Metformin Potentiates the Benefits of Dietary Restraint: A Metabolomic Study

 , , , , , , and
, , , , , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
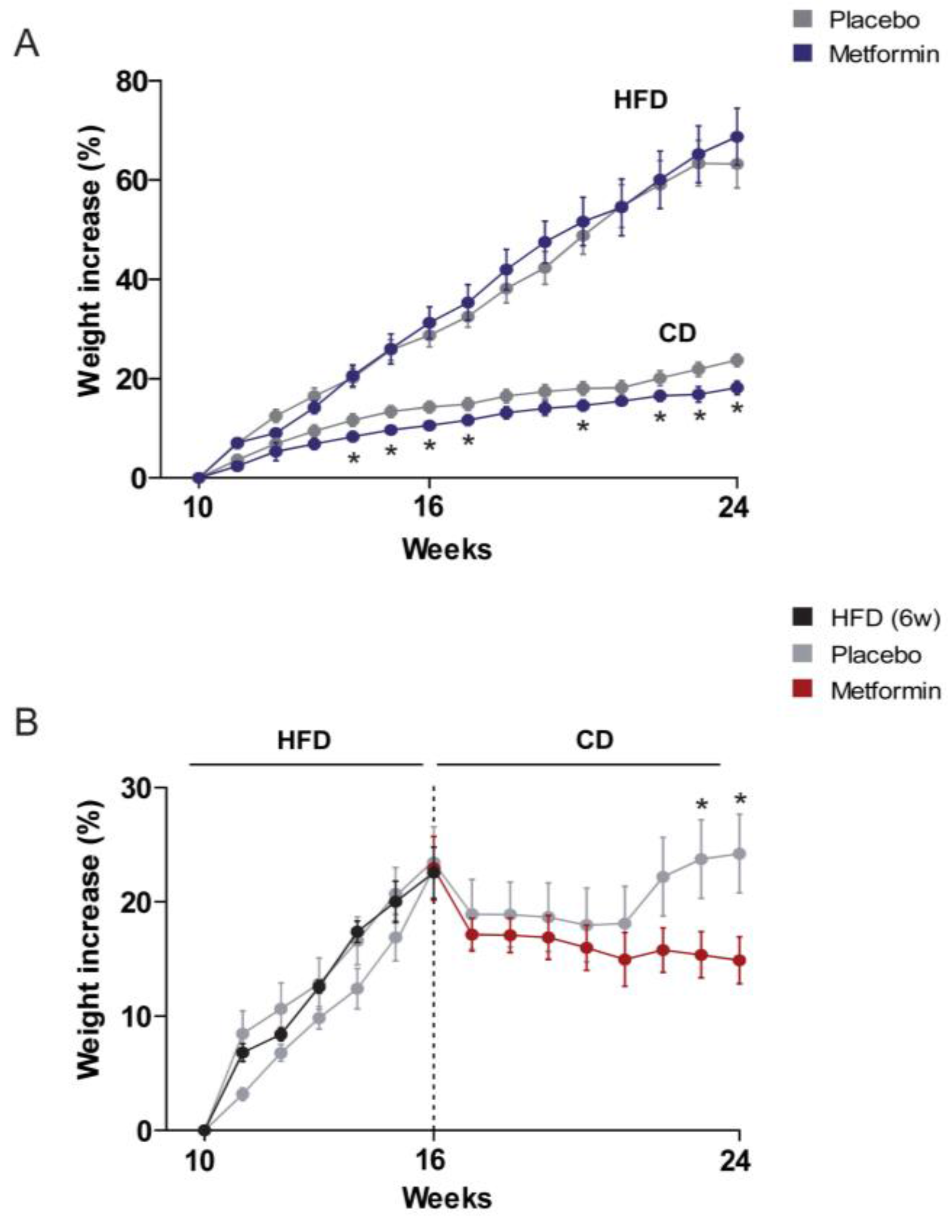
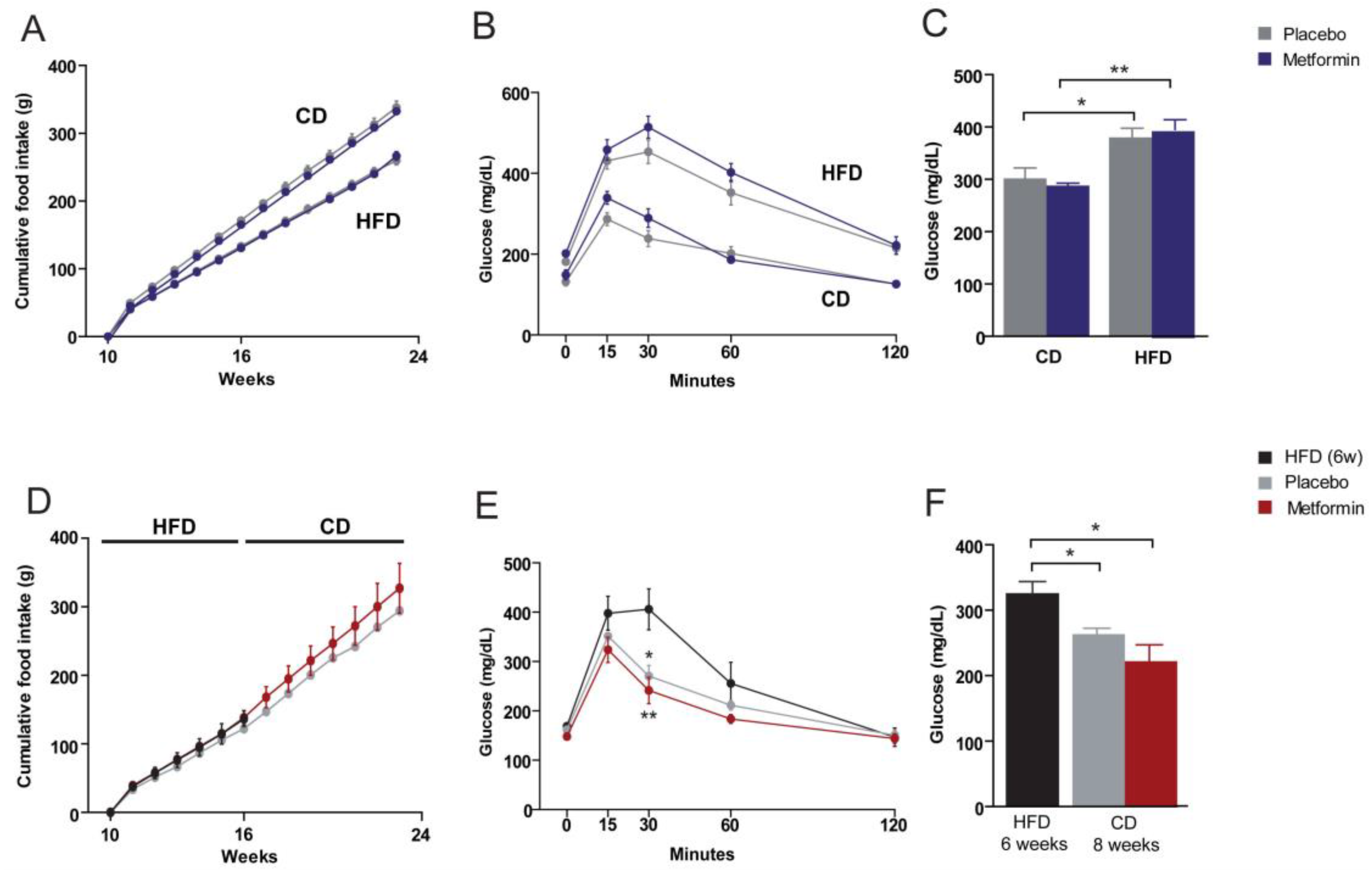
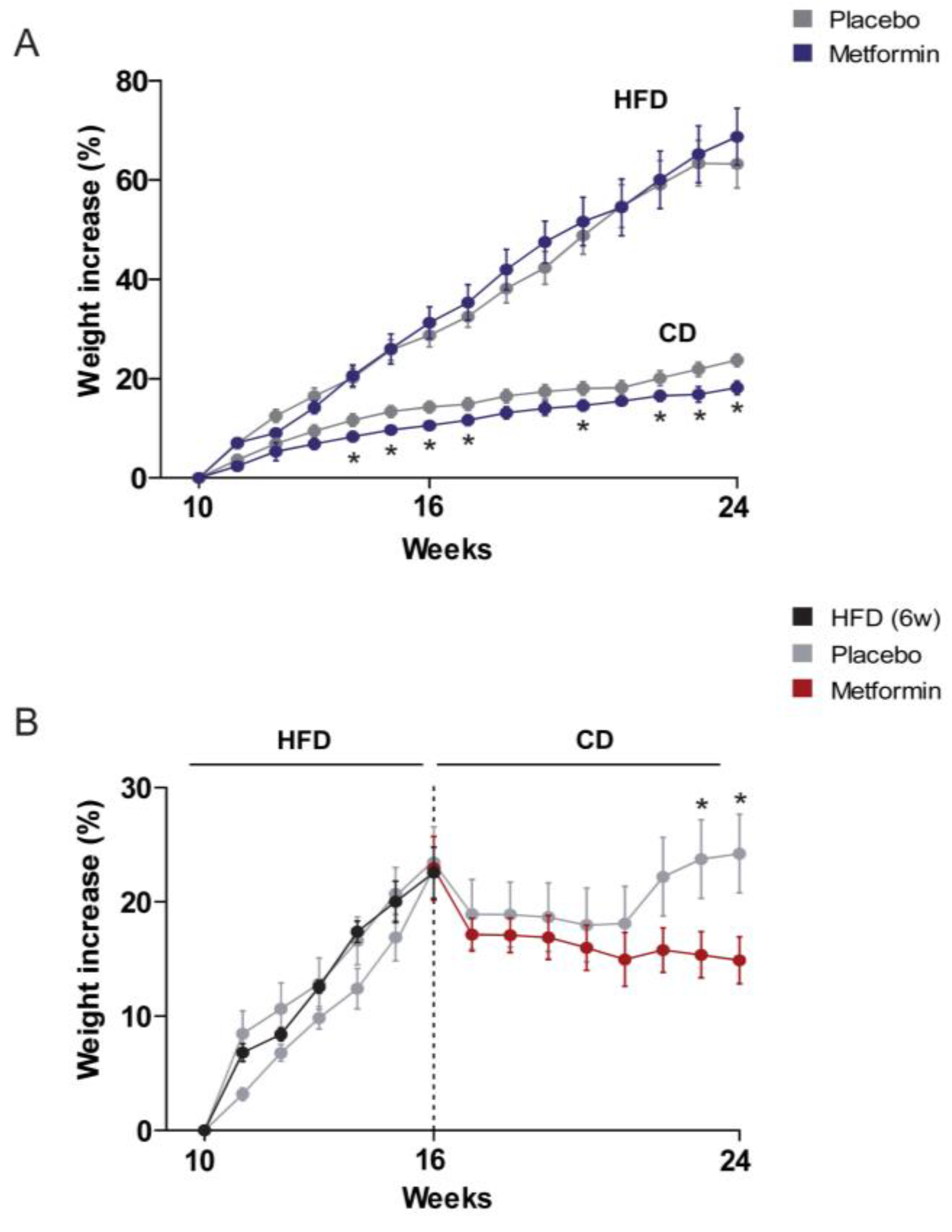
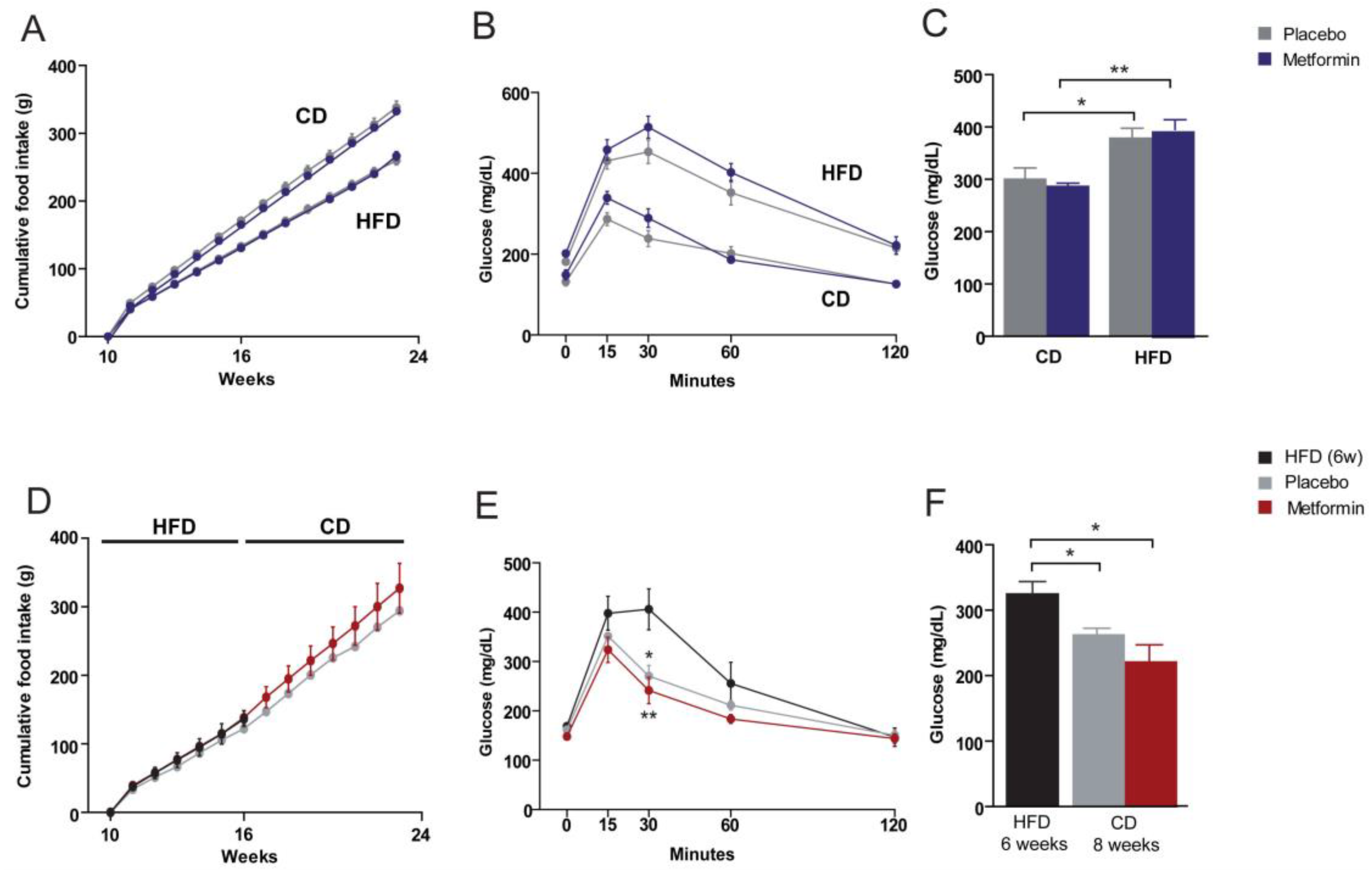
2.1. Effects on the Regulation of Body Weight and Food Intake
2.2. Effects on Glucose Homeostasis
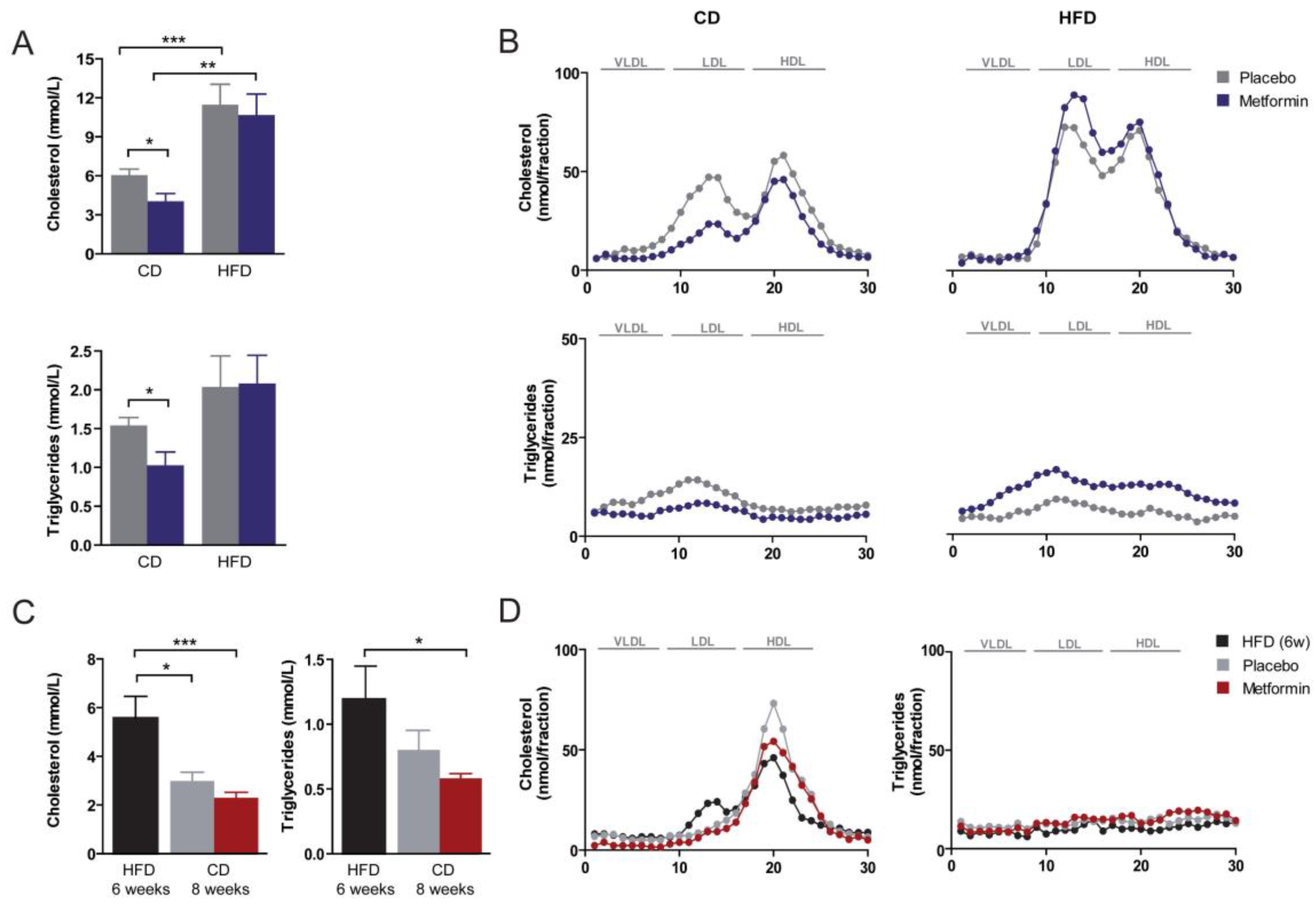
2.3. Effects on Hyperlipidemia
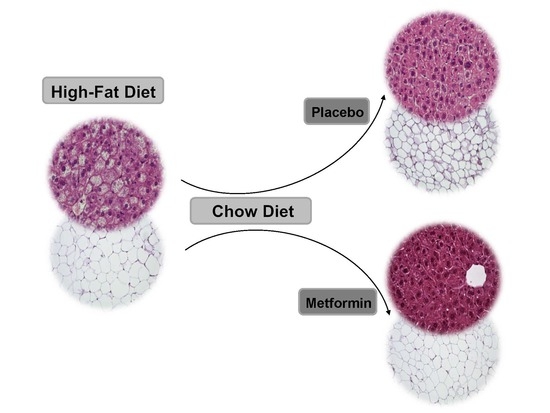
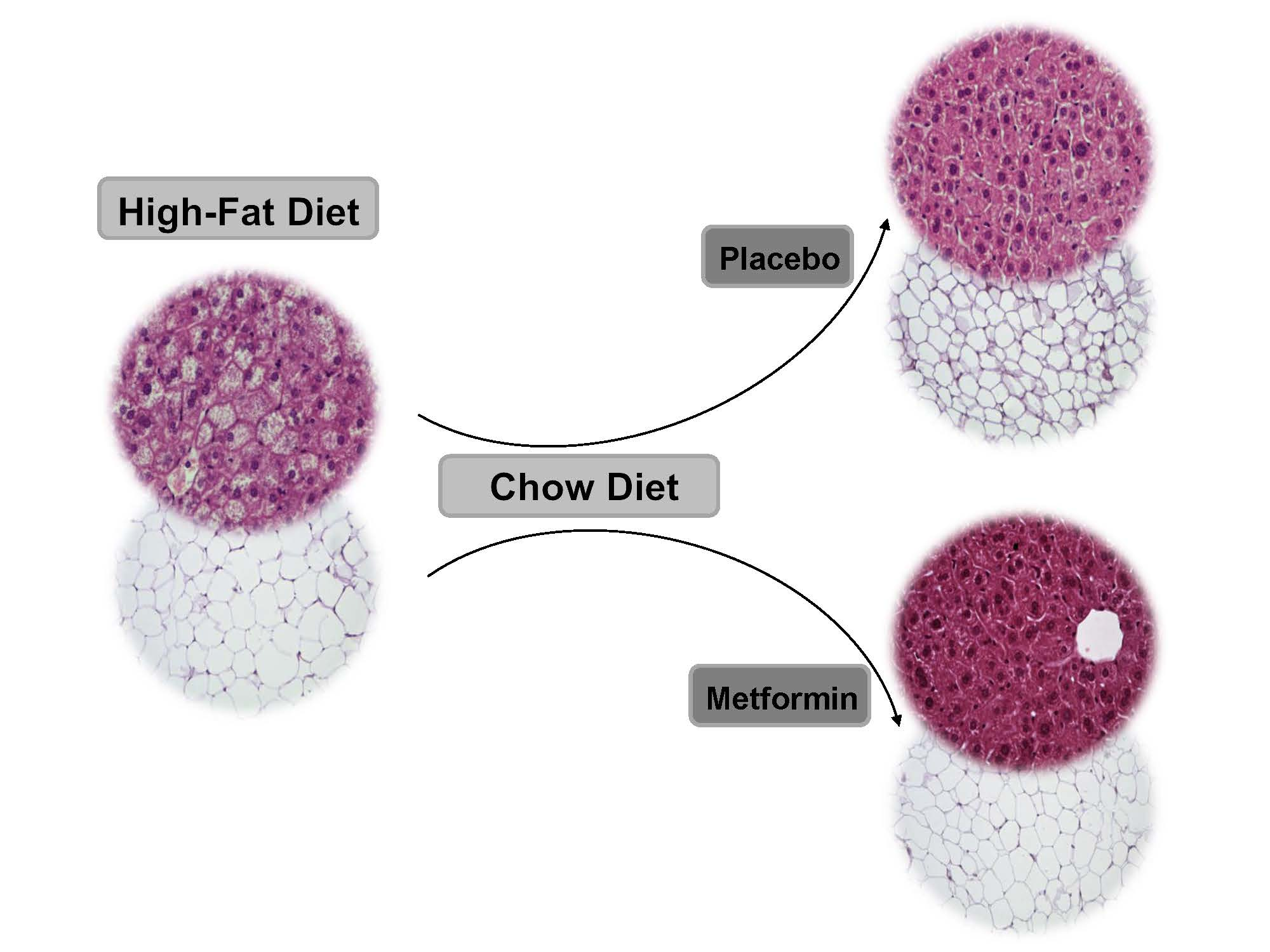
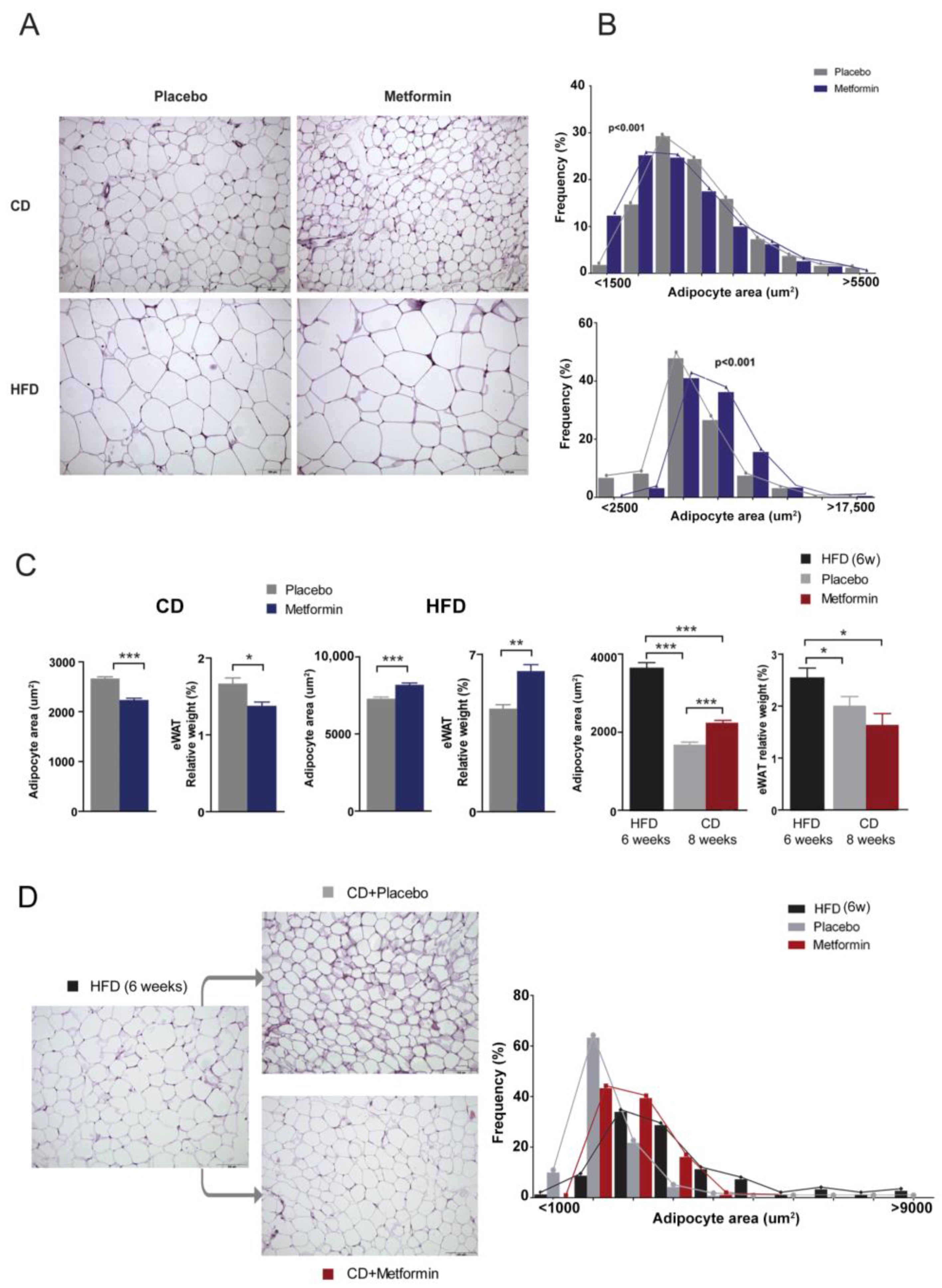
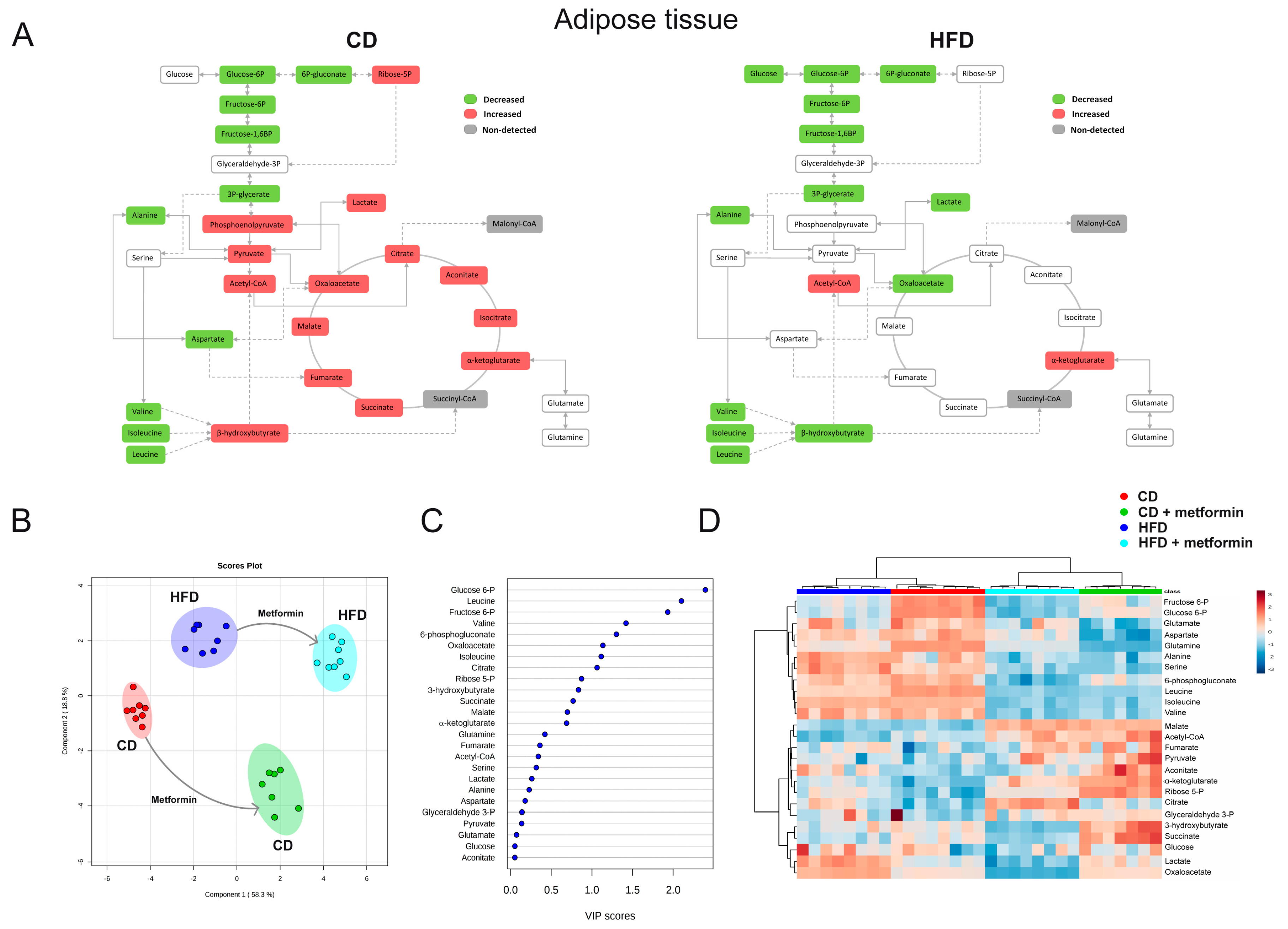
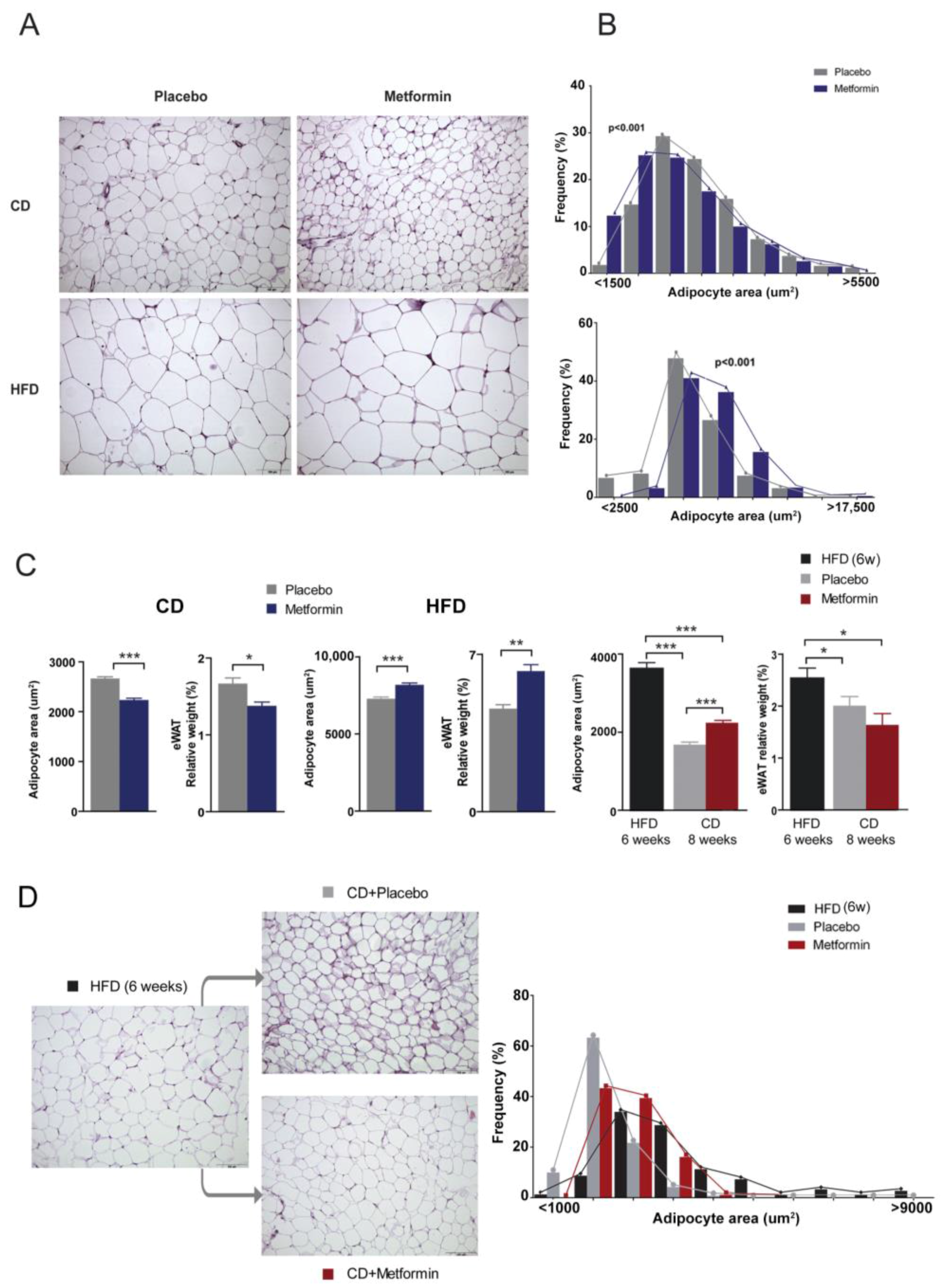
2.4. Effects on White Adipose Tissue
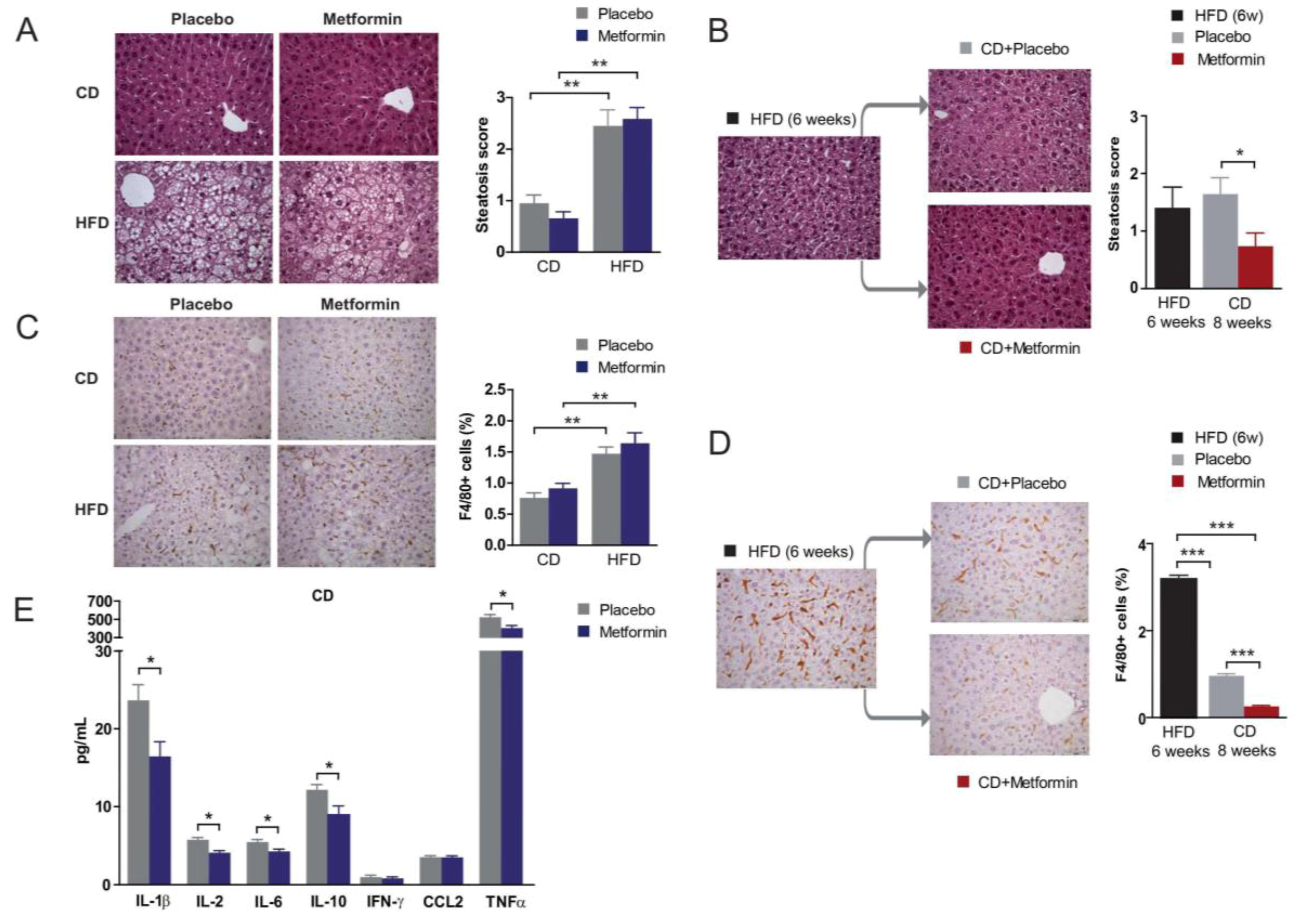
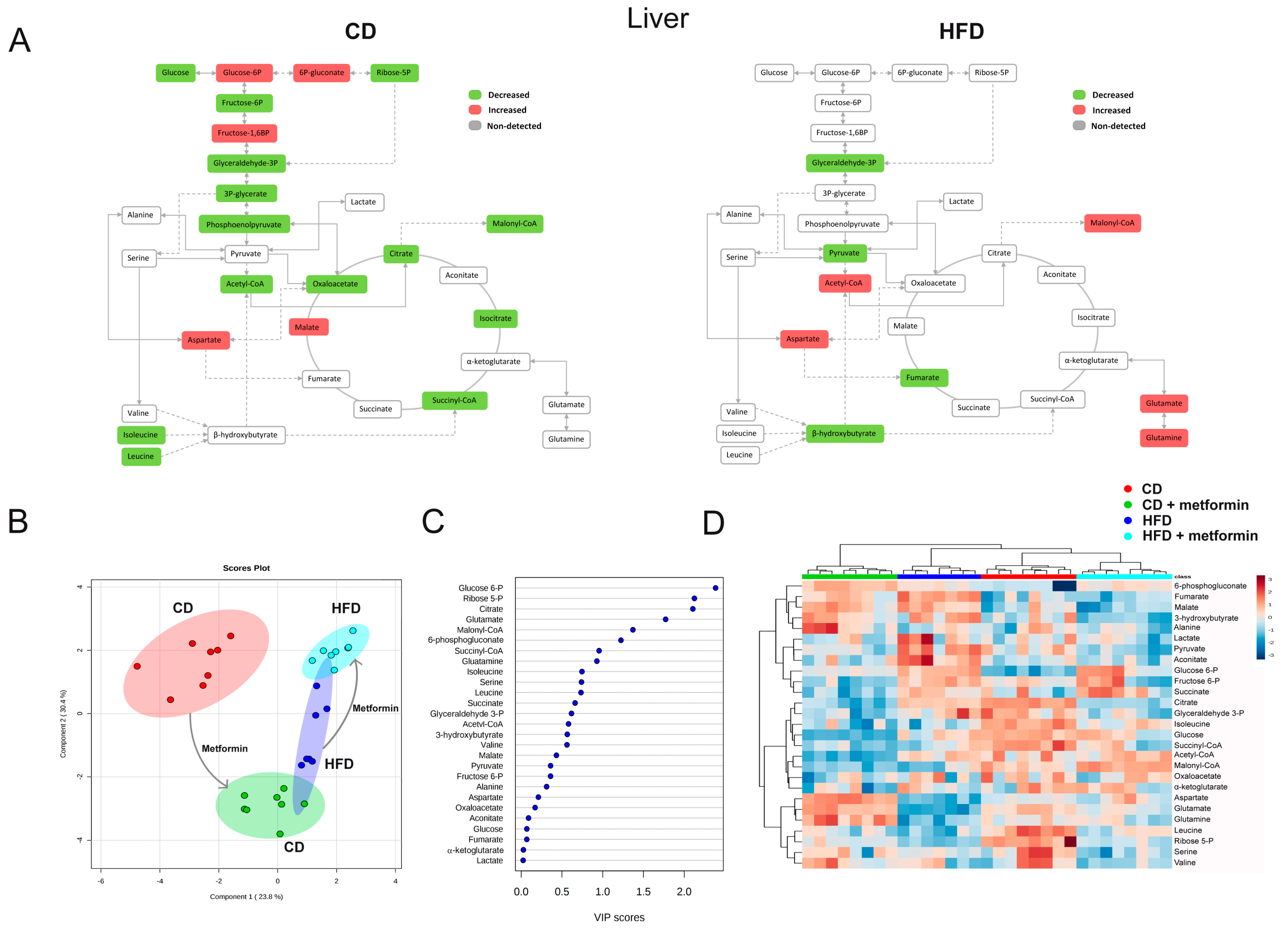
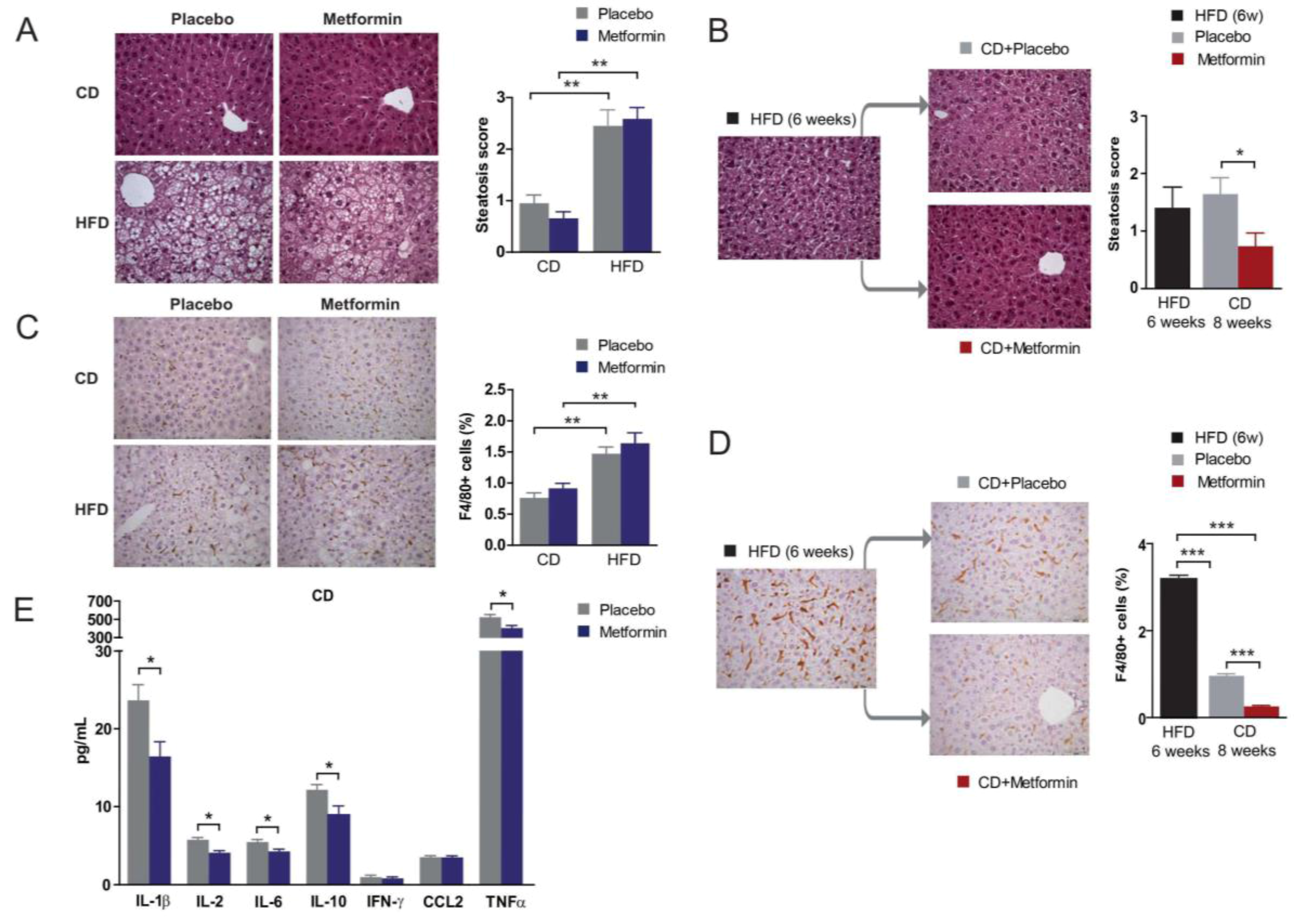
2.5. Effects on Hepatic Steatosis
2.6. Effects on Hepatic Inflammation
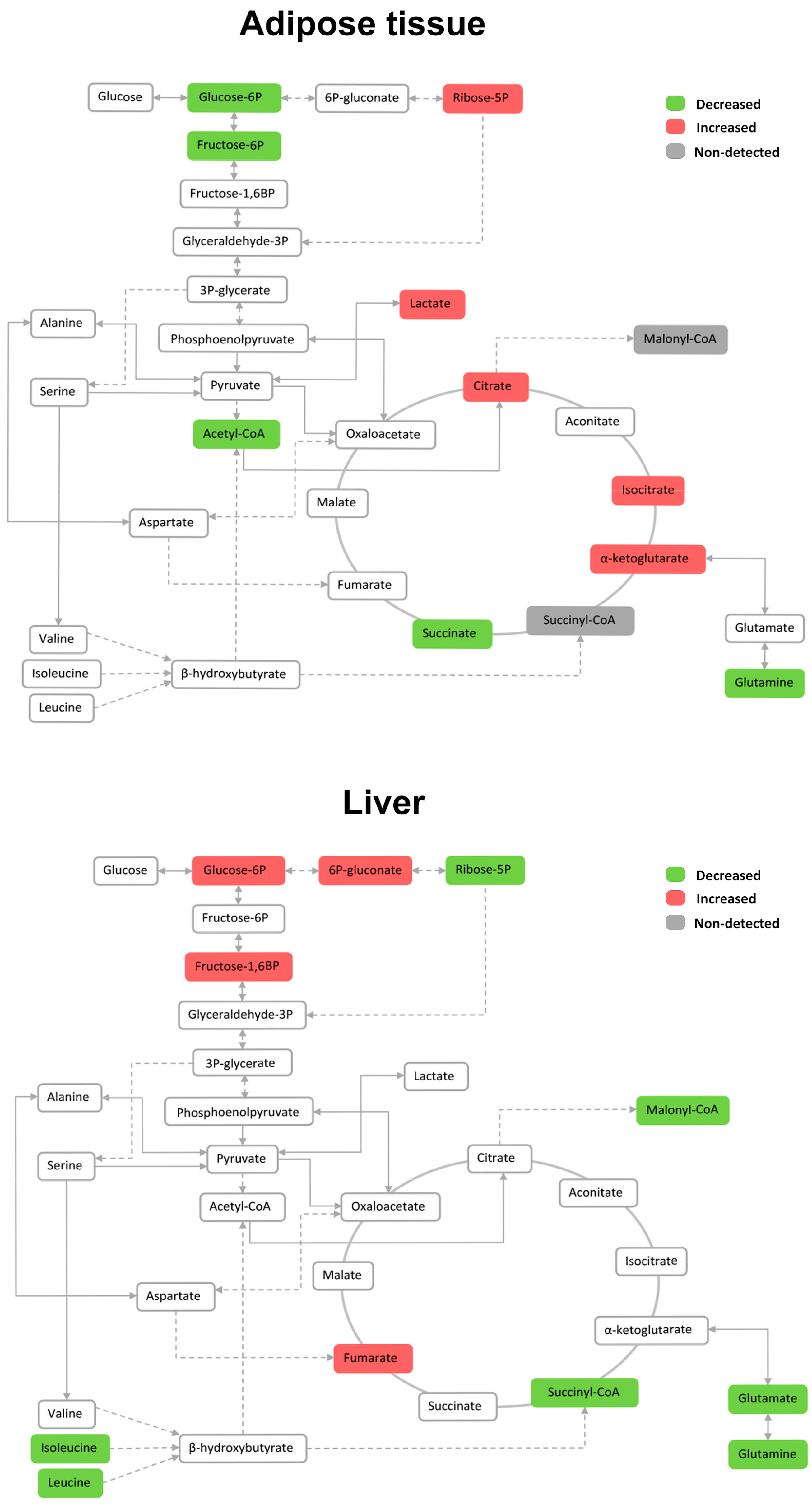
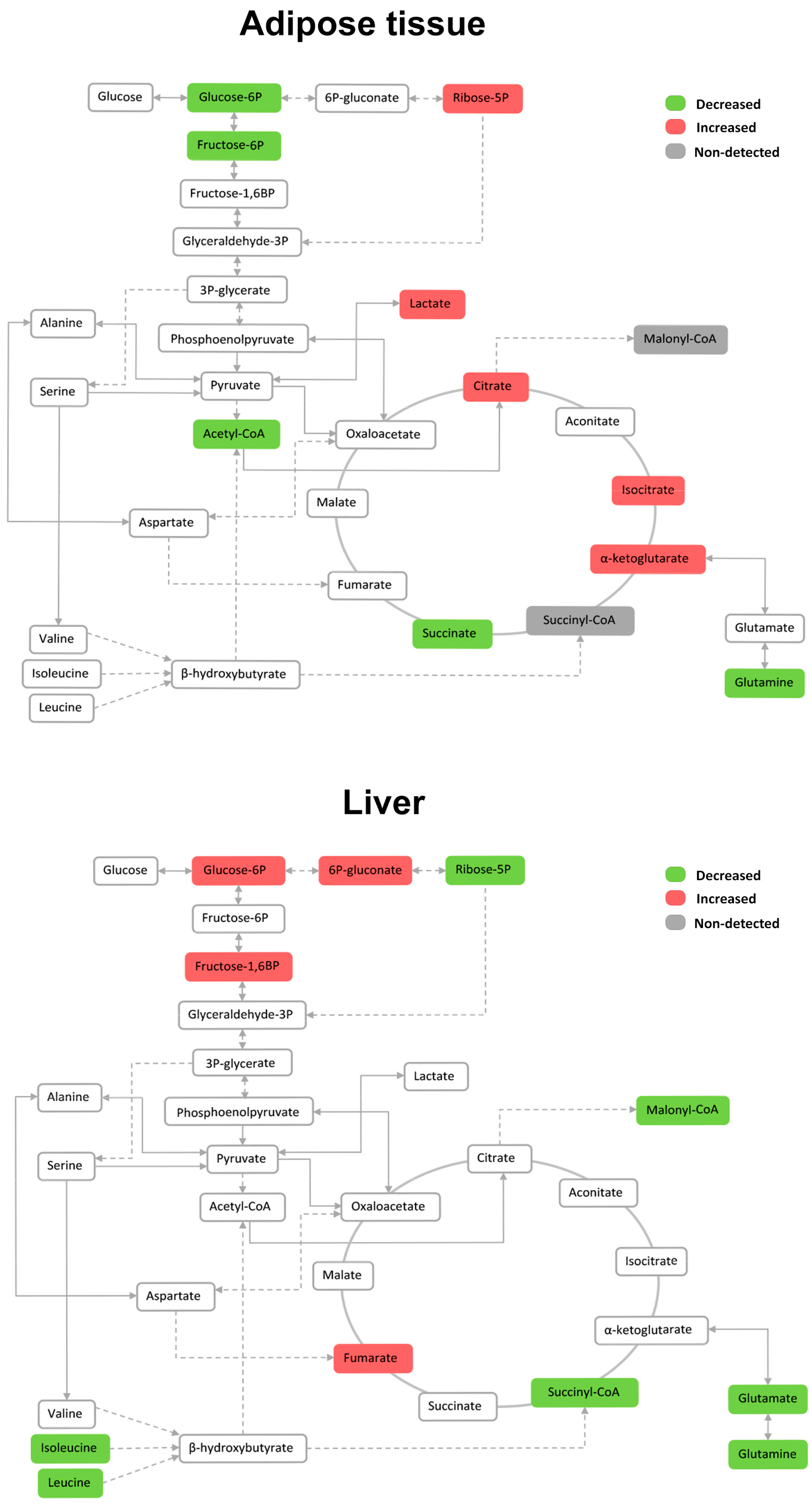
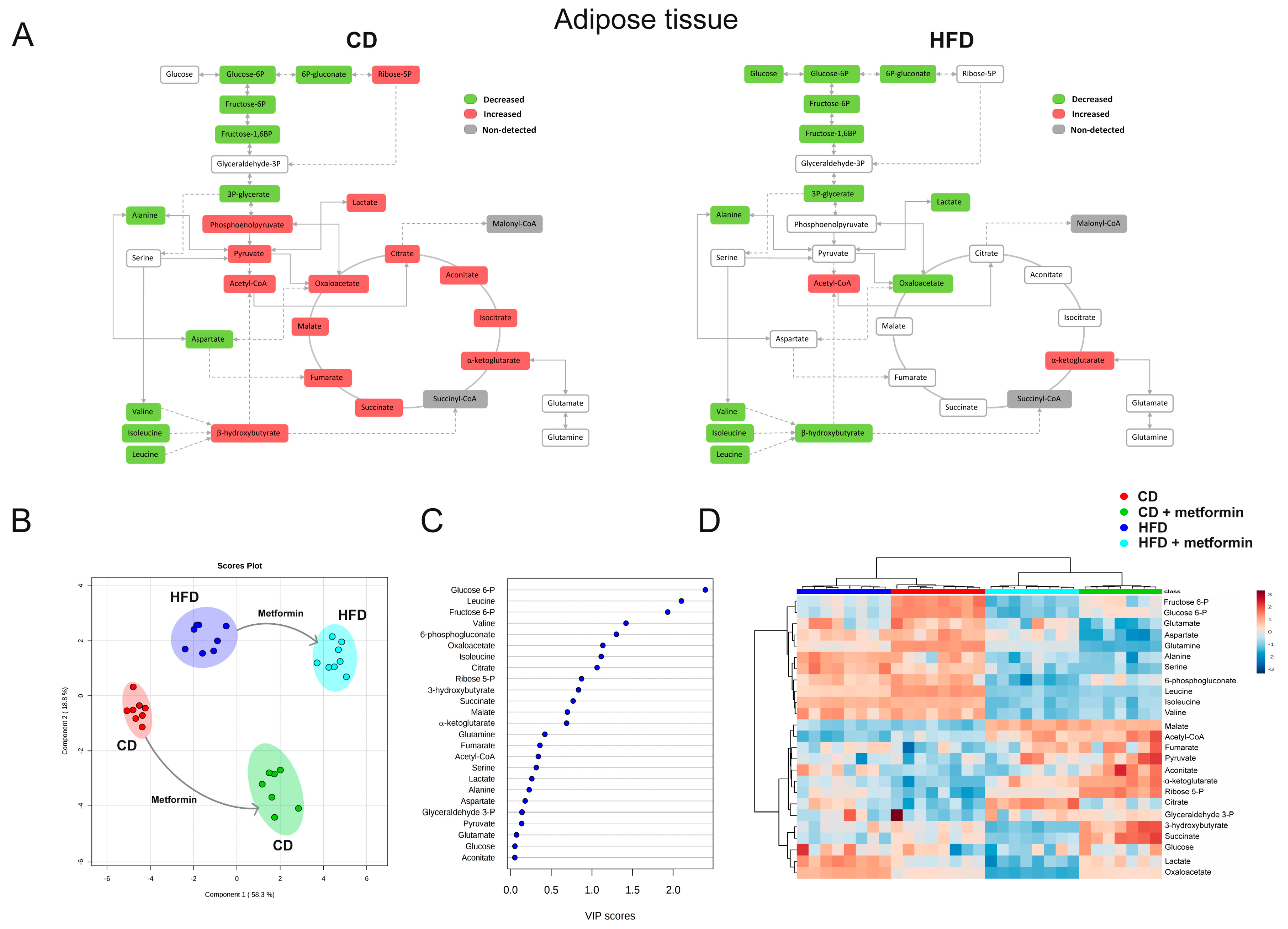
2.7. Effects on the Levels of Energy Metabolites in Hepatic and Adipose Tissue
3. Discussion
4. Materials and Methods
4.1. Animals, Animal Care and Dietary Details
4.2. Experimental Design and Metformin Provision
4.3. Sample Collection, Analytical Methods and Histological Analysis
4.4. Targeted Metabolomics
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hernández-Aguilera, A.; Fernández-Arroyo, S.; Cuyàs, E.; Luciano-Mateo, F.; Cabre, N.; Camps, J.; Lopez-Miranda, J.; Menendez, J.A.; Joven, J. Epigenetics and nutrition-related epidemics of metabolic diseases: Current perspectives and challenges. Food Chem. Toxicol. 