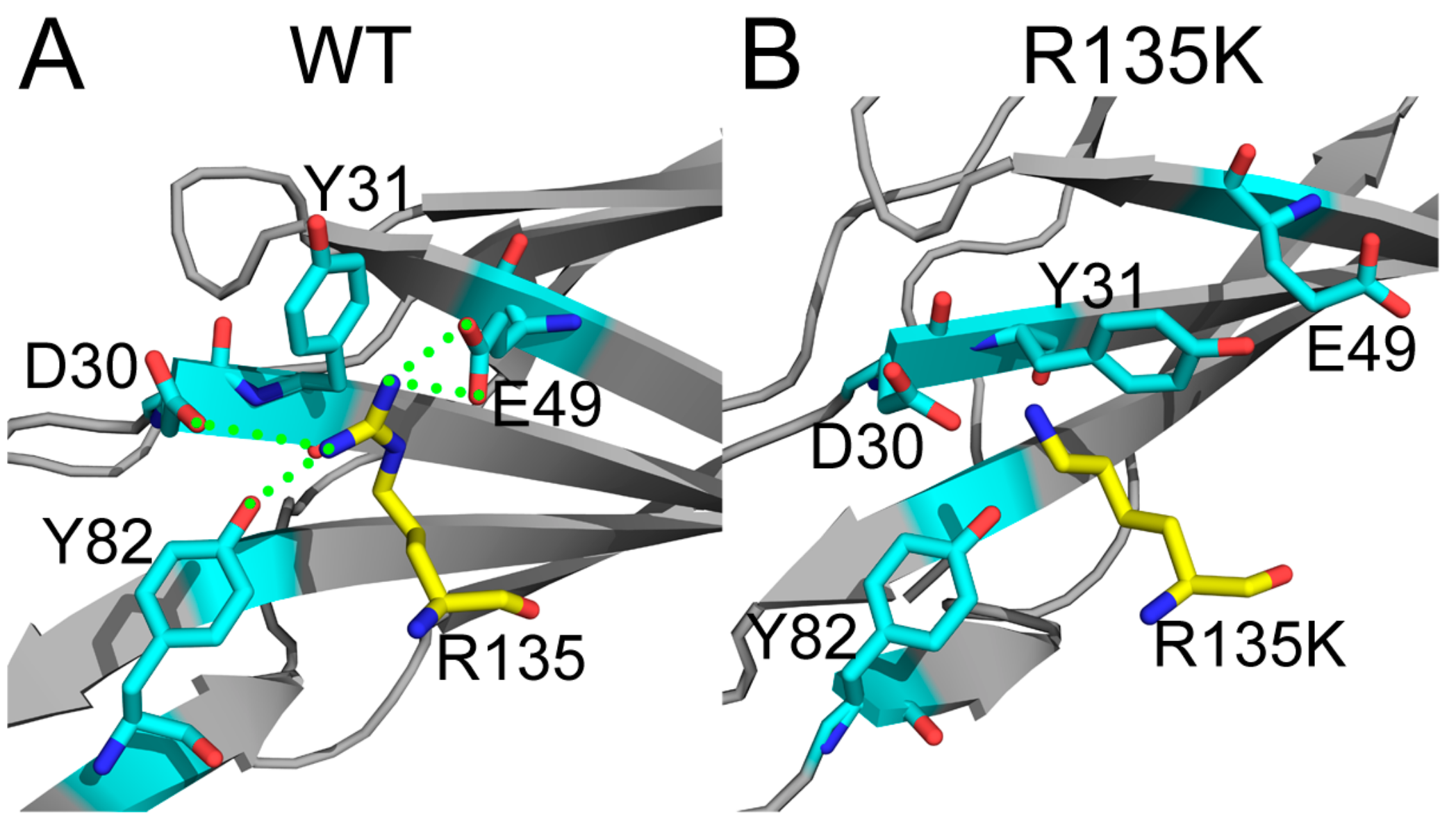
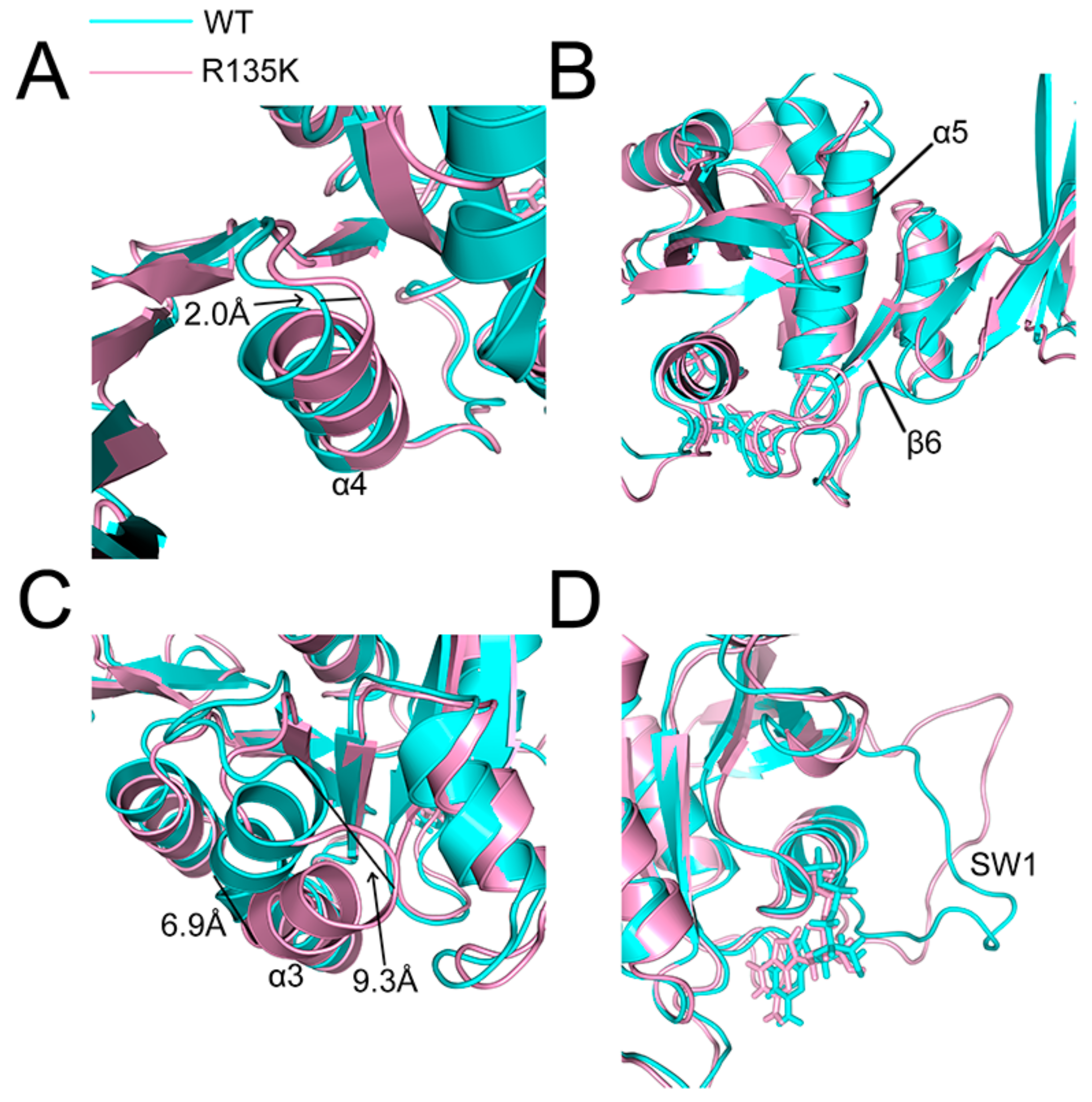
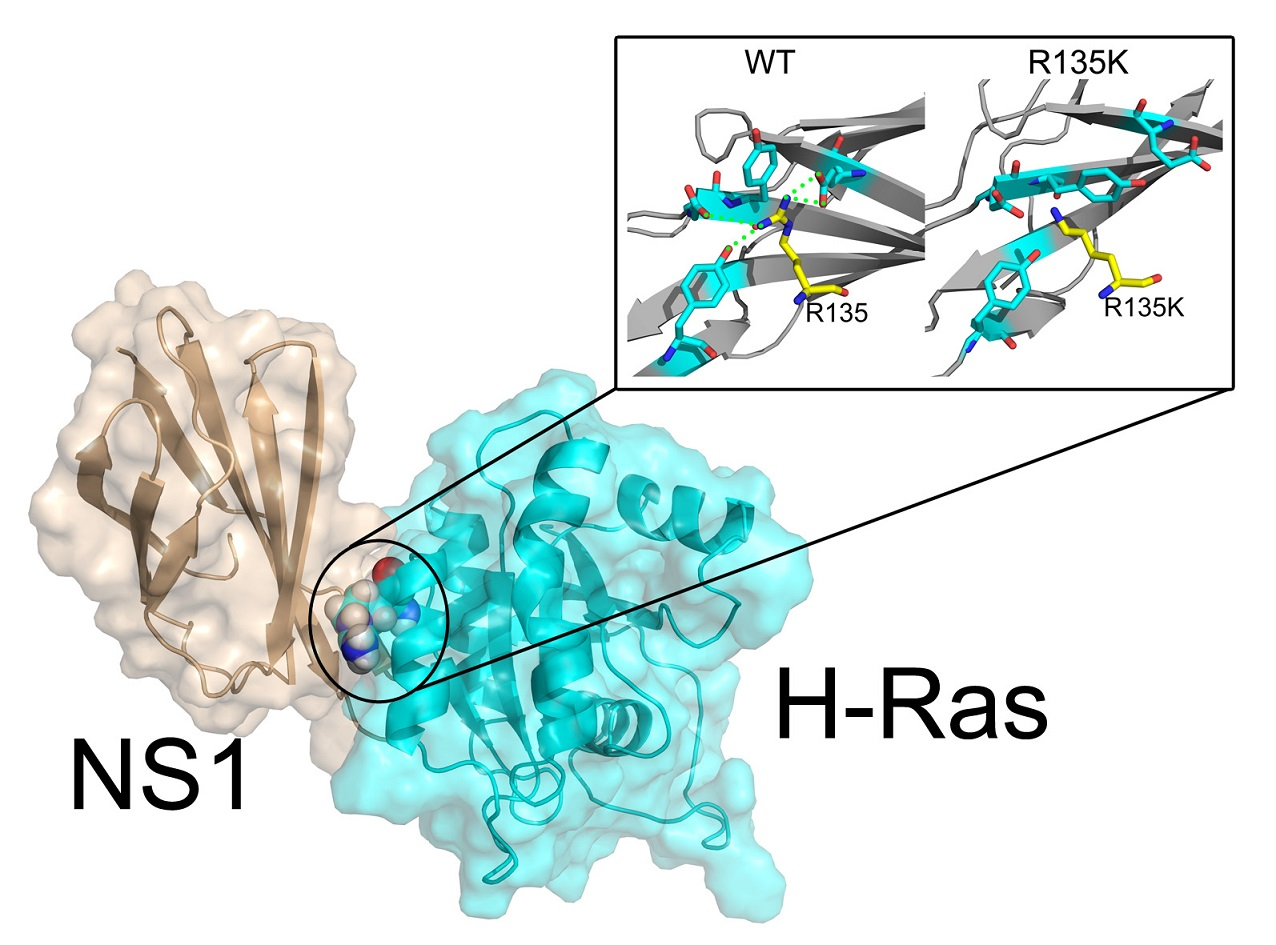
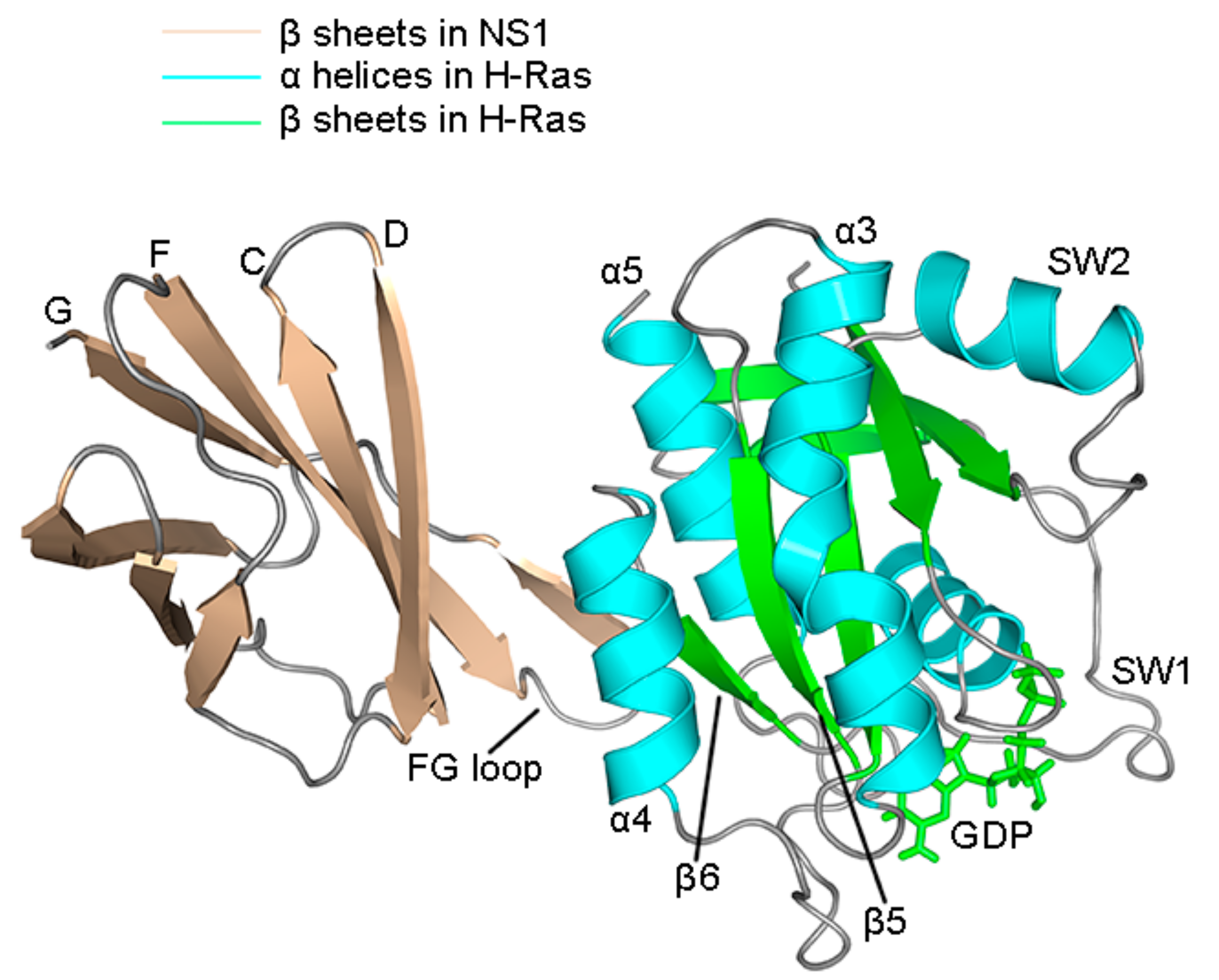
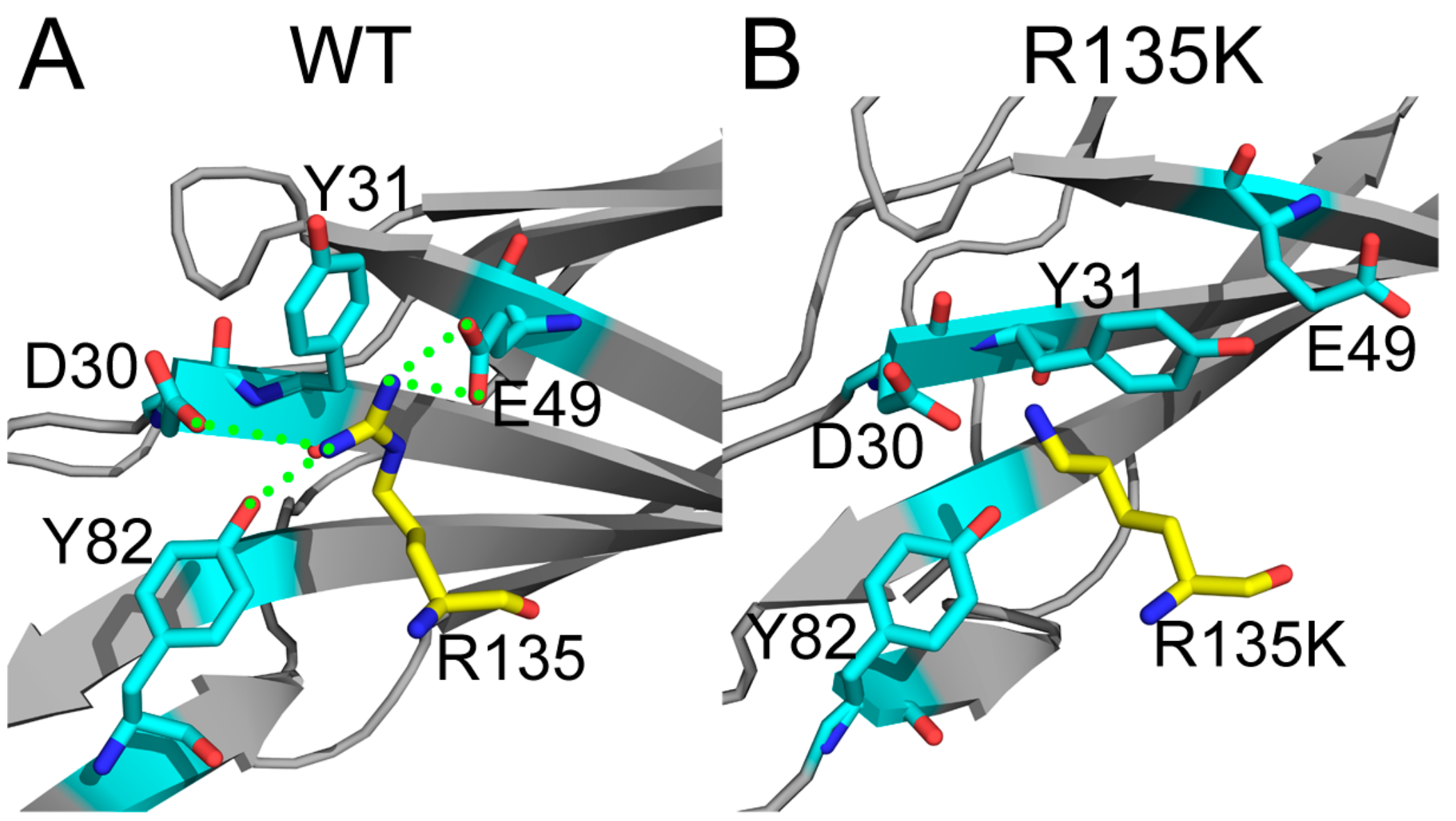
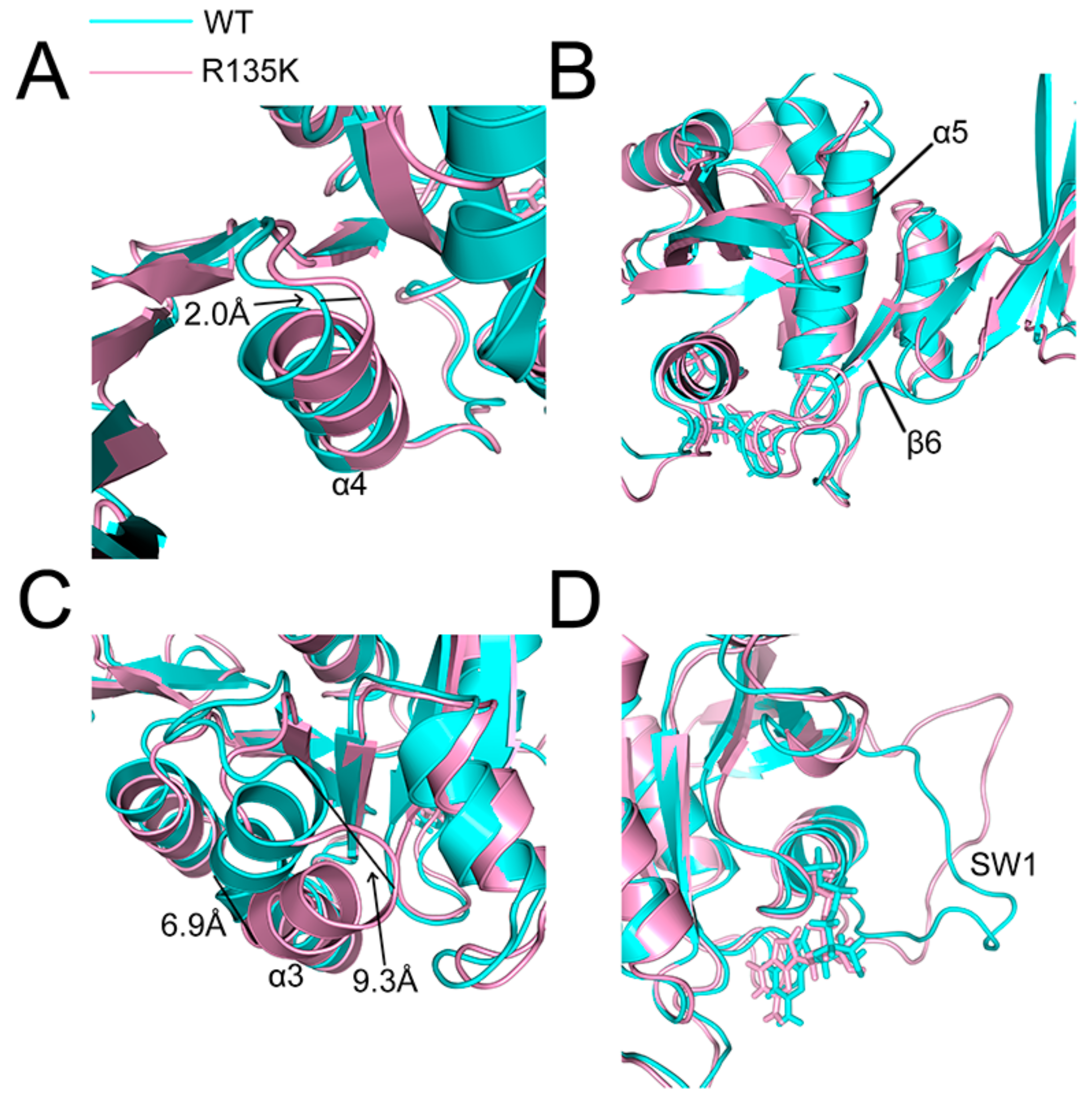
2.4. Superposition of the Wild Type and Mutation Structures
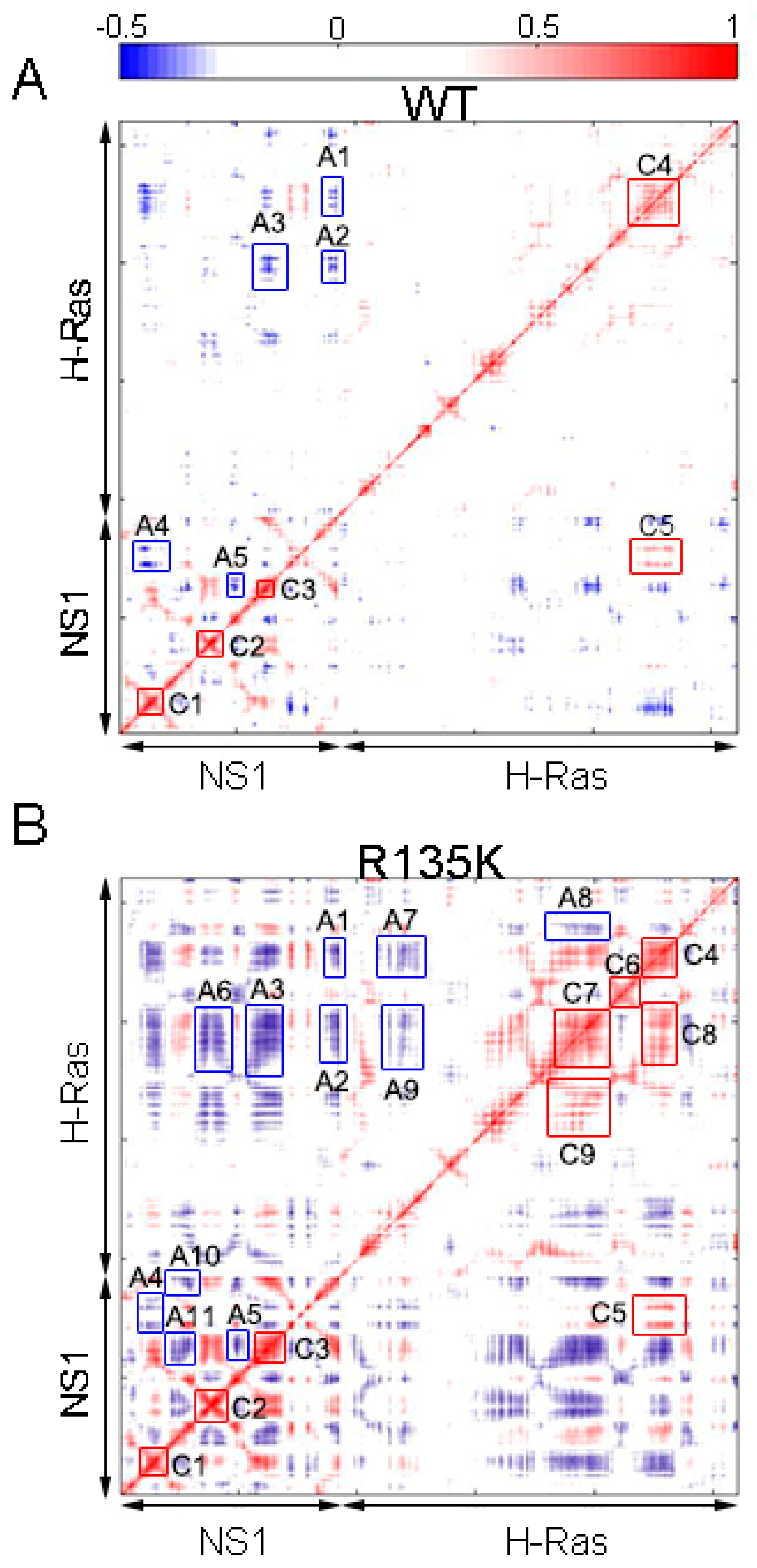
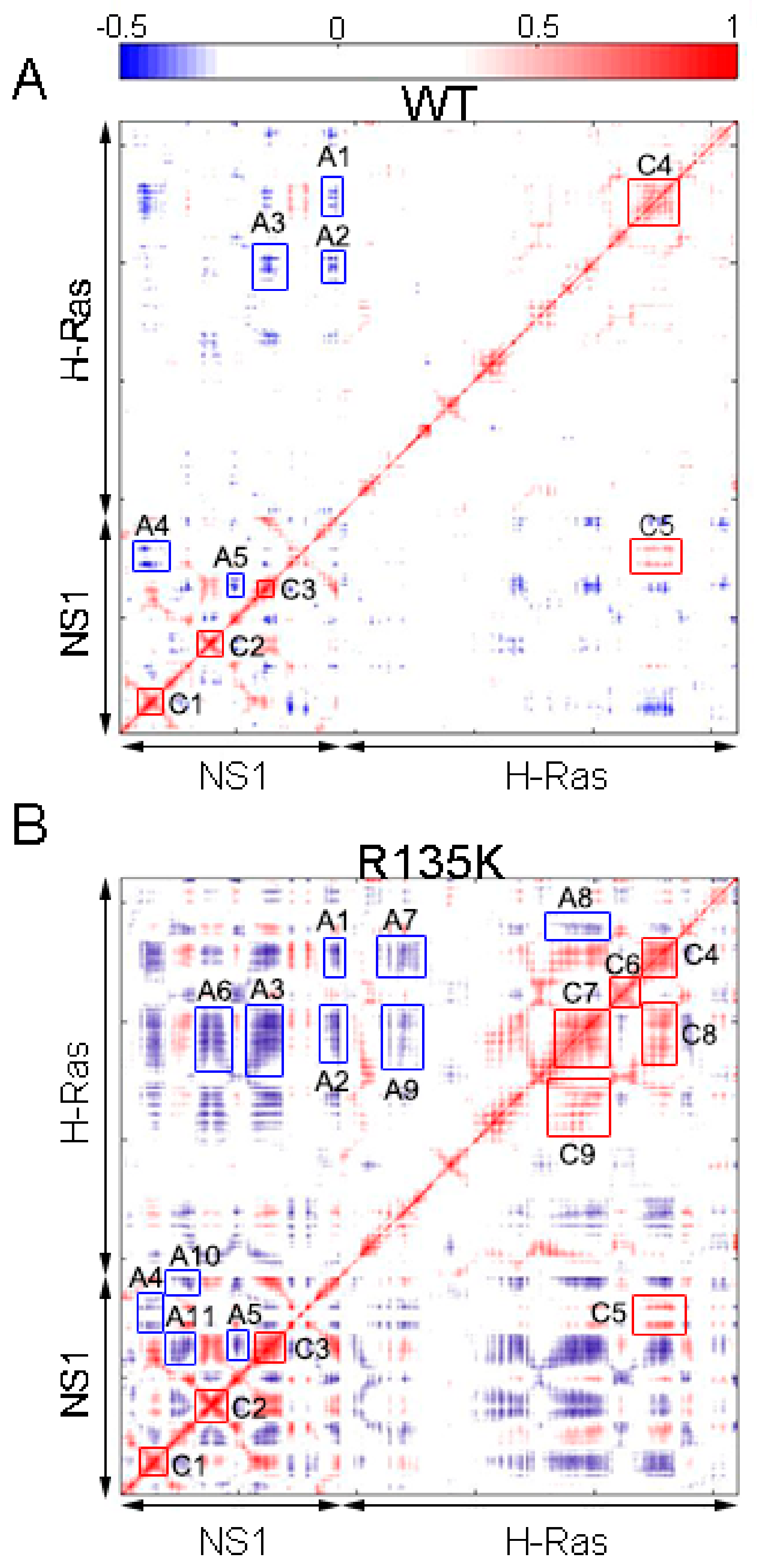
2.5. Allosteric Network Analysis
3. Materials and Methods
3.1. Construction of Simulation System
3.2. MD Simulations
3.3. Principal Component Analysis
3.4. Molecular Mechanics Generalized Born Surface Area Calculations
3.5. Dynamic Network Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cox, A.D.; Der, C.J. Ras history: The saga continues. Small GTPases 2010, 1, 2–27. [Google Scholar] [CrossRef] [PubMed]
- Drosten, M.; Dhawahir, A.; Sum, E.Y.; Urosevic, J.; Lechuga, C.G.; Esteban, L.M.; Castellano, E.; Guerra, C.; Santos, E.; Barbacid, M. Genetic analysis of Ras signalling pathways in cell proliferation, migration and survival. EMBO J. 2010, 29, 1091–1104. [Google Scholar] [CrossRef] [PubMed]
- Karnoub, A.E.; Weinberg, R.A. Ras oncogenes: Split personalities. Nat. Rev. Mol. Cell Biol. 2008, 9, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Young, A.; Lou, D.; McCormick, F. Oncogenic and wild-type Ras play divergent roles in the regulation of mitogen-activated protein kinase signaling. Cancer Discov. 2013, 3, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.J.; Nussinov, R. The free energy landscape in translational science: How can somatic mutations result in constitutive oncogenic activation? Phys. Chem. Chem. Phys. 2014, 16, 6332–6341. [Google Scholar] [CrossRef] [PubMed]
- Rajalingam, K.; Schreck, R.; Rapp, U.R.; Albert, S. Ras oncogenes and their downstream targets. Biochim. Biophys. Acta 2007, 1773, 1177–1195. [Google Scholar] [CrossRef] [PubMed]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed]
