The Complete Chloroplast Genome Sequences of the Medicinal Plant Forsythia suspensa (Oleaceae)

Abstract
:1. Introduction
2. Results and Discussions
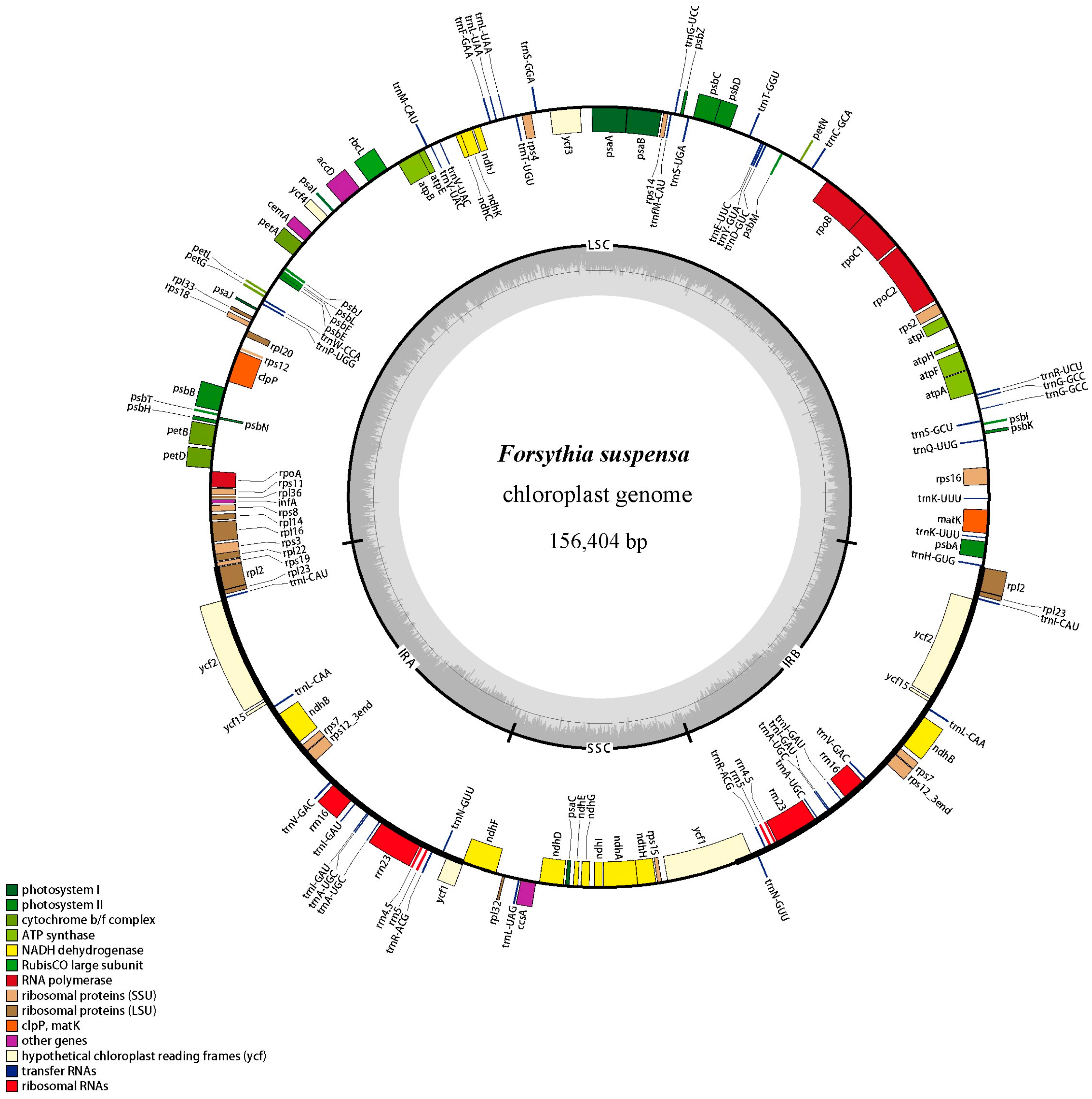
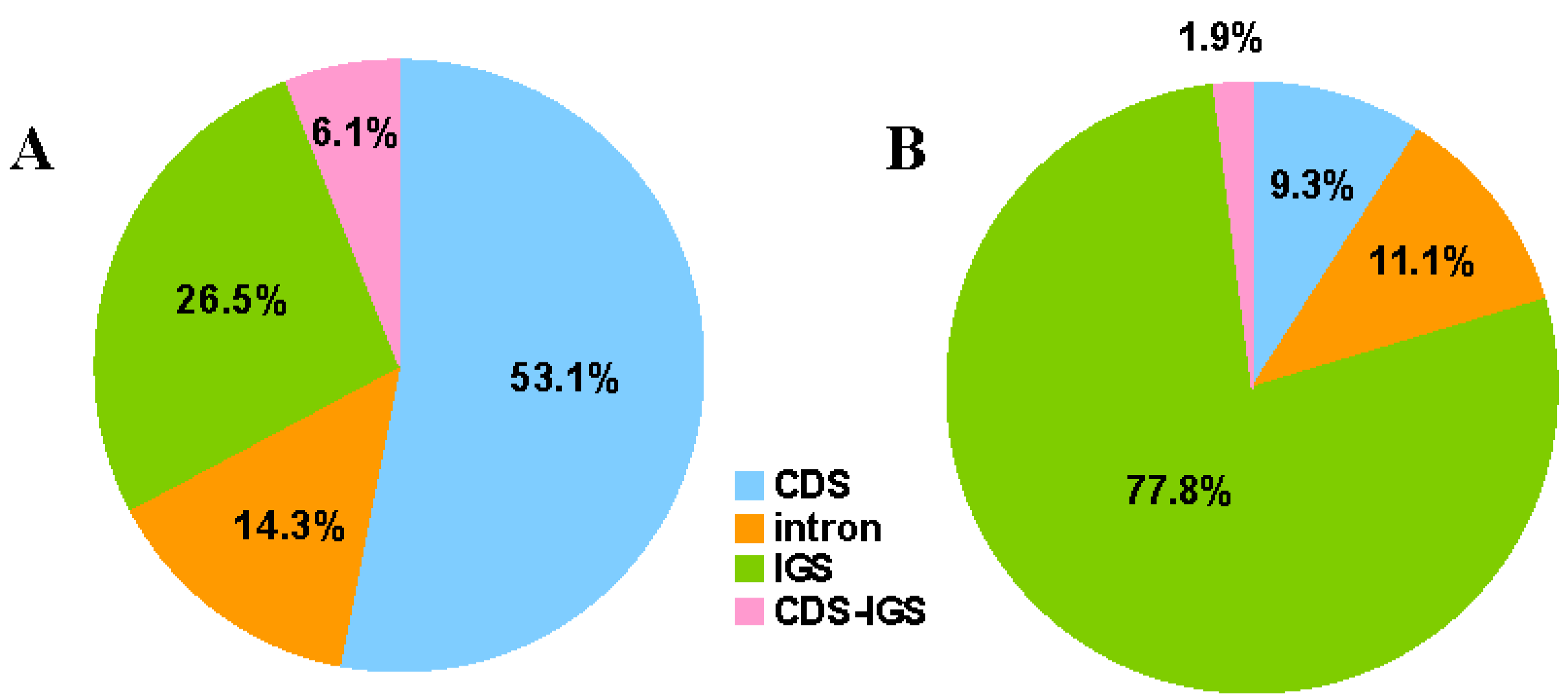
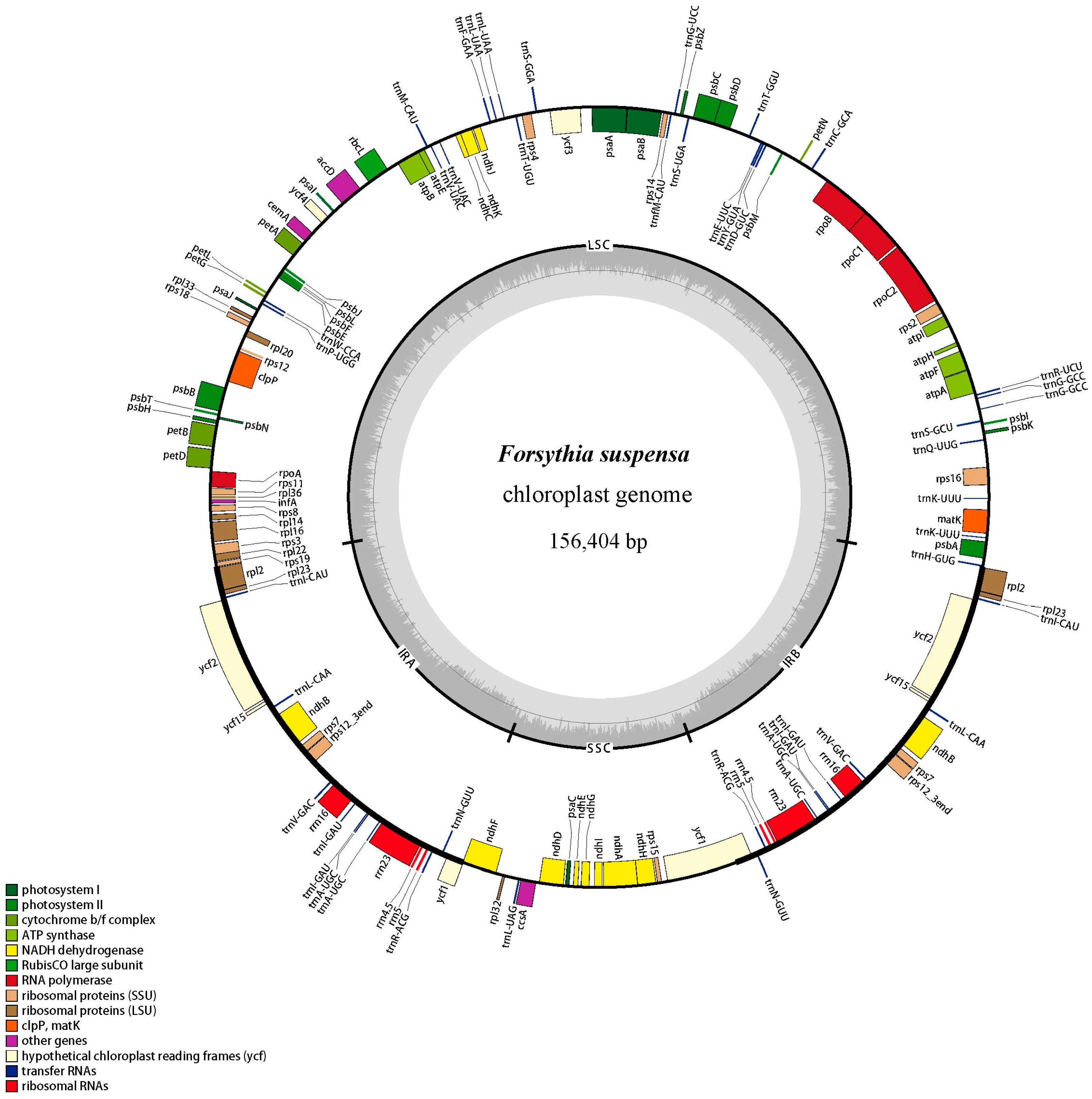
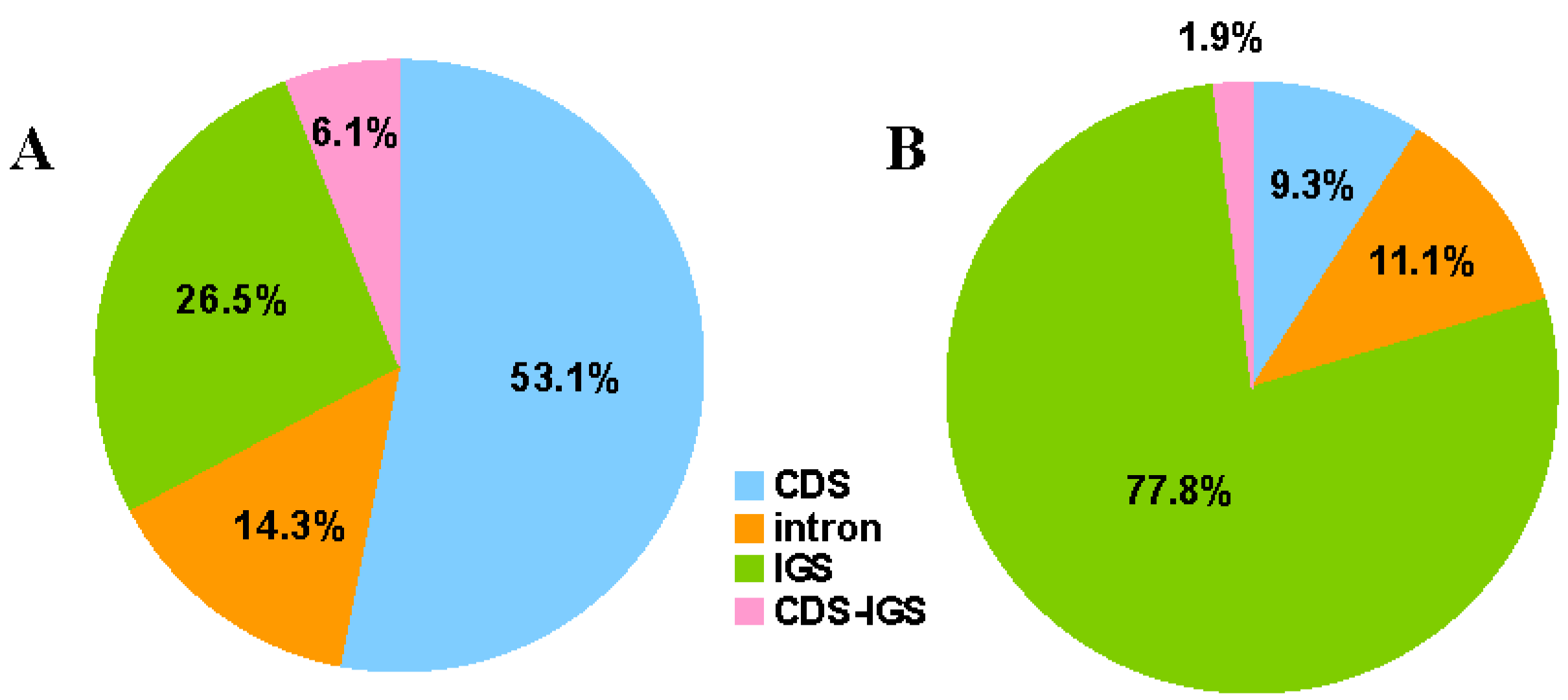
2.1. Genome Features
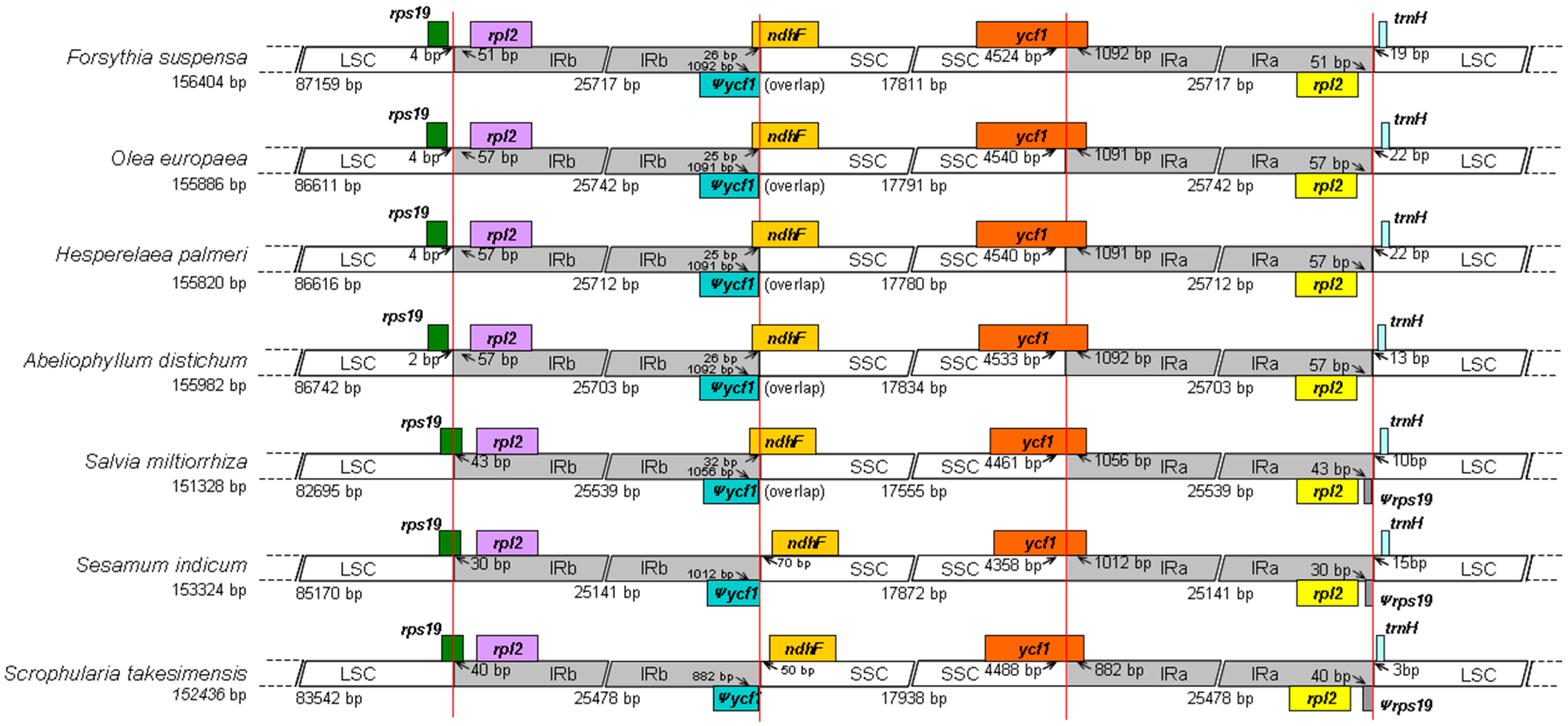
2.2. Comparison to Other Lamiales Species
2.3. Codon Usage Analysis
2.4. Repeats and Simple Sequence Repeats Analysis
2.5. Predicted RNA Editing Sites in the F. suspensa Chloroplast Genes
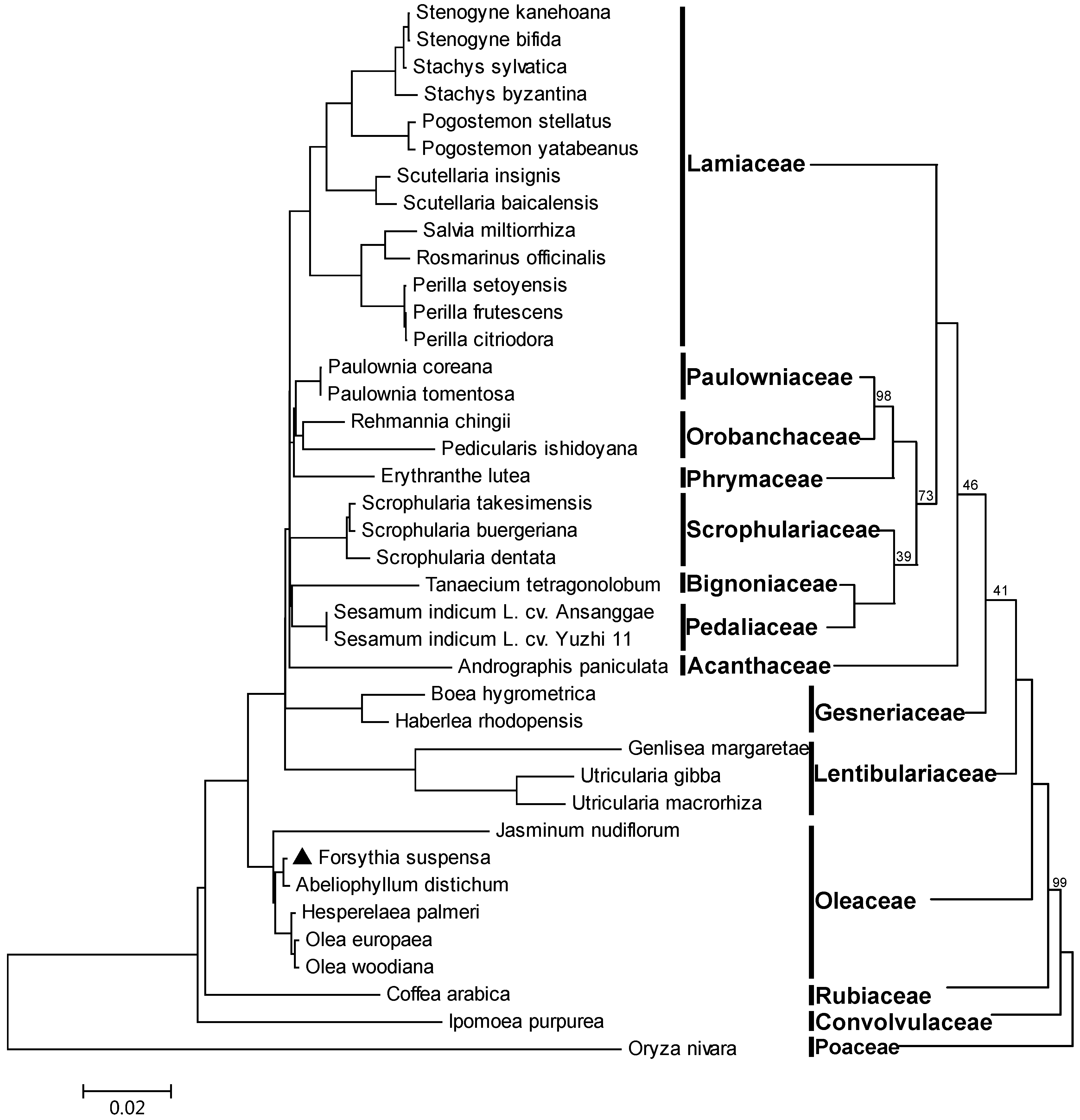
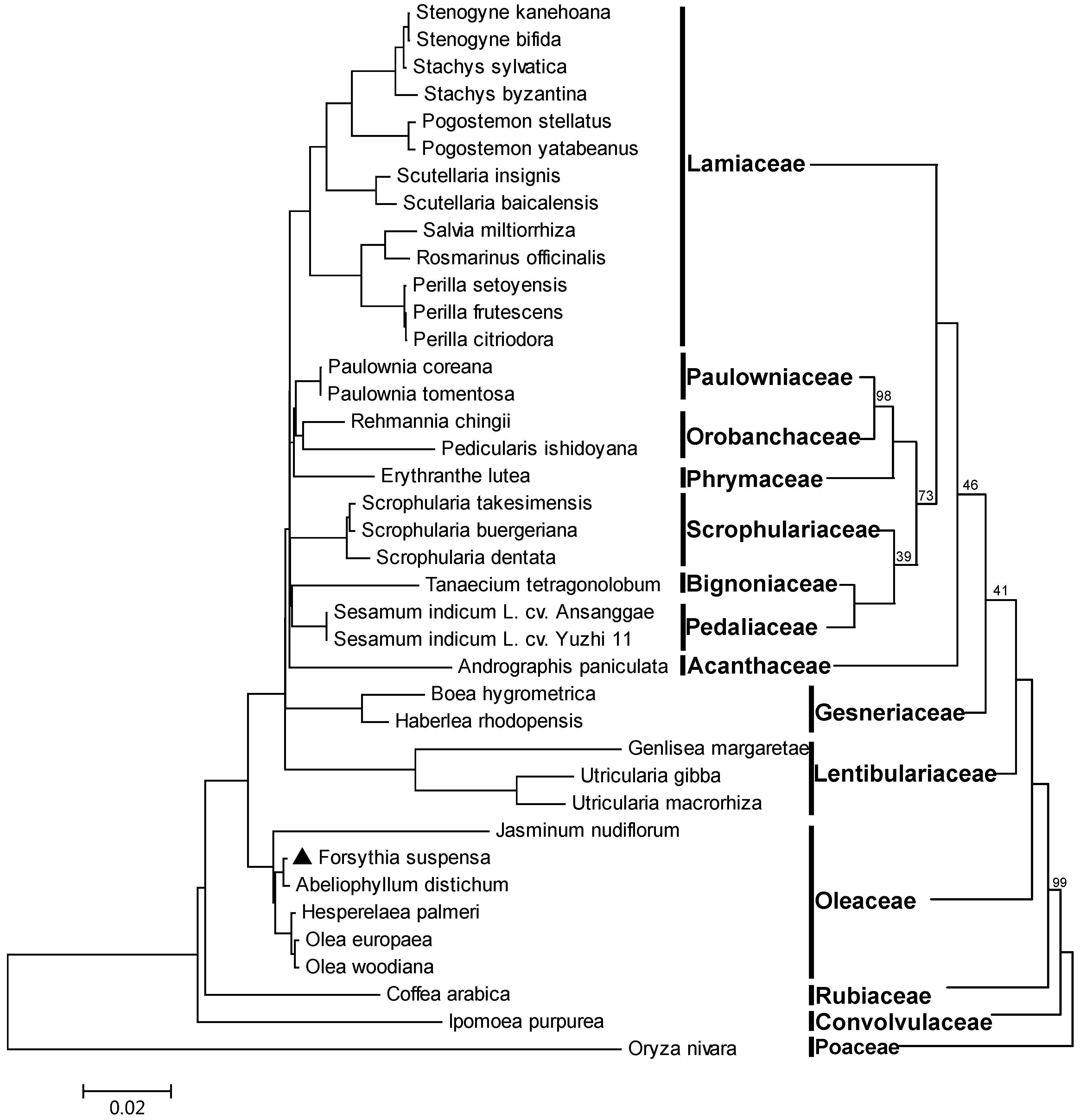
2.6. Phylogeny Reconstruction of Lamiales Based on Complete Chloroplast Genome Sequences
3. Materials and Methods
3.1. Plant Materials
3.2. DNA Library Preparation, Sequencing, and Genome Assembly
3.3. Genome Annotation and Comparative Genomics
3.4. Repeat Sequence Analyses
3.5. Codon Usage
3.6. Prediction of RNA Editing Sites
3.7. Phylogenomic Analyses
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhao, L.; Yan, X.; Shi, J.; Ren, F.; Liu, L.; Sun, S.; Shan, B. Ethanol extract of Forsythia suspensa root induces apoptosis of esophageal carcinoma cells via the mitochondrial apoptotic pathway. Mol. Med. Rep. 2015, 11, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Piao, X.L.; Jang, M.H.; Cui, J.; Piao, X. Lignans from the fruits of Forsythia suspensa. Bioorg. Med. Chem. Lett. 2008, 18, 1980–1984. [Google Scholar] [CrossRef] [PubMed]
- Kang, W.; Wang, J.; Zhang, L. α-glucosidase inhibitors from leaves of Forsythia suspense in Henan province. China J. Chin. Mater. Med. 2010, 35, 1156–1159. [Google Scholar]
- Bu, Y.; Shi, T.; Meng, M.; Kong, G.; Tian, Y.; Chen, Q.; Yao, X.; Feng, G.; Chen, H.; Lu, Z. A novel screening model for the molecular drug for diabetes and obesity based on tyrosine phosphatase Shp2. Bioorg. Med. Chem. Lett. 2011, 21, 874–878. [Google Scholar] [CrossRef] [PubMed]
- Wicke, S.; Schneeweiss, G.M.; dePamphilis, C.W.; Muller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Xiao, H.; Deng, C.; Xiong, L.; Yang, J.; Peng, C. The complete chloroplast genome sequences of the medicinal plant Pogostemon cablin. Int. J. Mol. Sci. 2016, 17, 820. [Google Scholar] [CrossRef] [PubMed]
- Shendure, J.; Ji, H. Next-generation DNA sequencing. Nat. Biotechnol. 2008, 26, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, K.; Ohme, M.; Tanaka, M.; Wakasugi, T.; Hayashida, N.; Matsubayashi, T.; Zaita, N.; Chunwongse, J.; Obokata, J.; Yamaguchi-Shinozaki, K.; et al. The complete nucleotide sequence of the tobacco chloroplast genome: Its gene organization and expression. EMBO J. 1986, 5, 2043–2049. [Google Scholar] [CrossRef] [PubMed]
- Ohyama, K.; Fukuzawa, H.; Kohchi, T.; Shirai, H.; Sano, T.; Sano, S.; Umesono, K.; Shiki, Y.; Takeuchi, M; Chang, Z.; et al. Chloroplast gene organization deduced from complete sequence of liverwort marchantia polymorpha chloroplast DNA. Nature 1986, 572–574. [Google Scholar] [CrossRef]
