A Versatile Chemo-Enzymatic Conjugation Approach Yields Homogeneous and Highly Potent Antibody-Drug Conjugates

 , and
, and
Abstract
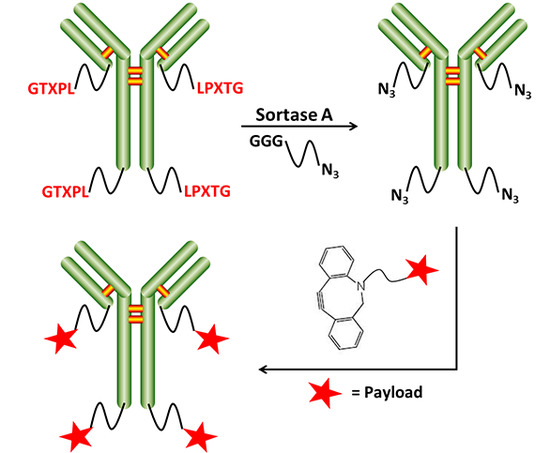
:
1. Introduction
2. Results and Discussion
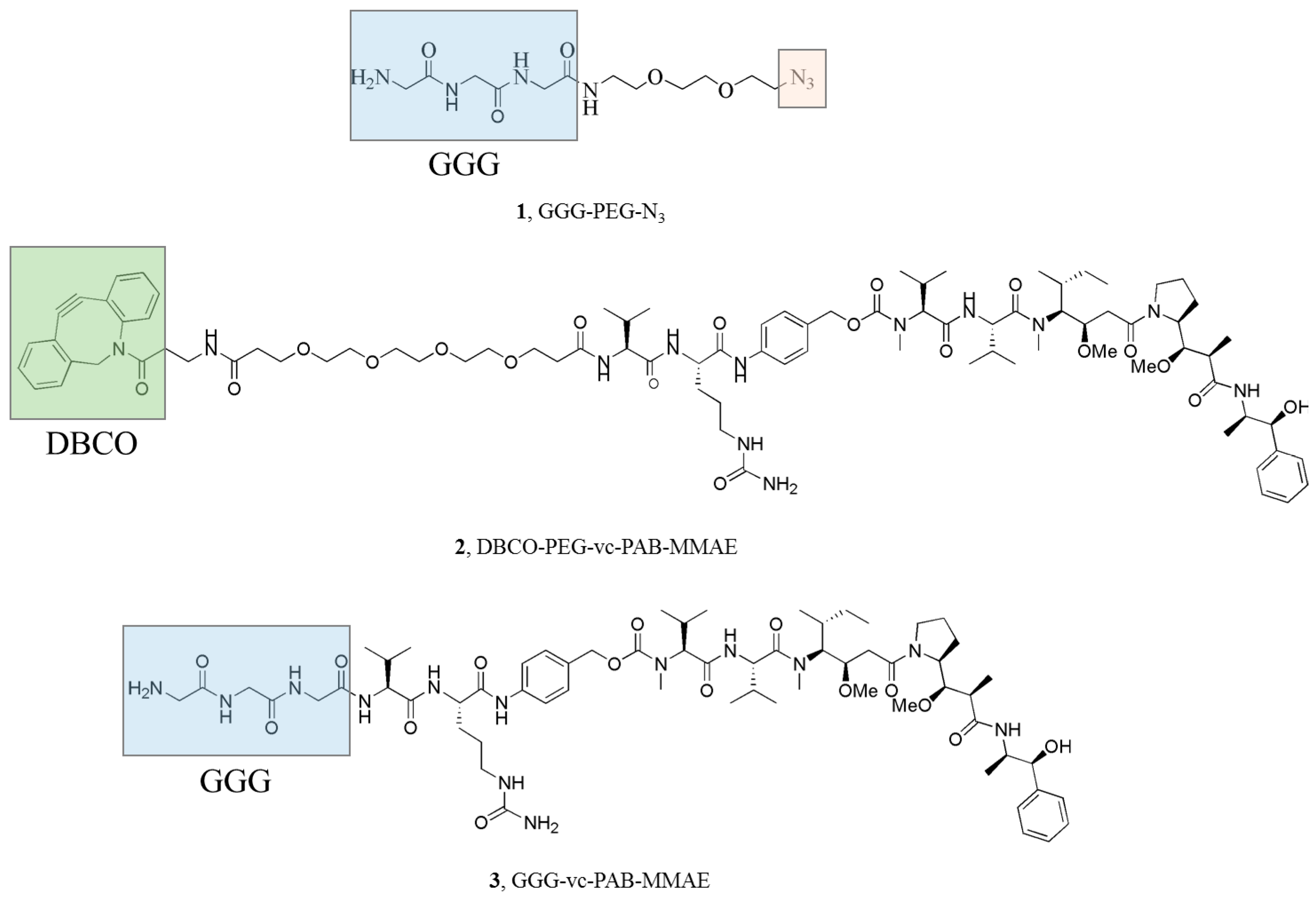
2.1. Preparation of Optimized OFA
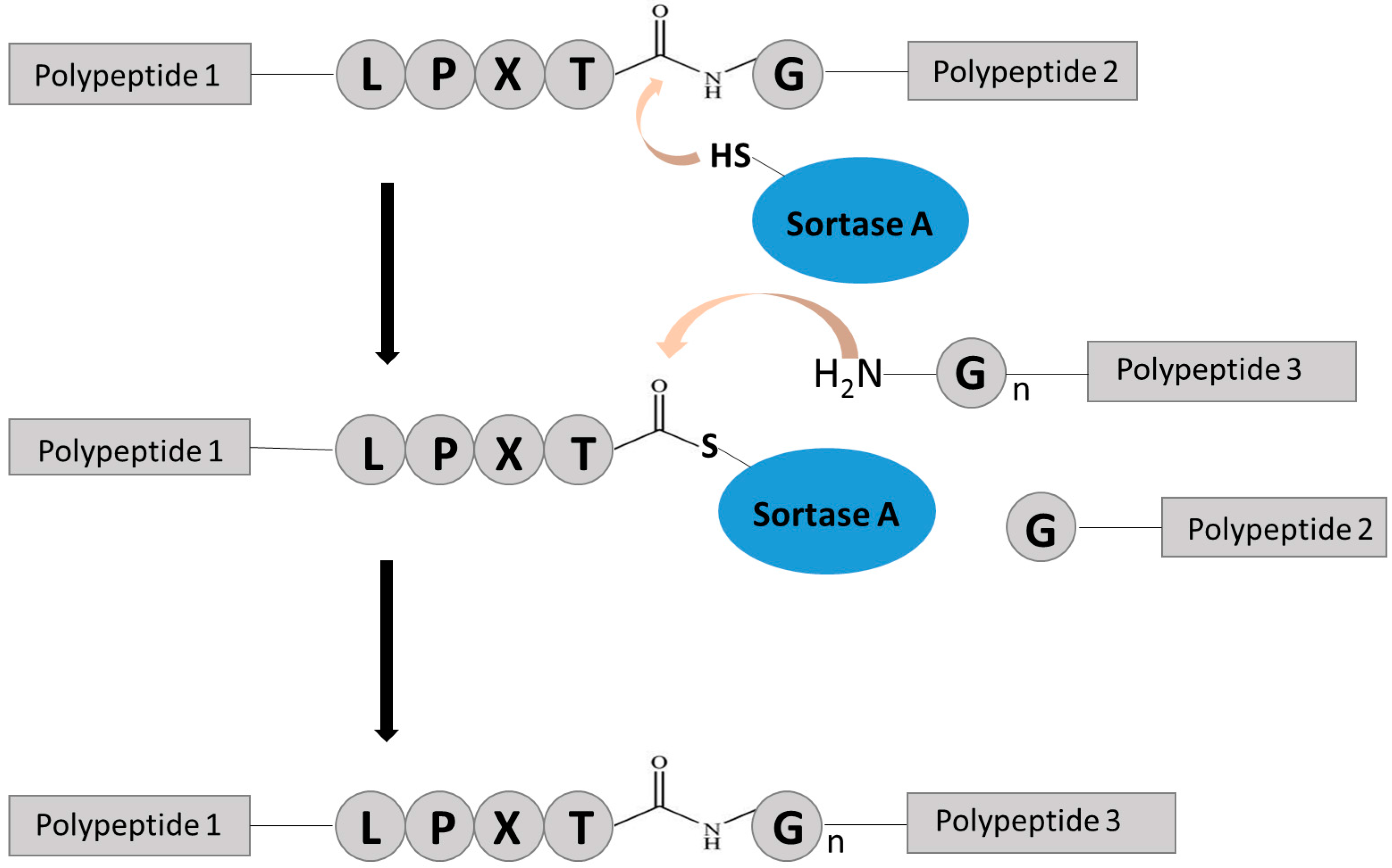
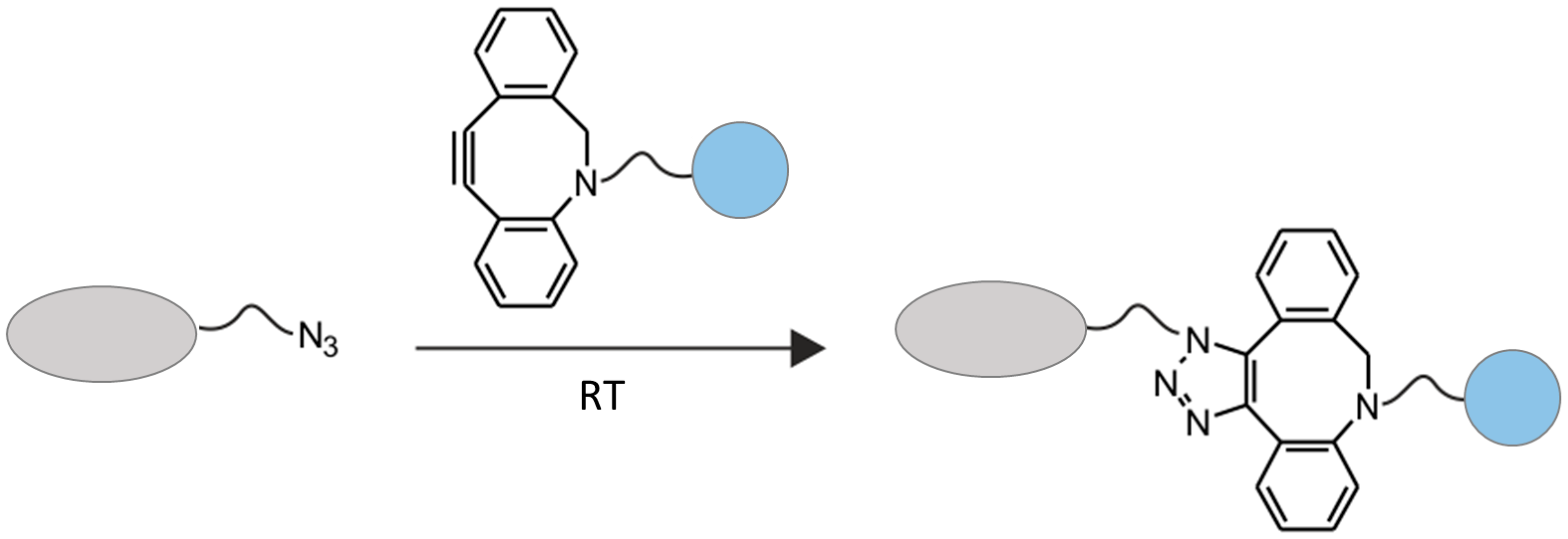
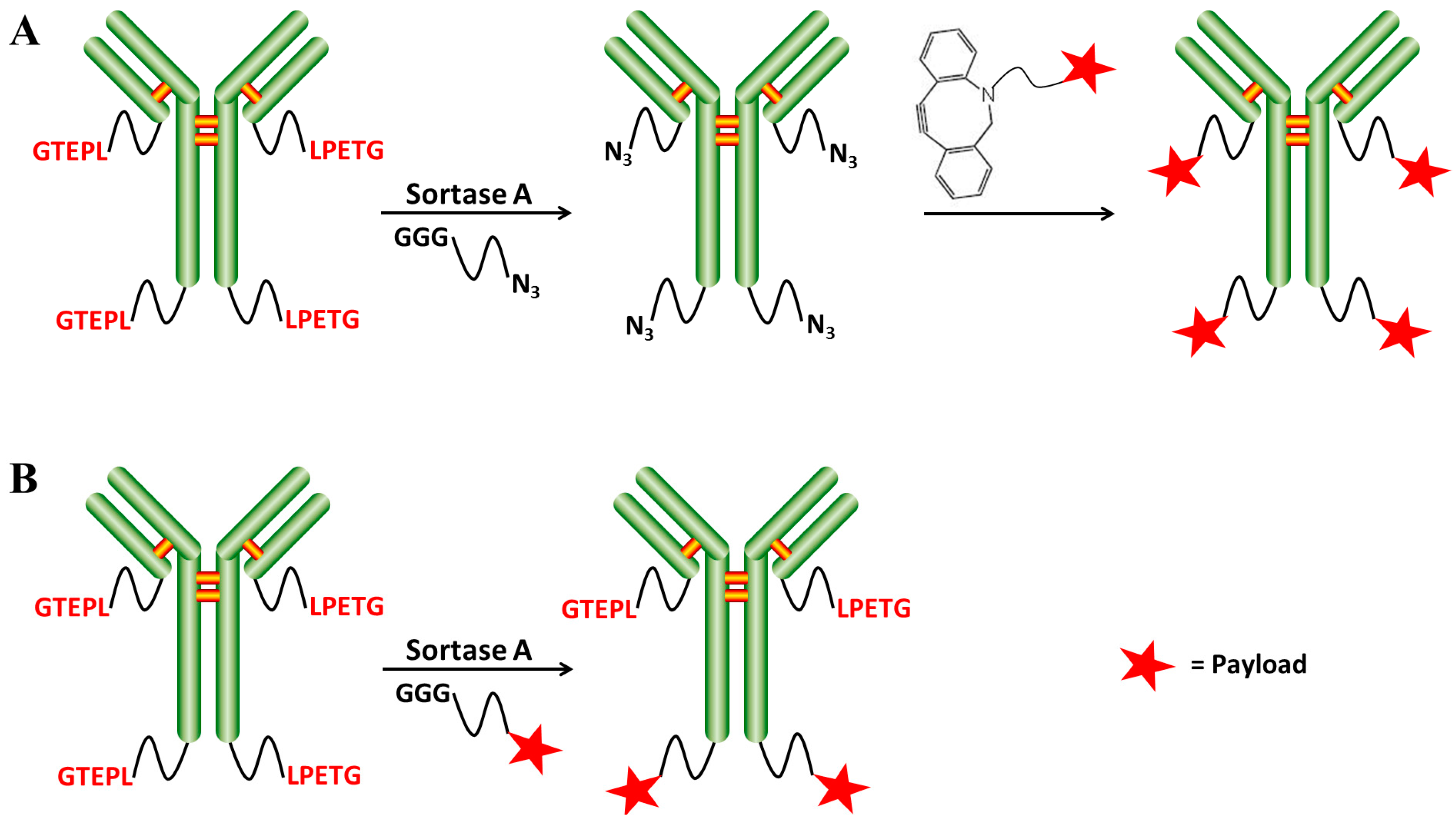
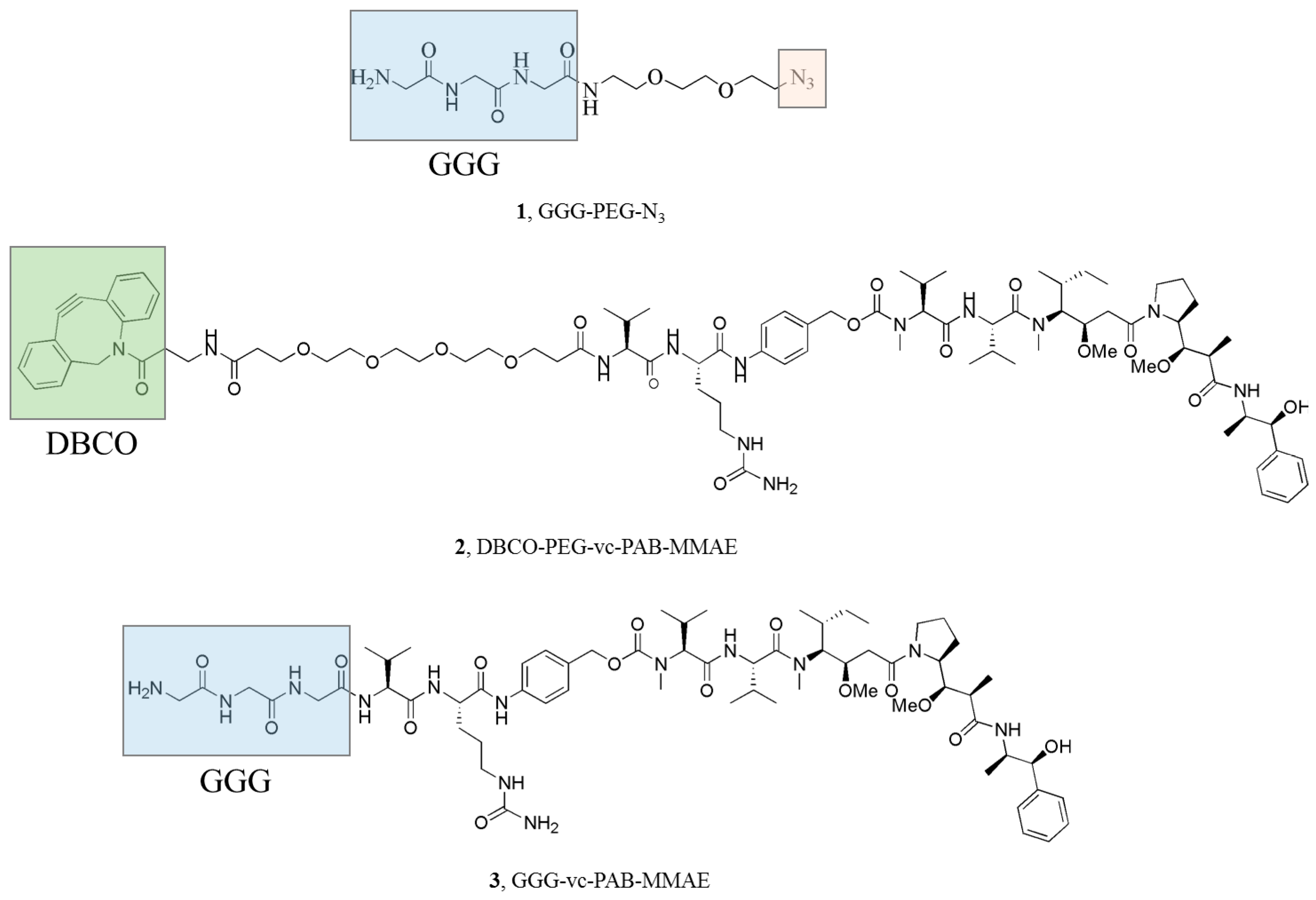
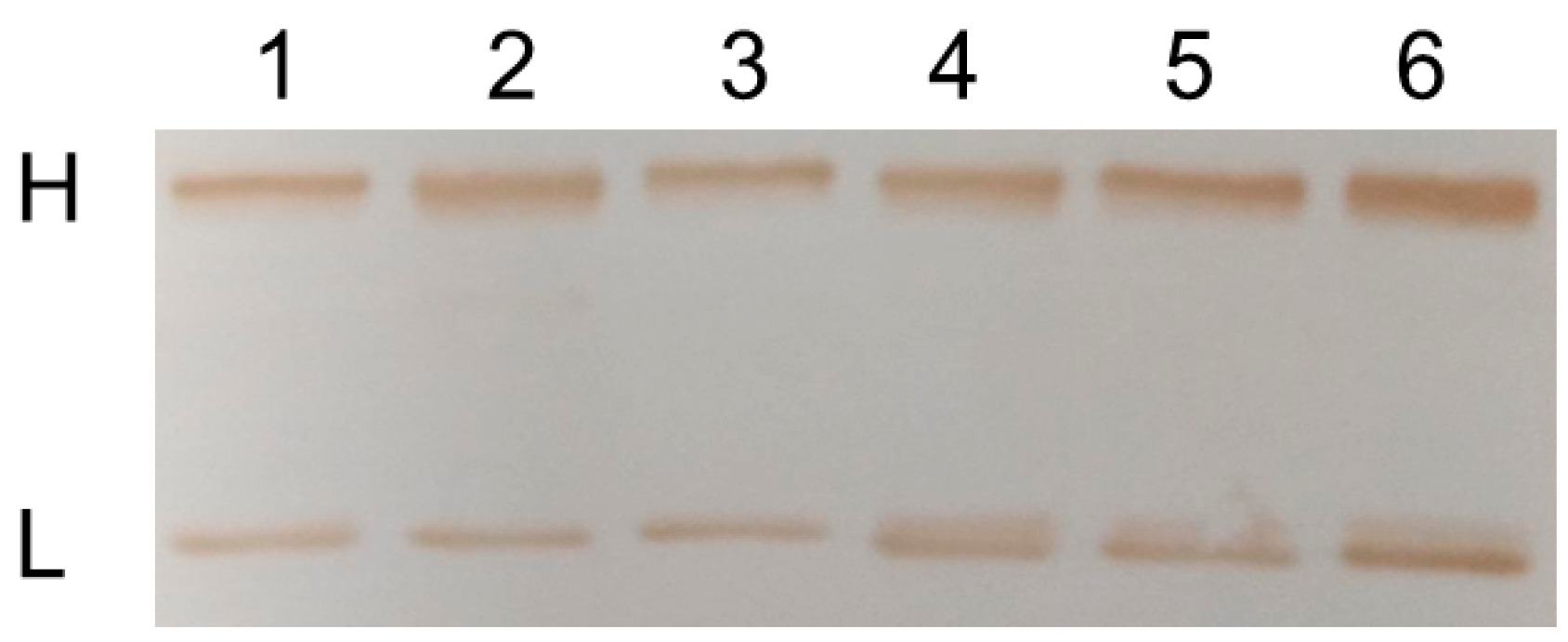
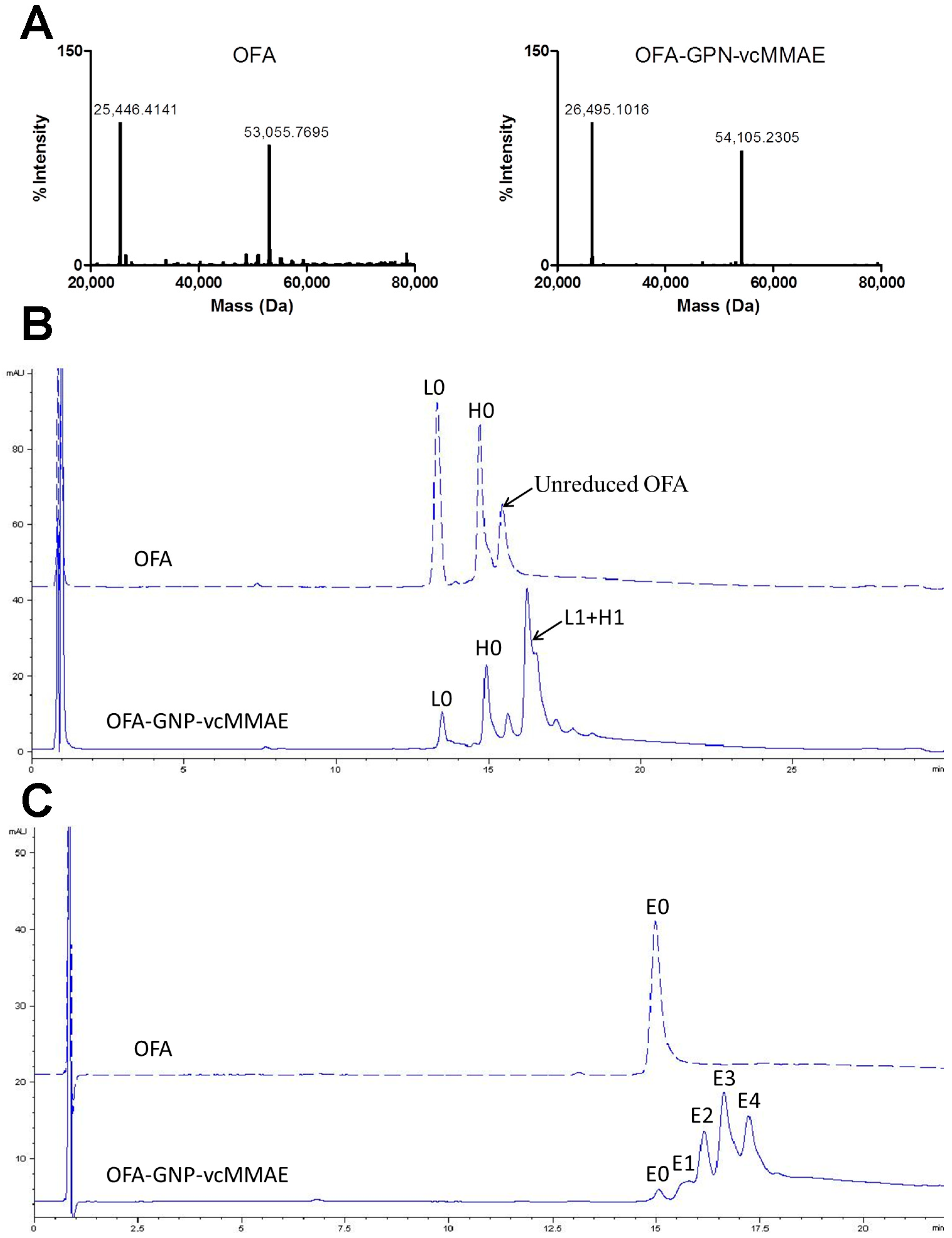
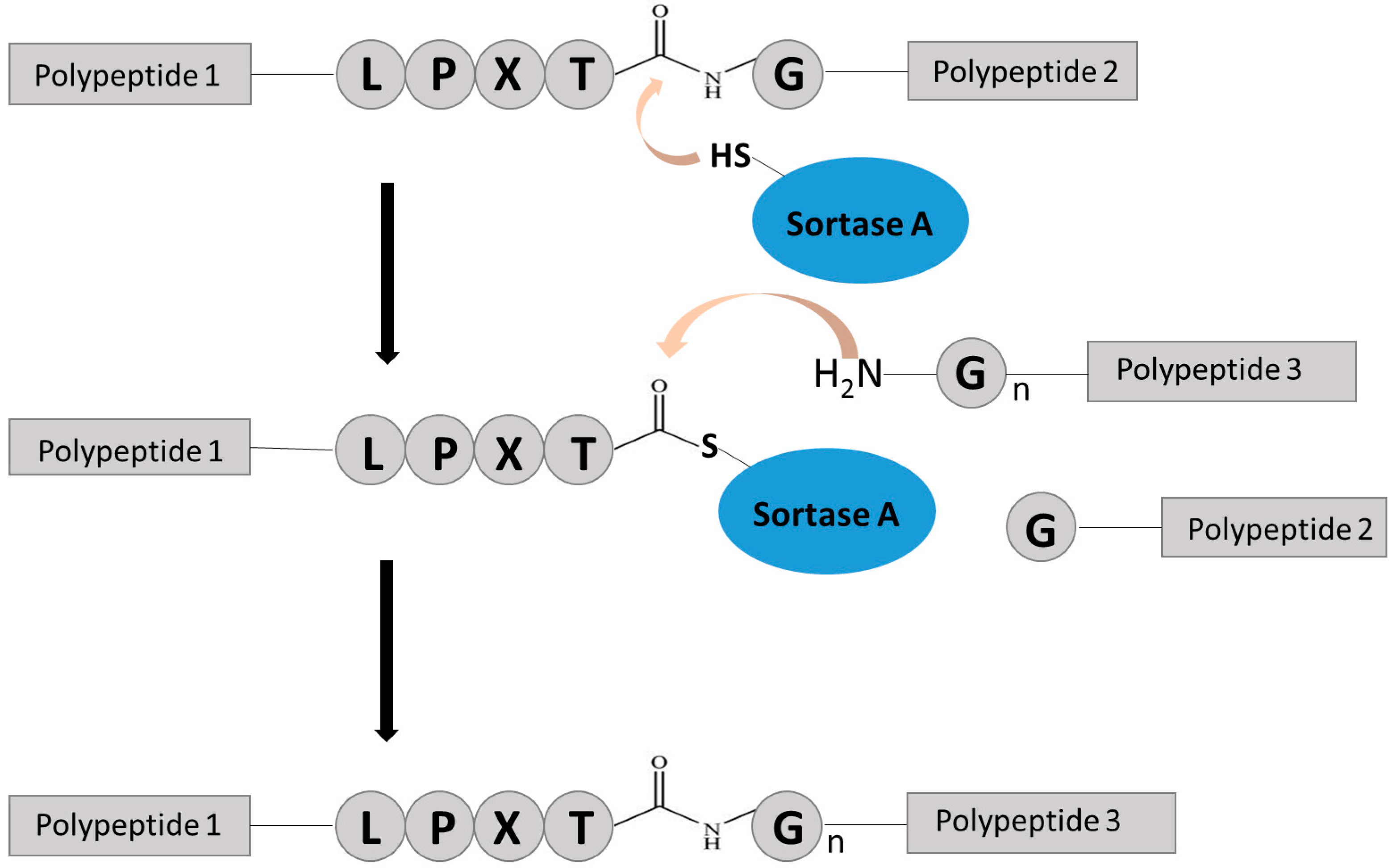
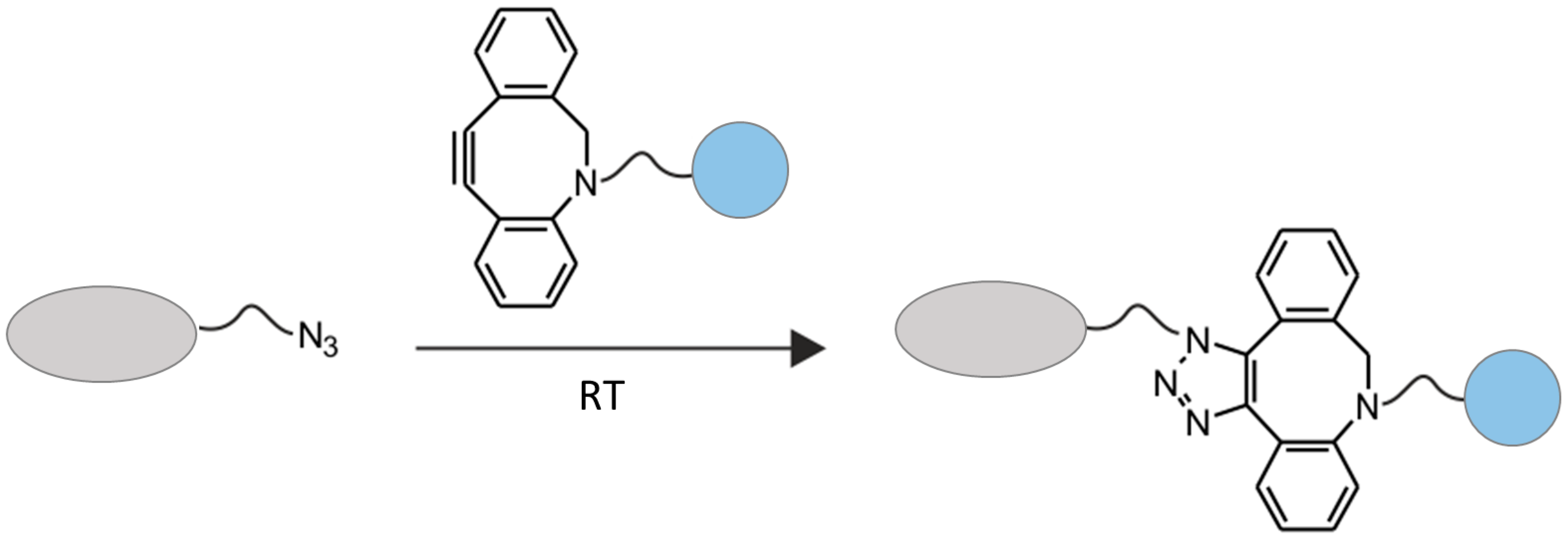
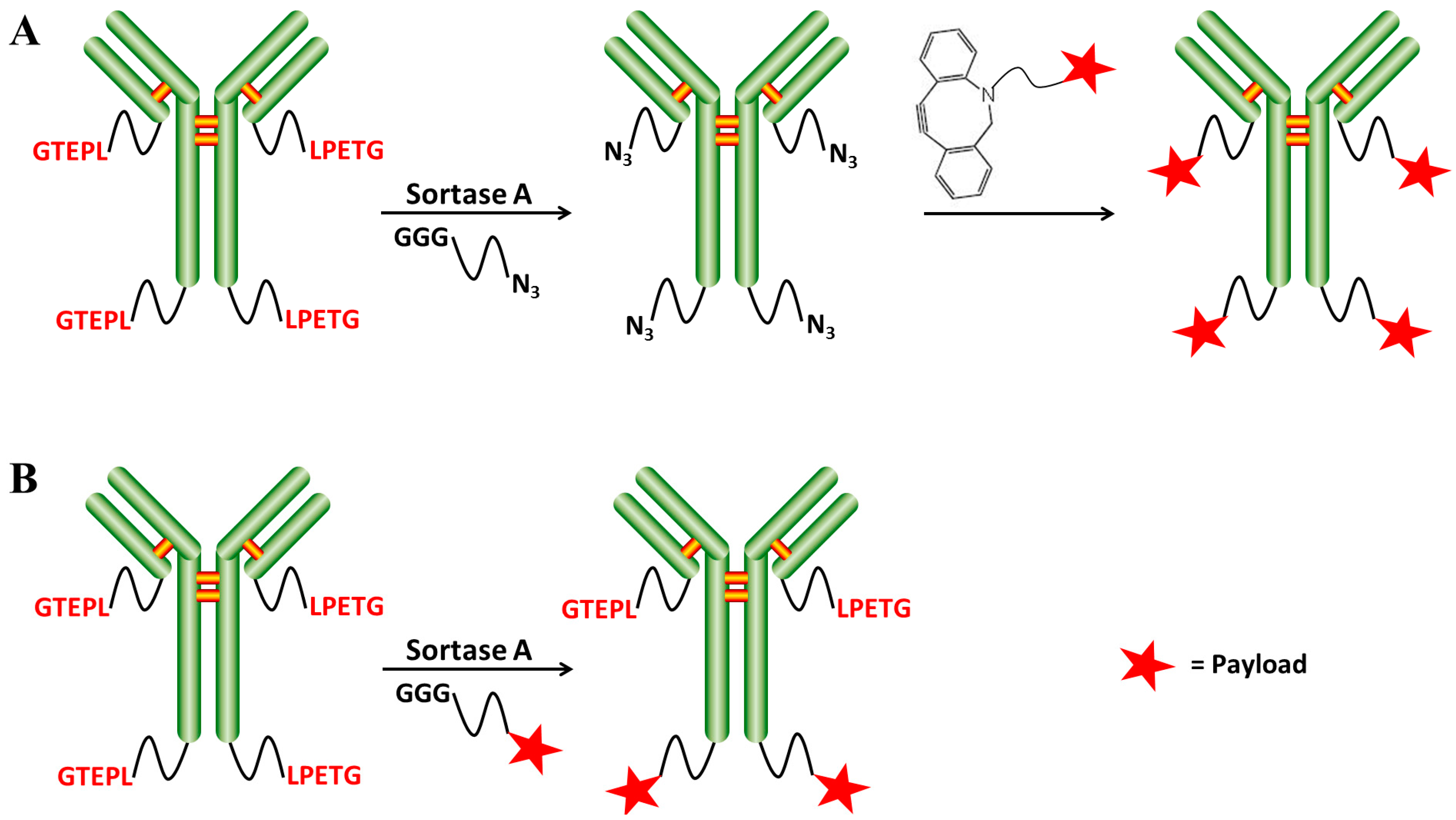
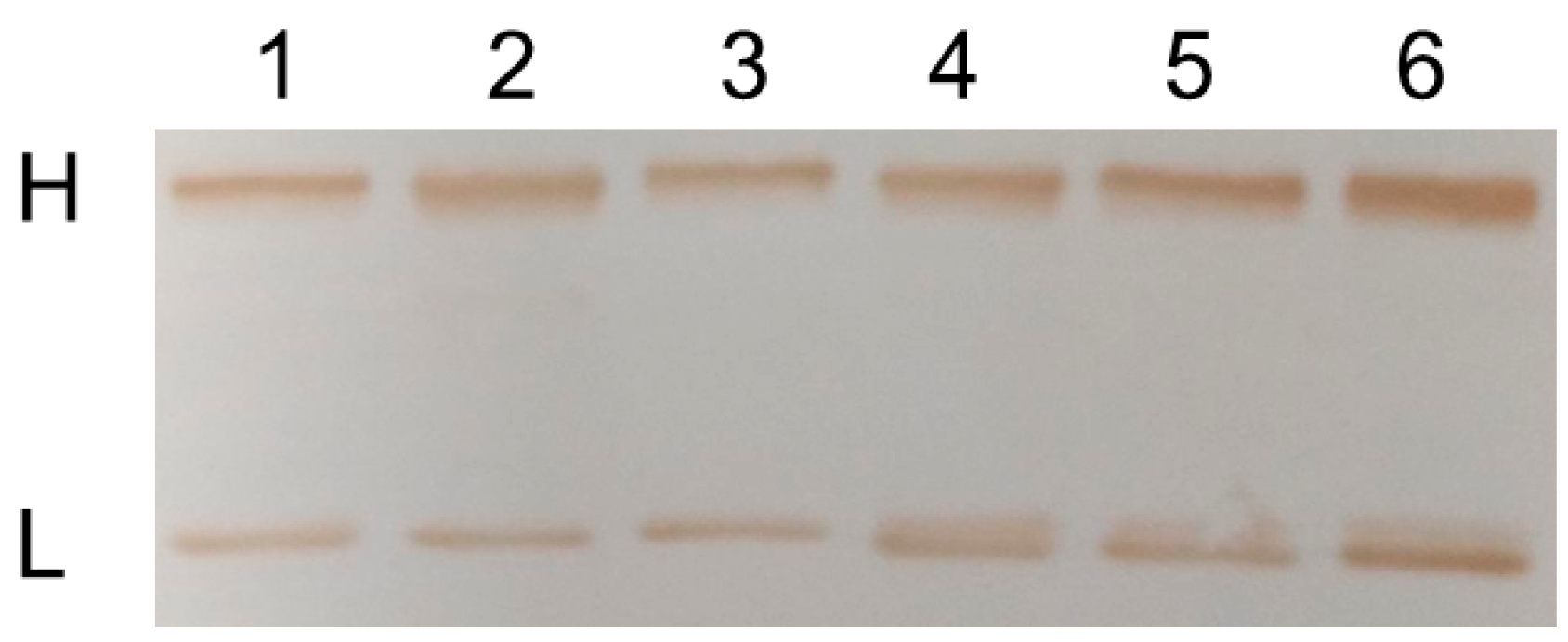
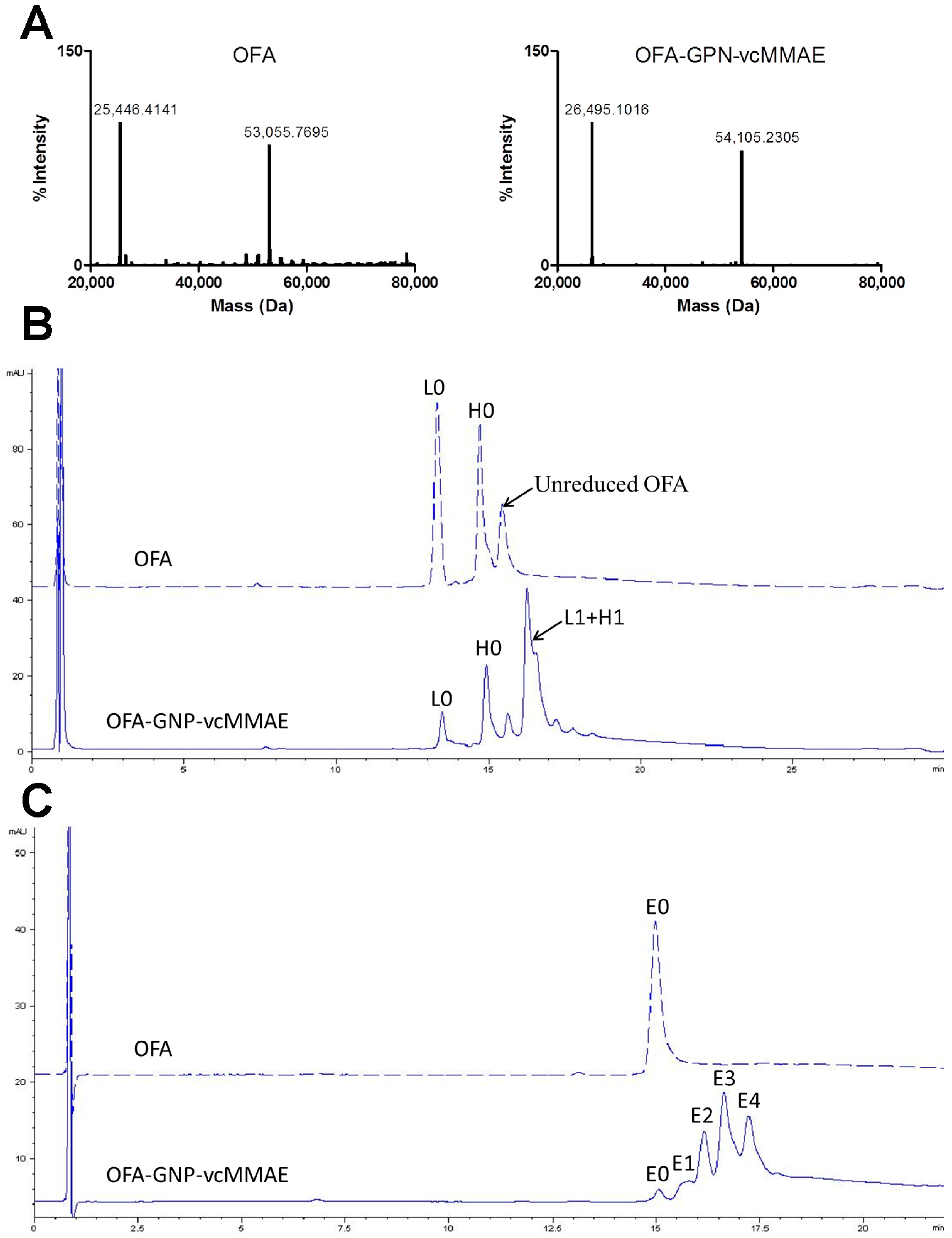
2.2. Chemo-Enzymatic Approach to Produce ADC and Its Characterization
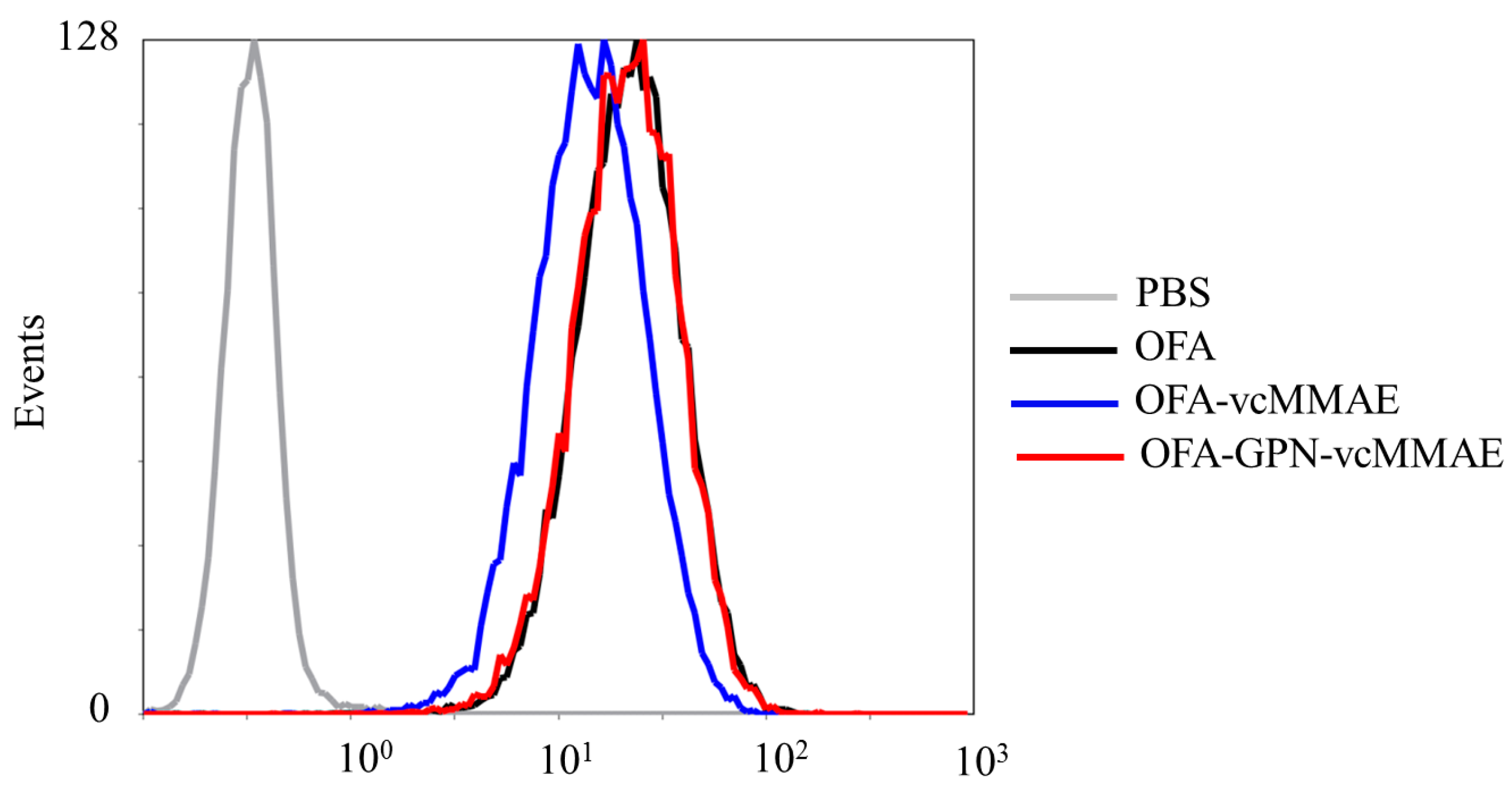
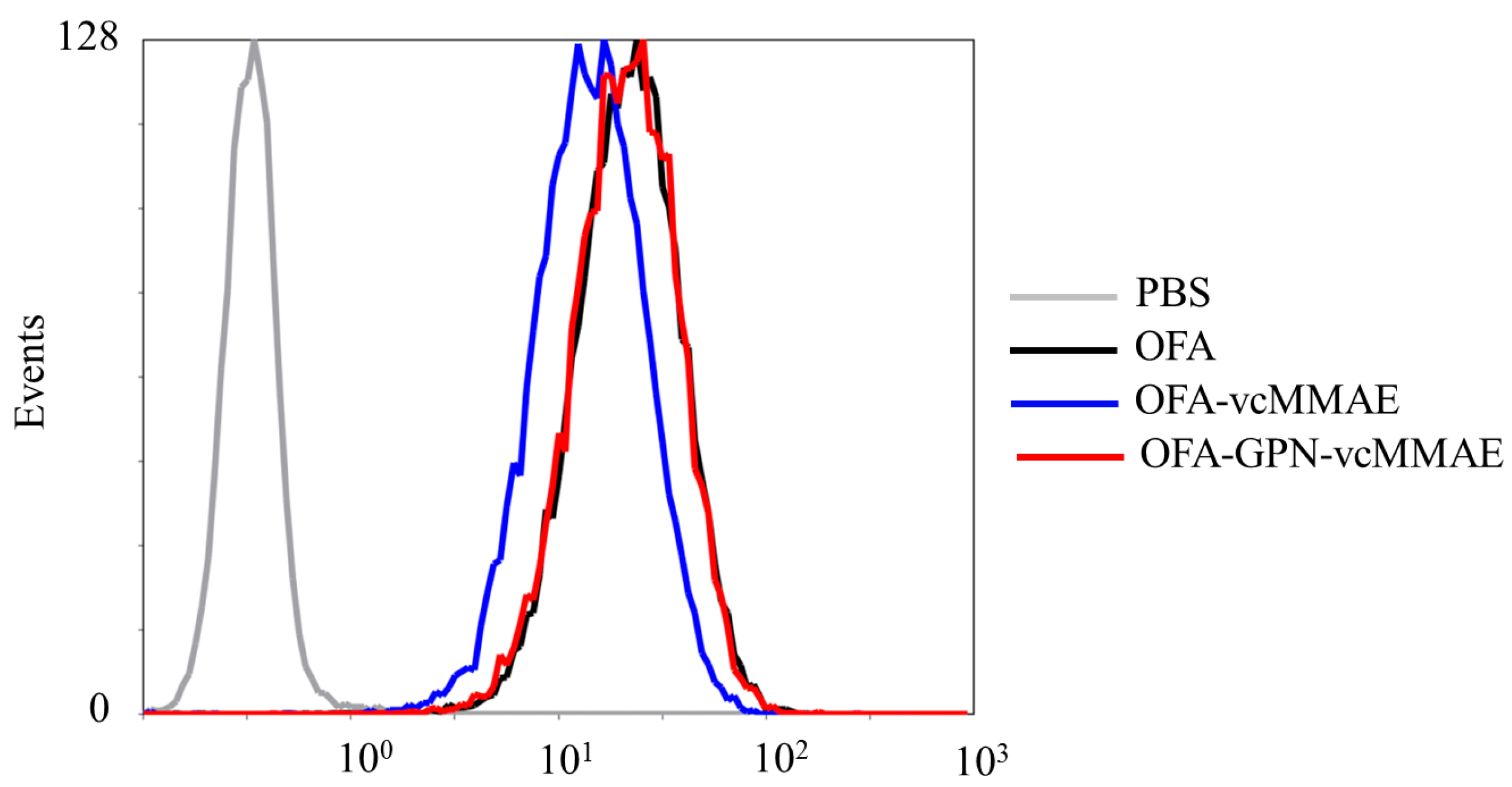
2.3. ADC Binding to CD20-Positive Cells
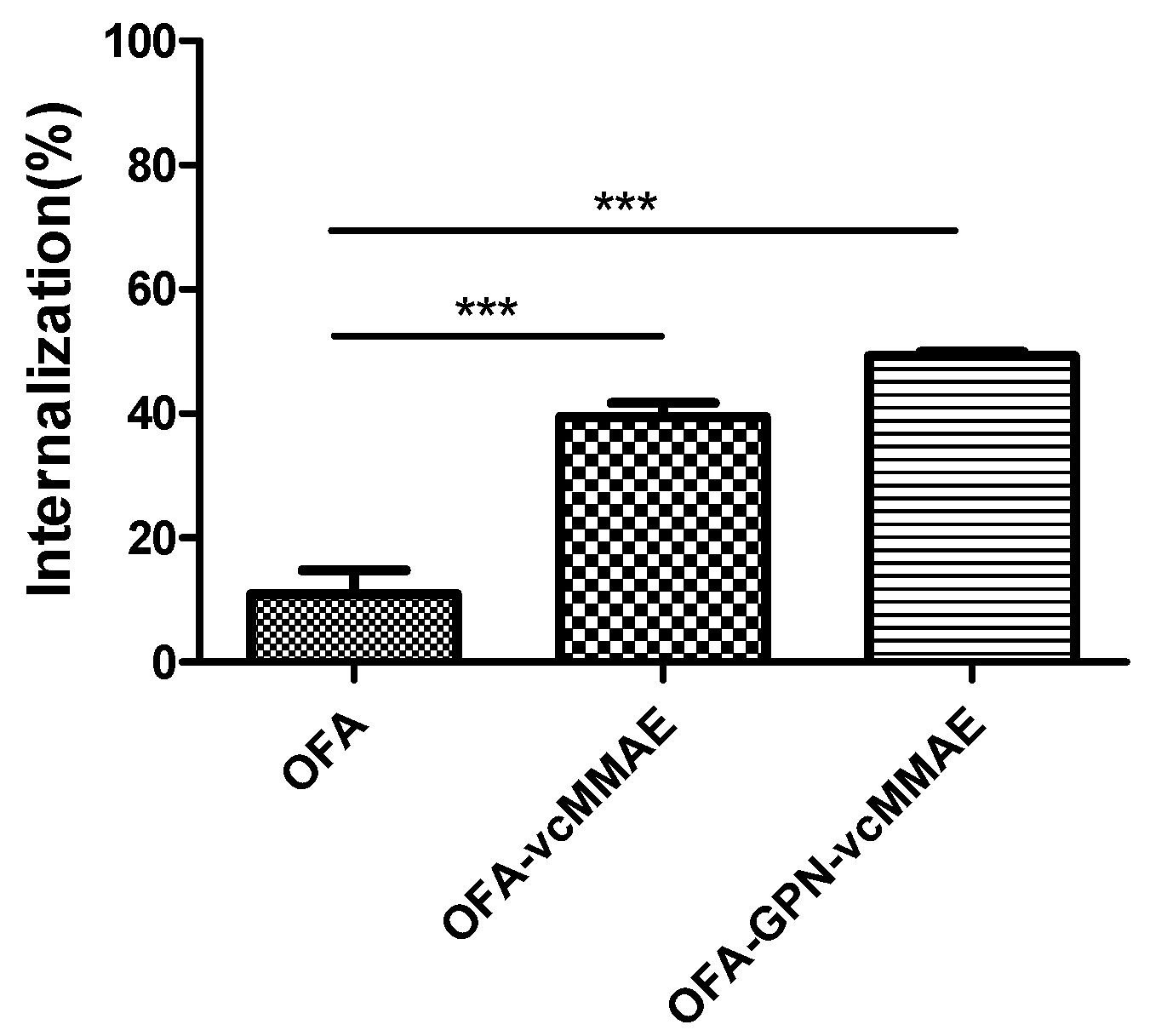
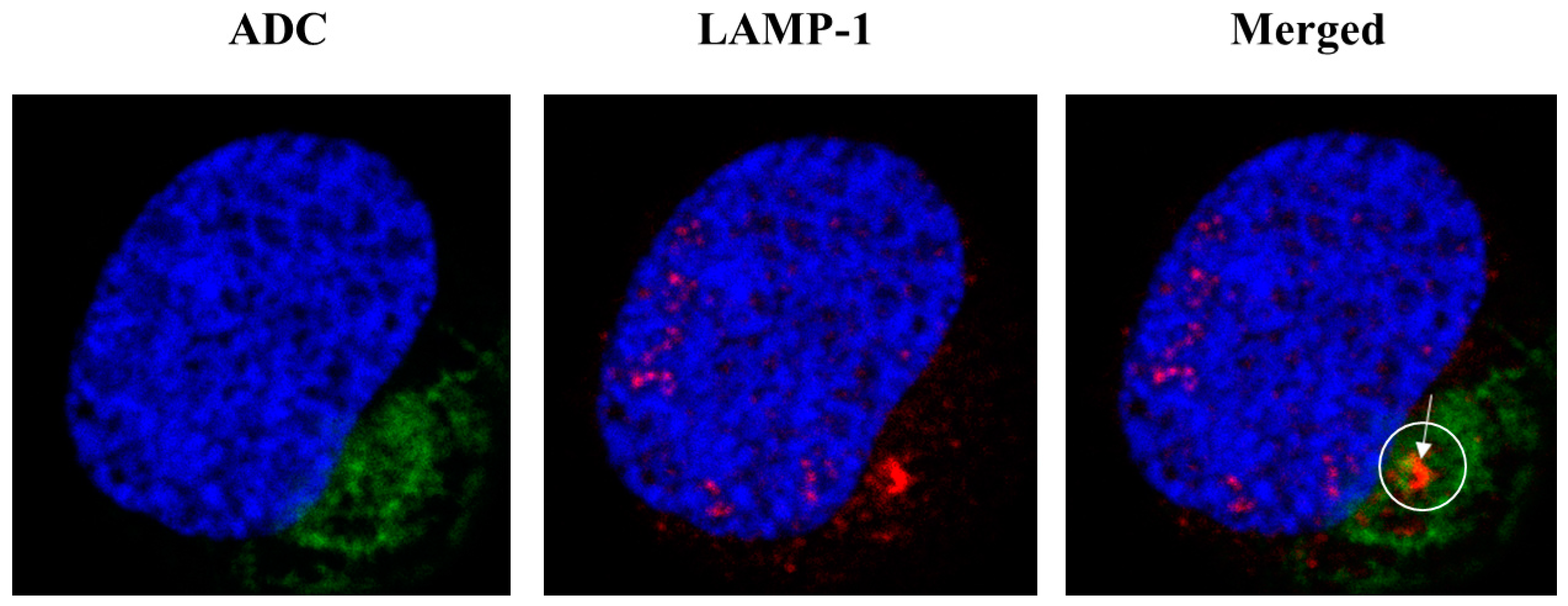
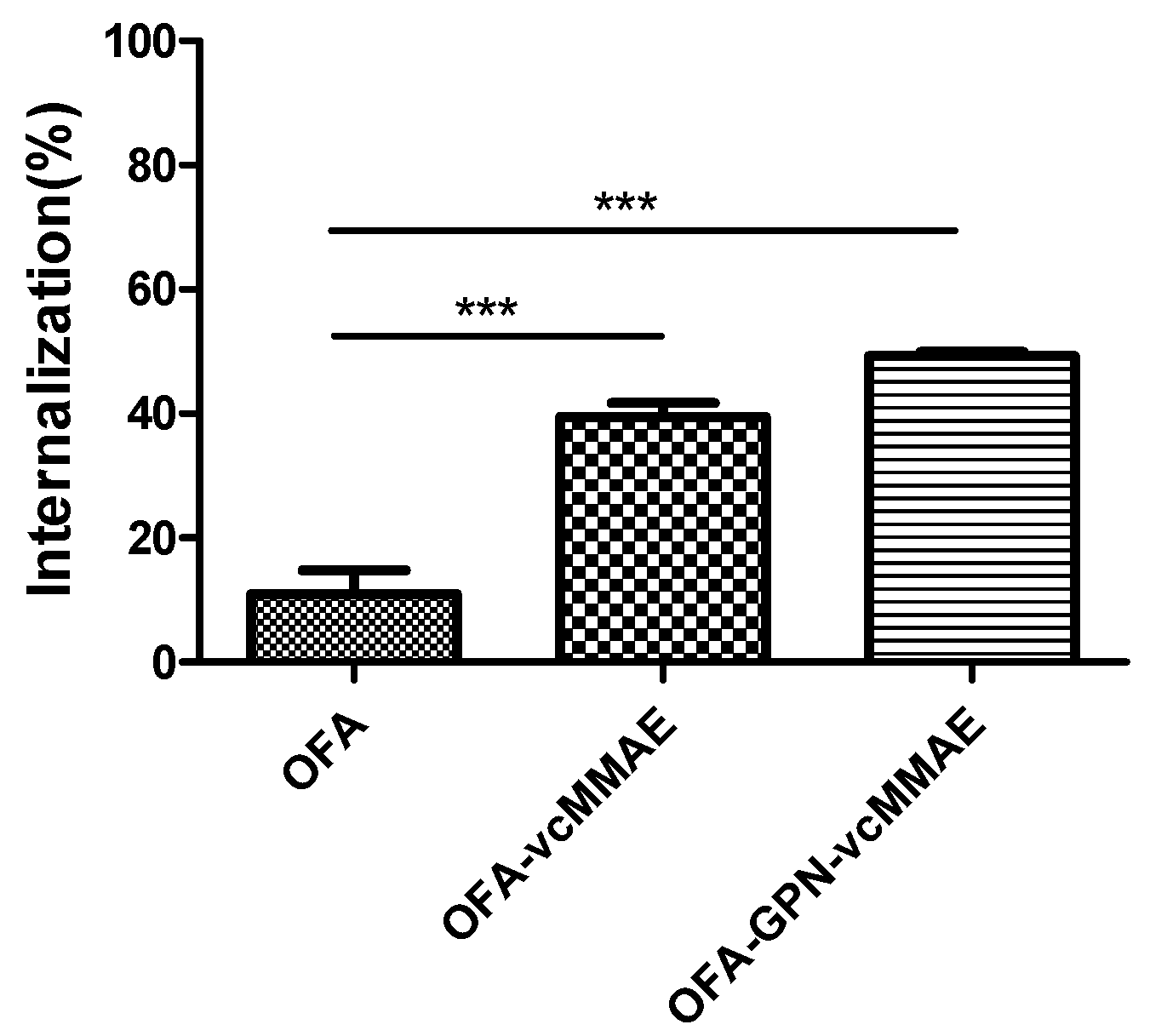
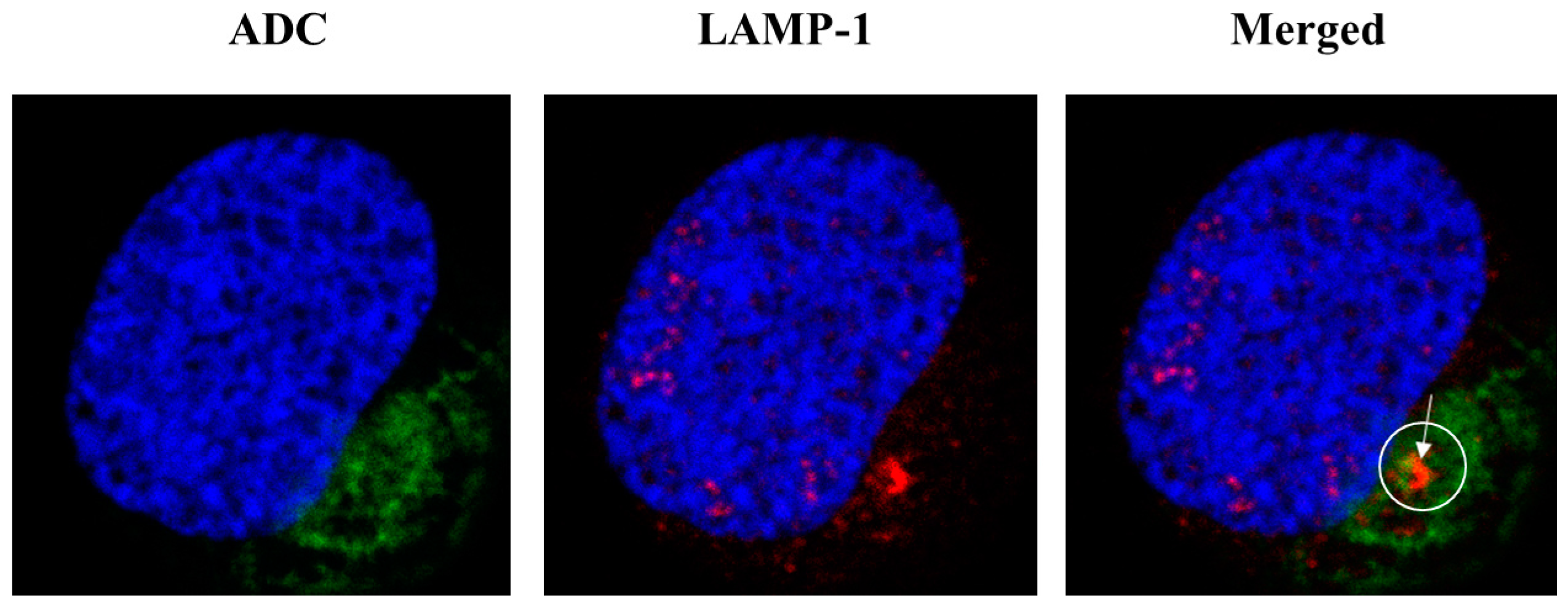
2.4. Internalization of ADC and Subcellular Localization
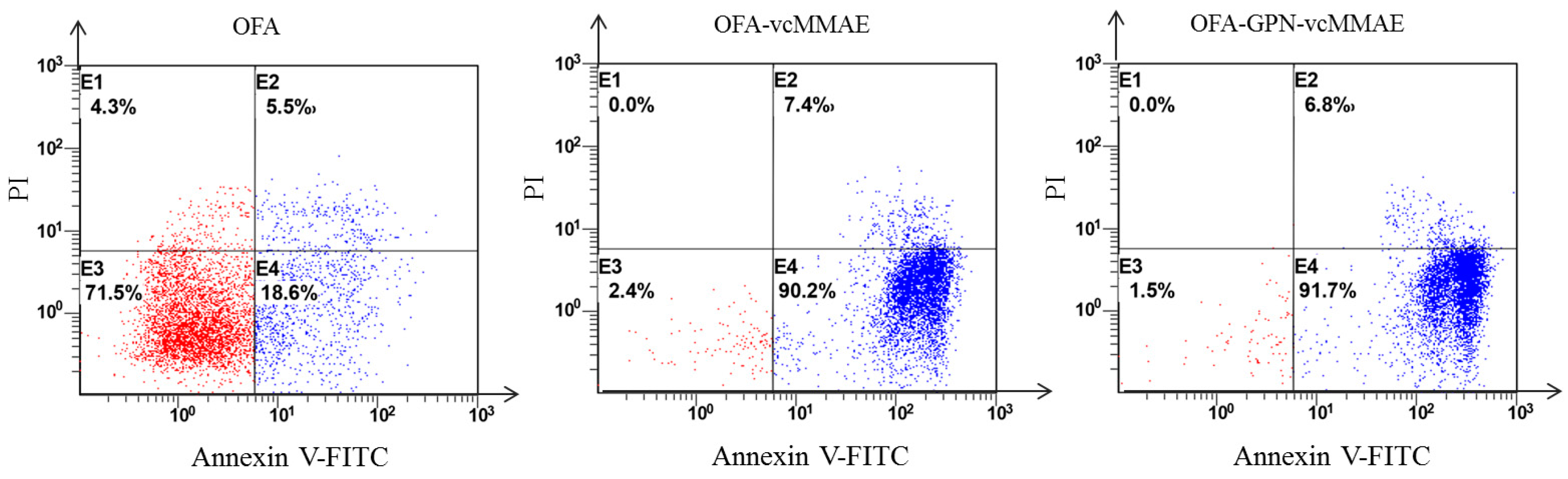
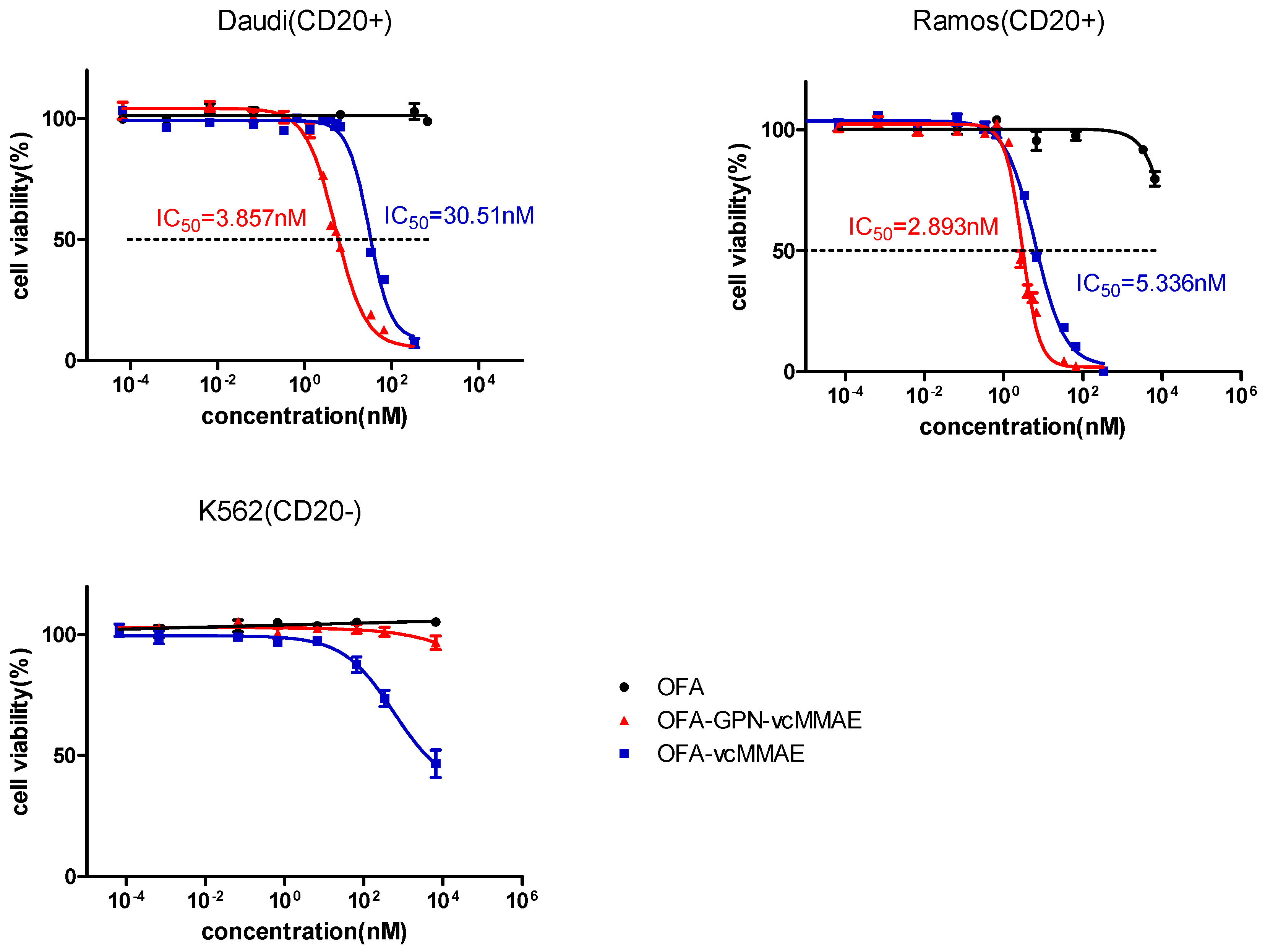
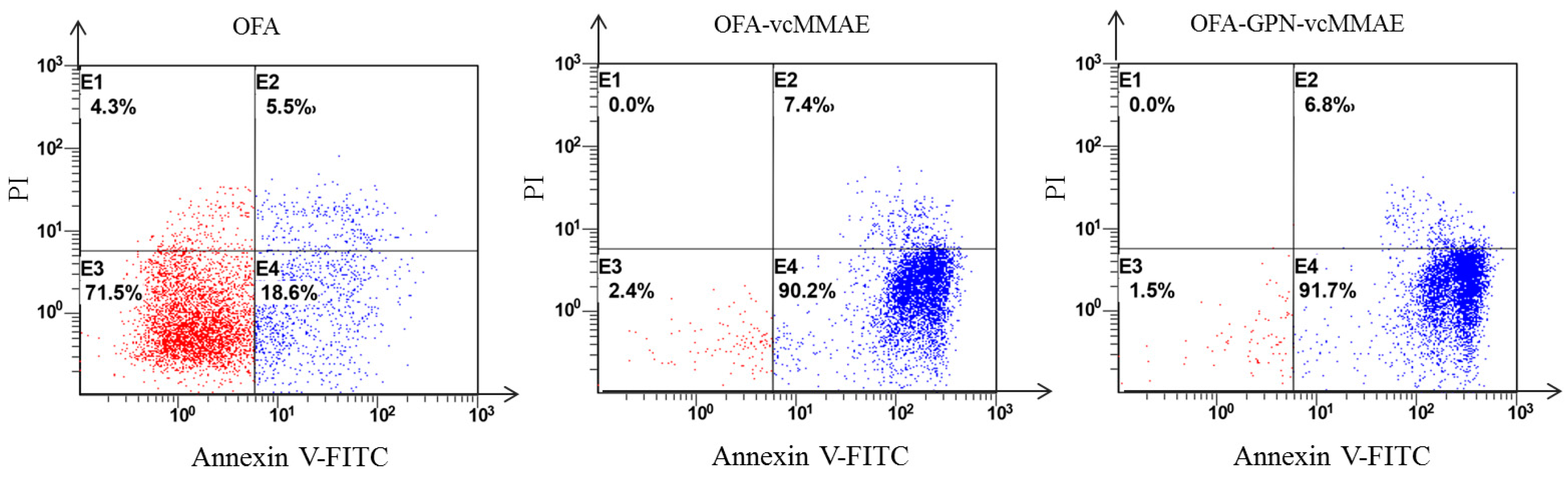
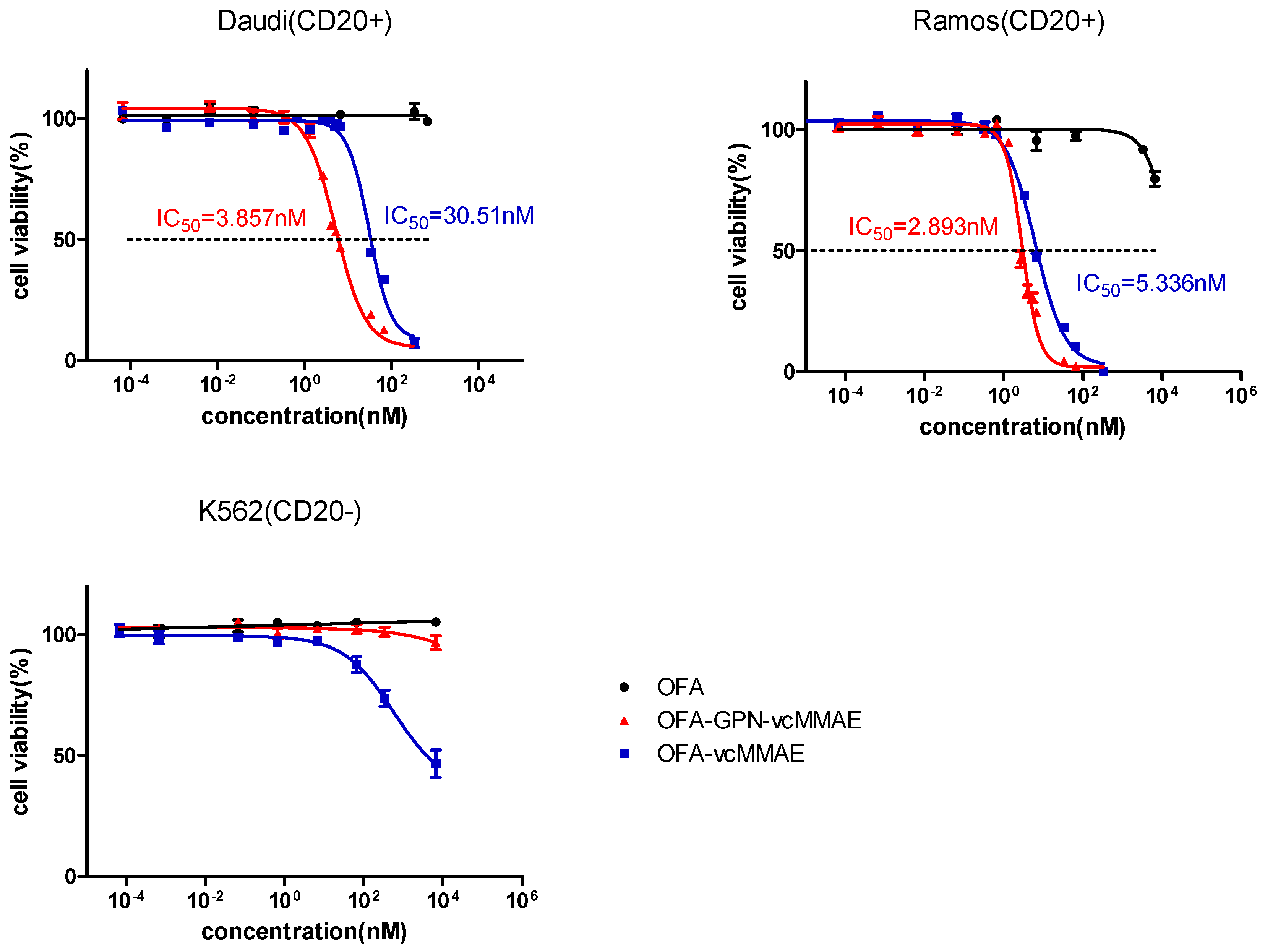
2.5. Apoptosis and In Vitro Cytotoxicity
