Relax, Cool Down and Scaffold: How to Restore Surface Expression of Folding-Deficient Mutant GPCRs and SLC6 Transporters

 , and
, and 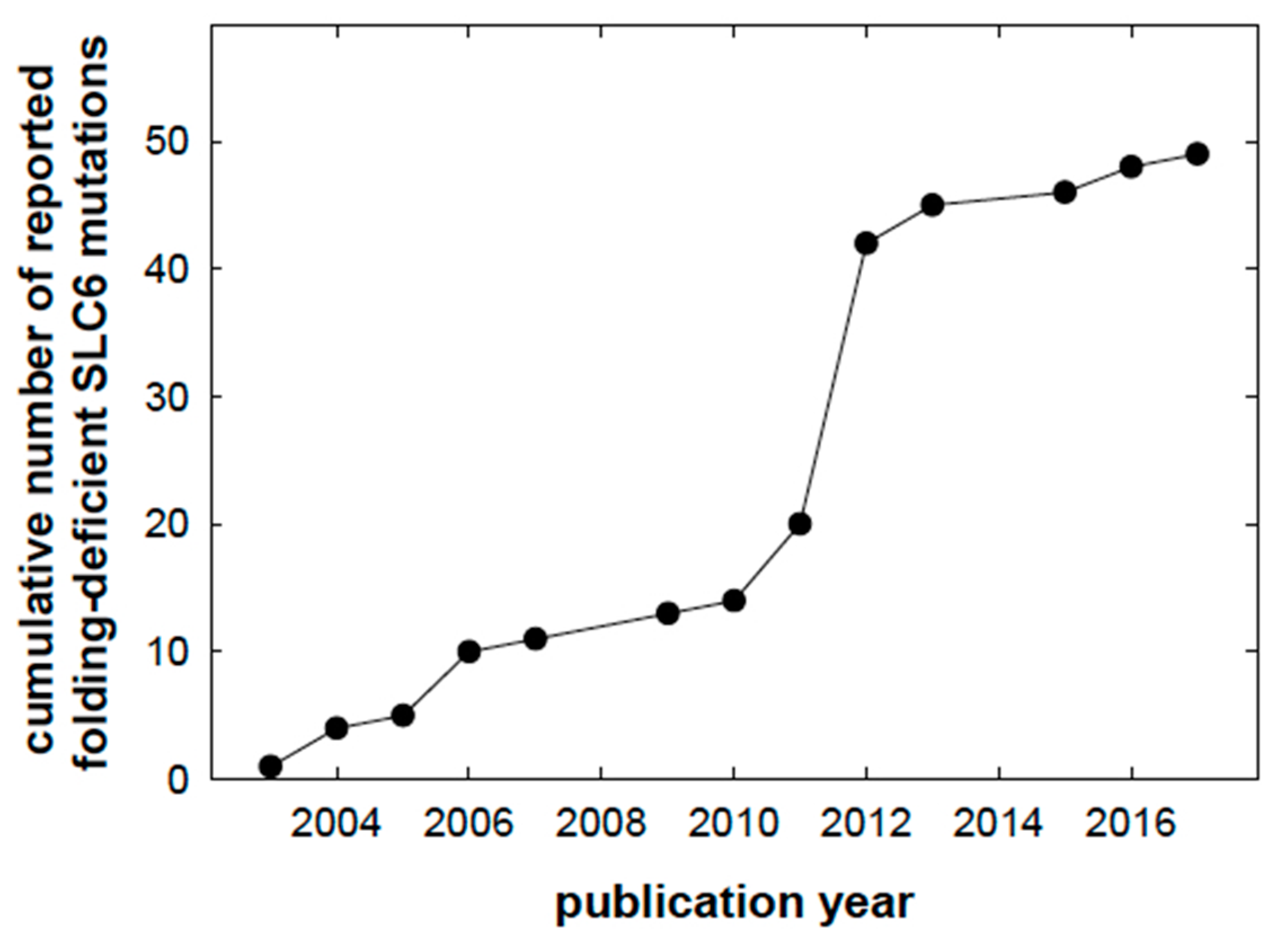{kind=link}
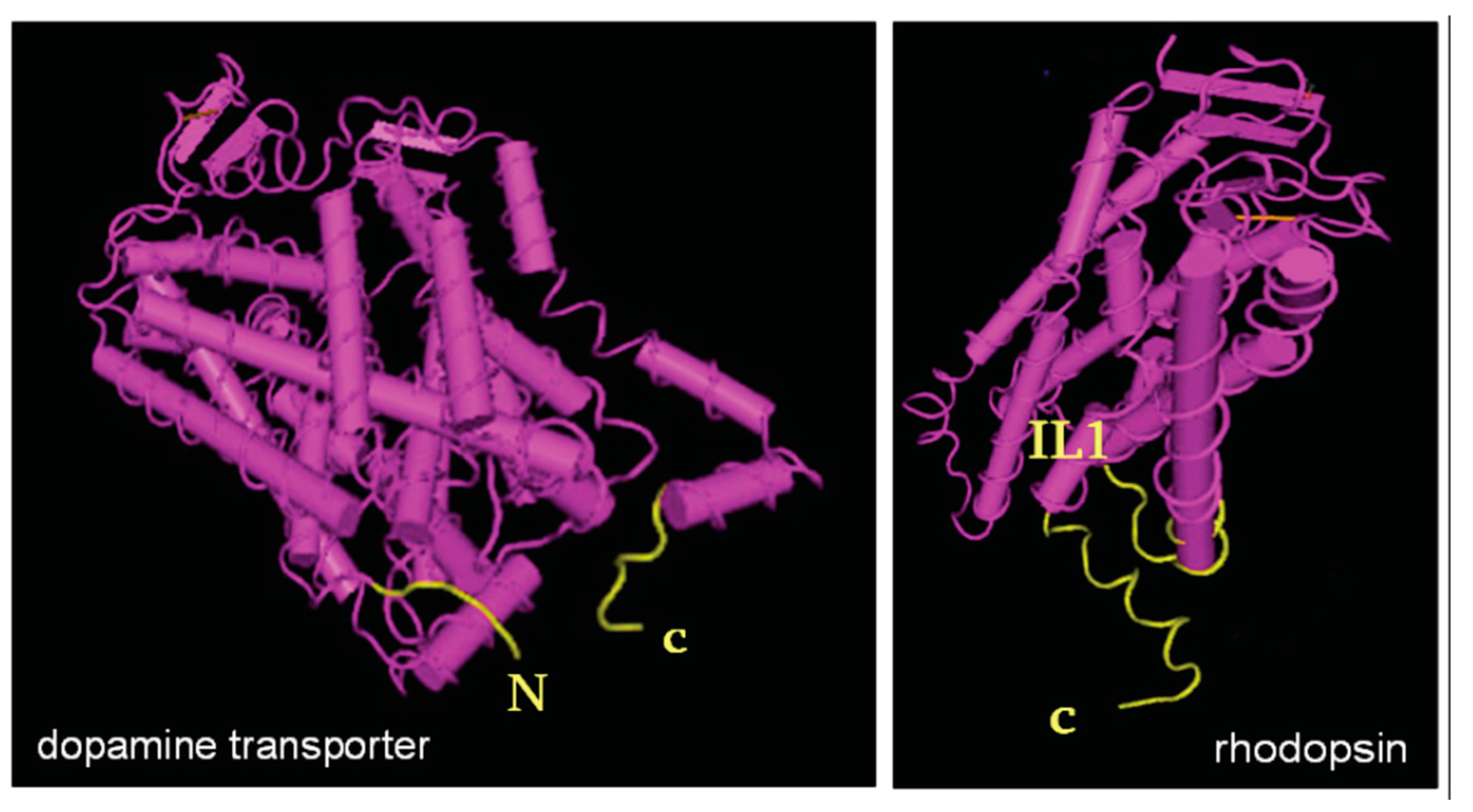{kind=link}
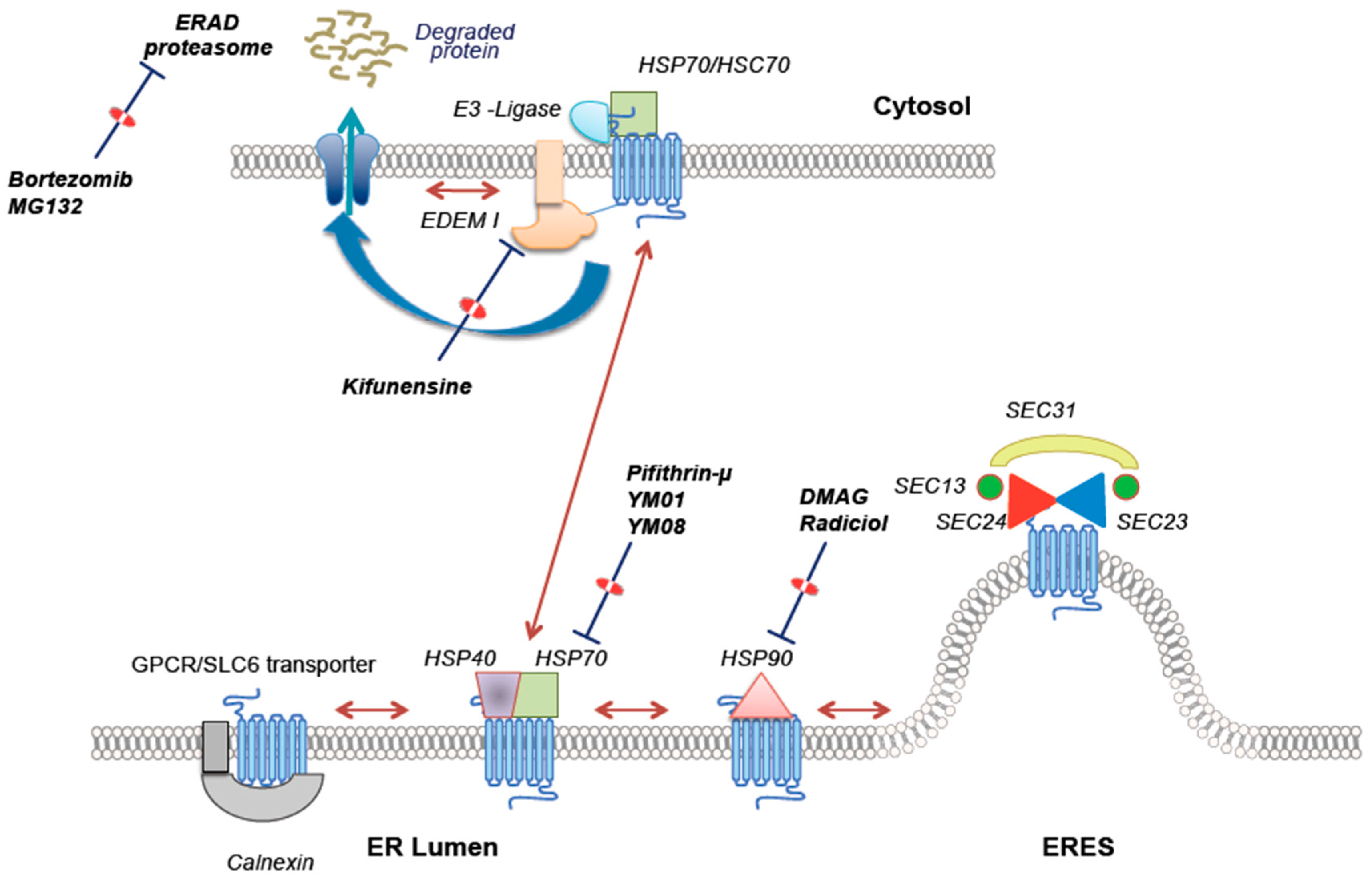{kind=link}
Abstract
:1. Introduction
2. The C-Terminus as a Folding Checkpoint
3. Remedying Folding Deficiency: Scaffolding vs. Relaxing Quality Control
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Morello, J.P.; Petäjä-Repo, U.E.; Bichet, D.G.; Bouvier, M. Pharmacological chaperones: A new twist on receptor folding. Trends Pharmacol. Sci. 2000, 21, 466–469. [Google Scholar] [CrossRef]
- Leidenheimer, N.J.; Ryder, K.G. Pharmacological chaperoning: A primer on mechanism and pharmacology. Pharmacol. Res. 2014, 83, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P.; Hughes, D.A.; Nicholls, K.; Bichet, D.G.; Giugliani, R.; Wilcox, W.R.; Feliciani, C.; Shankar, S.P.; Ezgu, F.; Amartino, H.; et al. Treatment of Fabry’s disease with the pharmacologic chaperone migalastat. N. Engl. J. Med. 2016, 375, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.; Burton, B.; Stack, J.H.; Straley, K.S.; Decker, C.J.; Miller, M.; McCartney, J.; Olson, E.R.; et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. USA 2011, 108, 18843–18848. [Google Scholar] [CrossRef] [PubMed]
- Alderman, E.L.; Coltart, D.J.; Wettach, G.E.; Harrison, D.C. Coronary artery syndromes after sudden propranolol withdrawal. Ann. Intern. Med. 1974, 81, 625–627. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.R.; Olson, H.G.; Amsterdam, E.A.; Mason, D.T. Propranolol-withdrawal rebound phenomenon. Exacerbation of coronary events after abrupt cessation of antianginal therapy. N. Engl. J. Med. 1975, 293, 416–418. [Google Scholar] [CrossRef] [PubMed]
- Aarons, R.D.; Nies, A.S.; Gal, J.; Hegstrand, L.R.; Molinoff, P.B. Elevation of β-adrenergic receptor density in human lymphocytes after propranolol administration. J. Clin. Investig. 1980, 65, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Nanoff, C.; Schütz, W. Characterization of the β-adrenoceptor blocking property of diprafenone in rats: Stereoselective interaction, subtype specificity, and sensitization. J. Cardiovasc. Pharmacol. 1991, 18, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Nasrollahi-Shirazi, S.; Sucic, S.; Yang, Q.; Freissmuth, M.; Nanoff, C. Comparison of the β-adrenergic receptor antagonists landiolol and esmolol: Receptor selectivity, partial agonism, and pharmacochaperoning actions. J. Pharmacol. Exp. Ther. 2016, 359, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Málaga-Diéguez, L.; Yang, Q.; Bauer, J.; Pankevych, H.; Freissmuth, M.; Nanoff, C. Pharmacochaperoning of the A1 adenosine receptor is contingent on the endoplasmic reticulum. Mol. Pharmacol. 2010, l77, 940–952. [Google Scholar] [CrossRef] [PubMed]
- Lester, H.A.; Miwa, J.M.; Srinivasan, R. Psychiatric drugs bind to classical targets within early exocytotic pathways: Therapeutic effects. Biol. Psychiatry 2012, 72, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Pauling, L.; Itano, H.A.; Singer, S.J.; Wells, I.C. Sickle cell anemia a molecular disease. Science 1949, 110, 543–548. [Google Scholar] [CrossRef] [PubMed]
- GBD 2015 Disease and Injury Incidence and Prevalence Collaborators Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1545–1602.
- Morello, J.P.; Salahpour, A.; Laperrière, A.; Bernier, V.; Arthus, M.F.; Lonergan, M.; Petäjä-Repo, U.; Angers, S.; Morin, D.; Bichet, D.G.; et al. Pharmacological chaperones rescue cell-surface expression and function of misfolded V2 vasopressin receptor mutants. J. Clin. Investig. 2000, 105, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.X.; Conn, P.M. Chaperoning G protein-coupled receptors: From cell biology to therapeutics. Endocr. Rev. 2014, 35, 602–647. [Google Scholar] [CrossRef] [PubMed]