- Maier, R.M.; Neckermann, K.; Igloi, G.L.; Kossel, H. Complete sequence of the maize chloroplast genome: Gene content, hotspots of divergence and fine tuning of genetic information by transcript editing. J. Mol. Biol. 1995, 251, 614–628. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Lee, S.C.; Lee, J.; Yu, Y.; Yang, K.; Choi, B.S.; Koh, H.J.; Waminal, N.E.; Choi, H.I.; Kim, N.H.; et al. Complete chloroplast and ribosomal sequences for 30 accessions elucidate evolution of Oryza AA genome species. Sci. Rep. 2015, 5, 15655. [Google Scholar] [CrossRef] [PubMed]
- Besnard, G.; Hernandez, P.; Khadari, B.; Dorado, G.; Savolainen, V. Genomic profiling of plastid DNA variation in the Mediterranean olive tree. BMC Plant Biol. 2011, 11, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.L.; Jansen, R.K.; Chumley, T.W.; Kim, K.J. Gene relocations within chloroplast genomes of Jasminum and Menodora (Oleaceae) are due to multiple, overlapping inversions. Mol. Biol. Evol. 2007, 24, 1161–1180. [Google Scholar] [CrossRef] [PubMed]
- Zedane, L.; Hong-Wa, C.; Murienne, J.; Jeziorski, C.; Baldwin, B.G.; Besnard, G. Museomics illuminate the history of an extinct, paleoendemic plant lineage (Hesperelaea, Oleaceae) known from an 1875 collection from Guadalupe island, Mexico. Biol. J. Linn. Soc. 2015, 117, 44–57. [Google Scholar] [CrossRef]
- Kim, H.-W.; Lee, H.-L.; Lee, D.-K.; Kim, K.-J. Complete plastid genome sequences of abeliophyllum distichum nakai (oleaceae), a Korea endemic genus. Mitochondrial DNA Part B 2016, 1, 596–598. [Google Scholar] [CrossRef]
- Sugiura, M. The chloroplast genome. Plant Mol. Biol. 1992, 19, 149–168. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Ma, P.F.; Li, D.Z. High-throughput sequencing of six bamboo chloroplast genomes: Phylogenetic implications for temperate woody bamboos (poaceae: Bambusoideae). PLoS ONE 2011, 6, e20596. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Xiong, G.; Li, P.; He, F.; Huang, Y.; Wang, K.; Li, Z.; Hua, J. Analysis of complete nucleotide sequences of 12 Gossypium chloroplast genomes: Origin and evolution of allotetraploids. PLoS ONE 2012, 7, e37128. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Shi, C.; Gao, L.Z. Plastid genome sequence of a wild woody oil species, prinsepia utilis, provides insights into evolutionary and mutational patterns of rosaceae chloroplast genomes. PLoS ONE 2013, 8, e73946. [Google Scholar] [CrossRef] [PubMed]
- Marechal, A.; Brisson, N. Recombination and the maintenance of plant organelle genome stability. New Phytol. 2010, 186, 299–317. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Liu, H.; Hu, J.; Liang, Y.; Liang, J.; Wuyun, T.; Tan, X. Five complete chloroplast genome sequences from diospyros: Genome organization and comparative analysis. PLoS ONE 2016, 11, e0159566. [Google Scholar] [CrossRef] [PubMed]
- Chumley, T.W.; Palmer, J.D.; Mower, J.P.; Fourcade, H.M.; Calie, P.J.; Boore, J.L.; Jansen, R.K. The complete chloroplast genome sequence of pelargonium x hortorum: Organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol. Biol. Evol. 2006, 23, 2175–2190. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zhang, X.; Liu, G.; Yin, Y.; Chen, K.; Yun, Q.; Zhao, D.; Al-Mssallem, I.S.; Yu, J. The complete chloroplast genome sequence of date palm (Phoenix dactylifera L.). PLoS ONE 2010, 5, e12762. [Google Scholar] [CrossRef] [PubMed]
