Whole Blood mRNA Expression-Based Prognosis of Metastatic Renal Cell Carcinoma

 ,
,
Abstract
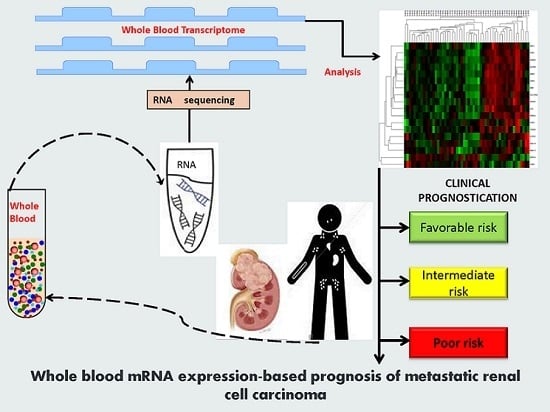
:
1. Introduction
2. Results
2.1. Patient Characteristics
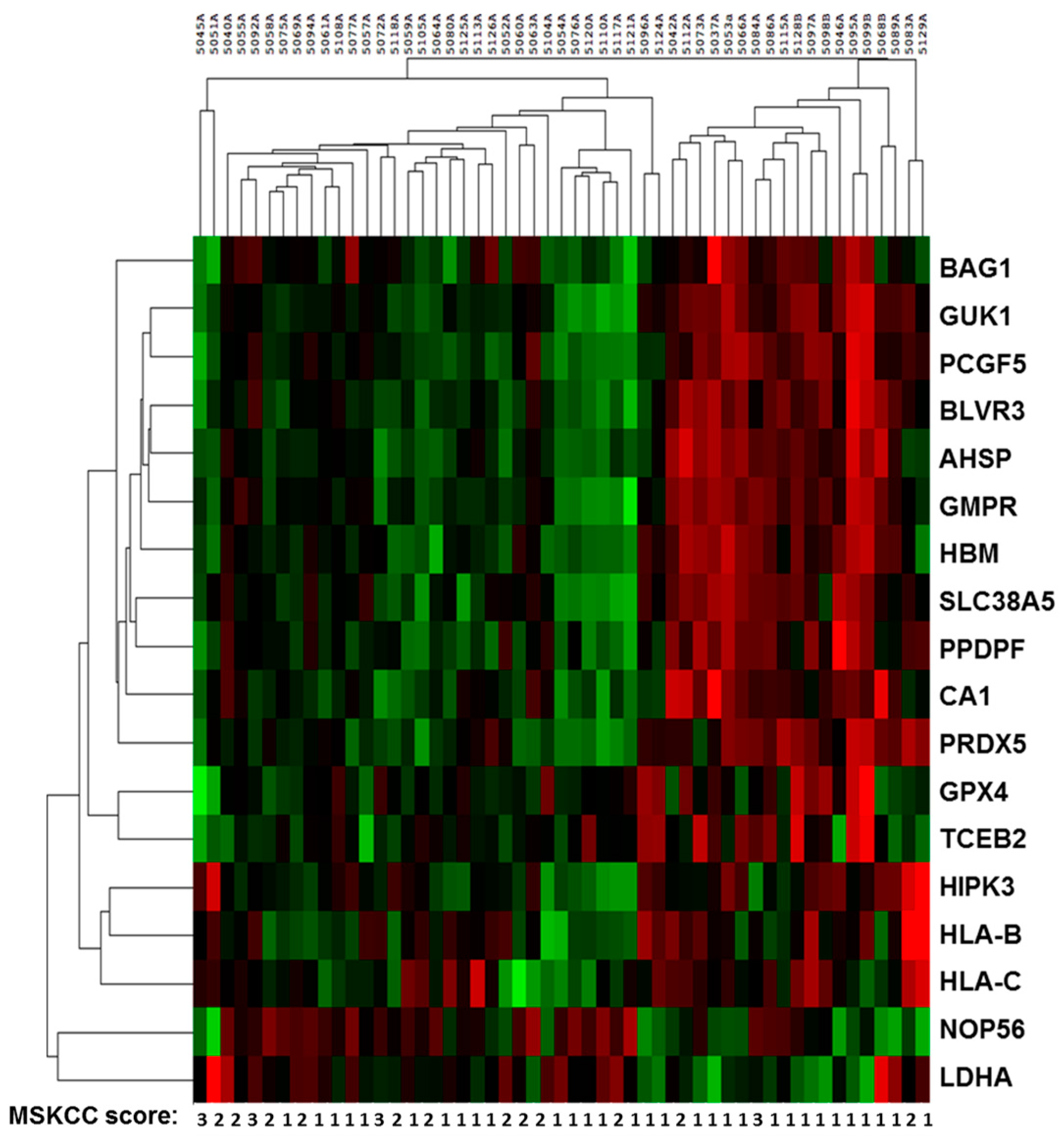
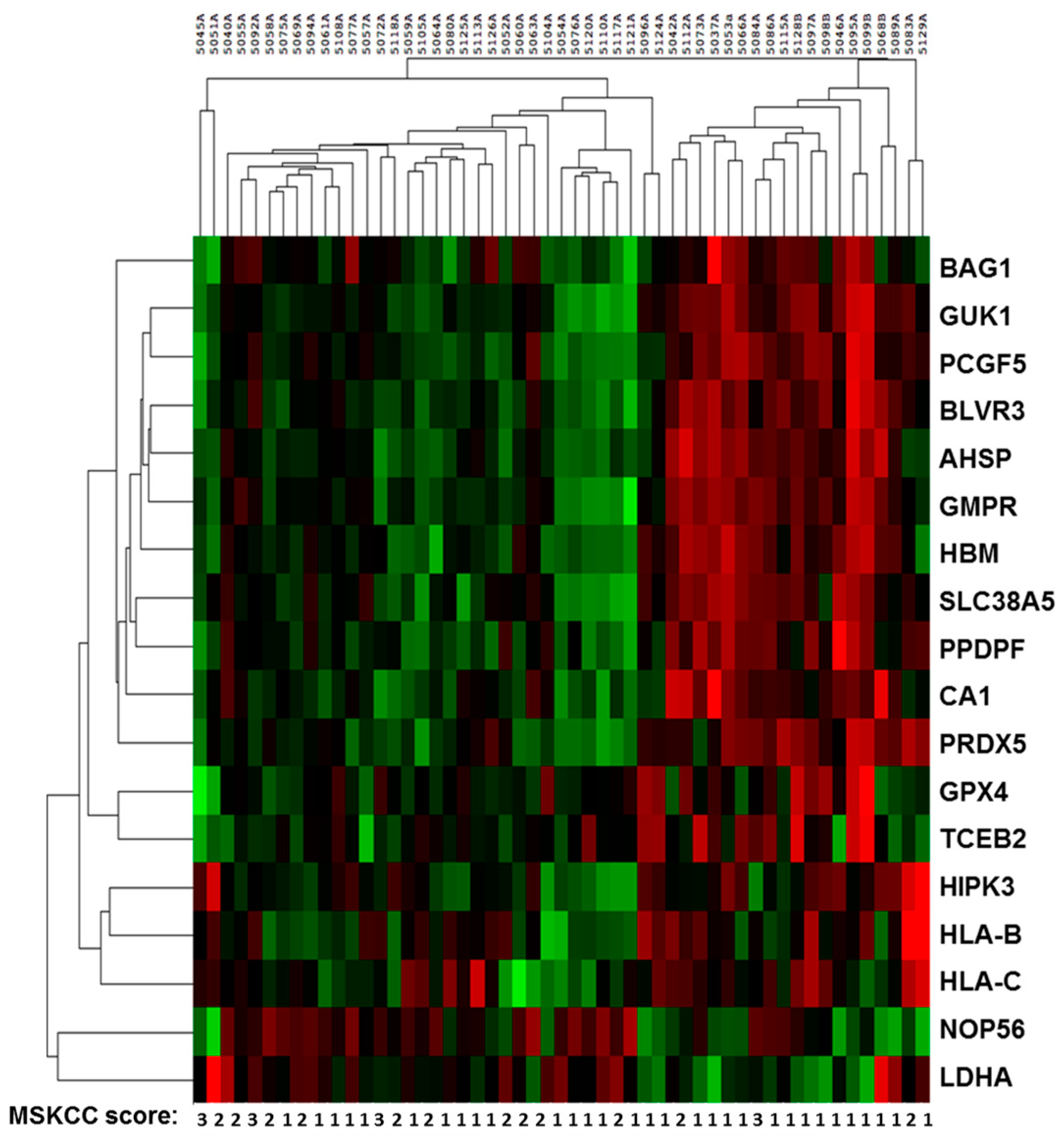
2.2. Sequencing Results of Discovery and Validation Sets
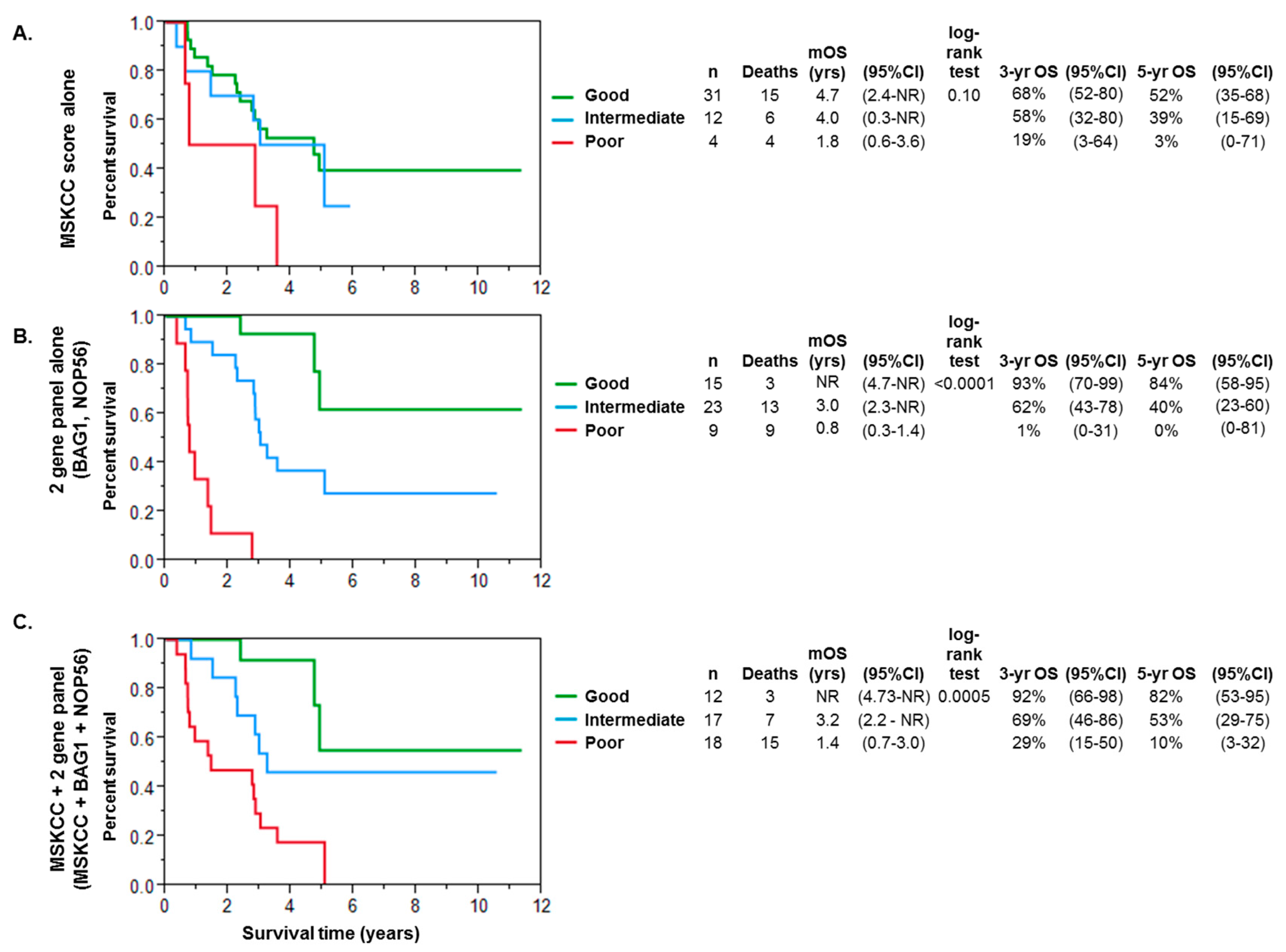
2.3. Multivariate Analysis and Modeling for Prognosis in Metastatic Renal Cell Cancer
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AUC | Area under the curve |
| CI | Confidence Interval |
| FDR | False discovery rate |
| FPKM | Fragments per kilobase of transcripts per million fragments mapped |
| HR | Hazard ratio |
| mCCRCC | Metastatic clear cell renal carcinoma |
| mRCC | Metastatic renal cell carcinoma |
| mRNA | Messenger ribonucleic acid |
| MSKCC | Memorial Sloan Kettering Cancer Center |
| OS | Overall survival |
| RCC | Renal cell carcinoma |
| RNA | Ribonucleic acid |
| ROC | Receiver operating characteristic curve |
References
- Choueiri, T.K.; Motzer, R.J. Systemic therapy for metastatic renal-cell carcinoma. N. Engl. J. Med. 2017, 376, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Mazumdar, M.; Bacik, J.; Berg, W.; Amsterdam, A.; Ferrara, J. Survival and prognostic stratification of 670 patients with advanced renal cell carcinoma. J. Clin. Oncol. 1999, 17, 2530. [Google Scholar] [CrossRef] [PubMed]
- McDermott, D.F. Immunotherapy of metastatic renal cell carcinoma. Cancer 2009, 115, 2298–2305. [Google Scholar] [CrossRef] [PubMed]
- Cardi, G.; Heaney, J.A.; Schned, A.R.; Phillips, D.M.; Branda, M.T.; Ernstoff, M.S. T-cell receptor ζ-chain expression on tumor-infiltrating lymphocytes from renal cell carcinoma. Cancer Res. 1997, 57, 3517–3519. [Google Scholar] [PubMed]