2016, 96, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Schaffer, B.E.; Brunet, A. AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends Cell Biol. 2016, 26, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Massucci, F.A.; Wheeler, J.; Beltrán-Debón, R.; Joven, J.; Sales-Pardo, M.; Guimerà, R. Inferring propagation paths for sparsely observed perturbations on complex networks. Sci. Adv. 2016, 2, e1501638. [Google Scholar] [CrossRef] [PubMed]
- Mattison, J.A.; Roth, G.S.; Beasley, T.M.; Tilmont, E.M.; Handy, A.M.; Herbert, R.L.; Longo, D.L.; Allison, D.B.; Young, J.E.; et al. Impact of caloric restriction on health and survival in rhesus monkeys from the NIA study. Nature 2012, 489, 318–321. [Google Scholar] [CrossRef] [PubMed]
- Roth, G.S.; Ingram, D.K. Manipulation of health span and function by dietary caloric restriction mimetics. Ann. N. Y. Acad. Sci. 2016, 1363, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.K.; Marcinko, K.; Desjardins, E.M.; Lally, J.S.; Ford, R.J.; Steinberg, G.R. Treatment of nonalcoholic fatty liver disease: Role of AMPK. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E730–E740. [Google Scholar] [CrossRef] [PubMed]
- Barzilai, N.; Crandall, J.P.; Kritchevsky, S.B.; Espeland, M.A. Metformin as a Tool to Target Aging. Cell Metab. 2016, 23, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Joven, J. One-carbon metabolism: An aging-cancer crossroad for the gerosuppressant metformin. Aging 2012, 4, 894–898. [Google Scholar] [CrossRef] [PubMed]
- Corominas-Faja, B.; Quirantes-Piné, R.; Oliveras-Ferraros, C.; Vazquez-Martin, A.; Cufí, S.; Martin-Castillo, B.; Micol, V.; Joven, J.; Segura-Carretero, A.; Menendez, J.A. Metabolomic fingerprint reveals that metformin impairs one-carbon metabolism in a manner similar to the antifolate class of chemotherapy drugs. Aging 2012, 4, 480–498. [Google Scholar] [CrossRef] [PubMed]