- Lei, W.; Ni, D.; Wang, Y.; Shao, J.; Wang, X.; Yang, D.; Wang, J.; Chen, H.; Liu, C. Intraspecific and heteroplasmic variations, gene losses and inversions in the chloroplast genome of Astragalus membranaceus. Sci. Rep. 2016, 6, 21669. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wuyun, T.N.; Du, H.; Wang, D.; Cao, D. Complete chloroplast genome sequences of Eucommia ulmoides: Genome structure and evolution. Tree Genet. Genomes 2016, 12, 12. [Google Scholar] [CrossRef]
- Ermolaeva, M.D. Synonymous codon usage in bacteria. Curr. Issues Mol. Biol. 2001, 3, 91–97. [Google Scholar] [PubMed]
- Wong, G.K.; Wang, J.; Tao, L.; Tan, J.; Zhang, J.; Passey, D.A.; Yu, J. Compositional gradients in gramineae genes. Genome Res. 2002, 12, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Xue, Q. Codon usage in the chloroplast genome of rice (Oryza sativa L. ssp. japonica). Acta Agron. Sin. 2004, 30, 1220–1224. [Google Scholar]
- Zhou, M.; Long, W.; Li, X. Analysis of synonymous codon usage in chloroplast genome of Populus alba. For. Res. 2008, 19, 293–297. [Google Scholar] [CrossRef]
- Zhou, M.; Long, W.; Li, X. Patterns of synonymous codon usage bias in chloroplast genomes of seed plants. For. Sci. Pract. 2008, 10, 235–242. [Google Scholar] [CrossRef]
- Morton, B.R. The role of context-dependent mutations in generating compositional and codon usage bias in grass chloroplast DNA. J. Mol. Evol. 2003, 56, 616–629. [Google Scholar] [CrossRef] [PubMed]
- Nazareno, A.G.; Carlsen, M.; Lohmann, L.G. Complete chloroplast genome of tanaecium tetragonolobum: The first bignoniaceae plastome. PLoS ONE 2015, 10, e0129930. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Tang, P.; Li, Z.; Li, D.; Liu, Y.; Huang, H. The first complete chloroplast genome sequences in actinidiaceae: Genome structure and comparative analysis. PLoS ONE 2015, 10, e0129347. [Google Scholar] [CrossRef] [PubMed]
- Ni, L.; Zhao, Z.; Dorje, G.; Ma, M. The complete chloroplast genome of ye-xing-ba (Scrophularia dentata; Scrophulariaceae), an alpine Tibetan herb. PLoS ONE 2016, 11, e0158488. [Google Scholar] [CrossRef] [PubMed]
- Cavalier-Smith, T. Chloroplast evolution: Secondary symbiogenesis and multiple losses. Curr. Biol. 2002, 12, R62–R64. [Google Scholar] [CrossRef]
- Xue, J.; Wang, S.; Zhou, S.L. Polymorphic chloroplast microsatellite loci in nelumbo (nelumbonaceae). Am. J. Bot. 2012, 99, e240–e244. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Gui, S.; Zhu, Z.; Wang, X.; Ke, W.; Ding, Y. Genome-wide identification of SSR and snp markers based on whole-genome re-sequencing of a Thailand wild sacred lotus (nelumbo nucifera). PLoS ONE 2015, 10, e0143765. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Song, J.; Gao, H.; Zhu, Y.; Xu, J.; Pang, X.; Yao, H.; Sun, C.; Li, X.; Li, C.; et al. The complete chloroplast genome sequence of the medicinal plant salvia miltiorrhiza. PLoS ONE 2013, 8, e57607. [Google Scholar] [CrossRef] [PubMed]
