Controlling the Molecular Weight of Lignosulfonates by an Alkaline Oxidative Treatment at Moderate Temperatures and Atmospheric Pressure: A Size-Exclusion and Reverse-Phase Chromatography Study

Abstract
:1. Introduction
2. Results and Discussion
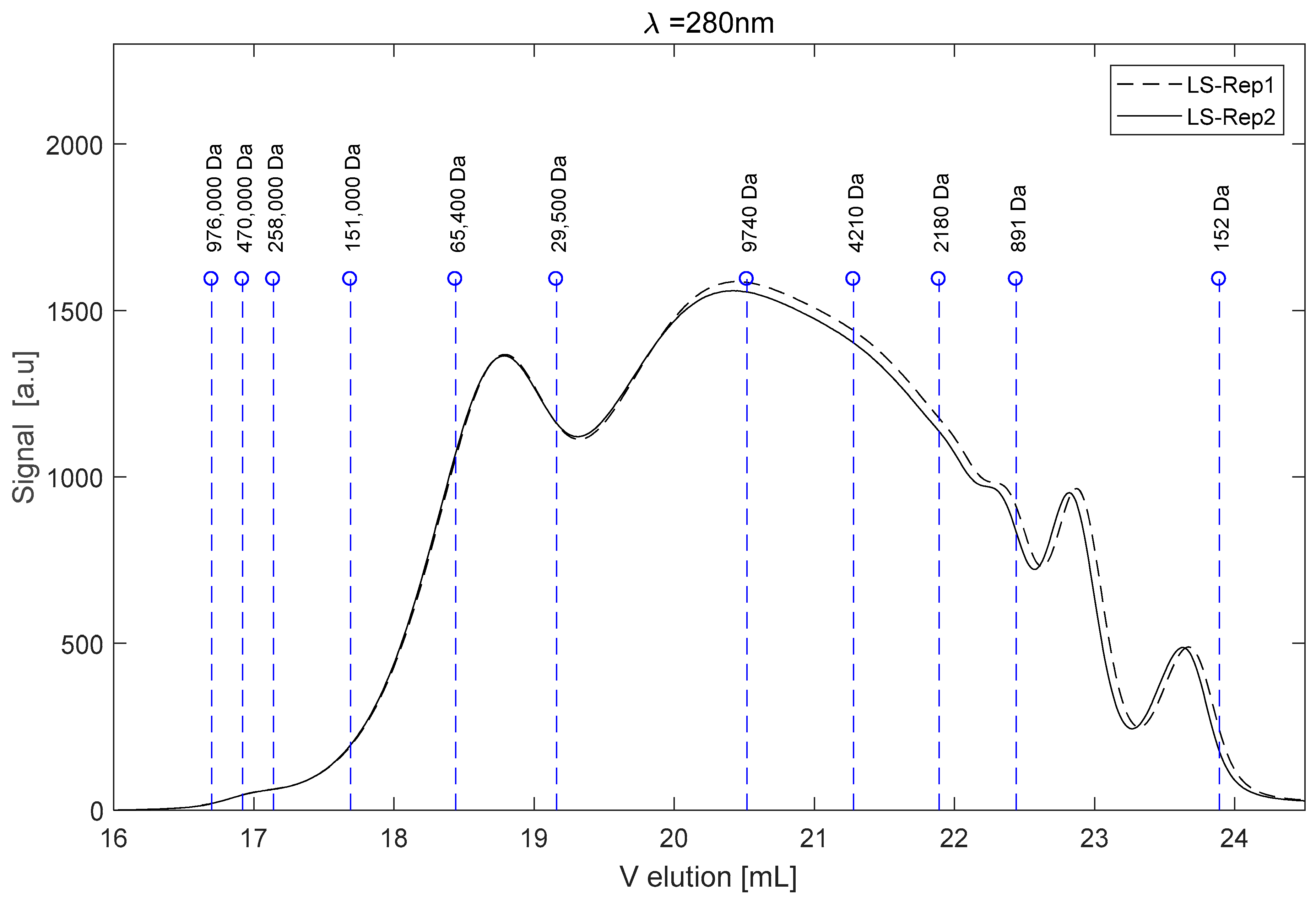