- Sahni, N.; Yi, S.; Taipale, M.; Bass, J.I.; Coulombe-Huntington, J.; Yang, F.; Peng, J.; Weile, J.; Karras, G.I.; Wang, Y.; et al. Widespread macromolecular interaction perturbations in human genetic disorders. Cell 2015, 161, 647–660. [Google Scholar] [CrossRef] [PubMed]
- Hahn, M.K.; Robertson, D.; Blakely, R.D. A mutation in the human norepinephrine transporter gene (SLC6A2) associated with orthostatic intolerance disrupts surface expression of mutant and wild-type transporters. J. Neurosci. 2003, 23, 4470–4478. [Google Scholar] [PubMed]
- Rosenberg, E.H.; Almeida, L.S.; Kleefstra, T.; de Grauw, R.S.; Yntema, H.G.; Bahi, N.; Moraine, C.; Ropers, H.H.; Fryns, J.P.; de Grauw, T.J.; et al. High prevalence of SLC6A8 deficiency in X-linked mental retardation. Am. J. Hum. Genet. 2004, 75, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Mancini, G.M.; Catsman-Berrevoets, C.E.; de Coo, I.F.; Aarsen, F.K.; Kamphoven, J.H.; Huijmans, J.G.; Duran, M.; van der Knaap, M.S.; Jakobs, C.; Salomons, G.S. Two novel mutations in SLC6A8 cause creatine transporter defect and distinctive X-linked mental retardation in two unrelated Dutch families. Am. J. Med. Genet. A 2005, 132, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.J.; Rosenberg, E.H.; Almeida, L.S.; Wood, T.C.; Jakobs, C.; Stevenson, R.E.; Schwartz, C.E.; Salomons, G.S. X-linked creatine transporter (SLC6A8) mutations in about 1% of males with mental retardation of unknown etiology. Hum. Genet. 2006, 119, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Lion-François, L.; Cheillan, D.; Pitelet, G.; Acquaviva-Bourdain, C.; Bussy, G.; Cotton, F.; Guibaud, L.; Gérard, D.; Rivier, C.; Vianey-Saban, C.; et al. High frequency of creatine deficiency syndromes in patients with unexplained mental retardation. Neurology 2006, 67, 1713–1714. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, E.H.; Martínez Muñoz, C.; Betsalel, O.T.; van Dooren, S.J.; Fernandez, M.; Jakobs, C.; de Grauw, T.J.; Kleefstra, T.; Schwartz, C.E.; Salomons, G.S. Functional characterization of missense variants in the creatine transporter gene (SLC6A8): Improved diagnostic application. Hum. Mutat. 2007, 28, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Alcaide, P.; Rodriguez-Pombo, P.; Ruiz-Sala, P.; Ferrer, I.; Castro, P.; Ruiz Martin, Y.; Merinero, B.; Ugarte, M. A new case of creatine transporter deficiency associated with mild clinical phenotype and a novel mutation in the SLC6A8 gene. Dev. Med. Child Neurol. 2010, 52, 215–217. [Google Scholar] [CrossRef] [PubMed]
- DesRoches, C.L.; Patel, J.; Wang, P.; Minassian, B.; Salomons, G.S.; Marshall, C.R.; Mercimek-Mahmutoglu, S. Estimated carrier frequency of creatine transporter deficiency in females in the general population using functional characterization of novel missense variants in the SLC6A8 gene. Gene 2015, 565, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Comeaux, M.S.; Wang, J.; Wang, G.; Kleppe, S.; Zhang, V.W.; Schmitt, E.S.; Craigen, W.J.; Renaud, D.; Sun, Q.; Wong, L.J. Biochemical, molecular, and clinical diagnoses of patients with cerebral creatine deficiency syndromes. Mol. Genet. Metab. 2013, 109, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Ardon, O.; Procter, M.; Mao, R.; Longo, N.; Landau, Y.E.; Shilon-Hadass, A.; Gabis, L.V.; Hoffmann, C.; Tzadok, M.; Heimer, G.; et al. Creatine transporter deficiency: Novel mutations and functional studies. Mol. Genet. Metab. Rep. 2016, 8, 20–23. [Google Scholar] [CrossRef] [PubMed]