- Kuang, D.Y.; Wu, H.; Wang, Y.L.; Gao, L.M.; Zhang, S.Z.; Lu, L. Complete chloroplast genome sequence of magnolia kwangsiensis (magnoliaceae): Implication for DNA barcoding and population genetics. Genome 2011, 54, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Freyer, R.; López, C.; Maier, R.M.; Martín, M.; Sabater, B.; Kössel, H. Editing of the chloroplast ndhb encoded transcript shows divergence between closely related members of the grass family (poaceae). Plant Mol. Biol. 1995, 29, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Kahlau, S.; Aspinall, S.; Gray, J.C.; Bock, R. Sequence of the tomato chloroplast DNA and evolutionary comparison of solanaceous plastid genomes. J. Mol. Evol. 2006, 63, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Chateigner Boutin, A.L.; Small, I. A rapid high-throughput method for the detection and quantification of RNA editing based on high-resolution melting of amplicons. Nucleic Acids Res. 2007, 35, e114. [Google Scholar] [CrossRef] [PubMed]
- Bock, R. Sense from nonsense: How the genetic information of chloroplasts is altered by RNA editing. Biochimie 2000, 82, 549–557. [Google Scholar] [CrossRef]
- Jiang, Y.; Yun, H.E.; Fan, S.L.; Jia-Ning, Y.U.; Song, M.Z. The identification and analysis of RNA editing sites of 10 chloroplast protein-coding genes from virescent mutant of Gossypium Hirsutum. Cotton Sci. 2011, 23, 3–9. [Google Scholar]
- Jansen, R.K.; Cai, Z.; Raubeson, L.A.; Daniell, H.; Depamphilis, C.W.; Leebens-Mack, J.; Muller, K.F.; Guisinger-Bellian, M.; Haberle, R.C.; Hansen, A.K.; et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Shi, C.; Liu, Y.; Mao, S.Y.; Gao, L.Z. Thirteen camellia chloroplast genome sequences determined by high-throughput sequencing: Genome structure and phylogenetic relationships. BMC Evol. Biol. 2014, 14, 151. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, S.; Yu, J.; Wang, L.; Zhou, S. A modified ctab protocol for plant DNA extraction. Chin. Bull. Bot. 2013, 48, 72–78. [Google Scholar]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. Spades: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped blast and psi-blast: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. Organellargenomedraw—A suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013, 41, W575–W581. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. Reputer: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Vieira Ldo, N.; Faoro, H.; Rogalski, M.; Fraga, H.P.; Cardoso, R.L.; de Souza, E.M.; de Oliveira Pedrosa, F.; Nodari, R.O.; Guerra, M.P. The complete chloroplast genome sequence of Podocarpus Lambertii: Genome structure, evolutionary aspects, gene content and SSR detection. PLoS ONE 2014, 9, e90618. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Hao, Z.; Xu, H.; Yang, L.; Liu, G.; Sheng, Y.; Zheng, C.; Zheng, W.; Cheng, T.; Shi, J. The complete chloroplast genome sequence of the relict woody plant Metasequoia glyptostroboides Hu et Cheng. Front. Plant Sci. 2015, 6, 447. [Google Scholar] [CrossRef] [PubMed]
- Thiel, T.; Michalek, W.; Varshney, R.K.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). TAG. Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Wright, F. The effective number of codons used in a gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Sharp, P.M.; Tuohy, T.M.; Mosurski, K.R. Codon usage in yeast: Cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 1986, 14, 5125–5143. [Google Scholar] [CrossRef] [PubMed]