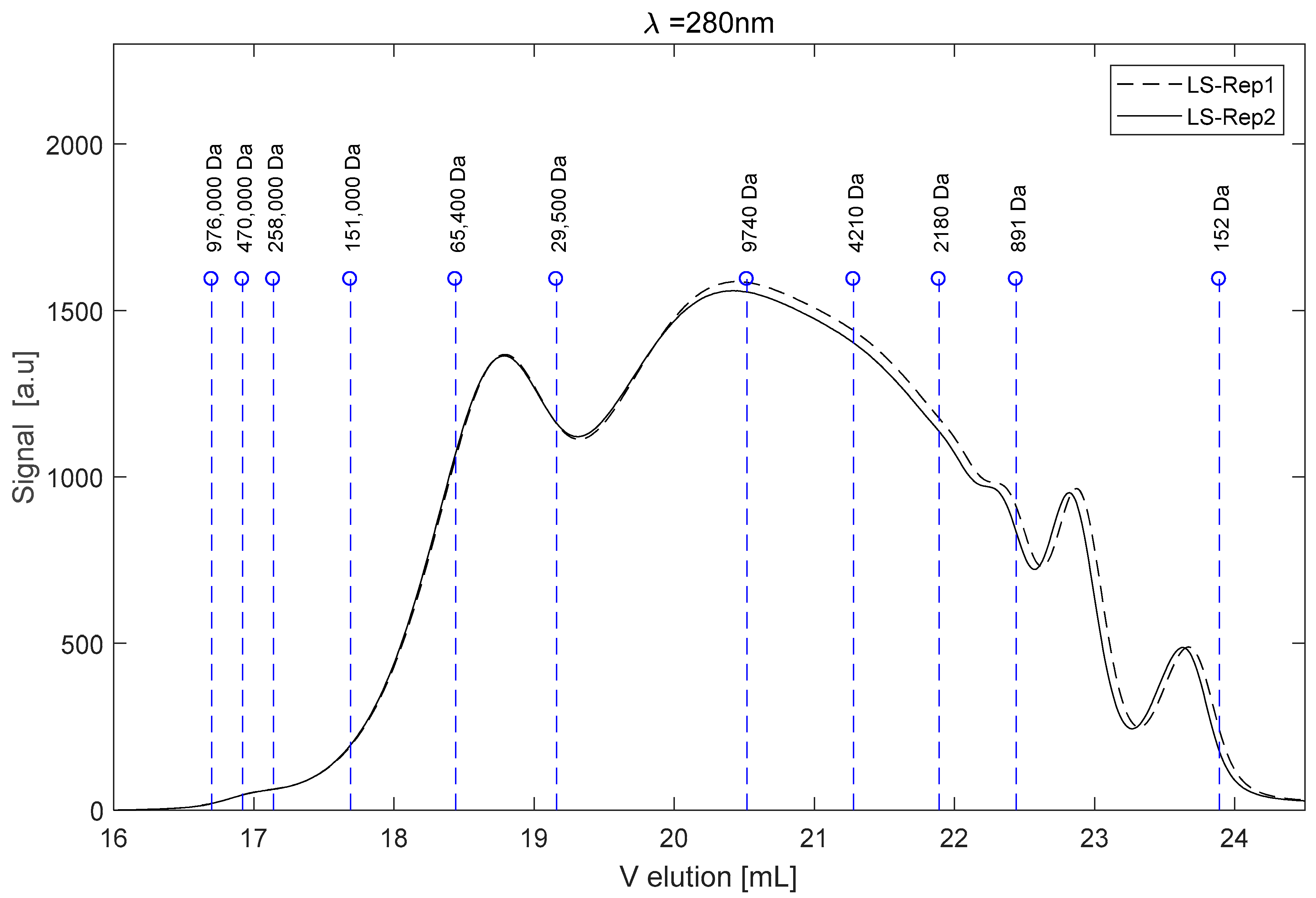
2.1. The Raw LS
2.2. Monitoring the LS Depolymerisation
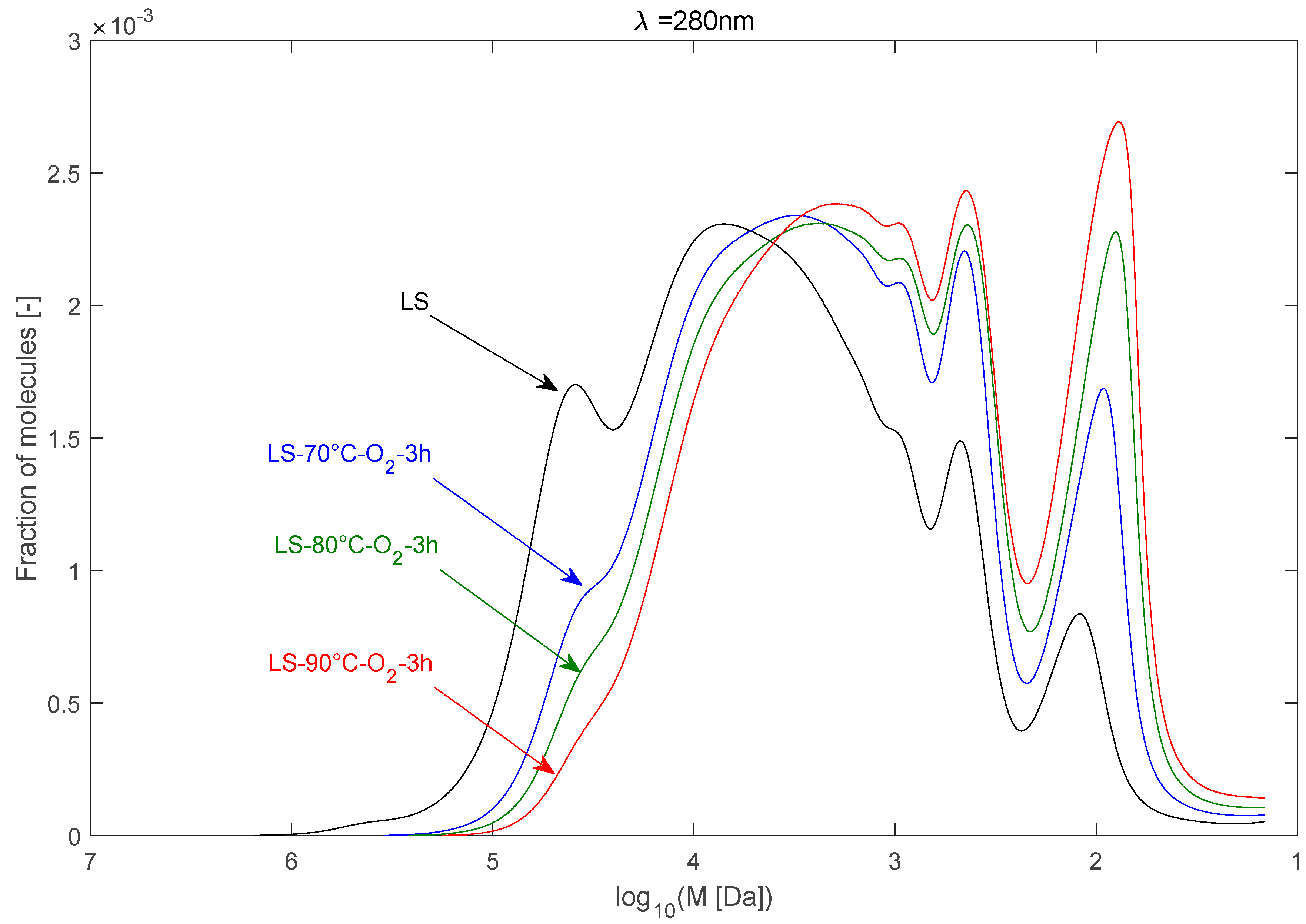
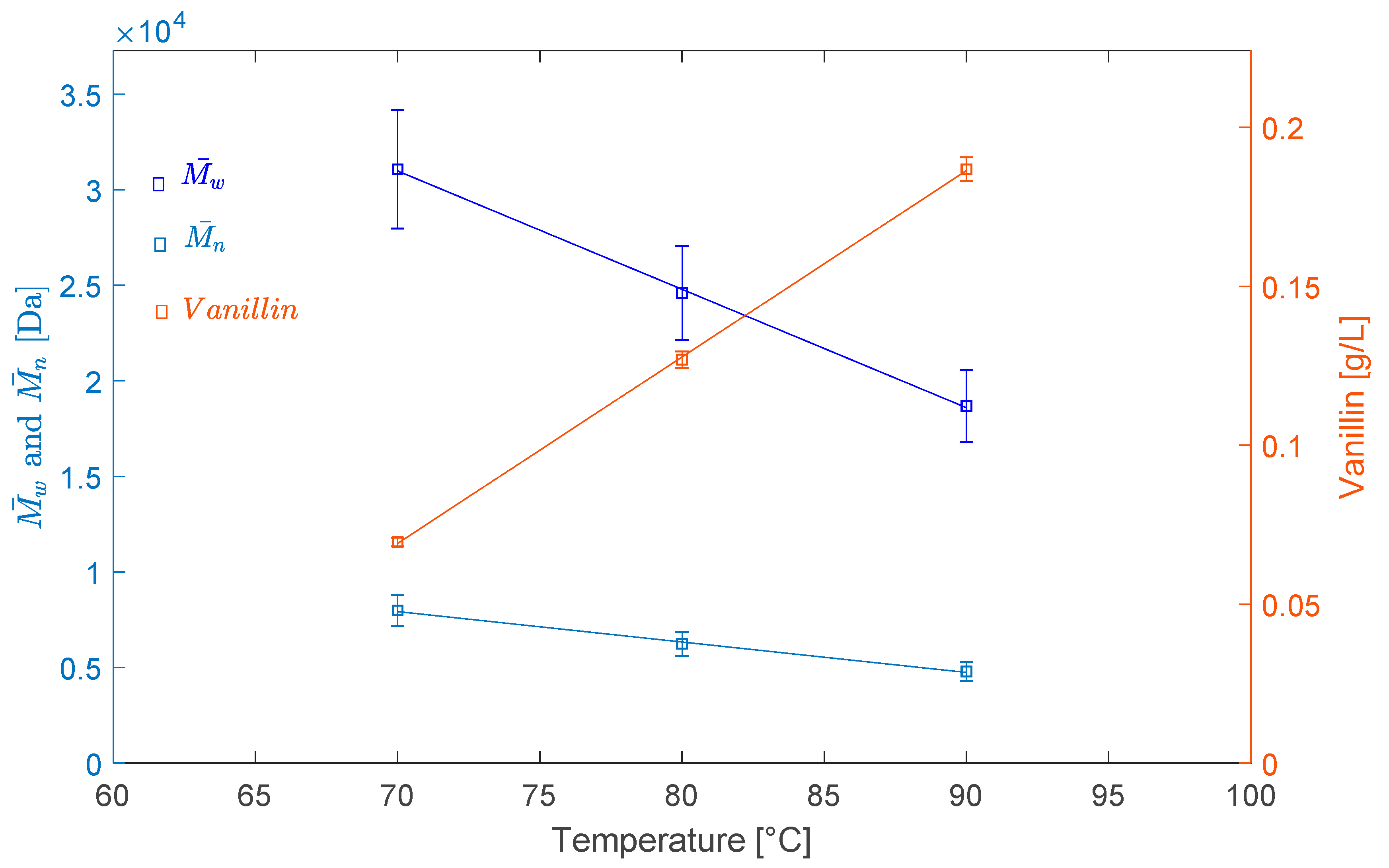
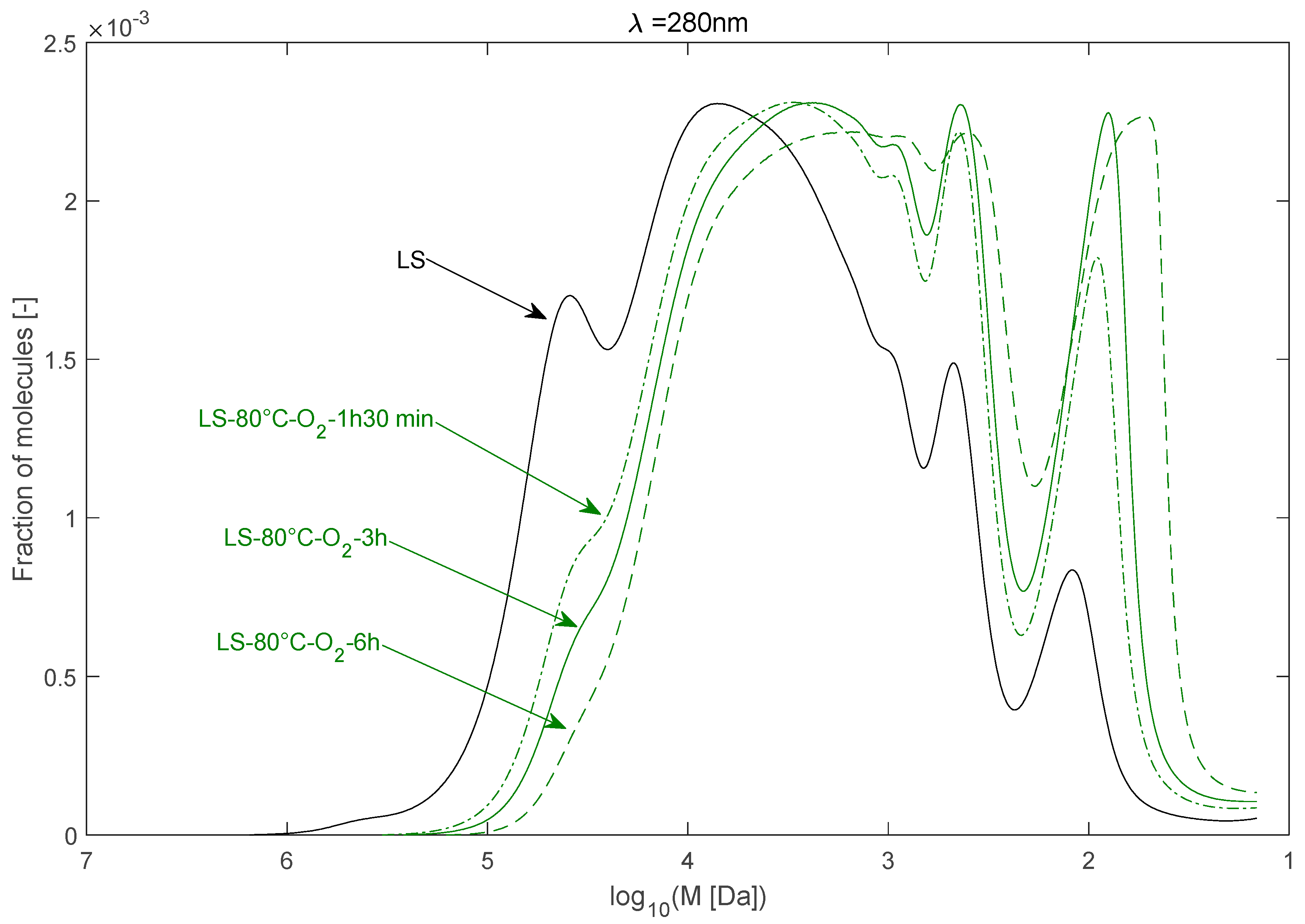
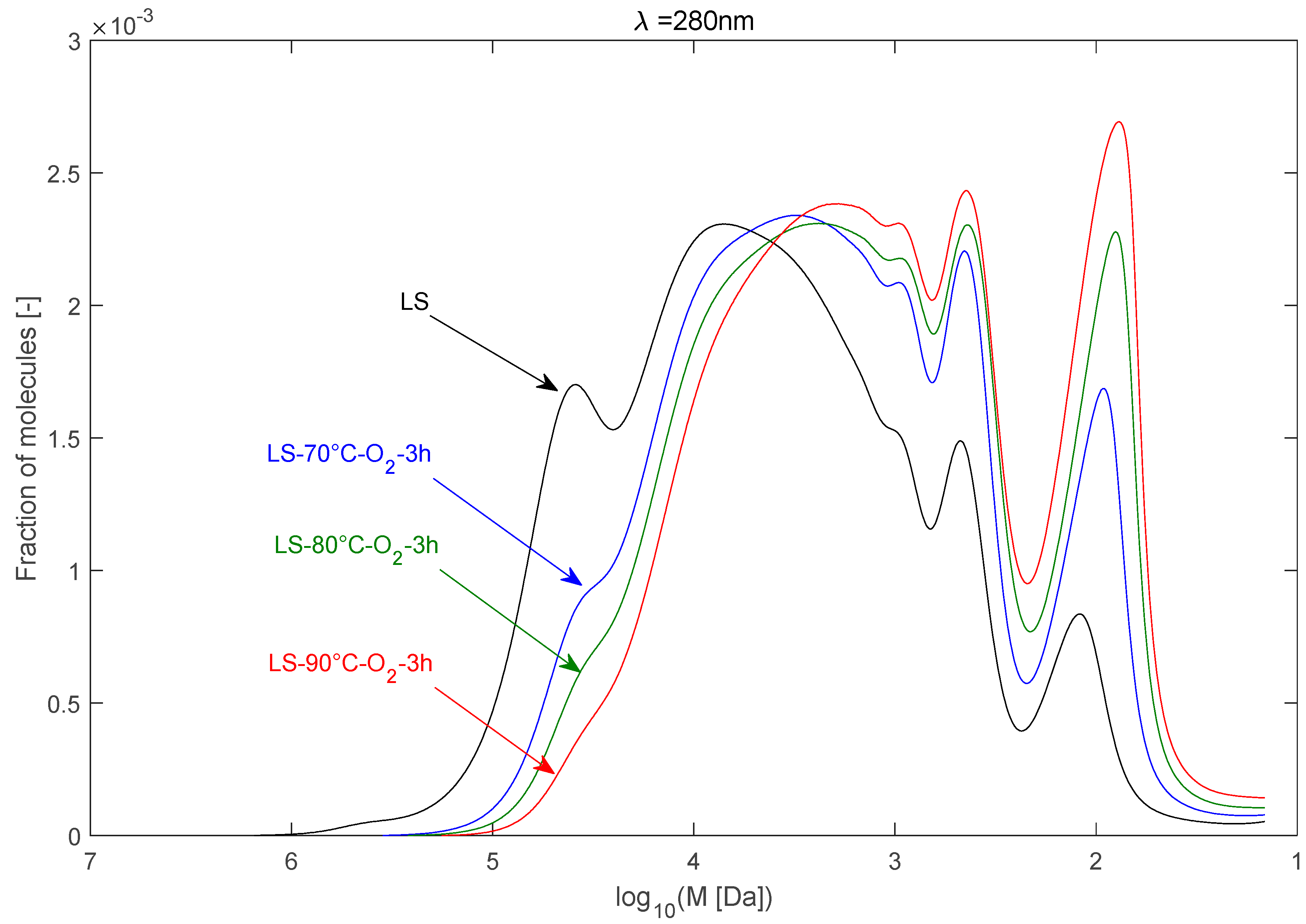
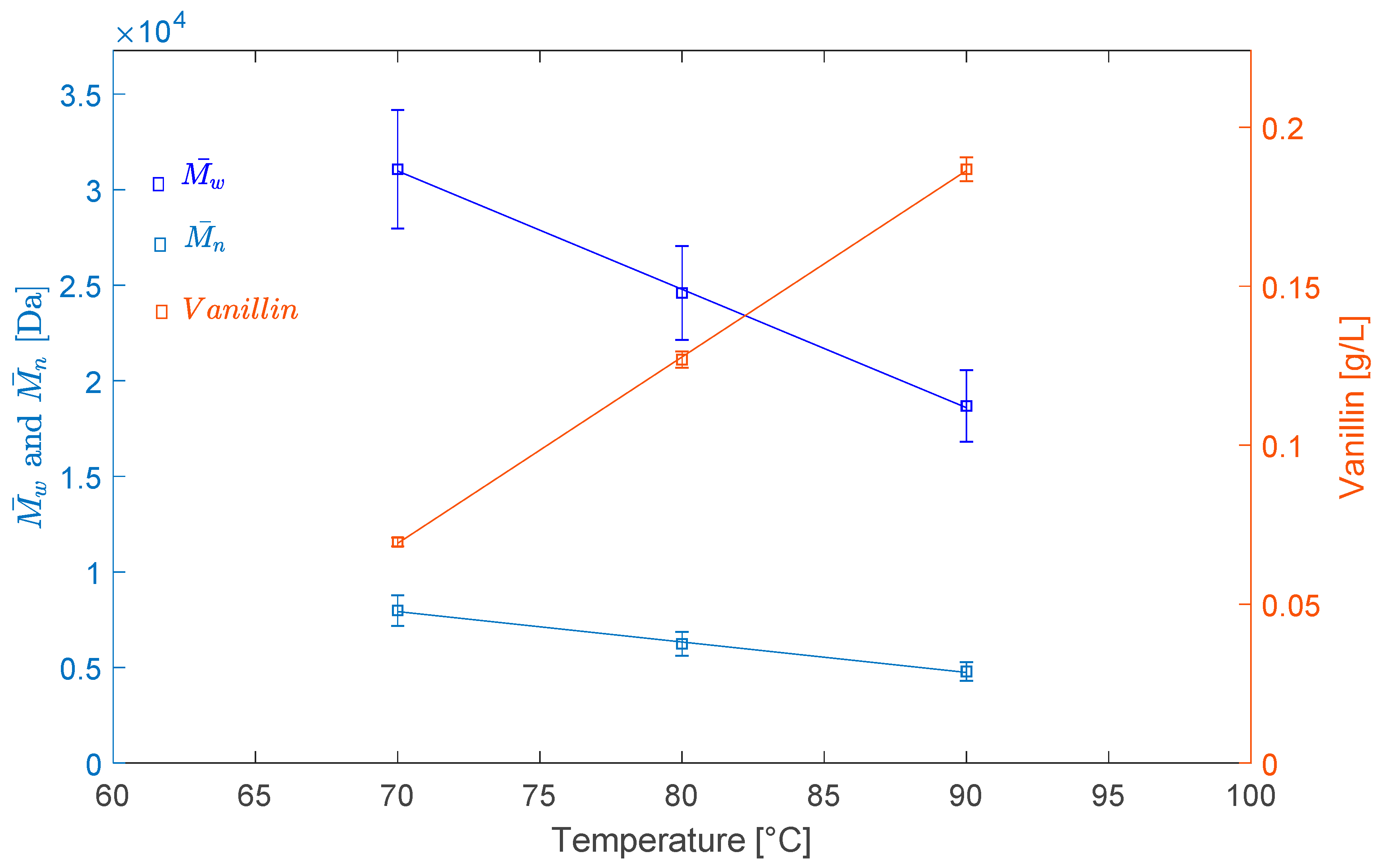