- Cardi, G.; Heaney, J.A.; Schned, A.R.; Ernstoff, M.S. Expression of fas (apo-1/cd95) in tumor-infiltrating and peripheral blood lymphocytes in patients with renal cell carcinoma1. Cancer Res. 1998, 58, 2078–2080. [Google Scholar] [PubMed]
- Ernstoff, M.S.; Crocenzi, T.S.; Seigne, J.D.; Crosby, N.A.; Cole, B.F.; Fisher, J.L.; Uhlenhake, J.C.; Mellinger, D.; Foster, C.; Farnham, C.J. Developing a rational tumor vaccine therapy for renal cell carcinoma: Immune yin and yang. Clin. Cancer Res. 2007, 13, 733s–740s. [Google Scholar] [CrossRef] [PubMed]
- Metzker, M.L. Sequencing technologies—The next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Silver, N.; Best, S.; Jiang, J.; Thein, S.L. Selection of housekeeping genes for gene expression studies in human reticulocytes using real-time pcr. BMC Mol. Biol. 2006, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Thellin, O.; Zorzi, W.; Lakaye, B.; de Borman, B.; Coumans, B.; Hennen, G.; Grisar, T.; Igout, A.; Heinen, E. Housekeeping genes as internal standards: Use and limits. J. Biotechnol. 1999, 75, 291–295. [Google Scholar] [CrossRef]
- Jung, M.; Ramankulov, A.; Roigas, J.; Johannsen, M.; Ringsdorf, M.; Kristiansen, G.; Jung, K. In search of suitable reference genes for gene expression studies of human renal cell carcinoma by real-time pcr. BMC Mol. Biol. 2007, 8, 47. [Google Scholar] [CrossRef] [PubMed]
- Budczies, J.; Klauschen, F.; Sinn, B.V.; Győrffy, B.; Schmitt, W.D.; Darb-Esfahani, S.; Denkert, C. Cutoff finder: A comprehensive and straightforward web application enabling rapid biomarker cutoff optimization. PLoS ONE 2012, 7, e51862. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Tian, Z.; Zhang, L.; Hou, S.; Hu, S.; Wu, J.; Jing, Y.; Sun, H.; Yu, F.; Zhao, L. Combined cell surface carbonic anhydrase 9 and cd147 antigens enable high-efficiency capture of circulating tumor cells in clear cell renal cell carcinoma patients. Oncotarget 2016, 7, 59877–59891. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.G.; Takayama, S.; Rapp, U.R.; Reed, J.C. Bcl-2 interacting protein, bag-1, binds to and activates the kinase raf-1. Proc. Natl. Acad. Sci. USA 1996, 93, 7063–7068. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Takeda, M.; Morimoto, R.I. Bag1–hsp70 mediates a physiological stress signalling pathway that regulates raf-1/erk and cell growth. Nat. Cell Biol. 2001, 3, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Adachi, M.; Sekiya, M.; Torigoe, T.; Takayama, S.; Reed, J.C.; Miyazaki, T.; Minami, Y.; Taniguchi, T.; Imai, K. Interleukin-2 (il-2) upregulates bag-1 gene expression through serine-rich region within il-2 receptor β c chain. Blood 1996, 88, 4118–4123. [Google Scholar] [PubMed]
- Clevenger, C.V.; Thickman, K.; Ngo, W.; Chang, W.P.; Takayama, S.; Reed, J.C. Role of bag-1 in the survival and proliferation of the cytokine-dependent lymphocyte lines, ba/f3 and nb2. Mol. Endocrinol. 1997, 11, 608–618. [Google Scholar] [CrossRef] [PubMed]
- Stepanov, G.A.; Filippova, J.A.; Komissarov, A.B.; Kuligina, E.V.; Richter, V.A.; Semenov, D.V. Regulatory role of small nucleolar rnas in human diseases. BioMed Res. Int. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Mannoor, K.; Liao, J.; Jiang, F. Small nucleolar rnas in cancer. Biochim. Biophys. Acta BBA Rev. Cancer 2012, 1826, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Van’t Veer, L.J.; Bernards, R. Enabling personalized cancer medicine through analysis of gene-expression patterns. Nature 2008, 452, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Twine, N.C.; Stover, J.A.; Marshall, B.; Dukart, G.; Hidalgo, M.; Stadler, W.; Logan, T.; Dutcher, J.; Hudes, G.; Dorner, A.J. Disease-associated expression profiles in peripheral blood mononuclear cells from patients with advanced renal cell carcinoma. Cancer Res. 2003, 63, 6069–6075. [Google Scholar] [PubMed]
- Showe, M.K.; Vachani, A.; Kossenkov, A.V.; Yousef, M.; Nichols, C.; Nikonova, E.V.; Chang, C.; Kucharczuk, J.; Tran, B.; Wakeam, E. Gene expression profiles in peripheral blood mononuclear cells can distinguish patients with non–small cell lung cancer from patients with nonmalignant lung disease. Cancer Res. 2009, 69, 9202–9210. [Google Scholar] [CrossRef] [PubMed]
- Aarøe, J.; Lindahl, T.; Dumeaux, V.; Sæbø, S.; Tobin, D.; Hagen, N.; Skaane, P.; Lönneborg, A.; Sharma, P.; Børresen-Dale, A.L. Gene expression profiling of peripheral blood cells for early detection of breast cancer. Breast Cancer Res. 2010, 12, R7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baine, M.J.; Chakraborty, S.; Smith, L.M.; Mallya, K.; Sasson, A.R.; Brand, R.E.; Batra, S.K. Transcriptional profiling of peripheral blood mononuclear cells in pancreatic cancer patients identifies novel genes with potential diagnostic utility. PLoS ONE 2011, 6, e17014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olmos, D.; Brewer, D.; Clark, J.; Danila, D.C.; Parker, C.; Attard, G.; Fleisher, M.; Reid, A.H.; Castro, E.; Sandhu, S.K. Prognostic value of blood mrna expression signatures in castration-resistant prostate cancer: A prospective, two-stage study. Lancet Oncol. 2012, 13, 1114–1124. [Google Scholar] [CrossRef]
- Ross, R.W.; Galsky, M.D.; Scher, H.I.; Magidson, J.; Wassmann, K.; Lee, G.S.M.; Katz, L.; Subudhi, S.K.; Anand, A.; Fleisher, M. A whole-blood rna transcript-based prognostic model in men with castration-resistant prostate cancer: A prospective study. Lancet Oncol. 2012, 13, 1105–1113. [Google Scholar] [CrossRef]