- Jeantet, M.; Tougeron, D.; Tachon, G.; Cortes, U.; Archambaut, C.; Fromont, G.; Karayan-Tapon, L. High Intra- and Inter-Tumoral Heterogeneity of RAS Mutations in Colorectal Cancer. Int. J. Mol. Sci. 2016, 17, 2015. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Tsai, C.J.; Jang, H. Independent and core pathways in oncogenic KRAS signaling. Expert Rev. Proteom. 2016, 13, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Tsai, C.J.; Chakrabarti, M.; Jang, H. A New View of Ras Isoforms in Cancers. Cancer Res. 2016, 76, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Muratcioglu, S.; Tsai, C.J.; Jang, H.; Gursoy, A.; Keskin, O. K-Ras4B/calmodulin/PI3Kα: A promising new adenocarcinoma-specific drug target? Expert Opin. Ther. Targets 2016, 20, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Jang, H.; Nussinov, R.; Zhang, J. The Structural Basis of Oncogenic Mutations G12, G13 and Q61 in Small GTPase K-Ras4B. Sci. Rep. 2016, 6, 21949. [Google Scholar] [CrossRef] [PubMed]
- Tomasini, P.; Serdjebi, C.; Khobta, N.; Metellus, P.; Ouafik, L.; Nanni, I.; Greillier, L.; Loundou, A.; Fina, F.; Mascaux, C.; et al. EGFR and KRAS Mutations Predict the Incidence and Outcome of Brain Metastases in Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2016, 17, 2132. [Google Scholar] [CrossRef] [PubMed]
- Olmedillas Lopez, S.; Garcia-Olmo, D.C.; Garcia-Arranz, M.; Guadalajara, H.; Pastor, C.; Garcia-Olmo, D. KRAS G12V Mutation Detection by Droplet Digital PCR in Circulating Cell-Free DNA of Colorectal Cancer Patients. Int. J. Mol. Sci. 2016, 17, 484. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Banerjee, A.; Jang, H.; Zhang, J.; Gaponenko, V.; Nussinov, R. GTP Binding and Oncogenic Mutations May Attenuate Hypervariable Region (HVR)-Catalytic Domain Interactions in Small GTPase K-Ras4B, Exposing the Effector Binding Site. J. Biol. Chem. 2015, 290, 28887–28900. [Google Scholar] [CrossRef] [PubMed]
- Csermely, P.; Korcsmaros, T.; Nussinov, R. Intracellular and intercellular signaling networks in cancer initiation, development and precision anti-cancer therapy: RAS acts as contextual signaling hub. Semin. Cell Dev. Biol. 2016, 58, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.H.; Gonda, M.A.; Ellis, R.W.; Scolnick, E.M.; Lowy, D.R. Human genome contains four genes homologous to transforming genes of Harvey and Kirsten murine sarcoma viruses. Proc. Natl. Acad. Sci. USA 1982, 79, 4848–4852. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Jang, H.; Nussinov, R.; Gaponenko, V. The disordered hypervariable region and the folded catalytic domain of oncogenic K-Ras4B partner in phospholipid binding. Curr. Opin. Struct. Biol. 2016, 36, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Mei, F.C.; Xie, J.; Cheng, X. Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. J. Biol. Chem. 2007, 282, 14048–14055. [Google Scholar] [CrossRef] [PubMed]
- Xing, H.R.; Cordon-Cardo, C.; Deng, X.; Tong, W.; Campodonico, L.; Fuks, Z.; Kolesnick, R. Pharmacologic inactivation of kinase suppressor of Ras-1 abrogates Ras-mediated pancreatic cancer. Nat. Med. 2003, 9, 1266–1268. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Fukamachi, K.; Tsuda, H.; Ito, K.; Ito, Y.; Ochiai, A. RAS oncogenic signal upregulates EZH2 in pancreatic cancer. Biochem. Biophys. Res. Commun. 2012, 417, 1074–1079. [Google Scholar] [CrossRef] [PubMed]
- Eser, S.; Schnieke, A.; Schneider, G.; Saur, D. Oncogenic KRAS signalling in pancreatic cancer. Br. J. Cancer 2014, 111, 817–822. [Google Scholar] [CrossRef] [PubMed]
- Pasca di Magliano, M.; Sekine, S.; Ermilov, A.; Ferris, J.; Dlugosz, A.A.; Hebrok, M. Hedgehog/Ras interactions regulate early stages of pancreatic cancer. Genes Dev. 2006, 20, 3161–3173. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Jang, H.; Gu, S.; Zhang, J.; Nussinov, R. Drugging Ras GTPase: A comprehensive mechanistic and signaling structural view. Chem. Soc. Rev. 2016, 45, 4929–4952. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Li, S.; Zhang, J. Harnessing allostery: A novel approach to drug discovery. Med. Res. Rev. 2014, 34, 1245–1285. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Nussinov, R. Druggable orthosteric and allosteric hot spots to target protein-protein interactions. Curr. Pharm. Des. 2014, 20, 1293–1301. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Jang, H.; Muratcioglu, S.; Gursoy, A.; Keskin, O.; Nussinov, R.; Zhang, J. Ras Conformational Ensembles, Allostery, and Signaling. Chem. Rev. 2016, 116, 6607–6665. [Google Scholar] [CrossRef] [PubMed]
