Impaired Osteogenesis of Disease-Specific Induced Pluripotent Stem Cells Derived from a CFC Syndrome Patient

Abstract
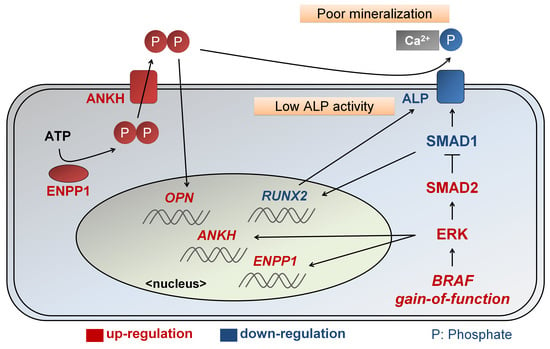
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
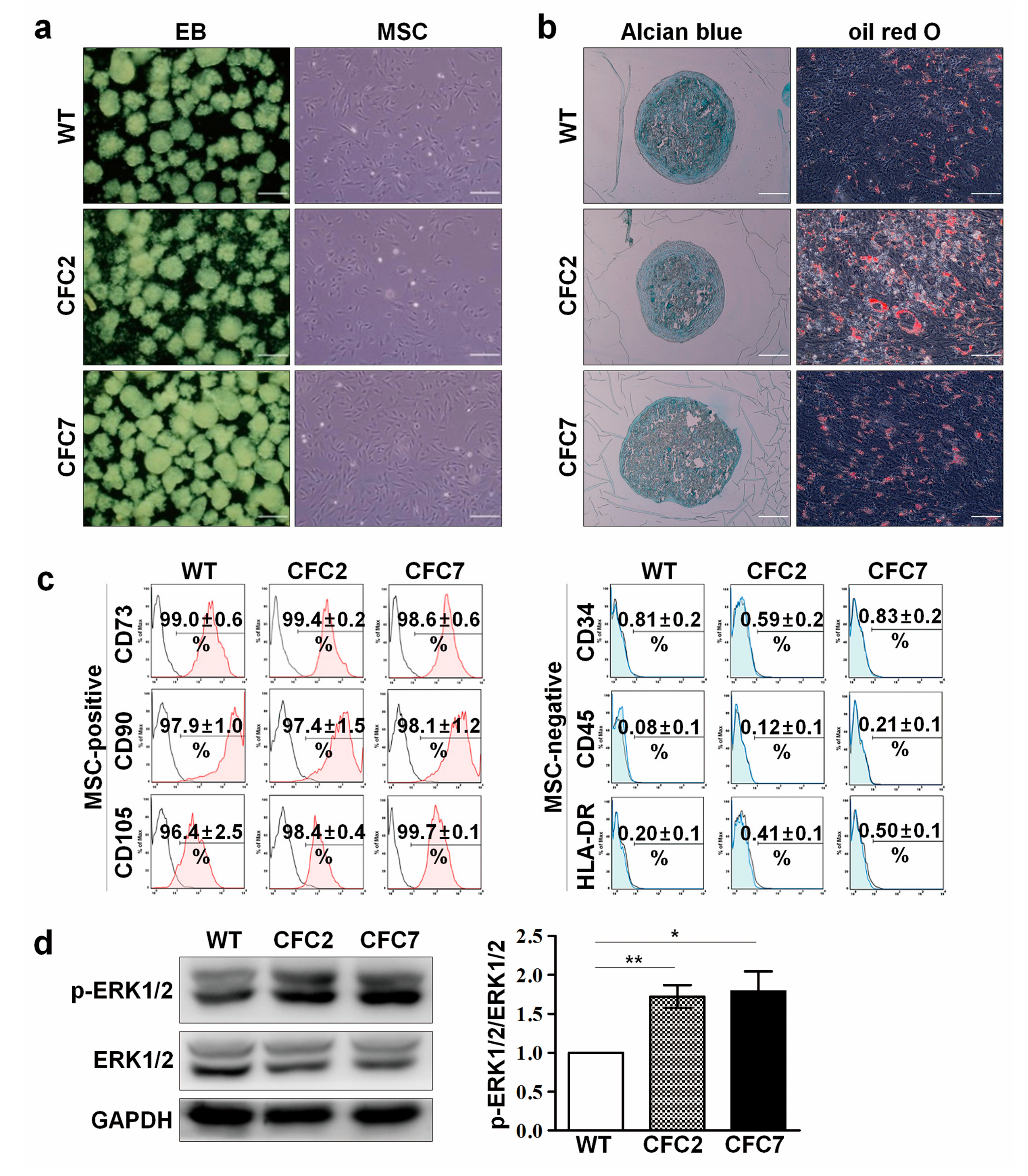
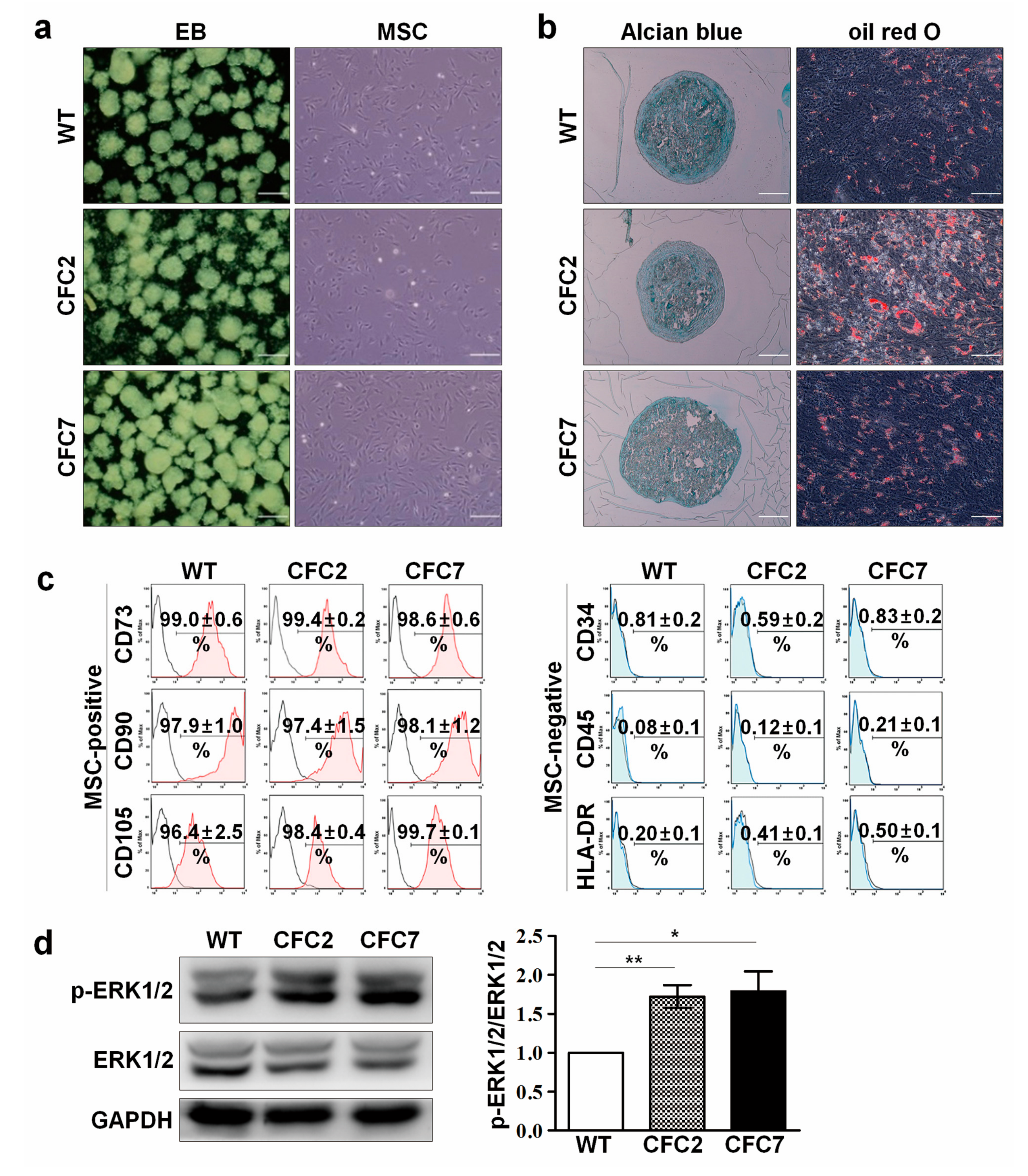
2.1. Generation and Characterization of CFC-iPSCs
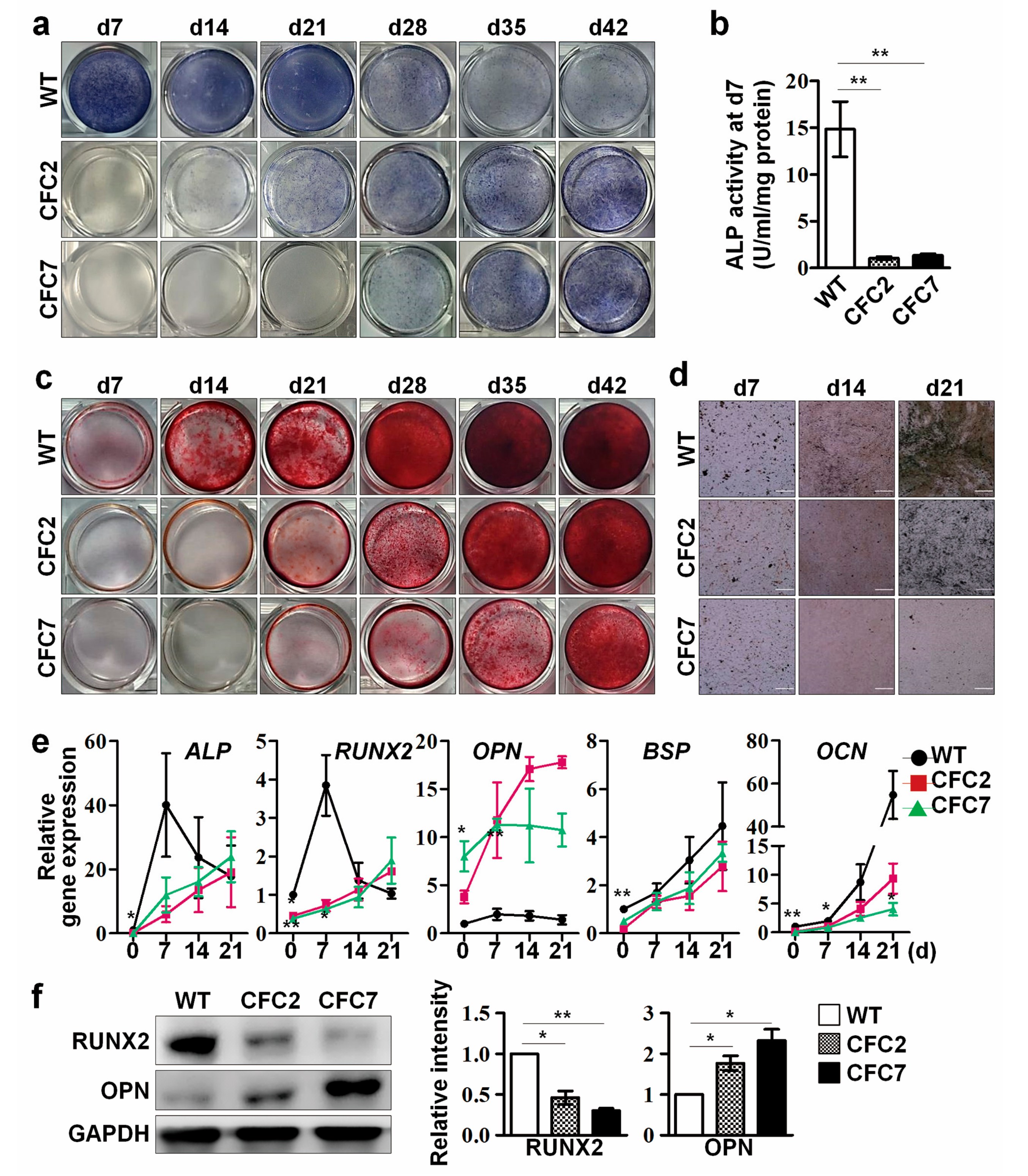
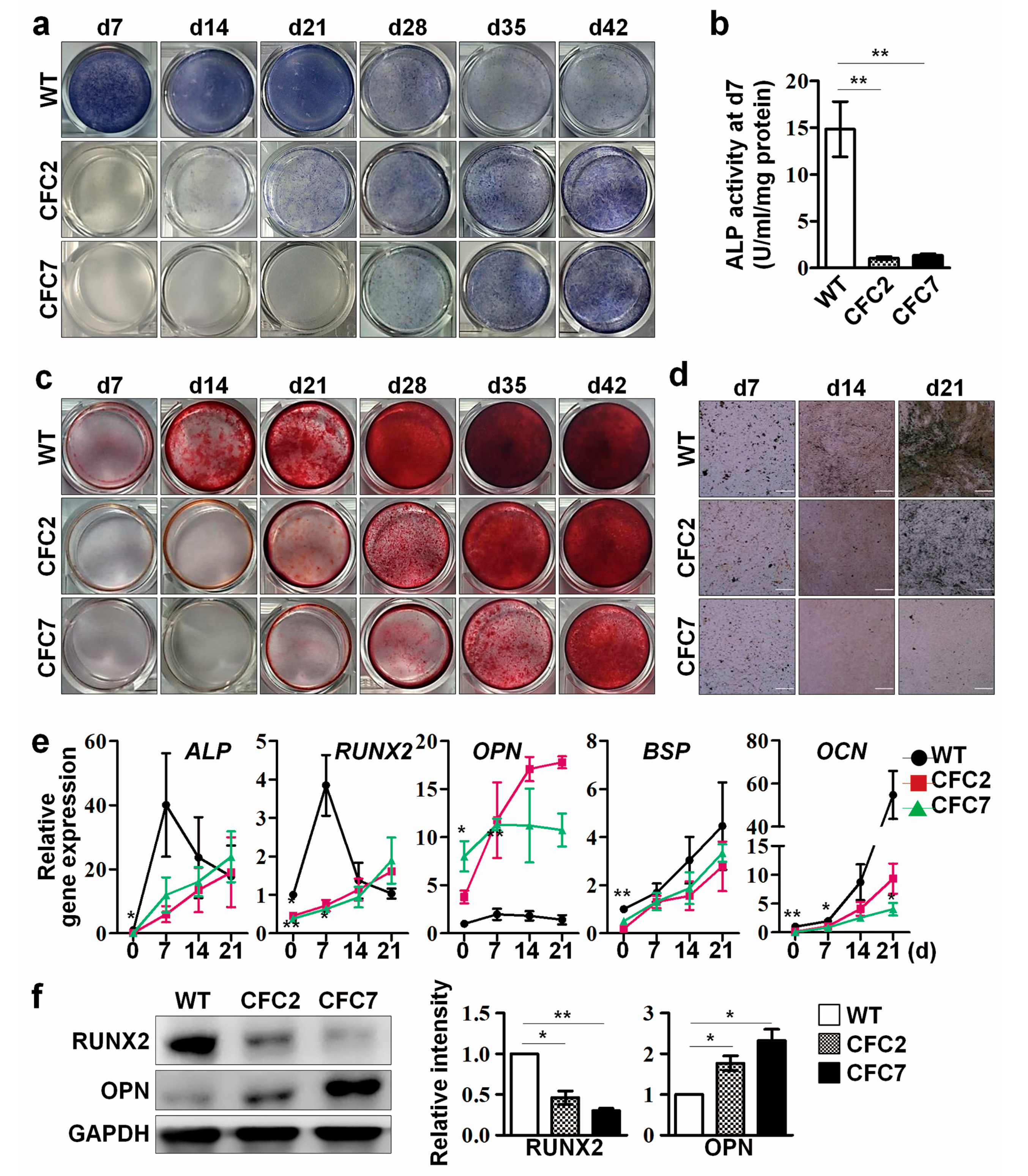
2.2. Impaired Osteogenesis of MSCs Derived from CFC-iPSCs In Vitro
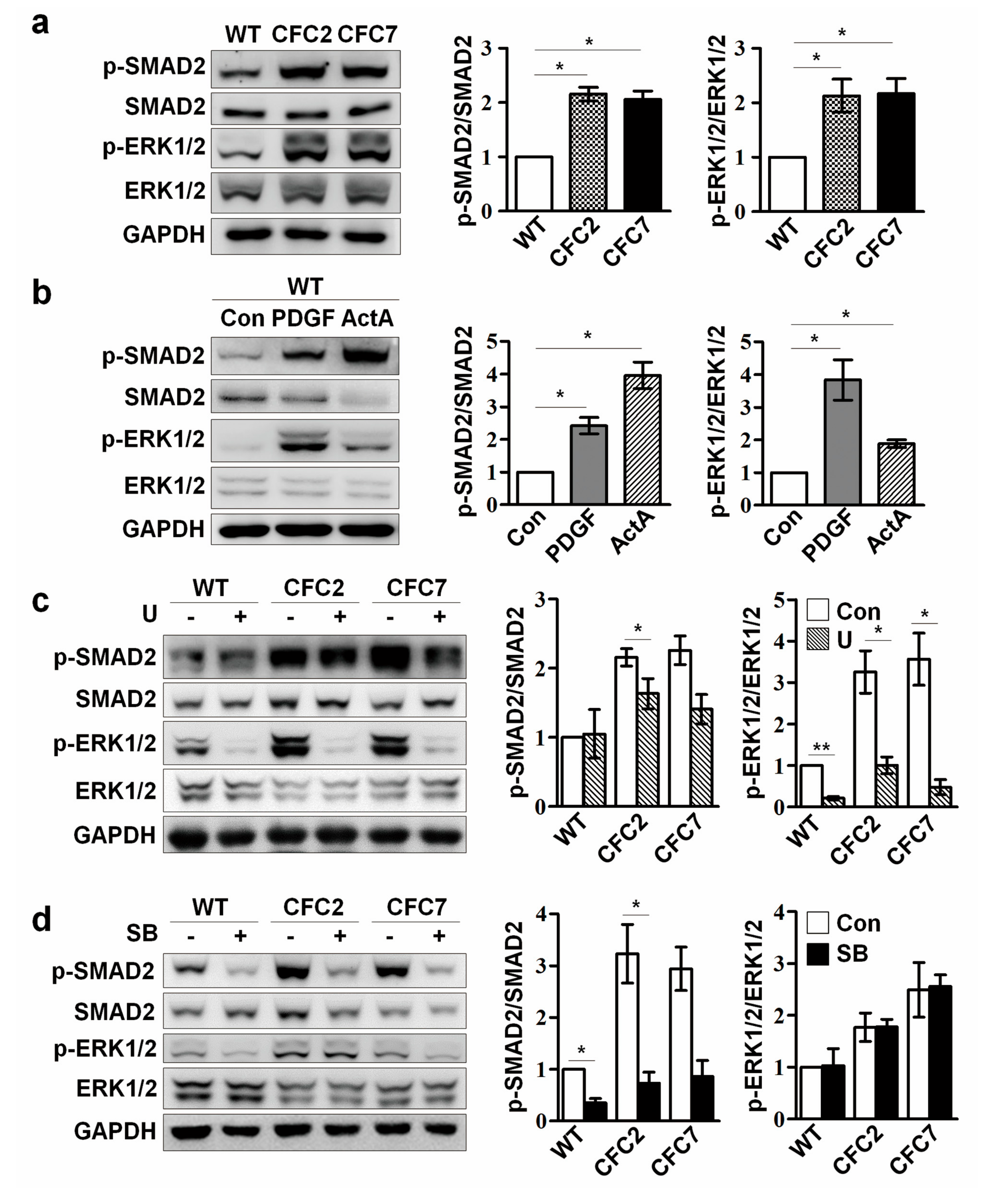
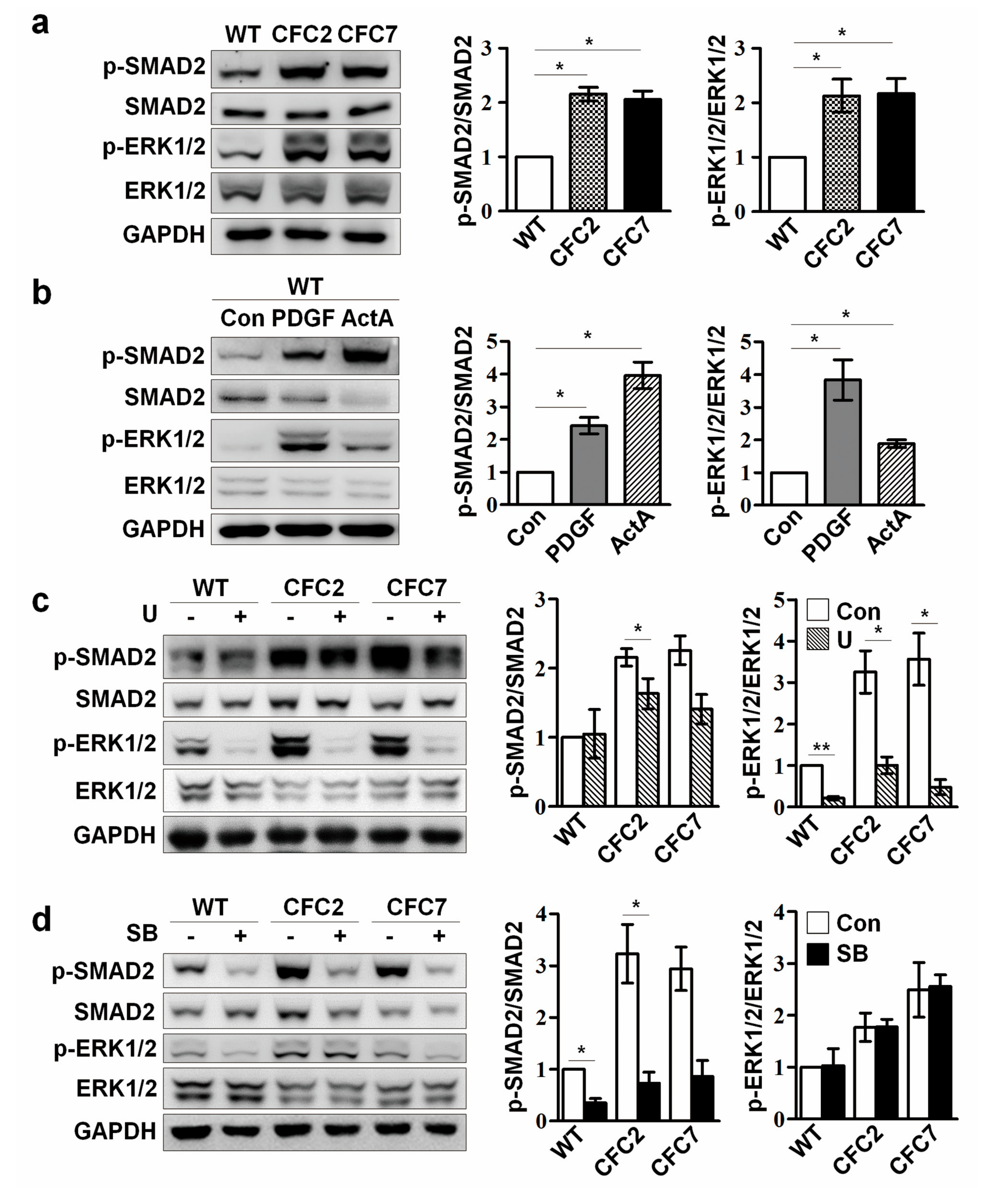
2.3. Enhanced ERK Signaling Activates SMAD2 Pathway in CFC-MSCs during Osteogenic Differentiation
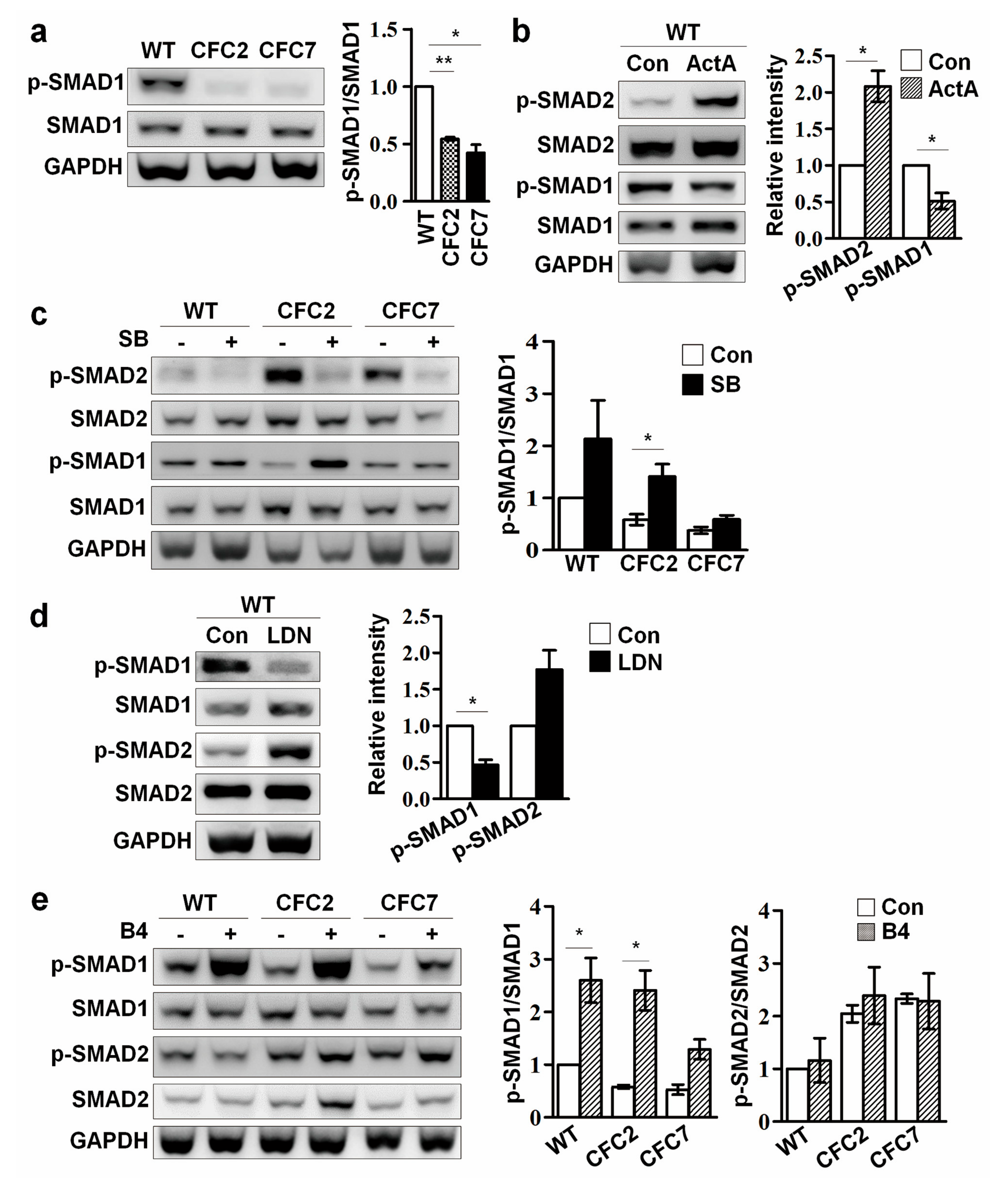
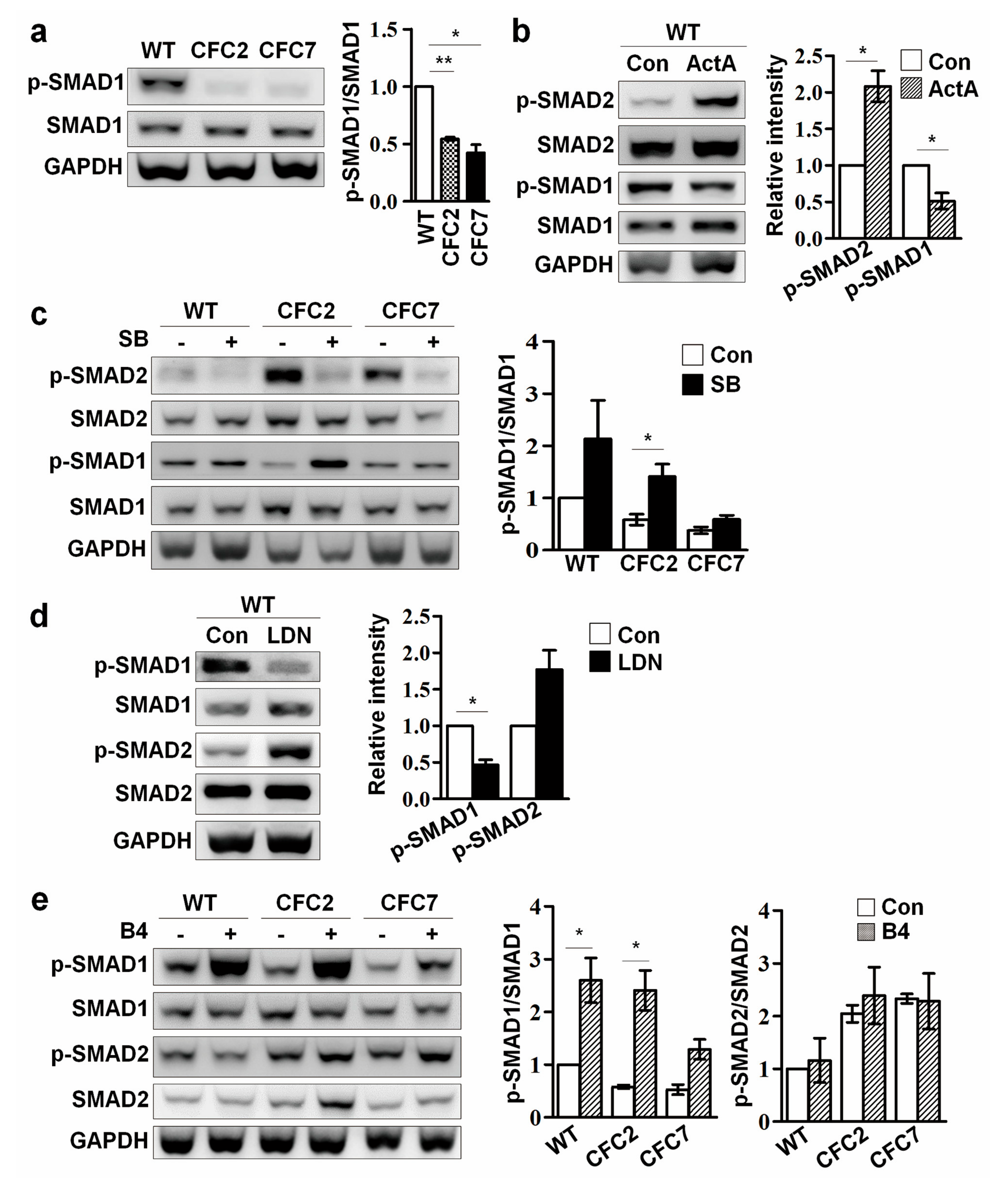
2.4. Enhanced SMAD2 Pathway Downregulates SMAD1 Pathway in CFC-MSCs during Osteogenic Differentiation
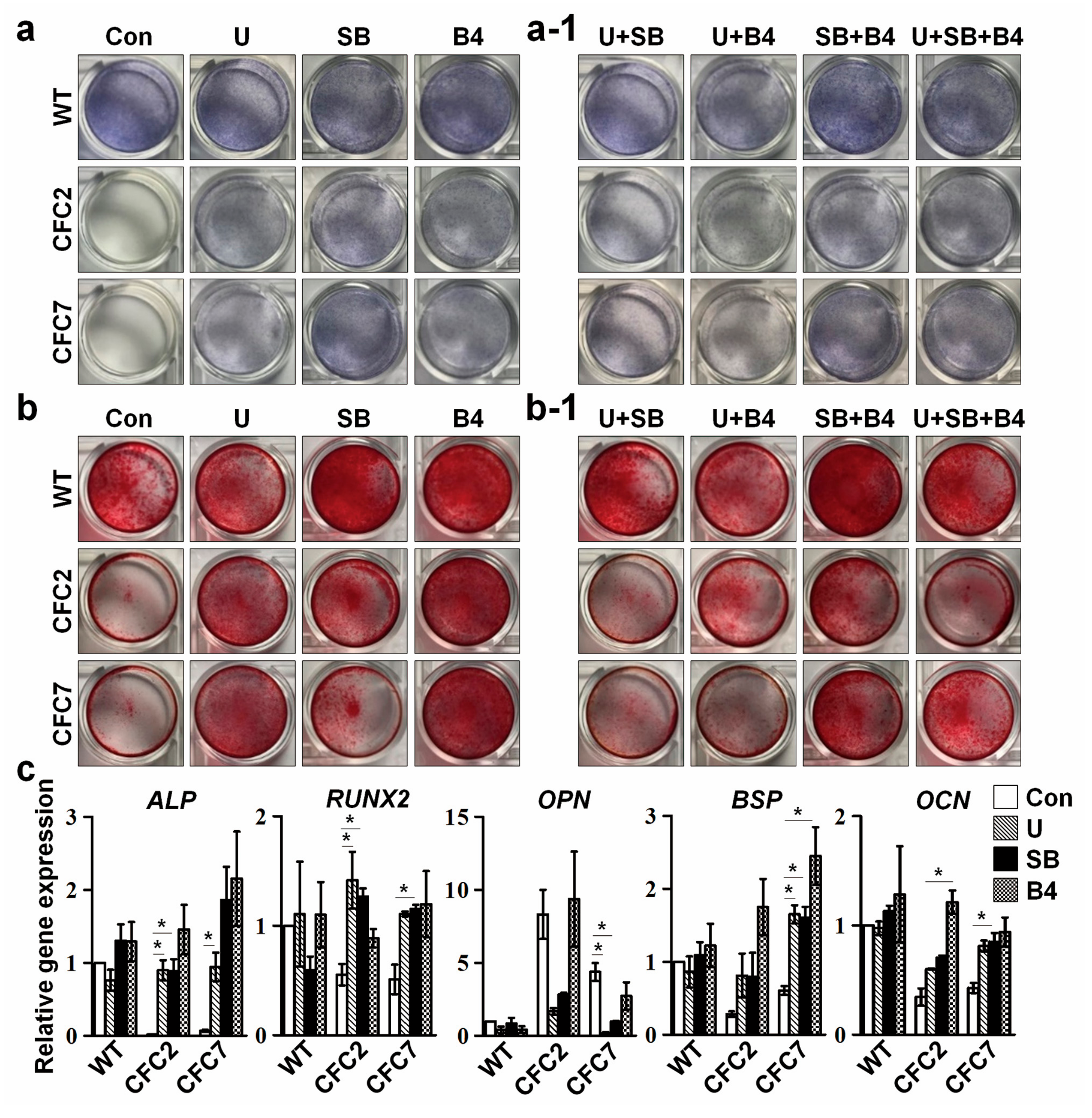
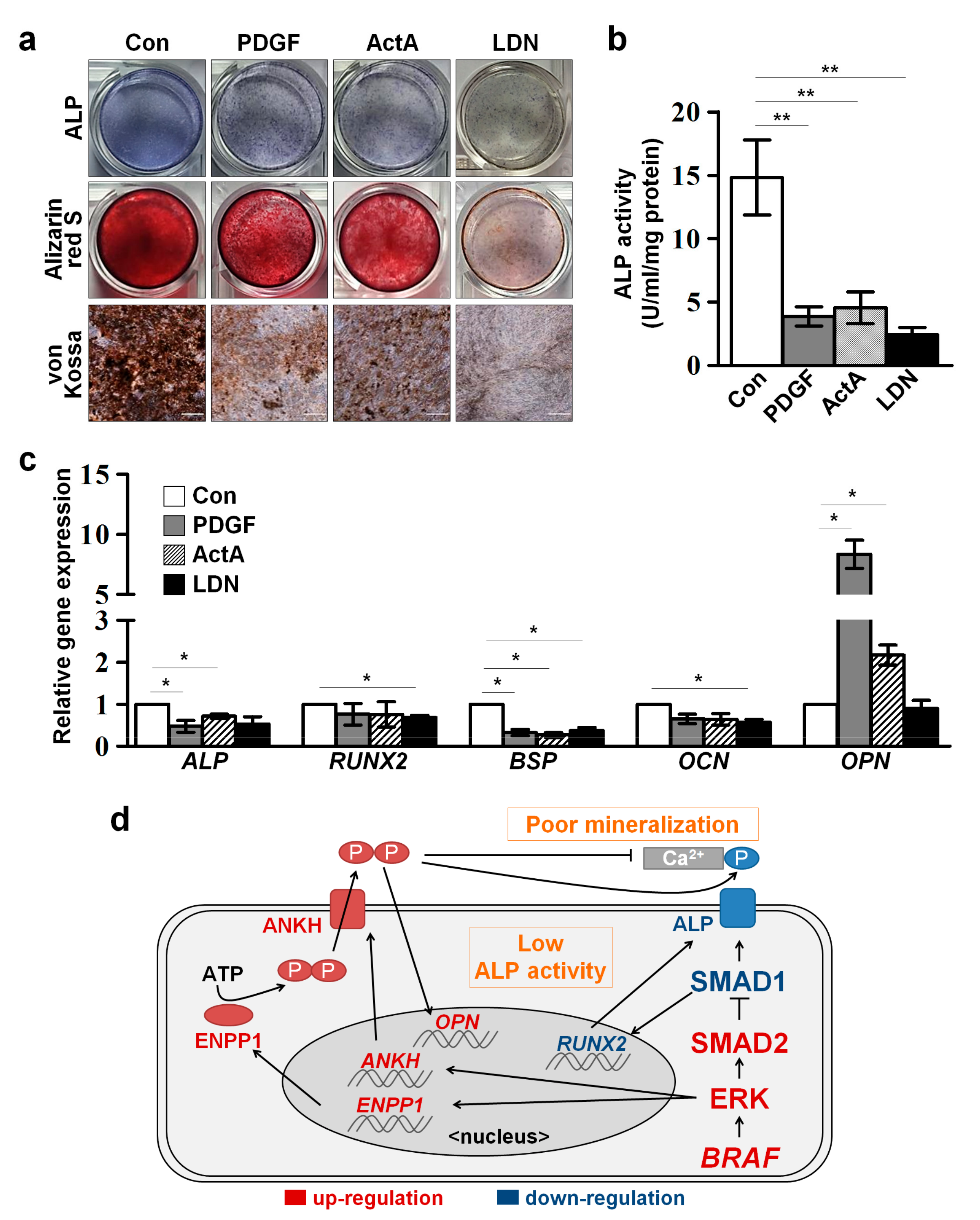
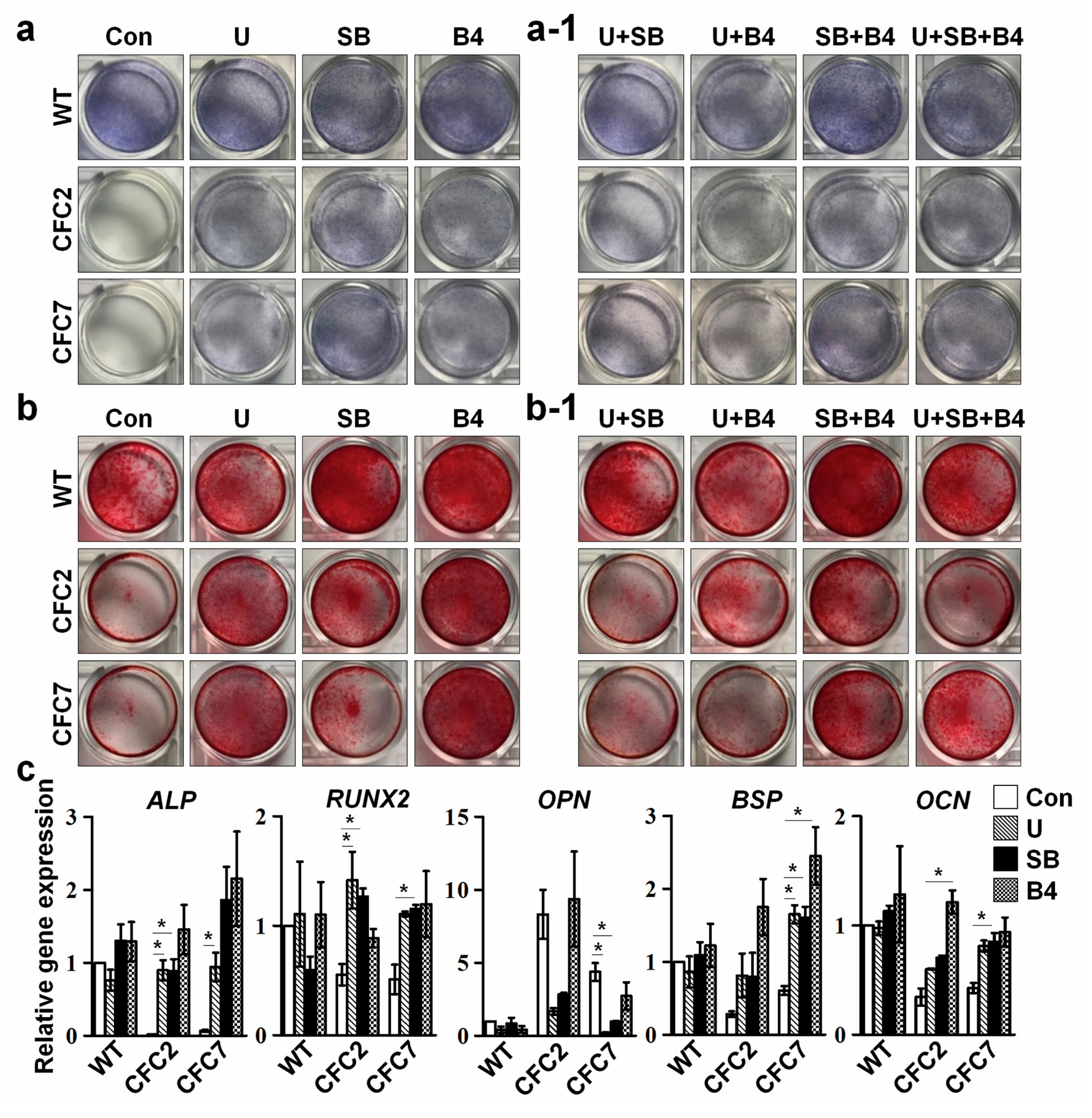
2.5. Defective Osteogenesis in CFC-MSCs Is Ameliorated by Modulation of ERK, SMAD2, and SMAD1 Signaling Pathways
3. Discussion
4. Materials and Methods
4.1. Ethical Statement
4.2. Generation and Characterization of Human iPSCs
4.3. Differentiation of iPSCs into MSCs
4.4. FACS Analysis
4.5. Differentiation of MSCs into Osteoblasts, Chondroblasts, and Adipocytes
4.6. RT-PCR and Quantitative RT-PCR
4.7. Western Blotting
4.8. Transfection of OPN Small Interfering RNA
4.9. Chemicals and Growth Factors Used for Regulating Various Signaling Pathways