2.2.1. Effect of Temperature
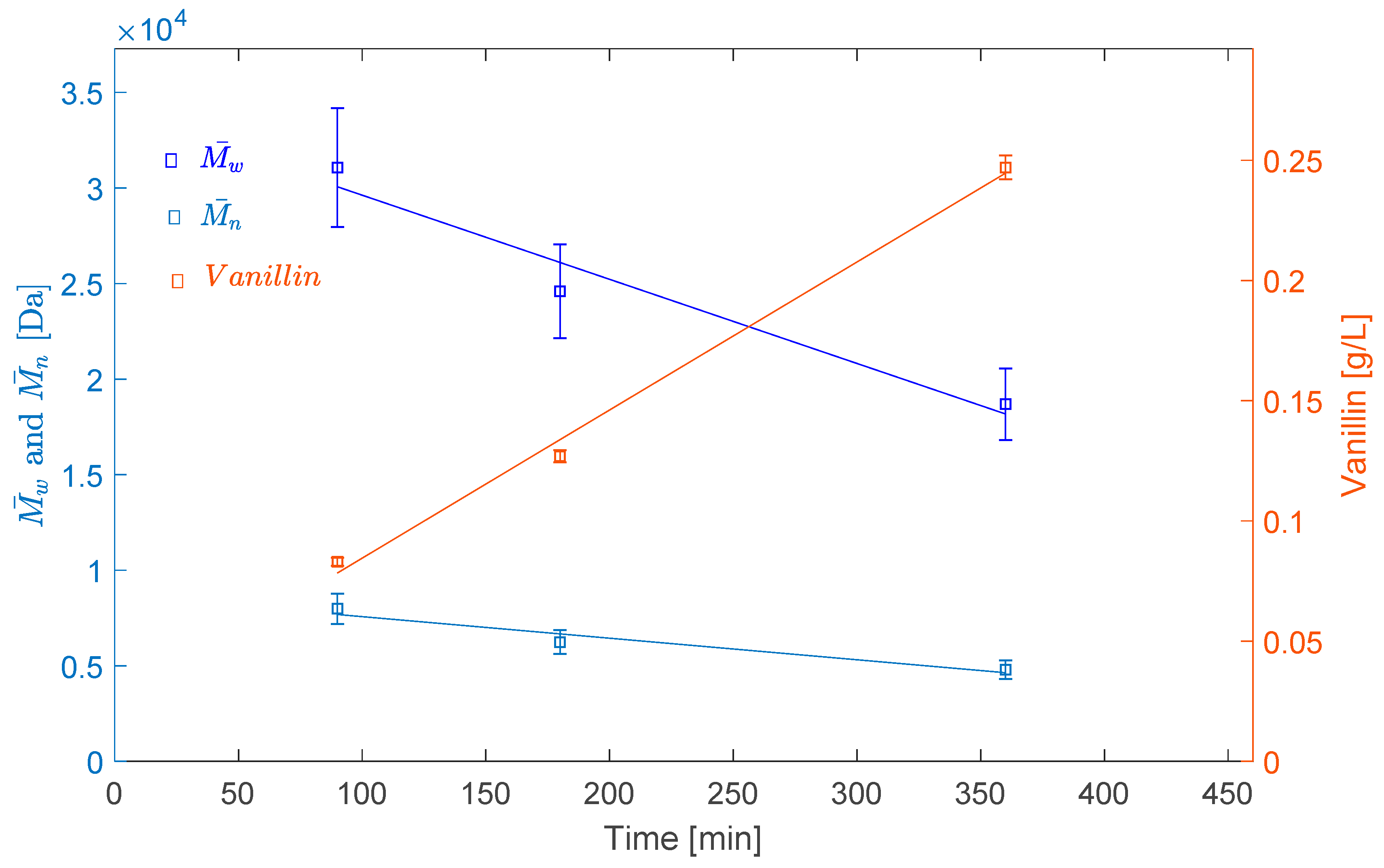
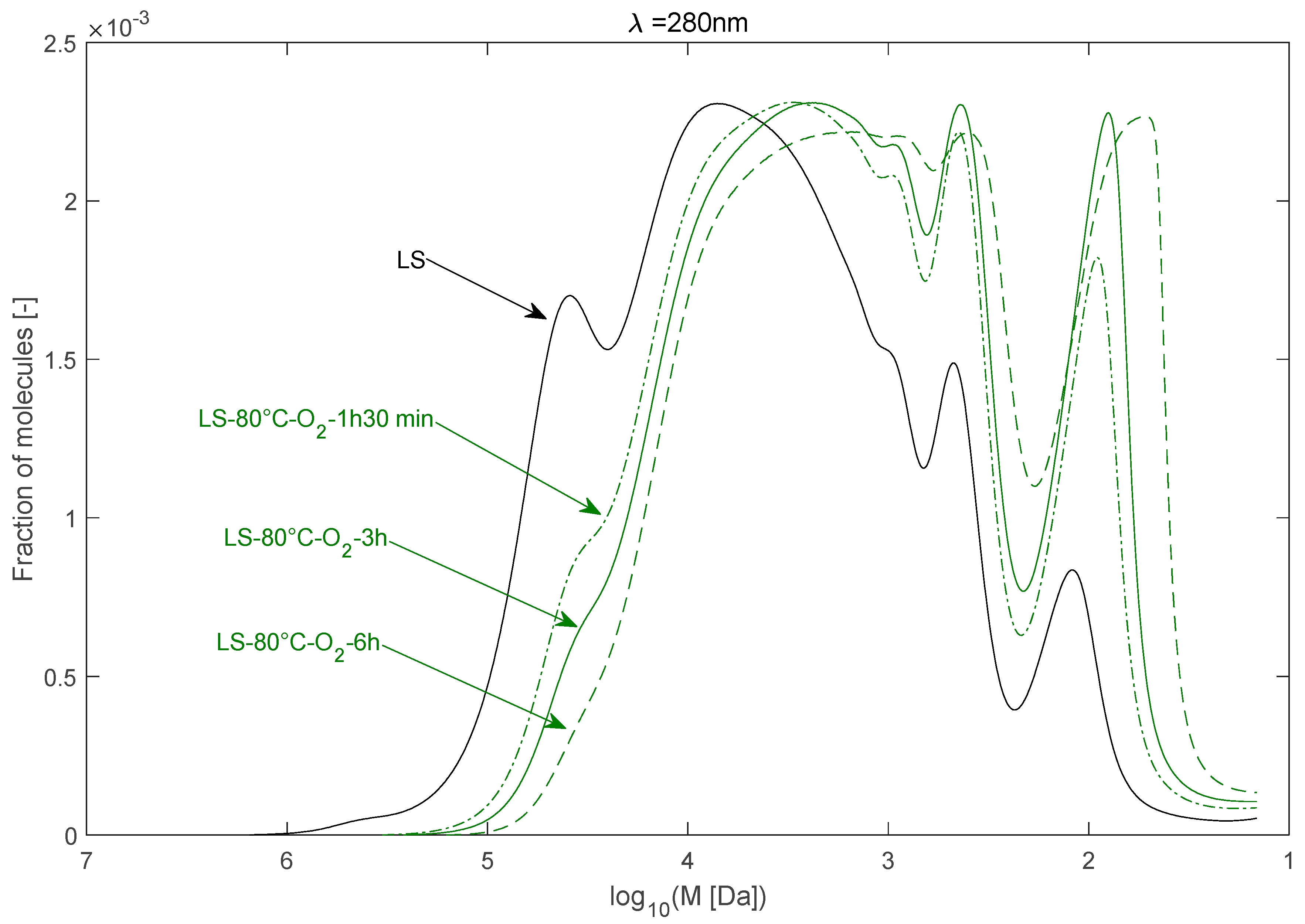
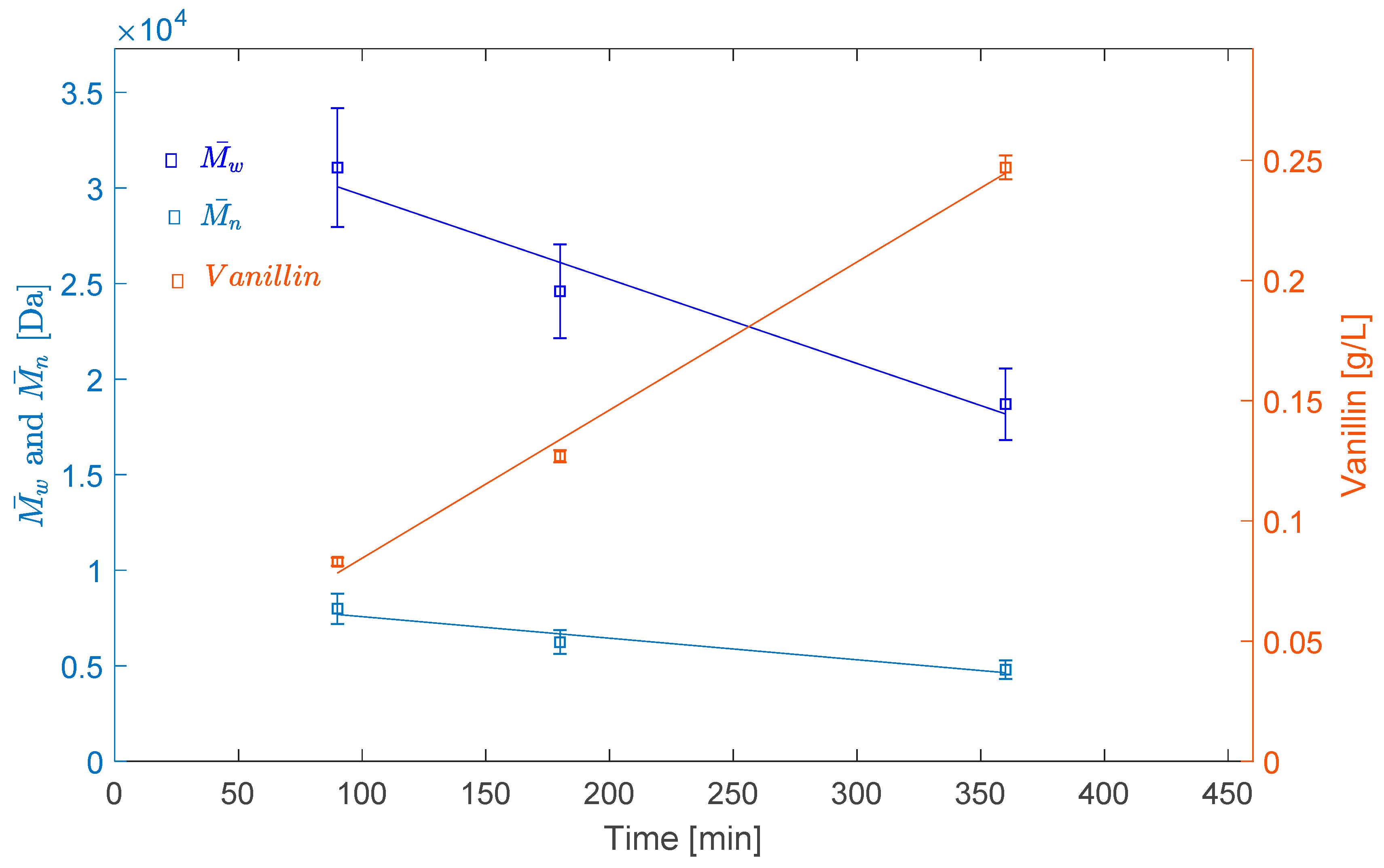
2.2.2. Effect of Reaction Time
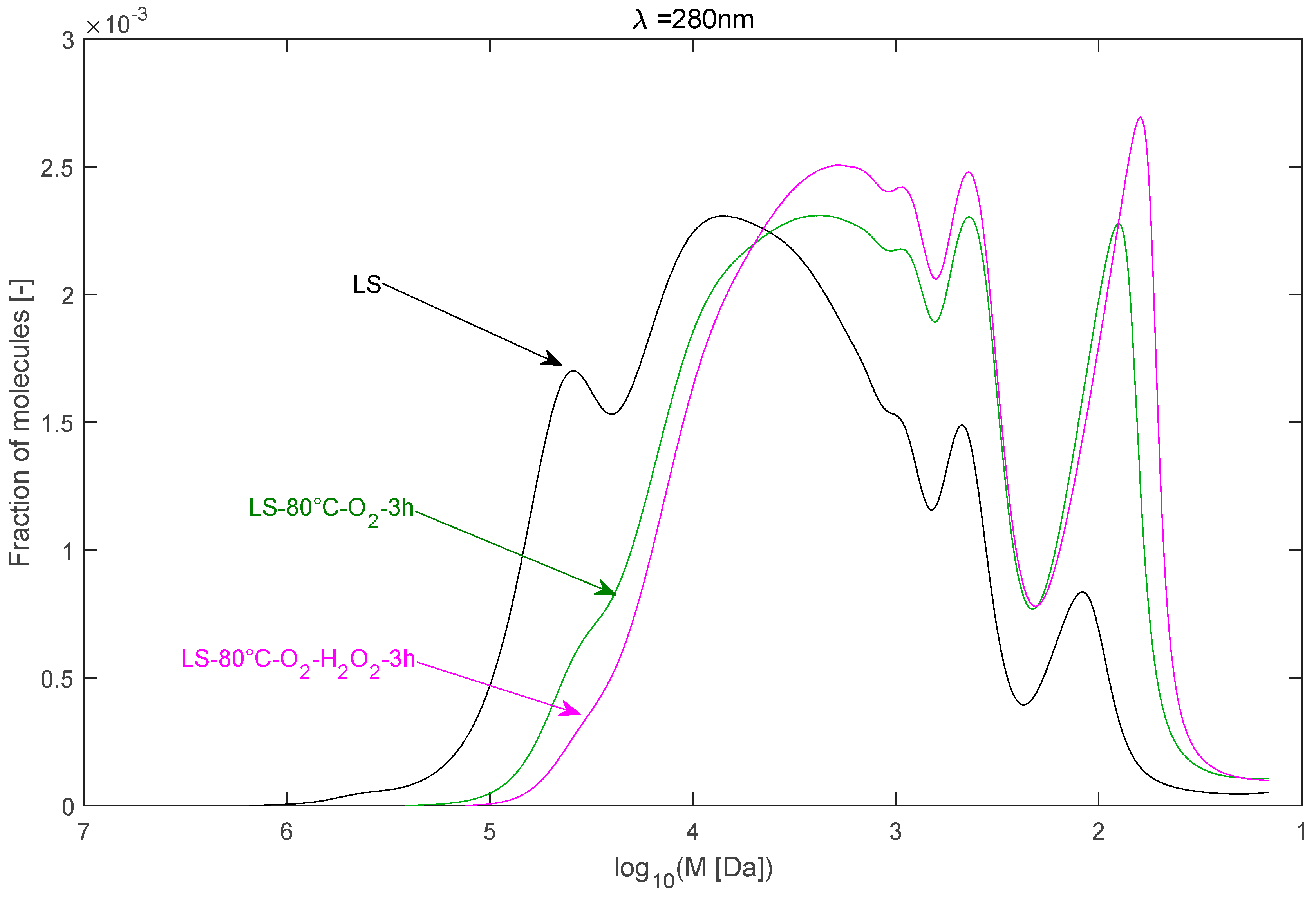
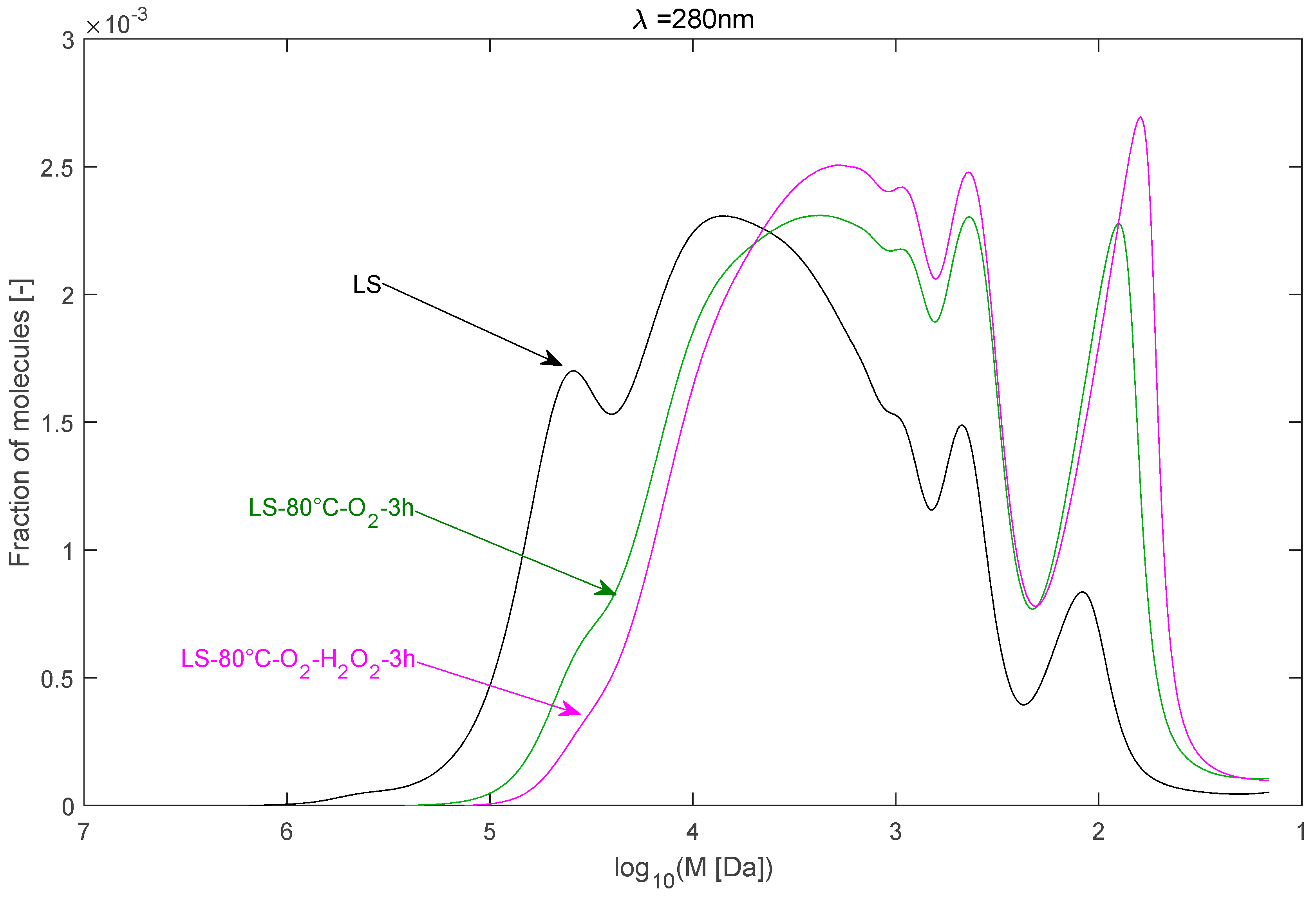
2.2.3. Effect of H2O2 as Co-Oxidant