- Viollet, B.; Guigas, B.; Sanz Garcia, N.; Leclerc, J.; Foretz, M.; Andreelli, F. Cellular and molecular mechanisms of metformin: An overview. Clin. Sci. 2012, 122, 253–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linden, M.A.; Lopez, K.T.; Fletcher, J.A.; Morris, E.M.; Meers, G.M.; Siddique, S.; Laughlin, M.H.; Sowers, J.R.; Thyfault, J.P.; Ibdah, J.A.; et al. Combining metformin therapy with caloric restriction for the management of type 2 diabetes and nonalcoholic fatty liver disease in obese rats. Appl. Physiol. Nutr. Metab. 2015, 40, 1038–1047. [Google Scholar] [PubMed]
- Schommers, P.; Thurau, A.; Bultmann-Mellin, I.; Guschlbauer, M.; Klatt, A.R.; Rozman, J.; Klingenspor, M.; de Angelis, M.H.; Alber, J.; Gründemann, D.; et al. Metformin causes a futile intestinal-hepatic cycle which increases energy expenditure and slows down development of a type 2 diabetes-like state. Mol. Metab. 2017, 6, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.L.; Xu, H.; Li, H.; Zhao, Y.; Hu, X.; Zhao, J.; Guo, X.; Guo, T.; Botchlett, R.; Qi, T.; et al. Metformin ameliorates hepatic steatosis and inflammation without altering adipose phenotype in diet-induced obesity. PLoS ONE 2014, 9, e91111. [Google Scholar] [CrossRef] [PubMed]
- De Vadder, F.; Kovatcheva-Datchary, P.; Goncalves, D.; Vinera, J.; Zitoun, C.; Duchampt, A.; Bäckhed, F.; Mithieux, G. Microbiota-generated metabolites promote metabolic benefits via gut-brain neural circuits. Cell 2014, 156, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Brunkwall, L.; Orho-Melander, M. The gut microbiome as a target for prevention and treatment of hyperglycaemia in type 2 diabetes: From current human evidence to future possibilities. Diabetologia 2017, 60, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Viollet, B. Metformin takes a new route to clinical efficacy. Nat. Rev. Endocrinol. 