4.10. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CFC | Cardiofaciocutaneous |
| ERK | Extracellular signal-regulated kinase |
| iPSC | Induced pluripotent stem cell |
| BRAF | Rapidly accelerated fibrosarcoma kinase B |
| qRT-PCR | Quantitative real-time polymerase chain reaction |
| MSC | Mesenchymal stem cell |
| WT | Wild-type |
| Ob | Osteoblast |
| HA | Hydroxyapatite |
| ALP | Alkaline phosphatase |
| PPi | Pyrophosphate |
| ENPP1 | Ectonucleotide pyrophasphatase/phosphodiesterase 1 |
| ANKH | Ankylosis human |
| OPN | Osteopontin |
| BSP | Bone sialoprotein |
| BMP | Bone morphogenetic protein |
| TGF-β | Transforming growth factor-β |
| RUNX2 | Runt-related transcription factor 2 |
| OCN | Osteocalcin |
| EB | Embryoid body |
| ActA | Activin A |
| SB | SB-431542 |
| LDN | LDN-193189 |
| PDGF | Platelet-derived growth factor-BB |
| U | U0126 |
| B4 | Bone morphogenic protein 4 |
| si-OPN | Osteopontin small interfering RNA |
| si-SCR | Scrambled small interfering RNA |
| FACS | fluorescence-activated cell sorting |
| α-MEM | α-minimum essential medium |
| FBS | Fetal bovine serum |
| SEM | Standard error of the mean |
References
- Aoki, Y.; Niihori, T.; Narumi, Y.; Kure, S.; Matsubara, Y. The RAS/MAPK syndromes: Novel roles of the RAS pathway in human genetic disorders. Hum. Mutat. 2008, 29, 992–1006. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Seger, R. The extracellular signal-regulated kinase: Multiple substrates regulate diverse cellular functions. Growth Factors 2006, 24, 21–44. [Google Scholar] [CrossRef] [PubMed]