- Betsalel, O.T.; Pop, A.; Rosenberg, E.H.; Fernandez-Ojeda, M.; Creatine Transporter Research Group; Jakobs, C.; Salomons, G.S. Detection of variants in SLC6A8 and functional analysis of unclassified missense variants. Mol. Genet. Metab. 2012, 105, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Uemura, T.; Ito, S.; Ohta, Y.; Tachikawa, M.; Wada, T.; Terasaki, T.; Ohtsuki, S. Abnormal N-glycosylation of a novel missense creatine transporter mutant, G561R, associated with cerebral creatine deficiency syndromes alters transporter activity and localization. Biol. Pharm. Bull. 2017, 40, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Rees, M.I.; Harvey, K.; Pearce, B.R.; Chung, S.K.; Duguid, I.C.; Thomas, P.; Beatty, S.; Graham, G.E.; Armstrong, L.; Shiang, R.; et al. Mutations in the gene encoding GlyT2 (SLC6A5) define a presynaptic componentof human startle disease. Nat. Genet. 2006, 38, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Giménez, C.; Pérez-Siles, G.; Martínez-Villarreal, J.; Arribas-González, E.; Jiménez, E.; Núñez, E.; de Juan-Sanz, J.; Fernández-Sánchez, E.; García-Tardón, N.; Ibáñez, I.; et al. A novel dominant hyperekplexia mutation Y705C alters trafficking and biochemical properties of the presynaptic glycine transporterGlyT2. J. Biol. Chem. 2012, 287, 28986–29002. [Google Scholar] [CrossRef] [PubMed]
- Kurian, M.A.; Zhen, J.; Cheng, S.Y.; Li, Y.; Mordekar, S.R.; Jardine, P.; Morgan, N.V.; Meyer, E.; Tee, L.; Pasha, S.; et al. Homozygous loss-of-function mutations in the gene encoding the dopamine transporter are associated with infantile parkinsonism-dystonia. J. Clin. Investig. 2009, 11, 1595–1603. [Google Scholar] [CrossRef] [PubMed]
- Kurian, M.A.; Li, Y.; Zhen, J.; Meyer, E.; Hai, N.; Christen, H.J.; Hoffmann, G.F.; Jardine, P.; von Moers, A.; Mordekar, S.R.; et al. Clinical and molecular characterisation of hereditary dopamine transporter deficiency syndrome: An observational cohort and experimental study. Lancet Neurol. 2011, 10, 54–62. [Google Scholar] [CrossRef]
- Ng, J.; Zhen, J.; Meyer, E.; Erreger, K.; Li, Y.; Kakar, N.; Ahmad, J.; Thiele, H.; Kubisch, C.; Rider, N.L.; et al. Dopamine transporter deficiency syndrome: Phenotypic spectrum from infancy to adulthood. Brain 2014, 137, 1107–1119. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.; Towne, M.C.; Pearl, P.L.; Pelletier, R.C.; Genetti, C.A.; Shi, J.; Beggs, A.H.; Agrawal, P.B.; Brownstein, C.A. SLC6A1 mutation and ketogenic diet in epilepsy with myoclonic-atonic seizures. Pediatr. Neurol. 2016, 64, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Cymer, F.; von Heijne, G.; White, S.H. Mechanisms of integral membrane protein insertion and folding. J. Mol. Biol. 2015, 427, 999–1022. [Google Scholar] [CrossRef] [PubMed]
- De Marothy, M.T.; Elofsson, A. Marginally hydrophobic transmembrane α-helices shaping membrane protein folding. Protein Sci. 2015, 24, 1057–1074. [Google Scholar] [CrossRef] [PubMed]
- Chiba, P.; Freissmuth, M.; Stockner, T. Defining the blanks—Pharmacochaperoning of SLC6 transporters and ABC transporters. Pharmacol. Res. 2014, 83, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Kasture, A.; Stockner, T.; Freissmuth, M.; Sucic, S. An unfolding story: Small molecules remedy misfolded monoamine transporters. Int. J. Biochem. Cell Biol. 2017, 92, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Freissmuth, M.; Stockner, T.; Sucic, S. SLC6 Transporter Folding Diseases and Pharmacochaperoning. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2018; in press (Epub 31 October 2017). [Google Scholar] [CrossRef]
- Pankevych, H.; Korkhov, V.; Freissmuth, M.; Nanoff, C. Truncation of the A1 adenosine receptor reveals distinct roles of the membrane-proximal carboxyl terminus in receptor folding and G protein coupling. J. Biol. Chem. 2003, 278, 30283–32093. [Google Scholar] [CrossRef] [PubMed]