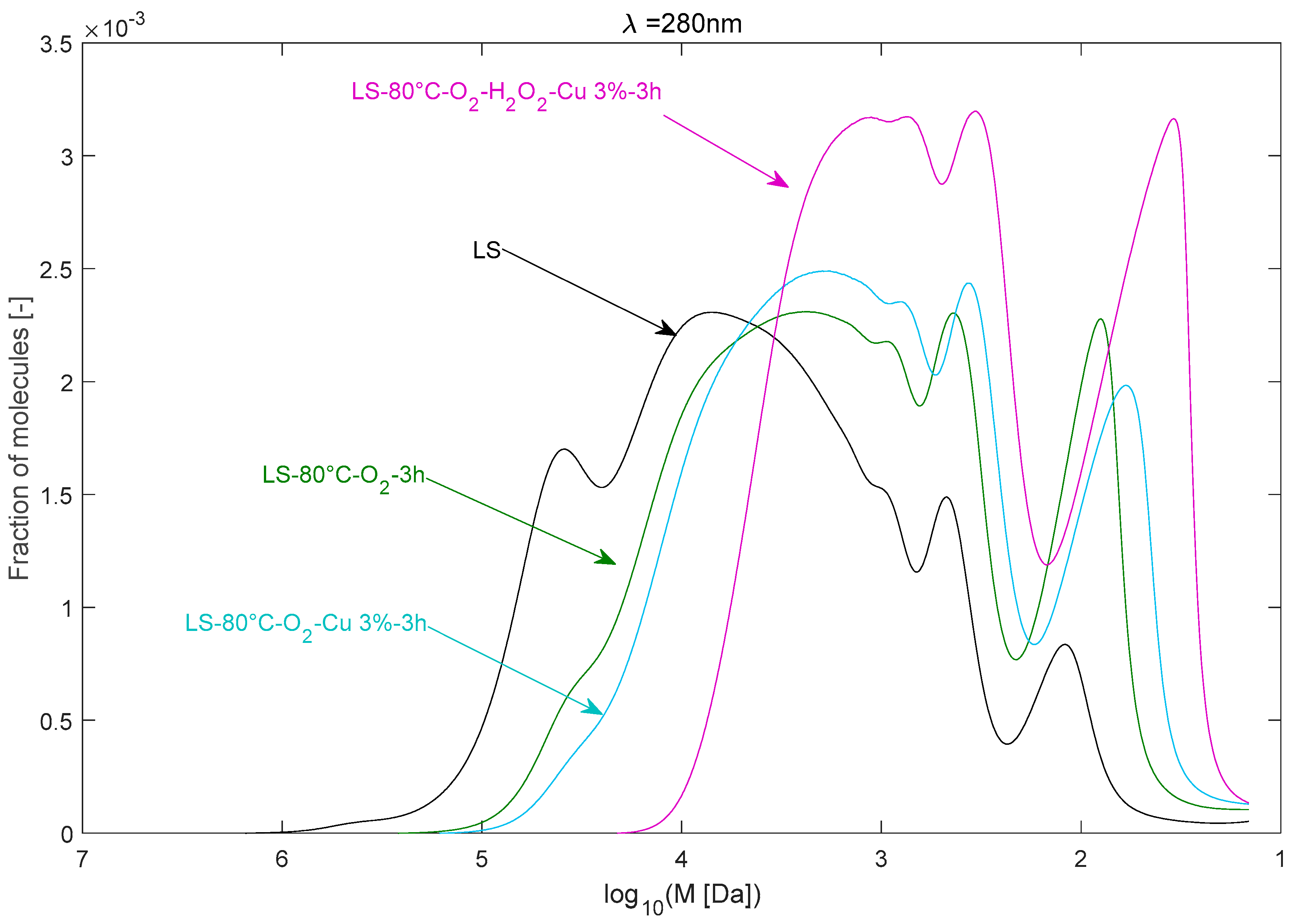
2.2.4. Effect of Cu2+ Catalyst in the Presence of O2
2.2.5. Effect of Cu2+ Catalyst in the Presence of the O2/H2O2 System
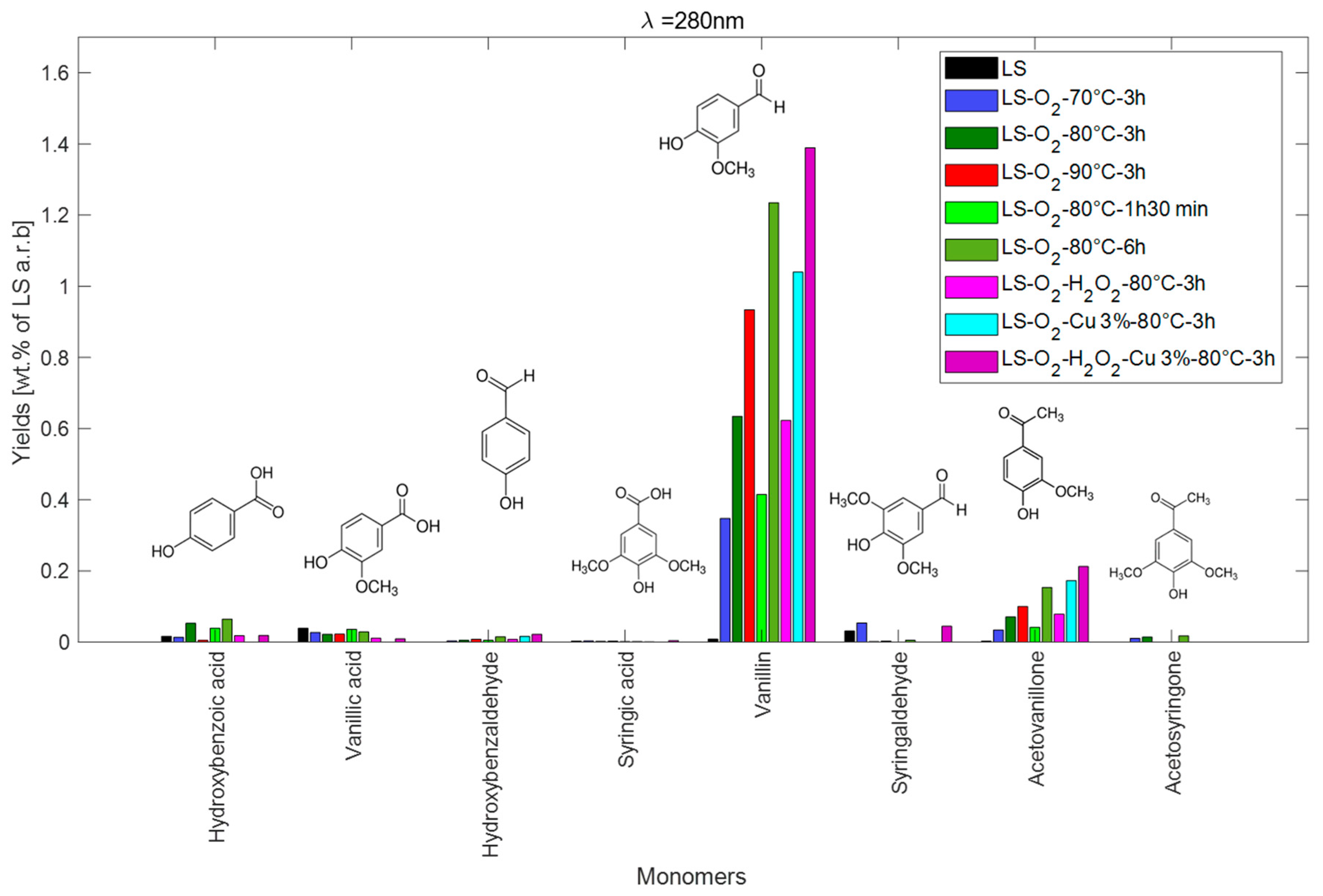
2.2.6. Summary of the Results
3. Material and Methods
3.1. Lignosulfonates
3.2. Lab-Scale Experimental Facility for LS Deploymerisation
- The reactor (1) which is a 100 mL Duran bottle with a special spare screw cap for GL 45 stirred reactor. This cap has three upper entries: one along the revolution axis of the bottle and two inclined, others making a 45° angle with this axis.