2015, 11, 390–392. [Google Scholar] [CrossRef] [PubMed]
- Pernicova, I.; Korbonits, M. Metformin—Mode of action and clinical implications for diabetes and cancer. Nat. Rev. Endocrinol. 2014, 10, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Saisho, Y. Metformin and Inflammation: Its Potential Beyond Glucose-lowering Effect. Endocr. Metab. Immune Disord. Drug Targets 2015, 15, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Domecq, J.P.; Prutsky, G.; Leppin, A.; Sonbol, M.B.; Altayar, O.; Undavalli, C.; Wang, Z.; Elraiyah, T.; Brito, J.P.; Mauck, K.F.; et al. Clinical review: Drugs commonly associated with weight change: A systematic review and meta-analysis. J. Clin. Endocrinol. Metab. 2015, 100, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017. [Google Scholar] [CrossRef] [PubMed]
- Luciano-Mateo, F.; Hernández-Aguilera, A.; Cabre, N.; Camps, J.; Fernández-Arroyo, S.; Lopez-Miranda, J.; Menendez, J.A.; Joven, J. Nutrients in Energy and One-Carbon Metabolism: Learning from Metformin Users. Nutrients 2017, 9, 121. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Ren, L.; Zhan, P.; Yang, H.; Chai, D.; Yu, Z. Metformin exerts glucose-lowering action in high-fat fed mice via attenuating endotoxemia and enhancing insulin signaling. Acta Pharmacol. Sin. 2016, 43, 1063–1075. [Google Scholar] [CrossRef] [PubMed]