- Burczynski, M.E.; Twine, N.C.; Dukart, G.; Marshall, B.; Hidalgo, M.; Stadler, W.M.; Logan, T.; Dutcher, J.; Hudes, G.; Trepicchio, W.L. Transcriptional profiles in peripheral blood mononuclear cells prognostic of clinical outcomes in patients with advanced renal cell carcinoma. Clin. Cancer Res. 2005, 11, 1181–1189. [Google Scholar] [CrossRef]
- Isaksson, H.S.; Sorbe, B.; Nilsson, T.K. Whole genome expression profiling of blood cells in ovarian cancer patients: Prognostic impact of the cyp1b1, mtss1, ncald, and nop14 genes. Oncotarget 2014, 5, 4040–4049. [Google Scholar] [CrossRef] [PubMed]
- Schou, J.; Jensen, B.; Nielsen, D.; Palshof, J.; Høgdall, E.; Yilmaz, M.; Pfeiffer, P.; Johansen, J.; Rossi, S. Prognostic mrna expression signatures in whole blood in patients with metastatic colorectal cancer treated with 3rd line cetuximab and irinotecan. Ann. Oncol. 2016, 27, 574P. [Google Scholar] [CrossRef]
- Dumeaux, V.; Olsen, K.S.; Nuel, G.; Paulssen, R.H.; Børresen-Dale, A.L.; Lund, E. Deciphering normal blood gene expression variation—The nowac postgenome study. PLoS Genet. 2010, 6, e1000873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debey-Pascher, S.; Hofmann, A.; Kreusch, F.; Schuler, G.; Schuler-Thurner, B.; Schultze, J.L.; Staratschek-Jox, A. Rna-stabilized whole blood samples but not peripheral blood mononuclear cells can be stored for prolonged time periods prior to transcriptome analysis. J. Mol. Diagn. 2011, 13, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Kohli, M.; Young, C.Y.; Tindall, D.J.; Nandy, D.; McKenzie, K.M.; Bevan, G.H.; Donkena, K.V. Whole blood defensin mrna expression is a predictive biomarker of docetaxel response in castration-resistant prostate cancer. Onco Targets Ther. 2015, 8, 1915. [Google Scholar] [PubMed]