- A magnetic stirrer with heating and ceramic heating plate (2) (IKA-C-MAG HS 7) to which is connected a contact thermometer ETS-D5 (3), enabling precise temperature control of the solution. This contact thermometer is introduced in the reactor via the vertical entry of the cap and is in direct contact with the solution. Hence, the temperature of the reacting medium is precisely controlled. The vertical entry is made hermetic ensuring no mass transfer through it.
- An oxygen supply system: The left inclined entry is connected to an O2 cylinder. The O2 flowrate is measured and controlled by a volumetric flowmeter (4) which is connected to this left entry via a special connector and flexible tubing. A small flexible tube is also introduced along this left entry. A standard pipette tip (50–1000 µL) is fixed to this tube, plunges into the solution and ensures the oxygen bubbling inside.
- A vapor condensation system (5): The right entry is connected to an Allihn condenser consisting of a long glass tube with a cool water jacket. A series of bulbs on the tube increases the surface area upon which the vapor constituents may condense, and hence ensures the reflux. An additional home-made electrical fan (6) is permanently blowing air on the lower part of the condenser to ameliorate the condensation.
3.3. LS Depolymerisation Experimental Conditions
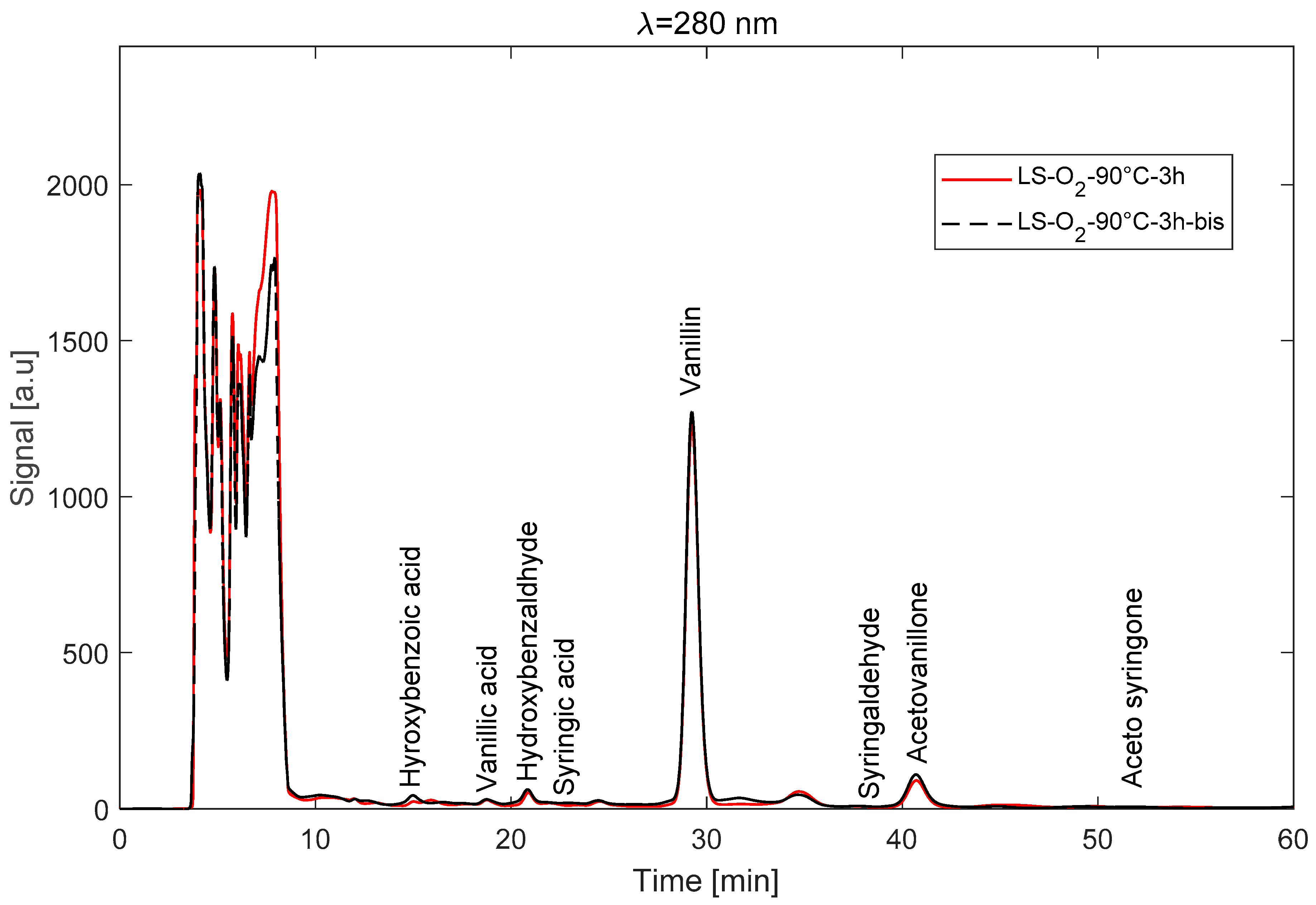
3.4. Unfolding of a Typical LS Depolymerisation Experiment
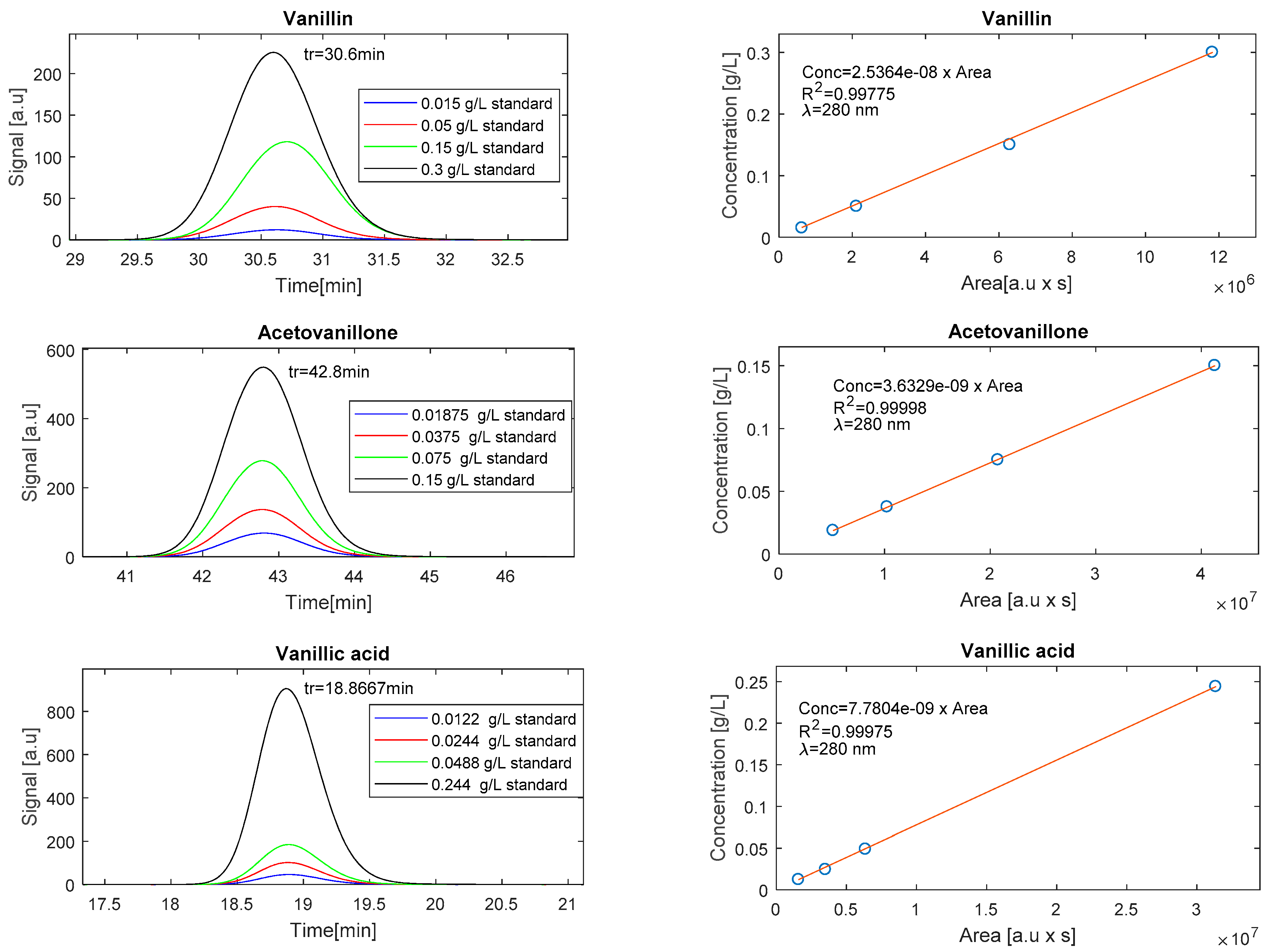
3.5. Monitoring the LS Depolymerisation: LS Molecular Weight Distribution and Monomer Concentration
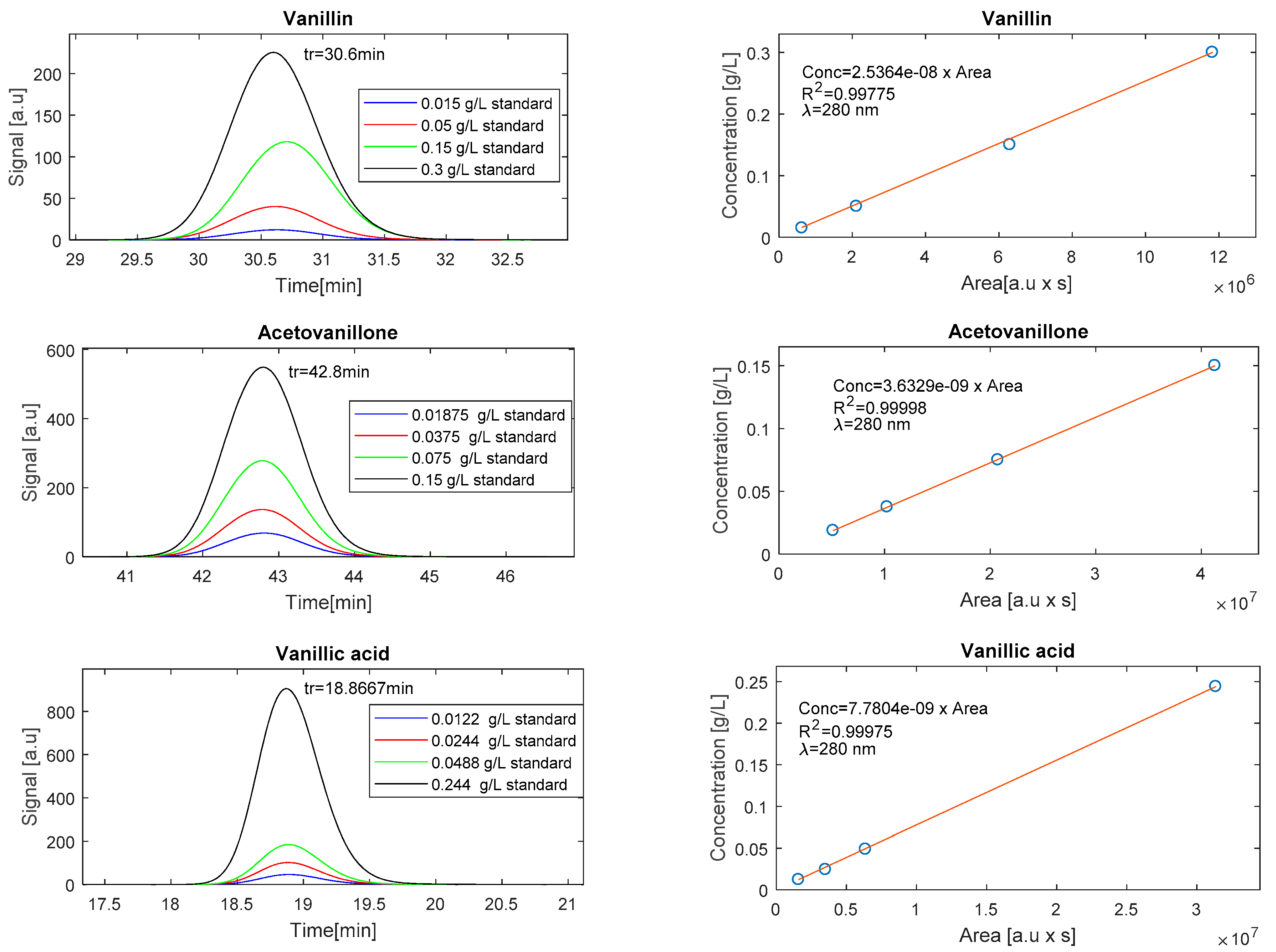
3.5.1. Operation in SEC Mode
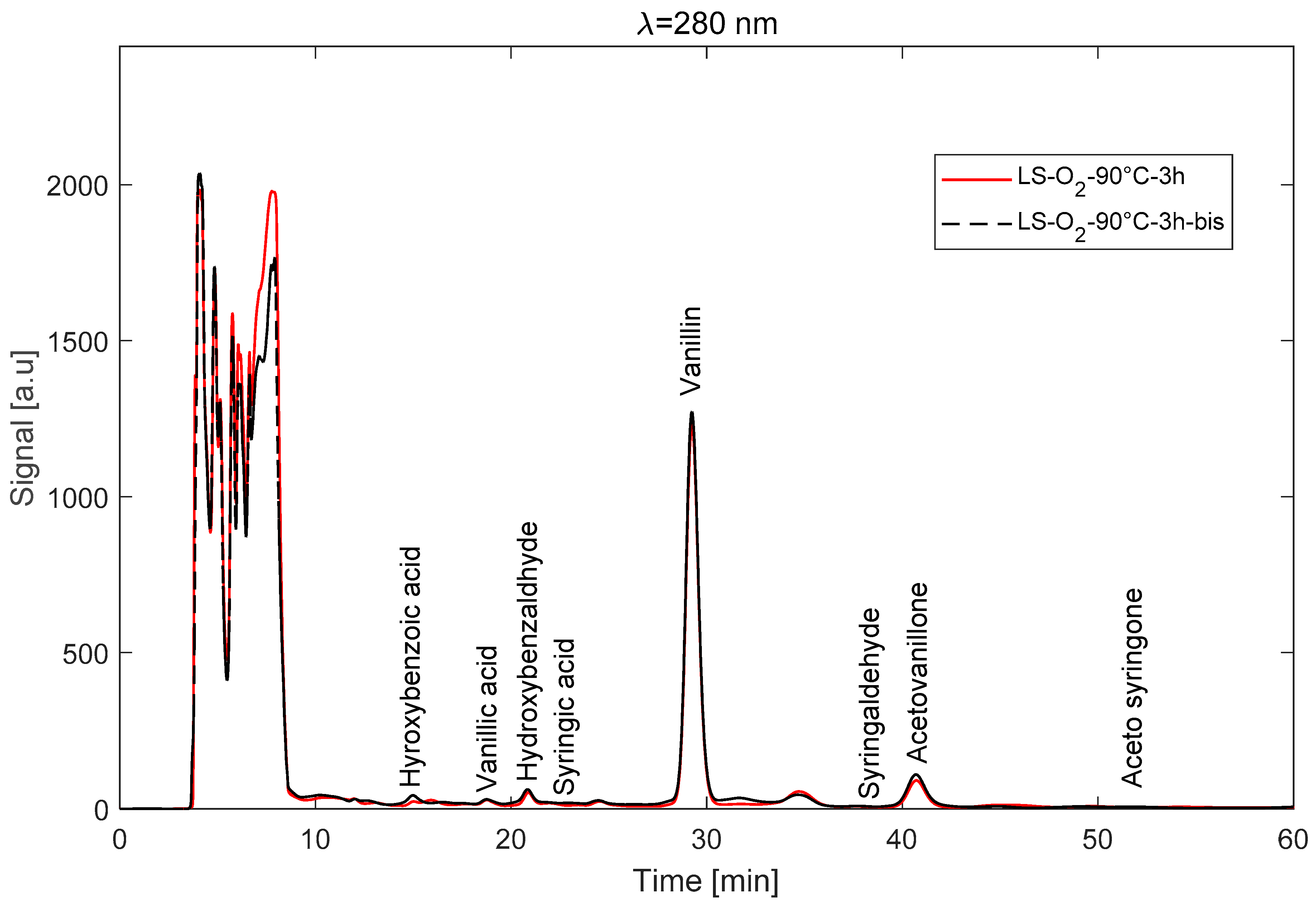
3.5.2. Operation in RPC Mode
4. Conclusions
Author Contributions
Conflicts of Interest
Appendix A

References
- Meadows, D.L.; Randers, J.; Meadows, D.H. Limits to Growth: The 30-Year Update, 3rd ed.; Chelsea Green Publishing: Hartford, VT, USA, 2004. [Google Scholar]
- Morin, E. Ecologiser l’homme: La Nature du Futur et le Futur de la Nature; Lemieux Editeur: Paris, France, 2016. [Google Scholar]
- Van Haveren, J.; Elinor, L.S.; Johan, S. Bulk chemicals from biomass. Biofuels Bioprod. Biorefin. 2012, 6, 246–256. [Google Scholar] [CrossRef]
- Aro, T.; Fatehi, P. Production and Application of Lignosulfonates and Sulfonated Lignin. ChemSusChem 2017, 10, 1861–1877. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Lu, H.; Hu, R.; Shupe, A.; Lin, L.; Liang, B. A sustainable woody biomass biorefinery. Biotechnol. Adv. 2012, 30, 785–810. [Google Scholar] [CrossRef] [PubMed]
- Renders, T.; van den Bosch, S.; Koelewijn, S.-F.; Schutyser, W.; Sels, B.F. Lignin-first biomass fractionation: The advent of active stabilisation strategies. Energy Environ. Sci. 2017, 7, 3419–3429. [Google Scholar] [CrossRef]
- Hu, L.; Pan, H.; Zhou, Y.; Zhang, M. Methods to improve lignin’s reactivity as a phenol substitute and as replacement for other phenolic compounds: A brief review. BioResources 2011, 6, 3515–3525. [Google Scholar]
- Meister, J.J. Modification of Lignin. J. Macromol. Sci. Part C Polym. Rev. 2002, 42, 235–289. [Google Scholar] [CrossRef]
- Yang, D.; Li, H.; Qin, Y.; Zhong, R.; Bai, M.; Qiu, X. Structure and Properties of Sodium Lignosulfonate with Different Molecular Weight Used as Dye Dispersant. J. Dispers. Sci. Technol. 2015, 36, 532–539. [Google Scholar] [CrossRef]
- Li, Q.; Serem, W.K.; Dai, W.; Yue, Y.; Naik, M.T.; Xie, S.; Karki, P.; Liu, L.; Sue, H.-J.; Liang, H.; et al. Molecular weight and uniformity define the mechanical performance of lignin-based carbon fiber. J. Mater. Chem. A 2017, 319, 908–909. [Google Scholar] [CrossRef]