- Jaganjac, M.; Aluraikhy, S.; Al-Khelaifi, F.; Al-Jaber, M.; Bashah, M.; Mazloum, N.A.; Zarkovic, K.; Zarkovic, N.; Waeg, G.; Kafienah, W.; et al. Combined metformin and insulin treatment reverses metabolically impaired omental adipogenensis and accumulation of 4-hydroxynonenal in obese diabetic patients. Redox Biol. 2017, 12, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Martin-Castillo, B.; Joven, J. Metformin and cancer: Quo vadis et cui bono? Oncotarget 2016, 7, 54096–54101. [Google Scholar] [CrossRef] [PubMed]
- Ota, T.; Takamura, T.; Kurita, S.; Matsuzawa, N.; Kita, Y.; Uno, M.; Akahori, H.; Misu, H.; Sakurai, M.; Zen, Y.; et al. Insulin resistance accelerates a dietary rat model of nonalcoholic steatohepatitis. Gastroenterology 2007, 132, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Liang, G.; Yao, Z.; Zhang, J.; Liu, R.; Chen, H.; Zhou, Y.; Wu, H.; Yang, B.; He, Q. Metformin prevents cancer metastasis by inhibiting M2-like polarization of tumor associated macrophages. Oncotarget 2015, 6, 36441–36455. [Google Scholar] [PubMed]
- Caër, C.; Rouault, C.; Le Roy, T.; Poitou, C.; Aron-Wisnewsky, J.; Torcivia, A.; Bichet, J.C.; Clément, K.; Guerre-Millo, M.; André, S. Immune cell-derived cytokines contribute to obesity-related inflammation, fibrogenesis and metabolic deregulation in human adipose tissue. Sci. Rep. 2017, 7, 3000. [Google Scholar] [CrossRef] [PubMed]
- Griss, T.; Vincent, E.E.; Egnatchik, R.; Chen, J.; Ma, E.H.; Faubert, B.; Viollet, B.; DeBerardinis, R.J.; Jones, R.G. Metformin Antagonizes Cancer Cell Proliferation by Suppressing Mitochondrial-Dependent Biosynthesis. PLoS Biol. 2015, 13, e1002309. [Google Scholar] [CrossRef] [PubMed]
- Cuyàs, E.; Fernández-Arroyo, S.; Alarcón, T.; Lupu, R.; Joven, J.; Menendez, J.A. Germline BRCA1 mutation reprograms breast epithelial cell metabolism towards mitochondrial-dependent biosynthesis: Evidence for metformin-based “starvation” strategies in BRCA1 carriers. Oncotarget 2016, 7, 52974–52992. [Google Scholar] [CrossRef] [PubMed]