- Farhan, H.; Korkhov, V.M.; Paulitschke, V.; Dorostkar, M.M.; Scholze, P.; Kudlacek, O.; Freissmuth, M.; Sitte, H.H. Two discontinuous segments in the carboxyl terminus are required for membrane targeting of the rat gamma-aminobutyric acid transporter-1 (GAT1). J. Biol. Chem. 2004, 279, 28553–28563. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.B.; Fjorback, A.W.; Wiborg, O. The C-terminus is critical for the functional expression of the human serotonin transporter. Biochemistry 2006, 45, 1331–1337. [Google Scholar] [CrossRef] [PubMed]
- El-Kasaby, A.; Just, H.; Malle, E.; Stolt-Bergner, P.C.; Sitte, H.H.; Freissmuth, M.; Kudlacek, O. Mutations in the carboxyl-terminal SEC24 binding motif of the serotonin transporter impair folding of the transporter. J. Biol. Chem. 2010, 285, 39201–39210. [Google Scholar] [CrossRef] [PubMed]
- Koban, F.; El-Kasaby, A.; Häusler, C.; Stockner, T.; Simbrunner, B.M.; Sitte, H.H.; Freissmuth, M.; Sucic, S. A salt bridge linking the first intracellular loop with the C terminus facilitates the folding of the serotonin transporter. J. Biol. Chem. 2015, 290, 13263–13278. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.Y. Transmembrane helical domain of the cannabinoid CB1 receptor. Biophys. J. 2009, 96, 3251–3262. [Google Scholar] [CrossRef] [PubMed]
- Nanoff, C.; Freissmuth, M. ER-bound steps in the biosynthesis of G protein-coupled receptors. Subcell. Biochem. 2012, 63, 1–21. [Google Scholar] [PubMed]
- Saito, H.; Kubota, M.; Roberts, R.W.; Chi, Q.; Matsunami, H. RTP family members induce functional expression of mammalian odorant receptors. Cell 2004, 119, 679–691. [Google Scholar] [CrossRef] [PubMed]
- Singer, D.; Camargo, S.M.; Huggel, K.; Romeo, E.; Danilczyk, U.; Kuba, K.; Chesnov, S.; Caron, M.G.; Penninger, J.M.; Verrey, F. Orphan transporter SLC6A18 is renal neutral amino acid transporter B0AT3. J. Biol. Chem. 2009, 284, 19953–19960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camargo, S.M.; Singer, D.; Makrides, V.; Huggel, K.; Pos, K.M.; Wagner, C.A.; Kuba, K.; Danilczyk, U.; Skovby, F.; Kleta, R.; et al. Tissue-specific amino acid transporter partners ACE2 and collectrin differentially interact with hartnup mutations. Gastroenterology 2009, 136, 872–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalczuk, S.; Bröer, A.; Tietze, N.; Vanslambrouck, J.M.; Rasko, J.E.; Bröer, S. A protein complex in the brush-border membrane explains a Hartnup disorder allele. FASEB J. 2008, 22, 2880–2887. [Google Scholar] [CrossRef] [PubMed]
- Milojevic, T.; Reiterer, V.; Stefan, E.; Korkhov, V.M.; Dorostkar, M.M.; Ducza, E.; Ogris, E.; Boehm, S.; Freissmuth, M.; Nanoff, C. The ubiquitin-specific protease Usp4 regulates the cell surface level of the A2A receptor. Mol. Pharmacol. 2006, 69, 1083–1094. [Google Scholar]
- Bergmayr, C.; Thurner, P.; Keuerleber, S.; Kudlacek, O.; Nanoff, C.; Freissmuth, M.; Gruber, C.W. Recruitment of a cytoplasmic chaperone relay by the A2A adenosine receptor. J. Biol. Chem. 2013, 288, 28831–28844. [Google Scholar] [CrossRef] [PubMed]
- Arslan, G.; Kull, B.; Fredholm, B.B. Anoxia redistributes adenosine A2A receptors in PC12 cells and increases receptor-mediated formation of cAMP. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2002, 365, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Korkhov, V.M.; Milan-Lobo, L.; Zuber, B.; Farhan, H.; Schmid, J.A.; Freissmuth, M.; Sitte, H.H. Peptide-based interactions with calnexin target misassembled membrane proteins into endoplasmic reticulum-derived multilamellar bodies. J. Mol. Biol. 2008, 378, 337–352. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Kusek, J.; Yang, Q.; Witek, M.; Gruber, C.W.; Nanoff, C.; Freissmuth, M. Chaperoning of the A1-adenosine receptor by endogenous adenosine—An extension of the retaliatory metabolite concept. Mol. Pharmacol. 2015, 87, 39–51. [Google Scholar] [CrossRef] [PubMed]