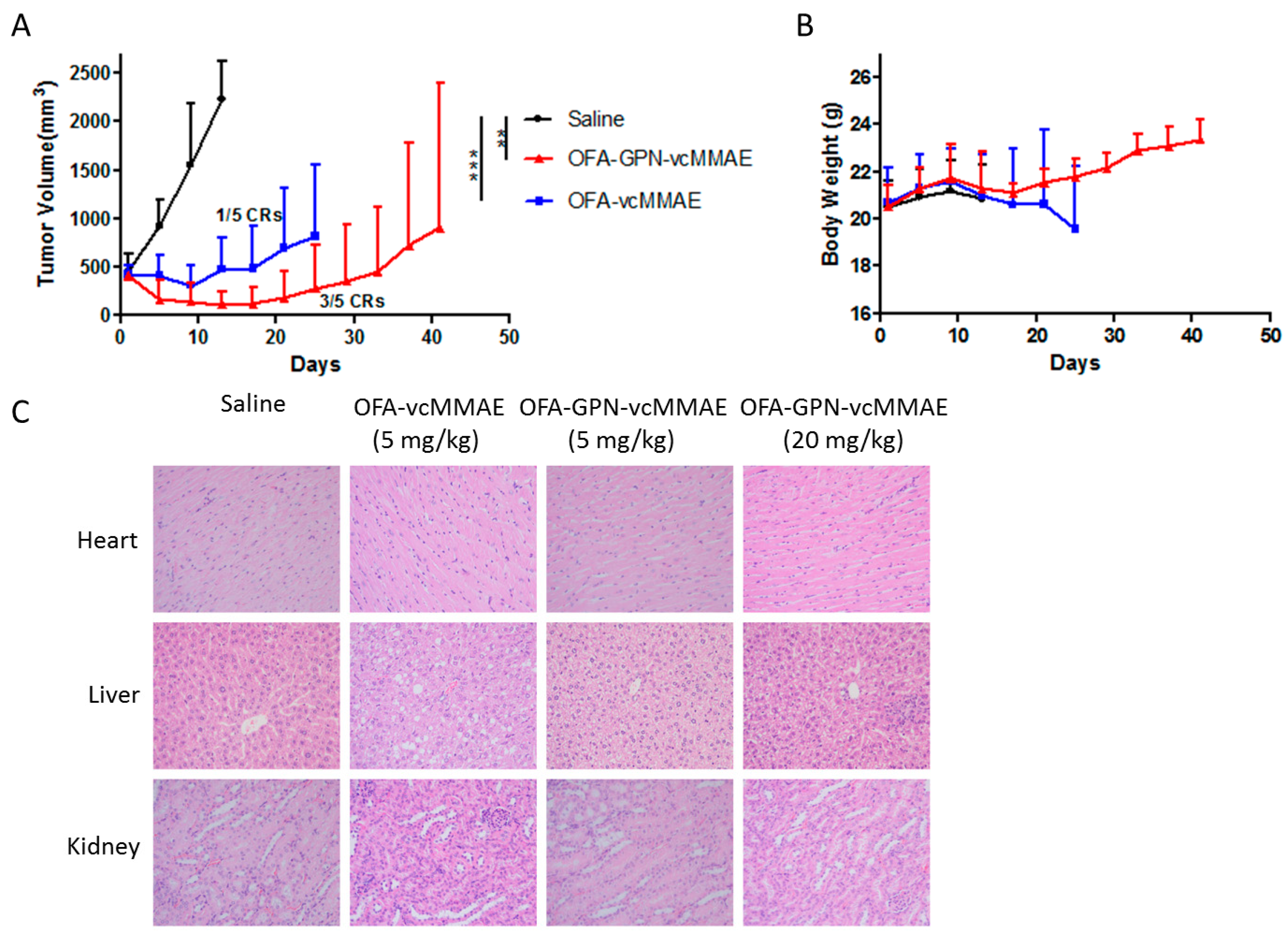
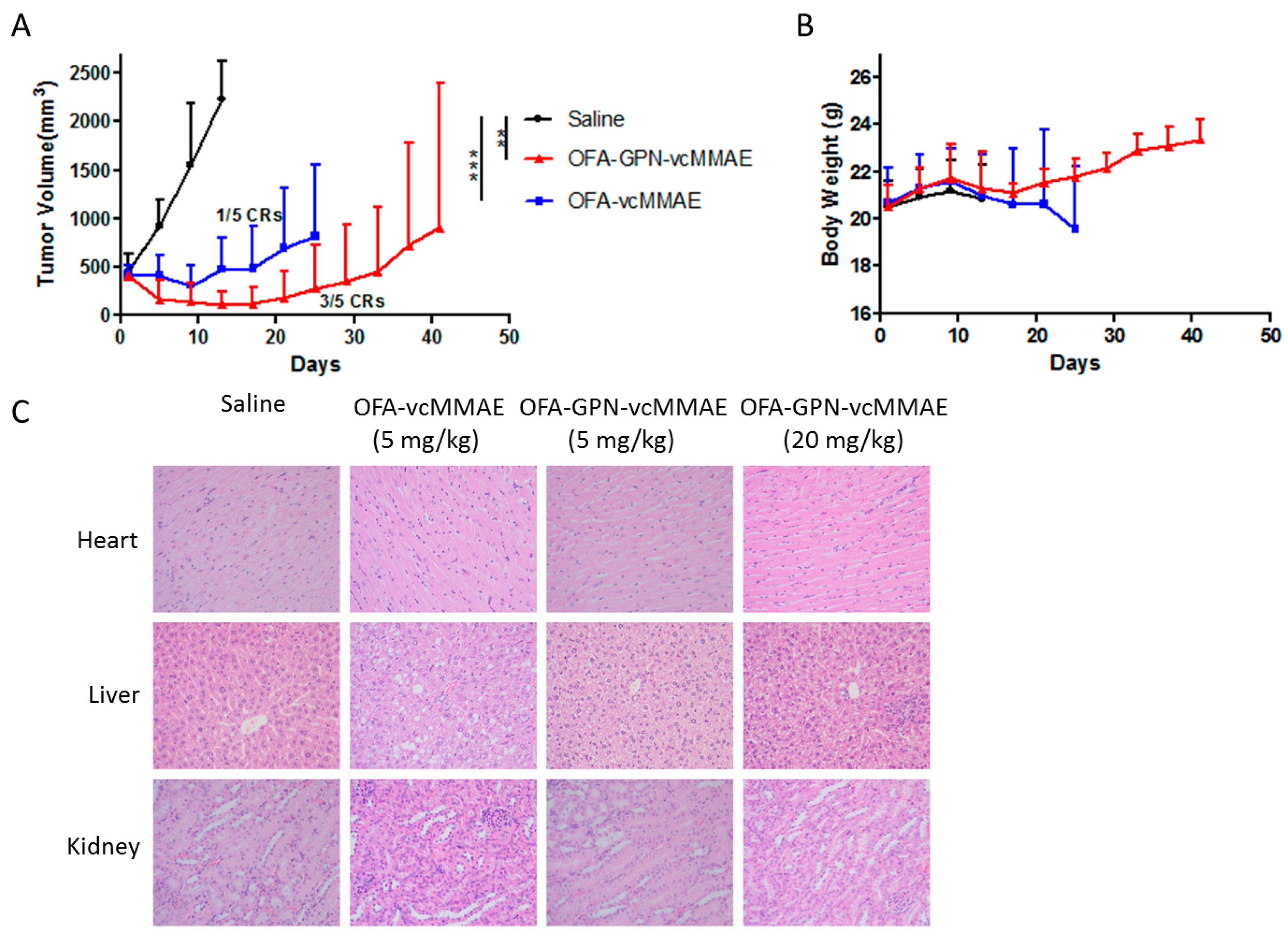
2.6. In vivo Antitumor Activity
3. Conclusions
4. Materials and Methods
4.1. Reagents
4.2. Cell Lines and Cell Culture
4.3. Expression and Purification of Recombinant SrtA
4.4. Production and Purification of Tagged Antibodies
4.5. SrtA-Mediated Conjugation
4.6. Click Chemistry Reaction
4.7. Western Blot
4.8. Liquid Chromatography-Mass Spectrometry (LC-MS)
4.9. Reversed Phase High Performance Liquid Chromatography (RP-HPLC)
4.10. Hydrophobic Interaction Chromatography (HIC)
4.11. Cell Binding
4.12. Cellular Internalization
4.13. Microscopy for ADC Trafficking
4.14. Assessment of Apoptosis
4.15. In Vitro Cytotoxicity
4.16. In Vivo Antitumor Activity
4.17. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ADC | Antibody-drug conjugate |
| OFA | Ofatumumab |
| SrtA | Sortase A |
| SPAAC | Strain-promoted azide-alkyne cycloaddition |
| DAR | Drug to antibody ratio |
| MMAE | Monomethyl auristatin E |
| FBS | Fetal bovine serum |
| PBS | Phosphate buffered saline |
| BSA | Bovine serum albumin |
| MFI | Mean fluorescence intensity |
| LAMP-1 | Lysosomal-associated membrane protein-1 |
References
- Younes, A.; Bartlett, N.L.; Leonard, J.P.; Kennedy, D.A.; Lynch, C.M.; Sievers, E.L.; Forero-Torres, A. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N. Eng. J. Med. 2010, 363, 1812–1821. [Google Scholar] [CrossRef] [PubMed]