- Rauen, K.A. The RASopathies. Annu. Rev. Genom. Hum. Genet. 2013, 14, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Jindal, G.A.; Goyal, Y.; Burdine, R.D.; Rauen, K.A.; Shvartsman, S.Y. RASopathies: Unraveling mechanisms with animal models. Dis. Model. Mech. 2015, 8, 769–782. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.F.; Neri, G.; Herrmann, J.P.; Blumberg, B.; Coldwell, J.G.; Miles, P.V.; Opitz, J.M. New Multiple Congenital-Anomalies Mental-Retardation Syndrome with Cardio-Facio-Cutaneous Involvement—The CFC Syndrome. Am. J. Med. Genet. 1986, 25, 413–427. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Viciana, P.; Tetsu, O.; Tidyman, W.E.; Estep, A.L.; Conger, B.A.; Cruz, M.S.; McCormick, F.; Rauen, K.A. Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science 2006, 311, 1287–1290. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Aoki, Y.; Kuriyama, S.; Kawame, H.; Okamoto, N.; Kurosawa, K.; Ohashi, H.; Mizuno, S.; Ogata, T.; Kure, S.; et al. Prevalence and clinical features of Costello syndrome and cardio-facio-cutaneous syndrome in Japan: Findings from a nationwide epidemiological survey. Am. J. Med. Genet. Part A 2012, 158, 1083–1094. [Google Scholar] [CrossRef] [PubMed]
- Niihori, T.; Aoki, Y.; Narumi, Y.; Neri, G.; Cave, H.; Verloes, A.; Okamoto, N.; Hennekam, R.C.M.; Gillessen-Kaesbach, G.; Wieczorek, D.; et al. Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat. Genet. 2006, 38, 294–296. [Google Scholar] [CrossRef] [PubMed]