- Mower, J.P. The prep suite: Predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 2009, 37, W253–W259. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Jia, L.; Li, Y. CURE-chloroplast: A chloroplast C-to-U RNA editing predictor for seed plants. BMC Bioinform. 2009, 10, 135. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D. Mafft multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. Mega7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category for Genes | Group of Gene | Name of Gene |
|---|---|---|
| Photosynthesis related genes | Rubisco | rbcL |
| Photosystem І | psaA, psaB, psaC, psaI, psaJ | |
| Assembly/stability of photosystem І | ycf3 *, ycf4 | |
| Photosystem ІІ | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | |
| ATP synthase | atpA, atpB, atpE, atpF *, atpH, atpI | |
| cytochrome b/f complex | petA, petB *, petD *, petG, petL, petN | |
| cytochrome c synthesis | ccsA | |
| NADPH dehydrogenase | ndhA *, ndhB *, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ | |
| Transcription and translation related genes | transcription | rpoA, rpoB, rpoC1 *, rpoC2 |
| ribosomal proteins | rps2, rps3, rps4, rps7, rps8, rps11, rps12 *, rps14, rps15, rps16 *, rps18, rps19, rpl2 *, rpl14, rpl16 *, rpl20, rpl22, rpl23, rpl32, rpl33, rpl36 | |
| translation initiation factor | infA | |
| RNA genes | ribosomal RNA | rrn5, rrn4.5, rrn16, rrn23 |
| transfer RNA | trnA-UGC *, trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnG-UCC *, trnG-GCC *, trnH-GUG, trnI-CAU, trnI-GAU *, trnK-UUU *, trnL-CAA, trnL-UAA *, trnL-UAG, trnfM-CAUI, trnM-CAU, trnN-GUU, trnP-UGG, trnQ-UUG, trnR-ACG, trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC, trnV-UAC *, trnW-CCA, trnY-GUA | |
| Other genes | RNA processing | matK |
| carbon metabolism | cemA | |
| fatty acid synthesis | accD | |
| proteolysis | clpP * | |
| Genes of unknown function | conserved reading frames | ycf1, ycf2, ycf15, ndhK |
| Gene | Location | Exon І (bp) | Intron І (bp) | Exon ІІ (bp) | Intron ІІ (bp) | Exon ІІІ (bp) |
|---|---|---|---|---|---|---|
| trnA-UGC | IR | 38 | 814 | 35 | ||
| trnG-GCC | LSC | 24 | 676 | 48 | ||
| trnI-GAU | IR | 42 | 942 | 35 | ||
| trnK-UUU | LSC | 38 | 2494 | 37 | ||
| trnL-UAA | LSC | 37 | 473 | 50 | ||
| trnV-UAC | LSC | 38 | 572 | 37 | ||
| rps12 * | LSC | 114 | - | 231 | 536 | 27 |
| rps16 | LSC | 40 | 864 | 227 | ||
| atpF | LSC | 144 | 705 | 411 | ||
| rpoC1 | LSC | 445 | 758 | 1619 | ||
| ycf3 | LSC | 129 | 714 | 228 | 737 | 153 |
| clpP | LSC | 69 | 815 | 291 | 642 | 228 |
| petB | LSC | 6 | 707 | 642 | ||
| petD | LSC | 8 | 713 | 475 | ||
| rpl16 | LSC | 9 | 865 | 399 | ||
| rpl2 | IR | 393 | 664 | 435 | ||
| ndhB | IR | 777 | 679 | 756 | ||
| ndhA | SSC | 555 | 1106 | 531 |
| Amino Acids | Codon | Number | RSCU | AA Frequency | Amino Acids | Codon | Number | RSCU | AA Frequency |
|---|---|---|---|---|---|---|---|---|---|
| Phe | UUU | 779 | 1.32 | 5.59% | Ser | UCU | 472 | 1.76 | 7.59% |
| UUC | 405 | 0.68 | UCC | 247 | 0.92 | ||||
| Leu | UUA | 720 | 1.93 | 10.56% | UCA | 307 | 1.15 | ||
| UUG | 451 | 1.21 | UCG | 152 | 0.57 | ||||
| CUU | 486 | 1.30 | AGU | 339 | 1.26 | ||||
| CUC | 129 | 0.35 | AGC | 91 | 0.34 | ||||
| CUA | 301 | 0.81 | Pro | CCU | 351 | 1.55 | 4.26% | ||
| CUG | 150 | 0.40 | CCC | 170 | 0.75 | ||||
| Ile | AUU | 890 | 1.47 | 8.57% | CCA | 269 | 1.19 | ||
| AUC | 377 | 0.62 | CCG | 113 | 0.50 | ||||
| AUA | 548 | 0.91 | Thr | ACU | 430 | 1.63 | 4.98% | ||
| Met | AUG | 495 | 1.00 | 2.34% | ACC | 201 | 0.76 | ||