- El-Kasaby, A.; Koban, F.; Sitte, H.H.; Freissmuth, M.; Sucic, S. A cytosolic relay of heat shock proteins HSP70–1A and HSP90β monitors the folding trajectory of the serotonin transporter. J. Biol. Chem. 2014, 289, 28987–29000. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, H.J.; Stevens, T.J.; Munro, S. A comprehensive comparison of transmembrane domains reveals organelle-specific properties. Cell 2010, 142, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Holthuis, J.C.; Menon, A.K. Lipid landscapes and pipelines in membrane homeostasis. Nature 2014, 510, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Kasture, A.; El-Kasaby, A.; Szöllösi, D.; Asjad, H.M.M.; Grimm, A.; Stockner, T.; Hummel, T.; Freissmuth, M.; Sucic, S. Functional rescue of a misfolded Drosophila melanogaster dopamine transporter mutant associated with a sleepless phenotype by pharmacological chaperones. J. Biol. Chem. 2016, 291, 20876–20890. [Google Scholar] [CrossRef] [PubMed]
- Asjad, H.M.M.; Kasture, A.; El-Kasaby, A.; Sackel, M.; Hummel, T.; Freissmuth, M.; Sucic, S. Pharmacochaperoning in a Drosophila model system rescues human dopamine transporter variants associated with infantile/juvenile parkinsonism. J. Biol. Chem. 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Filipeanu, C.M.; de Vries, R.; Danser, A.H.J.; Kapusta, D.R. Modulation of α2C adrenergic receptor temperature-sensitive trafficking by HSP90. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Meimaridou, E.; Gooljar, S.B.; Ramnarace, N.; Anthonypillai, L.; Clark, A.J.L.; Chapple, J.P. The cytosolic chaperone Hsc70 promotes traffic to the cell surface of intracellular retained melanocortin-4 receptor mutants. Mol. Endocrinol. 2011, 25, 1650–1660. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Génier, S.; Cartier, A.; Larrivée, J.F.; Stankova, J.; Young, J.C.; Parent, J.L. AG protein-coupled receptor and the intracellular synthase of its agonist functionally cooperate. J. Cell Biol. 2014, 204, 377–393. [Google Scholar] [CrossRef] [PubMed]
- Génier, S.; Degrandmaison, J.; Moreau, P.; Labrecque, P.; Hébert, T.E.; Parent, J.L. Regulation of GPCR expression through an interaction with CCT7, a subunit of the CCT/TRiC complex. Mol. Biol. Cell 2016, 27, 3800–3812. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wei, J.; Bowser, R.K.; Dong, S.; Xiao, S.; Zhao, Y. Molecular regulation of lysophosphatidic acid receptor 1 trafficking to the cell surface. Cell. Signal. 2014, 26, 2406–2411. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, B.F.; Needham, P.G.; Snyder, A.C.; Roy, A.; Khadem, S.; Brodsky, J.L.; Subramanya, A.R. Hsp70 and Hsp90 multichaperone complexes sequentially regulate thiazide-sensitive cotransporter endoplasmic reticulum-associated degradation and biogenesis. J. Biol. Chem. 2013, 288, 13124–13135. [Google Scholar] [CrossRef] [PubMed]
- Kampinga, H.H.; Craig, E.A. The HSP70 chaperone machinery. J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 2010, 11, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Chapple, J.P.; Cheetham, M.E. The chaperone environment at the cytoplasmic face of the endoplasmic reticulum can modulate rhodopsin processing and inclusion formation. J. Biol. Chem. 2003, 278, 19087–19094. [Google Scholar] [CrossRef] [PubMed]