- Malkov, V.A.; Serikawa, K.A.; Balantac, N.; Watters, J.; Geiss, G.; Mashadi-Hossein, A.; Fare, T. Multiplexed measurements of gene signatures in different analytes using the nanostring ncounter™ assay system. BMC Res. Notes 2009, 2, 80. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Variable | Category | Discovery Set (DS) | Validation Set (VS) |
|---|---|---|---|
| RNA seq | NanoString | ||
| n = 19 | n = 47 | ||
| Age (years) | 71.6 | 66.4 | |
| (Median, Interquartile range (IQR) | (63.4, 79.3) | (62.5, 75.4) | |
| Gender | Male | 6 | 13 |
| Female | 13 | 34 | |
| Histology | Clear cell | 16 | 47 |
| Chromophobe | 2 | ||
| Papillary | 1 | ||
| Fuhrman Grade (G) | G1 | 1 | |
| G2 | 6 | 17 | |
| G3 | 9 | 14 | |
| G4 | 3 | 9 | |
| Unspecified | 1 | 6 | |
| Sarcomatoid differentiation | Present | 0 | 5 |
| Absent | 19 | 42 | |
| Clinical Stage at Initial Diagnosis | I | 3 | 9 |
| II | 4 | 9 | |
| III | 8 | 10 | |
| IV | 3 | 19 | |
| Unspecified | 1 | ||
| MSKCC prognostic score | 1 | 7 | 31 |
| 2 | 8 | 12 | |
| 3 | 4 | 4 | |
| Duration of follow-up among survivors in years (median, range) | 4.28 | 3.6 | |
| (1.34–4.47) | (0.06–4.12) | ||
| Number of deaths | 11 | 25 | |
| Total lines of systemic therapy | 0 (active monitoring) | 3 | 7 |
| 1 | 4 | 14 | |
| 2 | 4 | 5 | |
| 3 | 3 | 4 | |
| 4 | 3 | 1 | |
| 5 or more | 2 | 3 | |
| Unknown | 13 | ||
| Univariate Analysis | Multivariate Analysis | ||||||
|---|---|---|---|---|---|---|---|
| Gene | Cutoff | Hazard Ratio (HR) | 95% CI | p-Value | HR | 95% CI | p-Value |
| AHSP | 977.9 | 0.51 | 0.23–1.13 | 0.091 | |||
| BAG1 | 223.1 | 0.22 | 0.09–0.55 | <0.0001 | 0.14 | 0.04–0.36 | <0.0001 |
| BLVRB | 4577 | 1.97 | 0.67–5.75 | 0.21 | |||