- Katz, J.; Janik, J.E.; Younes, A. Brentuximab Vedotin (SGN-35). Clin. Cancer Res. 2011, 17, 6428–6436. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, P.M.; Weiss, D.; Guardino, E.; Girish, S.; Sliwkowski, M.X. Trastuzumab emtansine: A unique antibody-drug conjugate in development for human epidermal growth factor receptor 2-positive cancer. Clin. Cancer Res. 2011, 17, 6437–6447. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.Y.; Dieras, V.; Guardino, E.; et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Eng. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Strop, P.; Liu, S.H.; Dorywalska, M.; Delaria, K.; Dushin, R.G.; Tran, T.T.; Ho, W.H.; Farias, S.; Casas, M.G.; Abdiche, Y.; et al. Location matters: Site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem. Biol. 2013, 20, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S.; et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008, 26, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Alley, S.C.; Benjamin, D.R.; Jeffrey, S.C.; Okeley, N.M.; Meyer, D.L.; Sanderson, R.J.; Senter, P.D. Contribution of linker stability to the activities of anticancer immunoconjugates. Bioconjug. Chem. 2008, 19, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, E.S.; Heibeck, T.H.; Gill, A.; Li, X.; Murray, C.J.; Madlansacay, M.R.; Tran, C.; Uter, N.T.; Yin, G.; Rivers, P.J.; et