- Moriya, M.; Inoue, S.; Miyagawa-Tomita, S.; Nakashima, Y.; Oba, D.; Niihori, T.; Hashi, M.; Ohnishi, H.; Kure, S.; Matsubara, Y.; et al. Adult mice expressing a Braf Q241R mutation on an ICR/CD-1 background exhibit a cardio-facio-cutaneous syndrome phenotype. Hum. Mol. Genet. 2015, 24, 7349–7360. [Google Scholar] [CrossRef] [PubMed]
- Soltanoff, C.S.; Chen, W.; Yang, S.; Li, Y.P. Signaling Networks that Control the Lineage Commitment and Differentiation of Bone Cells. Crit. Rev. Eukar. Gene Expr. 2009, 19, 1–46. [Google Scholar] [CrossRef]
- Terkeltaub, R.A. Inorganic pyrophosphate generation and disposition in pathophysiology. Am. J. Physiol.-Cell Physiol. 2001, 281, C1–C11. [Google Scholar] [PubMed]
- Johnson, K.; Goding, J.; Van Etten, D.; Sali, A.; Hu, S.I.; Farley, D.; Krug, H.; Hessle, L.; Millan, J.L.; Terkeltaub, R. Linked deficiencies in extracellular PP(i) and osteopontin mediate pathologic calcification associated with defective PC-1 and ANK expression. J. Bone Miner. Res. 2003, 18, 994–1004. [Google Scholar] [CrossRef] [PubMed]
- Addison, W.N.; Azari, F.; Sorensen, E.S.; Kaartinen, M.T.; Mckee, M.D. Pyrophosphate inhibits mineralization of osteoblast cultures by binding to mineral, up-regulating osteopontin, and inhibiting alkaline phosphatase activity. J. Biol. Chem. 2007, 282, 15872–15883. [Google Scholar] [CrossRef] [PubMed]
- Foster, B.L.; Soenjaya, Y.; Nociti, F.H.; Holm, E.; Zerfas, P.M.; Wimer, H.F.; Holdsworth, D.W.; Aubin, J.E.; Hunter, G.K.; Goldberga, H.A.; et al. Deficiency in Acellular Cementum and Periodontal Attachment in Bsp Null Mice. J. Dent. Res. 2013, 92, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.L.; Sharff, K.A.; Tang, N.; Song, W.X.; Luo, J.Y.; Luo, X.J.; Chen, J.; Bennett, E.; Reid, R.; Manning, D.; et al. Regulation of osteogenic differentiation during skeletal development. Front. Biosci. 