- Khosravi-Far, R.; Der, C.J. The Ras signal transduction pathway. Cancer Metastasis Rev. 1994, 13, 67–89. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Maiello, M.R.; D’Alessio, A.; Pergameno, M.; Normanno, N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: Role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin. Ther. Targets 2012, 16, S17–S27. [Google Scholar] [CrossRef] [PubMed]
- Chang, F.; Steelman, L.S.; Lee, J.T.; Shelton, J.G.; Navolanic, P.M.; Blalock, W.L.; Franklin, R.A.; McCubrey, J.A. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: Potential targeting for therapeutic intervention. Leukemia 2003, 17, 1263–1293. [Google Scholar] [CrossRef] [PubMed]
- Muratcioglu, S.; Chavan, T.S.; Freed, B.C.; Jang, H.; Khavrutskii, L.; Freed, R.N.; Dyba, M.A.; Stefanisko, K.; Tarasov, S.G.; Gursoy, A.; et al. GTP-Dependent K-Ras Dimerization. Structure 2015, 23, 1325–1335. [Google Scholar] [CrossRef] [PubMed]
- Nan, X.; Tamguney, T.M.; Collisson, E.A.; Lin, L.J.; Pitt, C.; Galeas, J.; Lewis, S.; Gray, J.W.; McCormick, F.; Chu, S. Ras-GTP dimers activate the Mitogen-Activated Protein Kinase (MAPK) pathway. Proc. Natl. Acad. Sci. USA 2015, 112, 7996–8001. [Google Scholar] [CrossRef] [PubMed]
- Sayyed-Ahmad, A.; Cho, K.J.; Hancock, J.F.; Gorfe, A.A. Computational Equilibrium Thermodynamic and Kinetic Analysis of K-Ras Dimerization through an Effector Binding Surface Suggests Limited Functional Role. J. Phys. Chem. B 2016, 120, 8547–8556. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.E.; Bar-Sagi, D.; Nassar, N. The structural basis for the transition from Ras-GTP to Ras-GDP. Proc. Natl. Acad. Sci. USA 2002, 99, 12138–12142. [Google Scholar] [CrossRef] [PubMed]
- Buhrman, G.; Kumar, V.S.; Cirit, M.; Haugh, J.M.; Mattos, C. Allosteric modulation of Ras-GTP is linked to signal transduction through RAF kinase. J. Biol. Chem. 2011, 286, 3323–3331. [Google Scholar] [CrossRef] [PubMed]