- Bermak, J.C.; Li, M.; Bullock, C.; Zhou, Q.Y. Regulation of transport of the dopamine D1 receptorby a new membrane-associated ER protein. Nat. Cell Biol. 2001, 3, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Seyer, P.; Vandermoere, F.; Cassier, E.; Bockaert, J.; Marin, P. Physical and functional interactions between the serotonin transporter and the neutral amino acid transporter ASCT2. Biochem. J. 2016, 473, 1953–1965. [Google Scholar] [CrossRef] [PubMed]
- Keuerleber, S.; Gsandtner, I.; Freissmuth, M. From cradle to twilight: The carboxyl terminus directs the fate of the A2A-adenosine receptor. Biochim. Biophys. Acta 2011, 1808, 1350–1357. [Google Scholar] [CrossRef] [PubMed]
- Farhan, H.; Reiterer, V.; Korkhov, V.M.; Schmid, J.A.; Freissmuth, M.; Sitte, H.H. Concentrative export from the endoplasmic reticulum of the γ-aminobutyric acid transporter 1 requires binding to SEC24D. J. Biol. Chem. 2007, 282, 7679–7689. [Google Scholar] [CrossRef] [PubMed]
- Sucic, S.; El-Kasaby, A.; Kudlacek, O.; Sarker, S.; Sitte, H.H.; Marin, P.; Freissmuth, M. The serotonin transporter is an exclusive client of the coat protein complex II (COPII) component SEC24C. J. Biol. Chem. 2011, 286, 16482–16490. [Google Scholar] [CrossRef] [PubMed]
- Sucic, S.; Koban, F.; El-Kasaby, A.; Kudlacek, O.; Stockner, T.; Sitte, H.H.; Freissmuth, M. Switching the clientele: A lysine residing in the C terminus of the serotonin transporter specifies its preference for the coat protein complex II component SEC24C. J. Biol. Chem. 2013, 288, 5330–5341. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Nichols, C.D.; Guo, J.; Huang, W.; Lambert, N.A.; Wu, G. A triple arg motif mediates α2B-adrenergic receptor interaction with Sec24C/D and export. Traffic 2012, 13, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Otsu, W.; Kurooka, T.; Otsuka, Y.; Sato, K.; Inaba, M. A new class of endoplasmic reticulum export signal ΦXΦXΦ for transmembrane proteins and its selective interaction with Sec24C. J. Biol. Chem. 2013, 288, 18521–18532. [Google Scholar] [CrossRef] [PubMed]
- Doly, S.; Marullo, S. Gatekeepers controlling GPCR export and function. Trends Pharmacol. Sci. 2015, 36, 636–644. [Google Scholar] [CrossRef] [PubMed]
- Doly, S.; Shirvani, H.; Gäta, G.; Meye, F.J.; Emerit, M.B.; Enslen, H.; Achour, L.; Pardo-Lopez, L.; Yang, S.K.; Armand, V.; et al. GABAB receptor cell-surface export is controlled by an endoplasmic reticulum gatekeeper. Mol. Psychiatry 2016, 21, 480–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauvageau, E.; Rochdi, M.D.; Oueslati, M.; Hamdan, F.F.; Percherancier, Y.; Simpson, J.C.; Pepperkok, R.; Bouvier, M. CNIH4 interacts with newly synthesized GPCR and controls their export from the endoplasmic reticulum. Traffic 2014, 15, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Jaquette, J.; Segaloff, D.L. Temperature sensitivity of some mutants of the lutropin/choriogonadotropin receptor. Endocrinology 1997, 138, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Jeyaraj, S.C.; Chotani, M.A.; Mitra, S.; Gregg, H.E.; Flavahan, N.A.; Morrison, K.J. Cooling evokes redistribution of α2C-adrenoceptors from Golgi to plasma membrane in transfected human embryonic kidney 293 cells. Mol. Pharmacol. 2001, 60, 1195–1200. [Google Scholar] [PubMed]
- Iiri, T.; Herzmark, P.; Nakamoto, J.M.; van Dop, C.; Bourne, H.R. Rapid GDP release from Gsα in patients with gain and loss of endocrine function. Nature 1994, 371, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Gentry, P.R.; Sexton, P.M.; Christopoulos, A. Novel Allosteric Modulators of G Protein-coupled Receptors. J. Biol. Chem. 2015, 290, 19478–19488. [Google Scholar] [CrossRef] [PubMed]
- White, E.; McKenna, J.; Cavanaugh, A.; Breitwieser, G.E. Pharmacochaperone-mediated rescue of calcium-sensing receptor loss-of-function mutants. Mol. Endocrinol. 2009, 23, 1115–1123. [Google Scholar] [CrossRef] [PubMed]