- Shao, Y.; Guizani, C.; Grosseau, P.; Beneventi, D.; Didier, C. Thermal characterization and kinetic analysis of microfibrillated cellulose/lignosulfonate blends. J. Anal. Appl. Pyrolysis 2017, 124, 25–34. [Google Scholar] [CrossRef]
- Santos, S.G.; Marques, A.P.; Lima, D.L.D.; Evtuguin, D.V.; Esteves, V.I. Kinetics of eucalypt lignosulfonate oxidation to aromatic aldehydes by oxygen in alkaline medium. Ind. Eng. Chem. Res. 2011, 50, 291–298. [Google Scholar] [CrossRef] [Green Version]
- Pacek, A.W.; Ding, P.; Garrett, M.; Sheldrake, G.; Nienow, A.W. Catalytic conversion of sodium lignosulfonate to vanillin: Engineering aspects. part 1. effects of processing conditions on vanillin yield and selectivity. Ind. Eng. Chem. Res. 2013, 52, 8361–8372. [Google Scholar] [CrossRef]
- Bjørsvik, H.-R.; Minisci, F. Fine Chemicals from Lignosulfonates. 1. Synthesis of Vanillin by Oxidation of Lignosulfonates. Org. Process Res. Dev. 1999, 3, 330–340. [Google Scholar] [CrossRef]
- El Mansouri, N.E.; Farriol, X.; Salvadó, J. Structural modification and characterization of lignosulfonate by a reaction in an alkaline medium for its incorporation into phenolic resins. J. Appl. Polym. Sci. 2006, 102, 3286–3292. [Google Scholar] [CrossRef]
- Hu, L.; Zhou, Y.; Liu, R.; Zhang, M.; Yang, X. Synthesis of foaming resol resin modified with oxidatively degraded lignosulfonate. Ind. Crops Prod. 2013, 44, 364–366. [Google Scholar] [CrossRef]
- Sumerskii, I.; Korntner, P.; Zinovyev, G.; Rosenau, T.; Potthast, A. Fast Track for Quantitative Isolation of Lignosulfonates from Spent Sulfite Liquors. RSC Adv. 2015, 5, 92732–92742. [Google Scholar] [CrossRef]
- Gierer, J.; Imsgard, F. The reactions of lignins with oxygen and hydrogen peroxide in alkaline media. Sven. Papperstidning 1977, 80, 510–518. [Google Scholar]
- Gierer, J.; Imsgard, F.; Norén, I.; Stilkerieg, B.; Christensen, A.; Schroll, G. Studies on the degradation of phenolic lignin units of the beta-aryl ether type with oxygen in alkaline media. Acta Chem. Scand. B 1977, 31, 561–572. [Google Scholar] [CrossRef]
- Imai, A.; Tomoda, I.; Yokoyama, T.; Matsumoto, Y.; Meshitsuka, G.; Tong, G. Application of the amount of oxygen consumption to the investigation of the oxidation mechanism of lignin during oxygen-alkali treatment. J. Wood Sci. 2008, 54, 62–67. [Google Scholar] [CrossRef]
- Wong, Z.; Chen, K.; Li, J. Formation of vanillin and syringaldehyde in an oxygen delignification process. BioResources 2010, 5, 1509–1516. [Google Scholar]
- Northey, R.A. A Review of Lignin Model Compound Reactions under Oxygen Bleaching Conditions. In Oxidative Delignification Chemistry Fundamentals and Catalysis; ACS Publications: Washington, DC, USA, 2001. [Google Scholar]
- Pinto, P.C.R.; da Silva, E.B.; Rodrigues, A.A.E. Lignin as Source of Fine Chemicals: Vanillin and Syringaldehyde. In Biomass Conversion. The Interface of Biotechnology, Chemistry and Materials Science; Springer: Berlin, Germany, 2012; p. 908. [Google Scholar]
- Tarabanko, V.E.; Hendogina, Y.V.; Petuhov, D.V.; Pervishina, E.P. On the Role of Retroaldol Reaction in the Process of Lignin Oxidation into Vanillin. Kinetics of the Vanillideneacetone Cleavage in Alkaline Media. React. Kinet. Catal. Lett. 2000, 69, 361–368. [Google Scholar] [CrossRef]
- Alunga, K.R.; Ye, Y.-Y.; Li, S.-R.; Wang, D.; Liu, Y.-Q. Catalytic oxidation of lignin–acetoderivatives: A potential new recovery route for value-added aromatic aldehydes from acetoderivatives. Catal. Sci. Technol. 2015, 5, 3746–3753. [Google Scholar] [CrossRef]
- Xiang, Q.; Lee, Y.Y. Oxidative cracking of precipitated hardwood lignin by hydrogen peroxide. Appl. Biochem. Biotechnol. 2000, 84–86, 153–162. [Google Scholar] [CrossRef]