al. Production of site-specific antibody-drug conjugates using optimized non-natural amino acids in a cell-free expression system. Bioconjug. Chem. 2014, 25, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Jeger, S.; Zimmermann, K.; Blanc, A.; Grünberg, J.; Honer, M.; Hunziker, P.; Struthers, H.; Schibli, R. Site-specific and stoichiometric modification of antibodies by bacterial transglutaminase. Angew. Chem. Int. Ed. Eng. 2010, 49, 9995–9997. [Google Scholar] [CrossRef] [PubMed]
- Drake, P.M.; Albers, A.E.; Baker, J.; Banas, S.; Barfield, R.M.; Bhat, A.S.; de Hart, G.W.; Garofalo, A.W.; Holder, P.; Jones, L.C.; et al. Aldehyde tag coupled with HIPS chemistry enables the production of ADCs conjugated site-specifically to different antibody regions with distinct in vivo efficacy and PK outcomes. Bioconjug. Chem. 2014, 25, 1331–1341. [Google Scholar] [CrossRef] [PubMed]
- Kornberger, P.; Skerra, A. Sortase-catalyzed in vitro functionalization of a HER2-specific recombinant Fab for tumor targeting of the plant cytotoxin gelonin. MAbs 2014, 6, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Popp, M.W.; Antos, J.M.; Grotenbreg, G.M.; Spooner, E.; Ploegh, H.L. Sortagging: A versatile method for protein labeling. Nat. Chem. Biol. 2007, 3, 707–708. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.Q.; Zhao, W.B.; Lai, J.; Ding, D.; Zhang, Q.; Yang, X.Y.; Huang, M.M.; Jin, S.J.; Xu, Y.C.; Zeng, S.; et al. Sortase A-generated highly potent anti-CD20-MMAE conjugates for efficient elimination of B-lineage lymphomas. Small 2017, 13. [Google Scholar] [CrossRef] [PubMed]
- Dennler, P.; Chiotellis, A.; Fischer, E.; Brégeon, D.; Belmant, C.; Gauthier, L.; Lhospice, F.; Romagne, F.; Schibli, R. Transglutaminase-based chemo-enzymatic conjugation approach yields homogeneous antibody-drug conjugates. Bioconjug. Chem. 2014, 25, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Tedder, T.F.; Engel, P. CD20: A regulator of cell-cycle progression of B lymphocytes. Immunol. Today 1994, 15, 450–454. [Google Scholar] [CrossRef]
- Dimasi, N.; Gao, C.; Fleming, R.; Woods, R.M.; Yao, X.T.; Shirinian, L.; Kiener, P.A.; Wu, H. The design and characterization of oligospecific antibodies for simultaneous targeting of multiple disease mediators. J. Mol. Biol. 2009, 393, 672–692. [Google Scholar] [CrossRef] [PubMed]
- Pritz, S.; Wolf, Y.; Kraetke, O.; Klose, J.; Bienert, M.; Beyermann, M. Synthesis of biologically active peptide nucleic acid-peptide conjugates by sortase-mediated ligation. J. Org. Chem. 2007, 72, 3909–3912. [Google Scholar] [CrossRef] [PubMed]
- Beerli, R.R.; Hell, T.; Merkel, A.S.; Grawunder, U. Sortase enzyme-mediated generation of site-specifically conjugated antibody dug conjugates with high in vitro and in vivo potency. PLoS ONE 2015, 10, e0131177. [Google Scholar] [CrossRef] [PubMed]