| Val | GUU | 423 | 1.48 | 5.41% | ACA | 324 | 1.23 | ||
| GUC | 126 | 0.44 | ACG | 100 | 0.38 | ||||
| GUA | 447 | 1.56 | Ala | GCU | 526 | 1.84 | 5.41% | ||
| GUG | 151 | 0.53 | GCC | 177 | 0.62 | ||||
| Tyr | UAU | 631 | 1.61 | 3.70% | GCA | 328 | 1.14 | ||
| UAC | 152 | 0.39 | GCG | 115 | 0.40 | ||||
| TER | UAA | 28 | 1.62 | 0.25% | Cys | UGU | 171 | 1.53 | 1.05% |
| UAG | 10 | 0.58 | UGC | 52 | 0.47 | ||||
| UGA | 14 | 0.81 | Arg | CGU | 275 | 1.30 | 6.00% | ||
| His | CAU | 404 | 1.58 | 2.42% | CGC | 90 | 0.42 | ||
| CAC | 108 | 0.42 | CGA | 284 | 1.34 | ||||
| Gln | CAA | 595 | 1.52 | 3.69% | CGG | 97 | 0.46 | ||
| CAG | 186 | 0.48 | Arg | AGA | 392 | 1.85 | |||
| Asn | AAU | 796 | 1.56 | 4.81% | AGG | 133 | 0.63 | ||
| AAC | 224 | 0.44 | Gly | GGU | 493 | 1.33 | 7.00% | ||
| Lys | AAA | 837 | 1.54 | 5.15% | GGC | 145 | 0.39 | ||
| AAG | 253 | 0.46 | GGA | 594 | 1.60 | ||||
| Asp | GAU | 690 | 1.59 | 4.09% | GGG | 251 | 0.68 | ||
| GAC | 176 | 0.41 | Glu | GAA | 866 | 1.54 | 5.32% | ||
| Trp | UGG | 386 | 1.00 | 1.82% | GAG | 262 | 0.46 |
| No. | Size/bp | Type # | Repeat 1 Start (Location) | Repeat 2 Start (Location) | Region |
|---|---|---|---|---|---|
| 1 | 30 | F | 10,814 (trnG-GCC *) | 38,746 (trnG-UCC) | LSC |
| 2 | 30 | F | 17,447 (rps2-rpoC2) | 17,448 (rps2-rpoC) | LSC |
| 3 | 30 | F | 44,547 (psaA-ycf3) | 44,550 (psaA-ycf3) | LSC |
| 4 | 30 | F | 45,978 (ycf3 intron2) | 101,338 (rps12_3end-trnV-GAC) | LSC, IRa |
| 5 | 30 | F | 91,923 (ycf2) | 91,965 (ycf2) | IRa |
| 6 | 30 | F | 110,167 (rrn4.5-rrn5) | 110,198 (rrn4.5-rrn5) | IRa |
| 7 | 30 | F | 133,335 (rrn5-rrn4.5) | 133,366 (rrn5-rrn4.5) | IRb |
| 8 | 30 | F | 149,178 (ycf2) | 149,214 (ycf2) | IRb |
| 9 | 30 | F | 149,196 (ycf2) | 149,214 (ycf2) | IRb |
| 10 | 30 | F | 151,568 (ycf2) | 151,610 (ycf2) | IRb |
| 11 | 32 | F | 9313 (trnS-GCU *) | 37,781 (psbC-trnS-UGA *) | LSC |
| 12 | 32 | F | 40,965 (psaB) | 43,189 (psaA) | LSC |
| 13 | 32 | F | 53,338 (ndhC-trnV-UAC) | 53,358 (ndhC-trnV-UAC) | LSC |
| 14 | 32 | F | 115,350 (ndhF-rpl32) | 115,378 (ndhF-rpl32) | SSC |
| 15 | 34 | F | 94,332 (ycf2) | 94,368 (ycf2) | IRa |
| 16 | 34 | F | 94,350 (ycf2) | 94,368 (ycf2) | IRa |
| 17 | 35 | F | 149,188(ycf2) | 149,206 (ycf2) | IRb |
| 18 | 39 | F | 45,966 (ycf3 intron2) | 101,326 (rps12_3end-trnV-GAC) | LSC, IRa |
| 19 | 39 | F | 45,966 (ycf3 intron2) | 122,604 (ndhA intron1) | LSC, SSC |
| 20 | 41 | F | 40,953 (psaB) | 43,177 (psaA) | LSC |
| 21 | 41 | F | 101,324 (rps12_3end-trnV-GAC) | 122,602 (ndhA intron) | IRa, SSC |
| 22 | 42 | F | 94,320 (ycf2) | 94,356 (ycf2) | IRa |
| 23 | 42 | F | 149,165 (ycf2) | 149,201 (ycf2) | IRb |
| 24 | 44 | F | 94,340 (ycf2) | 94,358 (ycf2) | IRa |
| 25 | 58 | F | 94,332 (ycf2) | 94,340 (ycf2) | IRa |
| 26 | 58 | F | 149,165 (ycf2) | 149,183 (ycf2) | IRb |
| 27 | 30 | P | 9315 (trnS-GCU *) | 47,653 (trnS-GGA) | LSC |
| 28 | 30 | P | 14,359 (atpF-atpH) | 14,359 (atpF-atpH) | LSC |
| 29 | 30 | P | 34,338 (trnT-GGU-psbD) | 34,338 (trnT-GGU-psbD) | LSC |
| 30 | 30 | P | 37,783 (psbC-trnS-UGA *) | 47,653 (trnS-GGA) | LSC |
| 31 | 30 | P | 45,978 (ycf3 intron2) | 142,195 (trnV-GAC-rps12_3end) | LSC, IRb |
| 32 | 30 | P | 91,923 (ycf2) | 151,568 (ycf2) | IRa, IRb |
| 33 | 30 | P | 91,965 (ycf2) | 151,610 (ycf2) | IRa, IRb |