- Satapati, S.; Sunny, N.E.; Kucejova, B.; Fu, X.; He, T.T.; Méndez-Lucas, A.; Shelton, J.M.; Perales, J.C.; Browning, J.D.; Burgess, S.C. Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. J. Lipid Res. 2012, 53, 1080–1092. [Google Scholar] [CrossRef] [PubMed]
- Satapati, S.; Kucejova, B.; Duarte, J.A.; Fletcher, J.A.; Reynolds, L.; Sunny, N.E.; He, T.; Nair, L.A.; Livingston, K.; Fu, X.; et al. Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J. Clin. Investig. 2015, 125, 4447–4462. [Google Scholar] [CrossRef] [PubMed]
- García-Heredia, A.; Riera-Borrull, M.; Fort-Gallifa, I.; Luciano-Mateo, F.; Cabré, N.; Hernández-Aguilera, A.; Joven, J.; Camps, J. Metformin administration induces hepatotoxic effects in paraoxonase-1-deficient mice. Chem. Biol. Interact. 2016, 249, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Herman, M.A.; She, P.; Peroni, O.D.; Lynch, C.J.; Kahn, B.B. Adipose tissue branched chain amino acid (BCAA) metabolism modulates circulating BCAA levels. J. Biol. Chem. 2010, 285, 11348–11356. [Google Scholar] [CrossRef] [PubMed]
- Green, C.R.; Wallace, M.; Divakaruni, A.S.; Phillips, S.A.; Murphy, A.N.; Ciaraldi, T.P.; Metallo, C.M. Branched-chain amino acid catabolism fuels adipocyte differentiation and lipogenesis. Nat. Chem. Biol. 2016, 12, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Karise, I.; Ornellas, F.; Barbosa-da-Silva, S.; Matsuura, C.; del Sol, M.; Aguila, M.B.; Mandarim-de-Lacerda, C.A. Liver and Metformin: Lessons of a fructose diet in mice. Biochim. Open 2017, 4, 19–30. [Google Scholar] [CrossRef]