2008, 13, 2001–2021. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Q.; Deng, C.X.; Li, Y.P. TGF-beta and BMP Signaling in Osteoblast Differentiation and Bone Formation. Int. J. Biol. Sci. 2012, 8, 272–288. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.S.; Serra, R. Deletion of Tgfbr2 in Prx1-cre expressing mesenchyme results in defects in development of the long bones and joints. Dev. Biol. 2007, 310, 304–316. [Google Scholar] [CrossRef] [PubMed]
- Yoon, B.S.; Ovchinnikov, D.A.; Yoshii, I.; Mishina, Y.; Behringer, R.R.; Lyons, K.M. Bmpr1a and Bmpr1b have overlapping functions and are essential for chondrogenesis in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 5062–5067. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, A.; Tsuji, K.; Cox, K.; Harfe, B.D.; Rosen, V.; Tabin, C.J. Genetic analysis of the roles of BMP2, BMP4, and BMP7 in limb patterning and skeletogenesis. PLoS. Genet. 2006, 2, 2116–2130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, K.M.; Kim, S.K.; Kim, D.; Choi, J.Y.; Im, I.; Hwang, K.S.; Kim, C.H.; Lee, B.H.; Yoo, H.W.; Han, Y.M. Enhanced SMAD1 Signaling Contributes to Impairments of Early Development in CFC-iPSCs. Stem Cells 2015, 33, 1447–1455. [Google Scholar] [CrossRef] [PubMed]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Mornet, E. Hypophosphatasia. Best Pract. Res. Clin. Rheumatol. 