- Grant, B.J.; Lukman, S.; Hocker, H.J.; Sayyah, J.; Brown, J.H.; McCammon, J.A.; Gorfe, A.A. Novel allosteric sites on Ras for lead generation. PLoS ONE 2011, 6, e25711. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Tsai, C.J. The design of covalent allosteric drugs. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 249–267. [Google Scholar] [CrossRef] [PubMed]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [PubMed]
- Spencer-Smith, R.; Koide, A.; Zhou, Y.; Eguchi, R.R.; Sha, F.; Gajwani, P.; Santana, D.; Gupta, A.; Jacobs, M.; Herrero-Garcia, E.; et al. Inhibition of RAS function through targeting an allosteric regulatory site. Nat. Chem. Biol. 2017, 13, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, S.; Wang, H.; Ding, K.; Wang, J.; Pan, L.; Lu, Y.; Liu, Q.; Zhang, C. Interactions of benzotriazole UV stabilizers with human serum albumin: Atomic insights revealed by biosensors, spectroscopies and molecular dynamics simulations. Chemosphere 2016, 144, 1050–1059. [Google Scholar] [CrossRef] [PubMed]
- Ding, K.; Zhang, H.; Wang, H.; Lv, X.; Pan, L.; Zhang, W.; Zhuang, S. Atomic-scale investigation of the interactions between tetrabromobisphenol A, tetrabromobisphenol S and bovine trypsin by spectroscopies and molecular dynamics simulations. J. Hazard. Mater. 2015, 299, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Alred, E.J.; Scheele, E.G.; Berhanu, W.M.; Hansmann, U.H. Stability of Iowa mutant and wild type Abeta-peptide aggregates. J. Chem. Phys. 2014, 141, 175101. [Google Scholar] [CrossRef] [PubMed]
- Clausen, R.; Ma, B.; Nussinov, R.; Shehu, A. Mapping the Conformation Space of Wildtype and Mutant H-Ras with a Memetic, Cellular, and Multiscale Evolutionary Algorithm. PLoS Comput. Biol. 2015, 11, e1004470. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Li, Y.; Tian, S.; Wang, J.; Hou, T. P-loop conformation governed crizotinib resistance in G2032R-mutated ROS1 tyrosine kinase: Clues from free energy landscape. PLoS Comput. Biol. 2014, 10, e1003729. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Jang, H.; Tsai, C.J. The structural basis for cancer treatment decisions. Oncotarget 2014, 5, 7285–7302. [Google Scholar] [CrossRef] [PubMed]