| CA1 | 3438 | 0.63 | 0.28–1.41 | 0.25 | |||
| GMPR | 4093 | 0.58 | 0.26–1.32 | 0.19 | |||
| GPX4 | 1923 | 2.57 | 1.1–6.02 | 0.024 | |||
| GUK1 | 8740 | 1.72 | 0.64–4.64 | 0.27 | |||
| HBM | 8640 | 0.48 | 0.16–1.42 | 0.18 | |||
| HIPK3 | 2652 | 1.95 | 0.8–4.8 | 0.14 | |||
| HLAB | 36350 | 2.32 | 1.05–5.11 | 0.032 | |||
| HLAC | 9550 | 2.88 | 0.86–9.67 | 0.074 | |||
| LDHA | 1626 | 1.76 | 0.8–3.88 | 0.16 | |||
| NOP56 | 545.3 | 0.23 | 0.1–0.53 | <0.0001 | 0.13 | 0.05–0.34 | <0.0001 |
| PCGF5 | 4361 | 0.42 | 0.14–1.24 | 0.11 | |||
| PPDPF | 2303 | 0.51 | 0.17–1.5 | 0.21 | |||
| PRDX5 | 2315 | 3.55 | 1.26–10.0 | 0.012 | |||
| SLC38A5 | 2109 | 0.35 | 0.1–1.16 | 0.073 | |||
| TCEB2 | 132 | 0.38 | 0.11–1.29 | 0.11 | |||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giridhar, K.V.; Sosa, C.P.; Hillman, D.W.; Sanhueza, C.; Dalpiaz, C.L.; Costello, B.A.; Quevedo, F.J.; Pitot, H.C.; Dronca, R.S.; Ertz, D.; et al. Whole Blood mRNA Expression-Based Prognosis of Metastatic Renal Cell Carcinoma. Int. J. Mol. Sci. 2017, 18, 2326. https://doi.org/10.3390/ijms18112326
Giridhar KV, Sosa CP, Hillman DW, Sanhueza C, Dalpiaz CL, Costello BA, Quevedo FJ, Pitot HC, Dronca RS, Ertz D, et al. Whole Blood mRNA Expression-Based Prognosis of Metastatic Renal Cell Carcinoma. International Journal of Molecular Sciences. 2017; 18(11):2326. https://doi.org/10.3390/ijms18112326
Chicago/Turabian StyleGiridhar, Karthik V., Carlos P. Sosa, David W. Hillman, Cristobal Sanhueza, Candace L. Dalpiaz, Brian A. Costello, Fernando J. Quevedo, Henry C. Pitot, Roxana S. Dronca, Donna Ertz, and et al. 2017. "Whole Blood mRNA Expression-Based Prognosis of Metastatic Renal Cell Carcinoma" International Journal of Molecular Sciences 18, no. 11: 2326. https://doi.org/10.3390/ijms18112326