- VanBrunt, M.P.; Shanebeck, K.; Caldwell, Z.; Johnson, J.; Thompson, P.; Martin, T.; Dong, H.; Li, G.; Xu, H.; D’Hooge, F.; et al. Genetically encoded azide containing amino acid in mammalian cells enables site-specific antibody-drug conjugates using click cycloaddition chemistry. Bioconjug. Chem. 2015, 26, 2249–2260. [Google Scholar] [CrossRef] [PubMed]
- Leung, M.K.; Hagemeyer, C.E.; Johnston, A.P.; Gonzales, C.; Kamphuis, M.M.; Ardipradja, K.; Such, G.K.; Peter, K.; Caruso, F. Bio-click chemistry: Enzymatic functionalization of PEGylated capsules for targeting applications. Angew. Chem. Int. Ed. Eng. 2012, 51, 7132–7136. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Isogawa, Y.; Tanaka, T.; Kondo, A. Streptavidin-hydrogel prepared by sortase A-assisted click chemistry for enzyme immobilization on an electrode. Biosens. Bioelectron. 2018, 99, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.H.; Zhang, Q.; Wang, H.B.; Zhang, Y.N.; Ding, D.; Pan, L.Q.; Miao, D.; Xu, S.; Zhang, C.; Luo, P.H.; et al. Preclinical studies of targeted therapies for CD20-positive B lymphoid malignancies by Ofatumumab conjugated with auristatin. Investig. New Drugs 2014, 32, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Law, C.L.; Cerveny, C.G.; Gordon, K.A.; Klussman, K.; Mixan, B.J.; Chace, D.F.; Meyer, D.L.; Doronina, S.O.; Siegall, C.B.; Francisco, J.A.; et