- Dror, R.O.; Dirks, R.M.; Grossman, J.P.; Xu, H.; Shaw, D.E. Biomolecular simulation: A computational microscope for molecular biology. Annu. Rev. Biophys. 2012, 41, 429–452. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, M.; Mortier, J.; Rakers, C.; Sydow, D.; Wolber, G. More than a look into a crystal ball: Protein structure elucidation guided by molecular dynamics simulations. Drug Discov. Today 2016, 21, 1799–1805. [Google Scholar] [CrossRef] [PubMed]
- Gkeka, P.; Evangelidis, T.; Pavlaki, M.; Lazani, V.; Christoforidis, S.; Agianian, B.; Cournia, Z. Investigating the structure and dynamics of the PIK3CA wild-type and H1047R oncogenic mutant. PLoS Comput. Biol. 2014, 10, e1003895. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Deng, R.; Jiang, H.; Song, H.; Li, S.; Shen, Q.; Huang, W.; Nussinov, R.; Yu, J.; Zhang, J. The Mechanism of ATP-Dependent Allosteric Protection of Akt Kinase Phosphorylation. Structure 2015, 23, 1725–1734. [Google Scholar] [CrossRef] [PubMed]
- Blacklock, K.; Verkhivker, G.M. Computational modeling of allosteric regulation in the hsp90 chaperones: A statistical ensemble analysis of protein structure networks and allosteric communications. PLoS Comput. Biol. 2014, 10, e1003679. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Huang, W.; Wang, Q.; Shen, Q.; Li, S.; Nussinov, R.; Zhang, J. The structural basis of ATP as an allosteric modulator. PLoS Comput. Biol. 2014, 10, e1003831. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Tian, S.; Zhou, S.; Li, Y.; Li, D.; Xu, L.; Shen, M.; Pan, P.; Hou, T. Revealing the favorable dissociation pathway of type II kinase inhibitors via enhanced sampling simulations and two-end-state calculations. Sci. Rep. 2015, 5, 8457. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Y.; Chen, X.E.; Zhang, Y.L. Insights into the key interactions between human protein phosphatase 5 and cantharidin using molecular dynamics and site-directed mutagenesis bioassays. Sci. Rep. 2015, 5, 12359. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Eargle, J.; Luthey-Schulten, Z. NetworkView: 3D display and analysis of protein. RNA interaction networks. Bioinformatics 2012, 28, 3000–3001. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- York, D.M.; Wlodawer, A.; Pedersen, L.G.; Darden, T.A. Atomic-level accuracy in simulations of large protein crystals. Proc. Natl. Acad. Sci. USA 1994, 91, 8715–8718. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E., 3rd; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Zhang, J. Designed covalent allosteric modulators: An emerging paradigm in drug discovery. Drug Discov. Today 2017, 22, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.Y.; Tolias, P. Recent advances in cancer drug discovery targeting RAS. Drug Discov. Today 2016, 21, 1915–1919. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Jang, H.; Zhang, J.; Nussinov, R. Inhibitors of Ras-SOS Interactions. Chemmedchem 2016, 11, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Deng, R.; Yang, X.; Shang, J.; Lu, S.; Zhao, Y.; Song, K.; Liu, X.; Zhang, Q.; Chen, Y.; et al. Peptidomimetic inhibitors of APC-Asef interaction block colorectal cancer migration. Nat. Chem. Biol. 2017, 13, 994–1001. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Items | WT | R135K | Energy Difference |
|---|---|---|---|
| ΔEvdW b | −68.36 (4.36) | −61.57 (5.49) | 6.79 |
| ΔEele c | −75.44 (31.88) | −37.60 (39.73) | 37.84 |
| ΔEGB d | 97.24 (28.18) | 68.16 (35.80) | −29.08 |
| ΔESURF e | −8.63 (0.57) | −7.46 (0.78) | 1.17 |
| ΔGgas | −143.80 (32.67) | −99.17 (40.27) | 44.63 |
| ΔGnonpolar | 97.37 (1.22) | 68.74 (10.14) | −28.63 |
| ΔGpolar | −8.76 (0.13) | −8.04 (0.12) | 0.72 |
| ΔGbinding | −55.19 (7.88) | −38.47 (8.56) | 16.72 |
| Number | H Bonds or Salt Bridges | Distance (Å) | |
| Hb1 | NS1-D30-OD2 | H-Ras-R135-NH2 | 3.75 |
| Hb2 | NS1-E49-OE1 | H-Ras-R135-NE | 2.73 |
| Hb3 | NS1-E49-OE2 | H-Ras-R135-NH1 | 2.89 |
| Hb4 | NS1-Y82-OH | H-Ras-R135-NH2 | 2.91 |
| Sb1 | NS1-D30-OD2 | H-Ras-R135-NH2 | 3.75 |