- Kadla, J.F.; Chang, H. The Reactions of Peroxides with Lignin and Lignin Model Compounds. In Oxidative Delignification Chemistry Fundamentals and Catalysis; no. 785.; ACS Publications: Washington, DC, USA, 2001. [Google Scholar]
- Halma, M.; Lachenal, D.; Marlin, N.; Deronzier, A.; Brochier, M.C.; Zarubin, M. H2O2 oxidation of lignin model dimers catalyzed by copper(II)—Phenanthroline. Ind. Crops Prod. 2015, 74, 514–522. [Google Scholar] [CrossRef]
- Villar, J.C.; Caperos, A.; García-Ochoa, F. Oxidation of hardwood kraft-lignin to phenolic derivatives with oxygen as oxidant. Wood Sci. Technol. 2001, 35, 245–255. [Google Scholar] [CrossRef]
- Sun, Y.; Fenster, M.; Yu, A.; Berry, R.M.; Argyropoulos, D.S. The effect of metal ions on the reaction of hydrogen peroxide with Kraft lignin model compounds. Can. J. Chem. 1999, 77, 667–675. [Google Scholar] [CrossRef]
- Kulik, F.; Wieber, J.; Pethica, B.; Zuman, P. Binding of copper(II) and zinc(II) ions on various lignins. J. Electroanal. Chem. 1986, 214, 331–342. [Google Scholar] [CrossRef]
- Mohan, D.; Pittman, C.U.; Steele, P.H. Single, binary and multi-component adsorption of copper and cadmium from aqueous solutions on Kraft lignin—A biosorbent. J. Colloid Interface Sci. 2006, 297, 489–504. [Google Scholar] [CrossRef] [PubMed]
- Baumberger, S.; Abaecherli, A.; Fasching, M.; Gellerstedt, G.; Gosselink, R.J.A.; Hortling, B.; Li, J.; Saake, B.; de Jong, E. Molar mass determination of lignins by size-exclusion chromatography: Towards standardisation of the method. Holzforschung 2007, 61, 459–468. [Google Scholar] [CrossRef]
- Azarpira, A.; Ralph, J.; Lu, F. Catalytic Alkaline Oxidation of Lignin and its Model Compounds: A Pathway to Aromatic Biochemicals. Bioenergy Res. 2014, 7, 78–86. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | (Da) | (Da) | (–) |
|---|---|---|---|
| LS | 90,357 | 16,172 | 5.59 |
| LS–70 °C–O2–3 h | 31,072 | 7983 | 3.89 |
| LS–80 °C–O2–3 h | 24,595 | 6236 | 3.94 |
| LS–90 °C–O2–3 h | 18,688 | 4793 | 3.90 |
| LS–80 °C–O2–1 h 30 min | 30,336 | 7759 | 3.91 |
| LS–80 °C–O2–6 h | 17,148 | 4581 | 3.74 |
| LS–80 °C–O2–H2O2–3 h | 16,003 | 4479 | 3.57 |
| LS–80 °C–O2–Cu 3%–3 h | 17,286 | 4549 | 3.80 |
| LS–80 °C–O2–H2O2–Cu 3%–3 h | 3583 | 1425 | 2.51 |
| Experiment Number | Experiment Name | Temperature (°C) | Duration (h) | Oxidant | Catalyst Cu (wt % on LS Basis) |
|---|---|---|---|---|---|
| 1 | LS–70 °C–O2–3 h | 70 | 3 | O2 | 0 |
| 2 | LS–80 °C–O2–3 h | 80 | 3 | O2 | 0 |
| 3 | LS–90 °C–O2–3 h | 90 | 3 | O2 | 0 |
| 4 | LS–80 °C–O2–1 h 30 min | 80 | 1.5 | O2 | 0 |
| 5 | LS–80 °C–O2–6 h | 80 | 6 | O2 | 0 |
| 6 | LS–80 °C–O2–H2O2–3 h | 80 | 3 | O2/H2O2 | 0 |
| 7 | LS–80 °C–O2–Cu 3%–3 h | 80 | 3 | O2 | 3 |
| 8 | LS–80 °C–O2–H2O2–Cu 3%–3 h | 80 | 3 | O2/H2O2 | 3 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guizani, C.; Lachenal, D. Controlling the Molecular Weight of Lignosulfonates by an Alkaline Oxidative Treatment at Moderate Temperatures and Atmospheric Pressure: A Size-Exclusion and Reverse-Phase Chromatography Study. Int. J. Mol. Sci. 2017, 18, 2520. https://doi.org/10.3390/ijms18122520
Guizani C, Lachenal D. Controlling the Molecular Weight of Lignosulfonates by an Alkaline Oxidative Treatment at Moderate Temperatures and Atmospheric Pressure: A Size-Exclusion and Reverse-Phase Chromatography Study. International Journal of Molecular Sciences. 2017; 18(12):2520. https://doi.org/10.3390/ijms18122520
Chicago/Turabian StyleGuizani, Chamseddine, and Dominique Lachenal. 2017. "Controlling the Molecular Weight of Lignosulfonates by an Alkaline Oxidative Treatment at Moderate Temperatures and Atmospheric Pressure: A Size-Exclusion and Reverse-Phase Chromatography Study" International Journal of Molecular Sciences 18, no. 12: 2520. https://doi.org/10.3390/ijms18122520