| 34 | 30 | P | 110,167 (rrn4.5-rrn5) | 133,335 (rrn5-rrn4.5) | IRa, IRb |
| 35 | 30 | P | 110,198 (rrn4.5-rrn5) | 133,366 (rrn5-rrn4.5) | IRa, IRb |
| 36 | 30 | P | 122,764 (ndhA intron1) | 122,766 (ndhA intron1) | SSC |
| 37 | 34 | P | 94,332 (ycf2) | 149,161 (ycf2) | IRa, IRb |
| 38 | 34 | P | 94,350 (ycf2) | 149,161 (ycf2) | IRa, IRb |
| 39 | 34 | P | 94,368 (ycf2) | 149,179 (ycf2) | IRa, IRb |
| 40 | 34 | P | 94,368 (ycf2) | 149,179 (ycf2) | IRa, IRb |
| 41 | 39 | P | 45,966 (ycf3 intron2) | 45,966 (ycf3 intron2) | LSC, IRb |
| 42 | 41 | P | 122,602 (ndhA intron1) | 142,198 (trnV-GAC–rps12_3end) | SSC, IRb |
| 43 | 42 | P | 94,320 (ycf2) | 149,165 (ycf2) | IRa, IRb |
| 44 | 42 | P | 94,356 (ycf2) | 149,201 (ycf2) | IRa, IRb |
| 45 | 44 | P | 77,475 (psbT-psbN) | 77,475 (psbT-psbN) | LSC |
| 46 | 44 | P | 94,340 (ycf2) | 149,161 (ycf2) | IRa, IRb |
| 47 | 44 | P | 94,358 (ycf2) | 149,179 (ycf2) | IRa, IRb |
| 48 | 58 | P | 94,332 (ycf2) | 149,165 (ycf2) | IRa, IRb |
| 49 | 58 | P | 94,340 (ycf2) | 149,183 (ycf2) | IRa, IRb |
| SSR Type # | SSR Sequence | Size | Start | SSR Location | Region |
|---|---|---|---|---|---|
| p1 | (A)10 | 10 | 31,855 | psbM-trnD-GUC | LSC |
| 10 | 31,992 | psbM-trnD-GUC | LSC | ||
| 10 | 38,025 | trnS-UGA-psbZ | LSC | ||
| 10 | 73,886 | clpP intron1 | LSC | ||
| 10 | 85,390 | rpl16 intron | LSC | ||
| (T)10 | 10 | 507 | trnH-GUG-psbA | LSC | |
| 10 | 9056 | psbK-psbI | LSC | ||
| 10 | 11,162 | trnR-UCU-atpA | LSC | ||
| 10 | 59,781 | rbcL-accD | LSC | ||
| 10 | 66,291 | petA-psbJ | LSC | ||
| 10 | 69,202 | petL-petG | LSC | ||
| (C)10 | 10 | 5236 | trnK-UUU-rps16 | LSC | |
| (T)11 | 11 | 19,678 | rpoC2 | LSC | |
| 11 | 50,871 | trnF-GAA-ndhJ | LSC | ||
| 11 | 61,662 | accD-psaI | LSC | ||
| 11 | 72,263 | rpl20-clpP | LSC | ||
| 11 | 74,741 | clpP intron2 | LSC | ||
| (T)12 | 12 | 20,216 | rpoC2 | LSC | |
| 12 | 81,254 | rpoA | LSC | ||
| 12 | 83,666 | rps8-rpl14 | LSC | ||
| (A)13 | 13 | 12,741 | atpA-atpF | LSC | |
| 13 | 46,877 | ycf3-trnS-GGA | LSC | ||
| (T)13 | 13 | 14,109 | atpF-atpH | LSC | |
| 13 | 34,486 | trnT-GGU-psbD | LSC | ||
| 13 | 37,645 | psbC-trnS-UGA | LSC | ||
| 13 | 86,860 | rpl22-rps19 | LSC | ||
| (T)14 | 14 | 48,630 | rps4-trnT-UGU | LSC | |
| (A)15 | 15 | 33,163 | trnE-UUC-trnT-GGU | LSC | |
| (A)16 | 16 | 46,618 | ycf3 intron2 | LSC | |
| (A)19 | 19 | 44,559 | psaA-ycf3 | LSC | |
| (T)19 | 19 | 117,928 | ndhD | SSC | |
| (A)20 | 20 | 29,957 | trnC-GCA-petN | LSC | |
| p2 | (AT)5 | 10 | 4646 | trnK-UUU-rps16 | LSC |
| 10 | 6558 | rps16-trnQ-UUG | LSC | ||
| 10 | 21,057 | rpoC2 | LSC | ||
| (TA)5 | 10 | 69,619 | trnW-CCA-trnP-UGG | LSC | |
| (TA)6 | 12 | 48,772 | rps4-trnT-UGU | LSC | |
| 12 | 49,291 | trnT-UGU-trnL-UAA | LSC | ||
| 12 | 69,931 | trnP-UGG-psaJ | LSC | ||
| p3 | (CCT)4 | 12 | 69,371 | petG-trnW-CCA | LSC |
| p4 | (AAAG)3 | 12 | 73,413 | clpP intron1 | LSC |
| (TCTT)3 | 12 | 31,191 | petN-psbM | LSC | |
| (TTTA)3 | 12 | 55,102 | trnM-CAU-atpE | LSC | |
| (AAAT)4 | 16 | 9284 | psbI-trnS-GCU | LSC | |
| p5 | (TCTAT)3 | 15 | 9458 | trnS-GCU-trnG-GCC | LSC |
| c | - | 23 | 17,456 | rps2-rpoC2 | LSC |
| - | 27 | 63,589 | ycf4-cemA | LSC | |
| - | 33 | 78,324 | petB intron | LSC | |
| - | 45 | 71,570 | rps18-rpl20 | LSC | |
| - | 59 | 38,501 | psbZ-trnG-UCC | LSC | |