- Janovick, J.A.; Maya-Núñez, G.; Ulloa-Aguirre, A.; Huhtaniemi, I.T.; Dias, J.A.; Verbost, P.; Conn, P.M. Increased plasma membrane expression of human follicle-stimulating hormone receptor by a small molecule thienopyr(im)idine. Mol. Cell. Endocrinol. 2009, 298, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Newton, C.L.; Whay, A.M.; McArdle, C.A.; Zhang, M.; van Koppen, C.J.; van de Lagemaat, R.; Segaloff, D.L.; Millar, R.P. Rescue of expression and signaling of human luteinizing hormone G protein-coupled receptor mutants with an allosterically binding small-molecule agonist. Proc. Natl. Acad. Sci. USA 2011, 108, 7172–7176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Generoso, S.F.; Giustiniano, M.; La Regina, G.; Bottone, S.; Passacantilli, S.; Di Maro, S.; Cassese, H.; Bruno, A.; Mallardo, M.; Dentice, M.; et al. Pharmacological folding chaperones act as allosteric ligands of Frizzled4. Nat. Chem. Biol. 2015, 11, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Freissmuth, M.; Selzer, E.; Marullo, S.; Schütz, W.; Strosberg, A.D. Expression of two human β-adrenergic receptors in Escherichia coli: Functional interaction with two forms of the stimulatory G protein. Proc. Natl. Acad. Sci. USA 1991, 88, 8548–8552. [Google Scholar] [CrossRef] [PubMed]
- Bertin, B.; Freissmuth, M.; Breyer, R.M.; Schütz, W.; Strosberg, A.D.; Marullo, S. Functional expression of the human serotonin 5-HT1A receptor in Escherichia coli. Ligand bindingproperties and interaction with recombinant G protein α-subunits. J. Biol. Chem. 1992, 267, 8200–8282. [Google Scholar] [PubMed]
- Jockers, R.; Linder, M.E.; Hohenegger, M.; Nanoff, C.; Bertin, B.; Strosberg, A.D.; Marullo, S.; Freissmuth, M. Species difference in the G protein selectivity of the human and bovine A1-adenosinereceptor. J. Biol. Chem. 1994, 269, 32077–32084. [Google Scholar] [PubMed]
- Beerepoot, P.; Lam, V.M.; Salahpour, A. Pharmacological chaperones of the dopamine trans-porter rescue dopamine transporter deficiency syndrome mutations in heterologous Cells. J. Biol. Chem. 2016, 291, 22053–22062. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, M.T.; Zhang, Y.W.; Campbell, S.D.; Rudnick, G. Ibogaine, a noncompetitive inhibitor of serotonin transport, acts by stabilizing the cytoplasm-facing state of the transporter. J. Biol. Chem. 2007, 282, 29441–29447. [Google Scholar] [CrossRef] [PubMed]
- Bulling, S.; Schicker, K.; Zhang, Y.W.; Steinkellner, T.; Stockner, T.; Gruber, C.W.; Boehm, S.; Freissmuth, M.; Rudnick, G.; Sitte, H.H.; et al. The mechanistic basis for noncompetitive ibogaine inhibition of serotonin and dopamine transporters. J. Biol. Chem. 2012, 287, 18524–18534. [Google Scholar] [CrossRef] [PubMed]
- Korkhov, V.M.; Holy, M.; Freissmuth, M.; Sitte, H.H. The conserved glutamate (Glu136) in transmembrane domain 2 of the serotonin transporter is required for the conformational switch in the transport cycle. J. Biol. Chem. 2006, 281, 13439–13448. [Google Scholar] [CrossRef] [PubMed]
- Bhat, S.; Hasenhuetl, P.S.; Kasture, A.; El-Kasaby, A.; Baumann, M.H.; Blough, B.E.; Sucic, S.; Sandtner, W.; Freissmuth, M. Conformational state interactions provide clues to the pharmacochaperone potential of serotonin transporter partial sub-strates. J. Biol. Chem. 2017, 292, 16773–16786. [Google Scholar] [CrossRef] [PubMed]