al. Efficient elimination of B-lineage lymphomas by anti-CD20-auristatin conjugates. Clin. Cancer Res. 2004, 10, 7842–7851. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.H.; Vaughan, A.T.; Ashton-Key, M.; Williams, E.L.; Dixon, S.V.; Chan, H.T.; Beers, S.A.; French, R.R.; Cox, K.L.; Davies, A.J.; et al. Fc gamma receptor IIb on target B cells promotes rituximab internalization and reduces clinical efficacy. Blood 2011, 118, 2530–2540. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, M.S.; Sanderson, R.J.; Gordon, K.A.; Andreyka, J.; Cerveny, C.G.; Yu, C.; Lewis, T.S.; Meyer, D.L.; Zabinski, R.F.; Doronina, S.O.; et al. Lysosomal trafficking and cysteine protease metabolism confer target-specific cytotoxicity by peptide-linked anti-CD30-auristatin conjugates. J. Biol. Chem. 2006, 281, 10540–10547. [Google Scholar] [CrossRef] [PubMed]
- Francisco, J.A.; Cerveny, C.G.; Meyer, D.L.; Mixan, B.J.; Klussman, K.; Chace, D.F.; Rejniak, S.X.; Gordon, K.A.; DeBlanc, R.; Toki, B.E.; et al. cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 2003, 102, 1458–1465. [Google Scholar] [CrossRef] [PubMed]
- Subedi, G.P.; Johnson, R.W.; Moniz, H.A.; Moremen, K.W.; Barb, A. High yield expression of recombinant human proteins with the transient transfection of HEK293 cells in suspension. J. Vis. Exp. 2015, e53568. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characterizations | Enzymatic ADC | Chemo-Enzymatic ADC |