| Sb2 | NS1-E49-OE1 | H-Ras-R135-NE | 2.73 |
| Sb3 | NS1-E49-OE1 | H-Ras-R135-NH1 | 3.49 |
| Sb4 | NS1-E49-OE2 | H-Ras-R135-NE | 3.67 |
| Sb5 | NS1-E49-OE2 | H-Ras-R135-NH1 | 2.89 |
| Number | Salt Bridges | Distance (Å) | |
| Sb1 | NS1-E49-OE1 | H-Ras-K135-NZ | 3.76 |
| Sb2 | NS1-E49-OE2 | H-Ras-K135-NZ | 3.96 |
| Residue | WT | R135K | Energy Difference b | |
|---|---|---|---|---|
| NS1 | D29 | −3.25 (2.59) | −1.56 (2.39) | 1.69 |
| Y30 | −3.02 (0.48) | −2.05 (0.77) | 0.97 | |
| K44 | 1.24 (0.65) | 1.62 (1.39) | 0.38 | |
| W72 | −6.02 (0.68) | −5.32 (0.73) | 0.70 | |
| W74 | −2.11 (0.91) | −1.52 (0.88) | 0.59 | |
| Y79 | −8.96 (0.87) | −8.04 (1.07) | 0.92 | |
| H-Ras–GDP | R128 | −3.40 (2.33) | −1.15 (2.38) | 2.25 |
| Q131 | −2.68 (1.36) | −2.01 (1.21) | 0.67 | |
| R135K | −10.56 (1.94) | −2.75 (3.27) | 7.81 | |
| I142 | −2.65 (0.76) | −2.34 (0.76) | 0.31 | |
| E143 | −1.70 (1.09) | −0.94 (1.29) | 0.76 |
| Pathway | Length a | Residue b | Subopt c | |||
|---|---|---|---|---|---|---|
| WT | R135K | WT | R135K | WT | R135K | |
| R135(K)-R128 | 38 | 60 | 3 | 8 | 12 | 1 |
| R135(K)-Q131 | 21 | 29 | 2 | 5 | 1 | 1 |
| R135(K)-I142 | 68 | 61 | 4 | 8 | 9 | 0 |
| R135(K)-E143 | 86 | 84 | 4 | 9 | 12 | 1 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ni, D.; Song, K.; Zhang, J.; Lu, S. Molecular Dynamics Simulations and Dynamic Network Analysis Reveal the Allosteric Unbinding of Monobody to H-Ras Triggered by R135K Mutation. Int. J. Mol. Sci. 2017, 18, 2249. https://doi.org/10.3390/ijms18112249
Ni D, Song K, Zhang J, Lu S. Molecular Dynamics Simulations and Dynamic Network Analysis Reveal the Allosteric Unbinding of Monobody to H-Ras Triggered by R135K Mutation. International Journal of Molecular Sciences. 2017; 18(11):2249. https://doi.org/10.3390/ijms18112249
Chicago/Turabian StyleNi, Duan, Kun Song, Jian Zhang, and Shaoyong Lu. 2017. "Molecular Dynamics Simulations and Dynamic Network Analysis Reveal the Allosteric Unbinding of Monobody to H-Ras Triggered by R135K Mutation" International Journal of Molecular Sciences 18, no. 11: 2249. https://doi.org/10.3390/ijms18112249