- Cufí, S.; Corominas-Faja, B.; Lopez-Bonet, E.; Bonavia, R.; Pernas, S.; López, I.Á.; Dorca, J.; Martínez, S.; López, N.B.; Fernández, S.D.; et al. Dietary restriction-resistant human tumors harboring the PIK3CA-activating mutation H1047R are sensitive to metformin. Oncotarget 2013, 4, 1484–1495. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Galluzzi, L.; Freije, J.M.; Madeo, F.; Kroemer, G. Metabolic Control of Longevity. Cell 2016, 166, 802–821. [Google Scholar] [CrossRef] [PubMed]
- Anisimov, V.N. Metformin: Do we finally have an anti-aging drug? Cell Cycle 2013, 12, 3483–3489. [Google Scholar] [CrossRef] [PubMed]
- Joven, J.; Rull, A.; Ferré, N.; Escolà-Gil, J.C.; Marsillach, J.; Coll, B.; Alonso-Villaverde, C.; Aragones, G.; Claria, J.; Camps, J. The results in rodent models of atherosclerosis are not interchangeable: The influence of diet and strain. Atherosclerosis 2007, 195, e85–e92. [Google Scholar] [CrossRef] [PubMed]
- Riera-Borrull, M.; Rodríguez-Gallego, E.; Hernández-Aguilera, A.; Luciano, F.; Ras, R.; Cuyàs, E.; Camps, J.; Segura-Carretero, A.; Menendez, J.A.; Joven, J.; et al. Exploring the Process of Energy Generation in Pathophysiology by Targeted Metabolomics: Performance of a Simple and Quantitative Method. J. Am. Soc. Mass Spectrom. 2016, 27, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Anisimov, V.N. Metformin for Prevention and Treatment of Colon Cancer: A Reappraisal of Experimental and Clinical Data. Curr. Drug Targets 2016, 17, 439–446. [Google Scholar] [CrossRef] [PubMed]
- García-Heredia, A.; Kensicki, E.; Mohney, R.P.; Rull, A.; Triguero, I.; Marsillach, J.; Tormos, C.; Mackness, B.; Mackness, M.; Shih, D.M.; et al. Paraoxonase-1 deficiency is associated with severe liver steatosis in mice fed a high-fat high-cholesterol diet: A metabolomic approach. J. Proteome Res. 2013, 12, 1946–1955. [Google Scholar] [CrossRef] [PubMed]