|---|---|---|
| Derivative payloads | 100-fold molar excess | 2 molar equivalent per azide group |
| DAR | 1.4 | 3.3 |
| Internalization (%) | 39.44 | 49.31 |
| IC50 on Ramos (nM) | 5.336 | 2.893 |
| IC50 on Daudi (nM) | 30.51 | 3.857 |
| Effect in vivo | *** p < 0.001 1 | ** p < 0.01 1 |
| Safety in vitro and in vivo | lower | higher |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.; Jin, S.; Zhao, W.; Liu, W.; Ding, D.; Zhou, J.; Chen, S. A Versatile Chemo-Enzymatic Conjugation Approach Yields Homogeneous and Highly Potent Antibody-Drug Conjugates. Int. J. Mol. Sci. 2017, 18, 2284. https://doi.org/10.3390/ijms18112284
Xu Y, Jin S, Zhao W, Liu W, Ding D, Zhou J, Chen S. A Versatile Chemo-Enzymatic Conjugation Approach Yields Homogeneous and Highly Potent Antibody-Drug Conjugates. International Journal of Molecular Sciences. 2017; 18(11):2284. https://doi.org/10.3390/ijms18112284
Chicago/Turabian StyleXu, Ying, Shijie Jin, Wenbin Zhao, Wenhui Liu, Ding Ding, Jie Zhou, and Shuqing Chen. 2017. "A Versatile Chemo-Enzymatic Conjugation Approach Yields Homogeneous and Highly Potent Antibody-Drug Conjugates" International Journal of Molecular Sciences 18, no. 11: 2284. https://doi.org/10.3390/ijms18112284