| - | 90 | 57,078 | atpB * | LSC |
| Gene | Codon Position | Amino Acid Position | Codon (Amino Acid) Conversion | Score |
|---|---|---|---|---|
| accD | 794 | 265 | uCg (S) => uUg (L) | 0.8 |
| 1403 | 468 | cCu (P) => cUu (L) | 1 | |
| atpA | 914 | 305 | uCa (S) => uUa (L) | 1 |
| atpF | 92 | 31 | cCa (P) => cUa(L) | 0.86 |
| atpI | 629 | 210 | uCa (S) => uUa (L) | 1 |
| ccsA | 71 | 24 | aCu (T) => aUu (I) | 1 |
| matK | 271 | 91 | Ccu (P) => Ucu (S) | 0.86 |
| 460 | 154 | Cac (H) => Uac (Y) | 1 | |
| 646 | 216 | Cau (H) => Uau (Y) | 1 | |
| 1180 | 394 | Cgg (R) => Ugg (W) | 1 | |
| 1249 | 417 | Cau (H) => Uau (Y) | 1 | |
| ndhA | 344 | 115 | uCa (S) => uUa (L) | 1 |
| 569 | 190 | uCa (S) => uUa (L) | 1 | |
| ndhB | 149 | 50 | uCa (S) => uUa (L) | 1 |
| 467 | 156 | cCa (P) => cUa (L) | 1 | |
| 586 | 196 | Cau (H) => Uau (Y) | 1 | |
| 611 | 204 | uCa (S) => uUa (L) | 0.8 | |
| 737 | 246 | cCa (P) => cUa (L) | 1 | |
| 746 | 249 | uCu (S) => uUu (F) | 1 | |
| 830 | 277 | uCa (S) => uUa (L) | 1 | |
| 836 | 279 | uCa (S) => uUa (L) | 1 | |
| 1292 | 431 | uCc (S) => uUc (F) | 1 | |
| 1481 | 494 | cCa (P) => cUa (L) | 1 | |
| ndhD | 2 | 1 | aCg (T) => aUg (M) | 1 |
| 47 | 16 | uCu (S) => uUu (F) | 0.8 | |
| 313 | 105 | Cgg (R) => Ugg (W) | 0.8 | |
| 878 | 293 | uCa (S) => uUa (L) | 1 | |
| 1298 | 433 | uCa (S) => uUa (L) | 0.8 | |
| 1310 | 437 | uCa (S) => uUa (L) | 0.8 | |
| ndhF | 290 | 97 | uCa (S) => uUa (L) | 1 |
| 671 | 224 | uCa (S) => uUa (L) | 1 | |
| ndhG | 314 | 105 | aCa (T) => aUa (I) | 0.8 |
| 385 | 129 | Cca (P) => Uca (S) | 0.8 | |
| petB | 418 | 140 | Cgg (R) => Ugg (W) | 1 |
| 611 | 204 | cCa (P) => cUa (L) | 1 | |
| petG | 94 | 32 | Cuu (L) => Uuu (F) | 0.86 |
| psbE | 214 | 72 | Ccu (P) => Ucu (S) | 1 |
| rpl2 | 596 | 199 | gCg (A) => gUg (V) | 0.86 |
| rpl20 | 308 | 103 | uCa (S) => uUa (L) | 0.86 |
| rpoA | 830 | 277 | uCa (S) => uUa (L) | 1 |
| rpoB | 338 | 113 | uCu (S) => uUu (F) | 1 |
| 551 | 184 | uCa (S) => uUa (L) | 1 | |
| 566 | 189 | uCg (S) => uUg (L) | 1 | |
| 1672 | 558 | Ccc (P) => Ucc (S) | 0.86 | |
| 2000 | 667 | uCu (S) => uUu (F) | 1 | |
| 2426 | 809 | uCa (S) => uUa (L) | 0.86 | |
| rpoC2 | 1792 | 598 | Cgu (R) => Ugu (C) | 0.86 |
| 2305 | 769 | Cgg (R) => Ugg (W) | 1 | |
| 3746 | 1249 | uCa (S) => uUa (L) | 0.86 | |
| rps2 | 248 | 83 | uCa (S) => uUa (L) | 1 |
| rps14 | 80 | 27 | uCa (S) => uUa (L) | 1 |
| 149 | 50 | cCa (P) => cUa (L) | 1 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, W.; Yu, H.; Wang, J.; Lei, W.; Gao, J.; Qiu, X.; Wang, J. The Complete Chloroplast Genome Sequences of the Medicinal Plant Forsythia suspensa (Oleaceae). Int. J. Mol. Sci. 2017, 18, 2288. https://doi.org/10.3390/ijms18112288
Wang W, Yu H, Wang J, Lei W, Gao J, Qiu X, Wang J. The Complete Chloroplast Genome Sequences of the Medicinal Plant Forsythia suspensa (Oleaceae). International Journal of Molecular Sciences. 2017; 18(11):2288. https://doi.org/10.3390/ijms18112288
Chicago/Turabian StyleWang, Wenbin, Huan Yu, Jiahui Wang, Wanjun Lei, Jianhua Gao, Xiangpo Qiu, and Jinsheng Wang. 2017. "The Complete Chloroplast Genome Sequences of the Medicinal Plant Forsythia suspensa (Oleaceae)" International Journal of Molecular Sciences 18, no. 11: 2288. https://doi.org/10.3390/ijms18112288