- Joven, J.; Espinel, E.; Rull, A.; Aragonès, G.; Rodríguez-Gallego, E.; Camps, J.; Micol, V.; Herranz-López, M.; Menéndez, J.A.; Borrás, I.; et al. Plant-derived polyphenols regulate expression of miRNA paralogs miR-103/107 and miR-122 and prevent diet-induced fatty liver disease in hyperlipidemic mice. Biochim. Biophys. Acta 2012, 1820, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Sinelnikov, I.V.; Han, B.; Wishart, D.S. MetaboAnalyst 3.0—Making metabolomics more meaningful. Nucleic Acids Res. 2015, 43, W251–W257. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riera-Borrull, M.; García-Heredia, A.; Fernández-Arroyo, S.; Hernández-Aguilera, A.; Cabré, N.; Cuyàs, E.; Luciano-Mateo, F.; Camps, J.; Menendez, J.A.; Joven, J. Metformin Potentiates the Benefits of Dietary Restraint: A Metabolomic Study. Int. J. Mol. Sci. 2017, 18, 2263. https://doi.org/10.3390/ijms18112263
Riera-Borrull M, García-Heredia A, Fernández-Arroyo S, Hernández-Aguilera A, Cabré N, Cuyàs E, Luciano-Mateo F, Camps J, Menendez JA, Joven J. Metformin Potentiates the Benefits of Dietary Restraint: A Metabolomic Study. International Journal of Molecular Sciences. 2017; 18(11):2263. https://doi.org/10.3390/ijms18112263
Chicago/Turabian StyleRiera-Borrull, Marta, Anabel García-Heredia, Salvador Fernández-Arroyo, Anna Hernández-Aguilera, Noemí Cabré, Elisabet Cuyàs, Fedra Luciano-Mateo, Jordi Camps, Javier A. Menendez, and Jorge Joven. 2017. "Metformin Potentiates the Benefits of Dietary Restraint: A Metabolomic Study" International Journal of Molecular Sciences 18, no. 11: 2263. https://doi.org/10.3390/ijms18112263