- Goloudina, A.R.; Demidov, O.N.; Garrido, C. Inhibition of HSP70: A challenging anti-cancer strategy. Cancer Lett. 2012, 325, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Neckers, L.; Workman, P. Hsp90 molecular chaperone inhibitors: Are we there yet? Clin. Cancer Res. 2012, 18, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Egan, M.E.; Glöckner-Pagel, J.; Ambrose, C.; Cahill, P.A.; Pappoe, L.; Balamuth, N.; Cho, E.; Canny, S.; Wagner, C.A.; Geibel, J.; et al. Calcium-pump inhibitors induce functional surface expression of ΔF508-CFTR protein in cystic fibrosis epithelial cells. Nat. Med. 2002, 8, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Grubb, B.R.; Gabriel, S.E.; Mengos, A.; Gentzsch, M.; Randell, S.H.; Van Heeckeren, A.M.; Knowles, M.R.; Drumm, M.L.; Riordan, J.R.; Boucher, R.C. SERCA pump inhibitors do not correct biosynthetic arrest of ΔF508 CFTR in cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 2006, 134, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Vij, N.; Fang, S.; Zeitlin, P.L. Selective inhibition of endoplasmic reticulum-associated degradation rescues ΔF508-cystic fibrosis transmembrane regulator and suppresses interleukin-8 levels: Therapeutic implications. J. Biol. Chem. 2006, 281, 17369–17378. [Google Scholar] [CrossRef] [PubMed]
- Petäjä-Repo, U.E.; Hogue, M.; Laperriere, A.; Bhalla, S.; Walker, P.; Bouvier, M. Newly synthesized human δ-opioid receptors retained in the endoplasmic reticulum are retrotranslocated to the cytosol, deglycosylated, ubiquitinated, and degraded by the proteasome. J. Biol. Chem. 2001, 276, 4416–4423. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Puhm, F.; Freissmuth, M.; Nanoff, C. Hyponatremia and V2 vasopressin receptor upregulation: A result of HSP90 inhibition. Cancer Chemother. Pharmacol. 2017, 80, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Kume, K.; Kume, S.; Park, S.K.; Hirsh, J.; Jackson, F.R. Dopamine is a regulator of arousal in the fruit fly. J. Neurosci. 2005, 25, 7377–7384. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.N.; Koh, K.; Yue, Z.; Joiner, W.J.; Sehgal, A. A genetic screen for sleep and circadian mutants reveals mechanisms underlying regulation of sleep in Drosophila. Sleep 2008, 31, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Assimon, V.A.; Gillies, A.T.; Rauch, J.N.; Gestwicki, J.E. Hsp70 protein complexes as drug targets. Curr. Pharm. Des. 2013, 19, 404–417. [Google Scholar] [CrossRef] [PubMed]
- Leu, J.I.; Pimkina, J.; Frank, A.; Murphy, M.E.; George, D.L. A small molecule inhibitor of inducible heat shock protein 70. Mol. Cell 2009, 36, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Schlecht, R.; Scholz, S.R.; Dahmen, H.; Wegener, A.; Sirrenberg, C.; Musil, D.; Bomke, J.; Eggenweiler, H.M.; Mayer, M.P.; Bukau, B. Functional analysis of Hsp70 inhibitors. PLoS ONE 2013, 8, e78443. [Google Scholar] [CrossRef]
- Janovick, J.A.; Stewart, M.D.; Jacob, D.; Martin, L.D.; Deng, J.M.; Stewart, C.A.; Wang, Y.; Cornea, A.; Chavali, L.; Lopez, S.; et al. Restoration of testis function in hypogonadotropic hypogonadal mice harboring a misfolded GnRHR mutant by pharmacoperone drug therapy. Proc. Natl. Acad. Sci. USA 2013, 110, 21030–21035. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asjad, H.M.M.; Nasrollahi-Shirazi, S.; Sucic, S.; Freissmuth, M.; Nanoff, C. Relax, Cool Down and Scaffold: How to Restore Surface Expression of Folding-Deficient Mutant GPCRs and SLC6 Transporters. Int. J. Mol. Sci. 2017, 18, 2416. https://doi.org/10.3390/ijms18112416
Asjad HMM, Nasrollahi-Shirazi S, Sucic S, Freissmuth M, Nanoff C. Relax, Cool Down and Scaffold: How to Restore Surface Expression of Folding-Deficient Mutant GPCRs and SLC6 Transporters. International Journal of Molecular Sciences. 2017; 18(11):2416. https://doi.org/10.3390/ijms18112416
Chicago/Turabian StyleAsjad, H.M. Mazhar, Shahrooz Nasrollahi-Shirazi, Sonja Sucic, Michael Freissmuth, and Christian Nanoff. 2017. "Relax, Cool Down and Scaffold: How to Restore Surface Expression of Folding-Deficient Mutant GPCRs and SLC6 Transporters" International Journal of Molecular Sciences 18, no. 11: 2416. https://doi.org/10.3390/ijms18112416