2008, 22, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.M.; Lee, E.H. Transcriptional regulatory cascades in Runx2-dependent bone development. Tissue Eng. Part B Rev. 2013, 19, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Boufker, H.I.; Lagneaux, L.; Najar, M.; Piccart, M.; Ghanem, G.; Body, J.J.; Journe, F. The Src inhibitor dasatinib accelerates the differentiation of human bone marrow-derived mesenchymal stromal cells into osteoblasts. BMC Cancer 2010, 10. [Google Scholar] [CrossRef] [PubMed]
- Erlebacher, A.; Derynck, R. Increased expression of TGF-beta 2 in osteoblasts results in an osteoporosis-like phenotype. J. Cell Biol. 1996, 132, 195–210. [Google Scholar] [CrossRef] [PubMed]
- Alliston, T.; Choy, L.; Ducy, P.; Karsenty, G.; Derynck, R. TGF-beta-induced repression of CBFA1 by Smad3 decreases cbfa1 and osteocalcin expression and inhibits osteoblast differentiation. EMBO J. 2001, 20, 2254–2272. [Google Scholar] [CrossRef] [PubMed]
- Phimphilai, M.; Zhoa, Z.R.; Boules, H.; Roca, H.; Franceschi, R.T. BMP signaling is required for RUNX2-dependent induction of the osteoblast phenotype. J. Bone Miner. Res. 2006, 21, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Papachroni, K.K.; Karatzas, D.N.; Papavassiliou, K.A.; Basdra, E.K.; Papavassiliou, A.G. Mechanotransduction in osteoblast regulation and bone disease. Trends Mol. Med. 2009, 15, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.H.; Liu, Z.Y.; Chen, Y.G. Regulation of TGF-beta signaling by Smad7. Acta Biochim. Biophys. Sin. 2009, 41, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Zhang, C.; Bollag, G.; Shokat, K.M.; Rosen, N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010, 464, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Harmey, D.; Hessle, L.; Narisawa, S.; Johnson, K.A.; Terkeltaub, R.; Millan, J.L. Concerted regulation of inorganic pyrophosphate and osteopontin by Akp2, Enpp1, and Ank: An integrated model of the pathogenesis of mineralization disorders. Am. J. Pathol. 2004, 164, 1199–1209. [Google Scholar] [CrossRef]
- Rahman, M.S.; Akhtar, N.; Jamil, H.M.; Banik, R.S.; Asaduzzaman, S.M. TGF-beta/BMP signaling and other molecular events: Regulation of osteoblastogenesis and bone formation. Bone Res. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Neptune, E.R.; Frischmeyer, P.A.; Arking, D.E.; Myers, L.; Bunton, T.E.; Gayraud, B.; Ramirez, F.; Sakai, L.Y.; Dietz, H.C. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat. Genet. 2003, 33, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Quarto, N.; Li, S.; Renda, A.; Longaker, M.T. Exogenous activation of BMP-2 signaling overcomes TGFbeta-mediated inhibition of osteogenesis in Marfan embryonic stem cells and Marfan patient-specific induced pluripotent stem cells. Stem Cells 2012, 30, 2709–2719. [Google Scholar] [CrossRef] [PubMed]
- Grafel, I.; Yang, T.; Alexander, S.; Homan, E.P.; Lietman, C.; Jiang, M.M.; Bertin, T.; Munivez, E.; Chen, Y.Q.; Dawson, B.; et al. Excessive transforming growth factor-beta signaling is a common mechanism in osteogenesis imperfecta. Nat. Med. 2014, 20, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, S.D.; Wu, X.H.; He, Y.Z.; Chen, S.; Yang, H.; Staser, K.W.; Wang, J.P.; Zhang, P.; Jiang, C.; Yokota, H.; et al. Hyperactive Transforming Growth Factor-beta 1 Signaling Potentiates Skeletal Defects in a Neurofibromatosis Type 1 Mouse Model. J. Bone Miner. Res. 2013, 28, 2476–2489. [Google Scholar] [CrossRef] [PubMed]
- Itoh, F.; Asao, H.; Sugamura, K.; Heldin, C.H.; Ten Dijke, P.; Itoh, S. Promoting bone morphogenetic protein signaling through negative regulation of inhibitory Smads. EMBO J. 2001, 20, 4132–4142. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Hayashi, M.; Komiya, S.; Imamura, T.; Miyazono, K. Endogenous TGF-beta signaling suppresses maturation of osteoblastic mesenchymal cells. EMBO J. 2004, 23, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Tidyman, W.E.; Rauen, K.A. The RASopathies: Developmental syndromes of Ras/MAPK pathway dysregulation. Curr. Opin. Genet. Dev. 2009, 19, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, D.A.; Yang, F.C. The Musculoskeletal Phenotype of the RASopathies. Am. J. Med. Genet. Part C Semin. Med. Genet. 2011, 157, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Colbert, M.; Krenz, M.; Molkentin, J.D.; Hahn, H.S.; Dorn, G.W.; Robbins, J. Mediating ERK1/2 signaling rescues congenital heart defects in a mouse model of Noonan syndrome. J. Clin. Investig. 2007, 117, 2123–2132. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Simpson, J.; Hong, J.H.; Kim, K.H.; Thavarajah, N.K.; Backx, P.H.; Neel, B.G.; Araki, T. MEK-ERK pathway modulation ameliorates disease phenotypes in a mouse model of Noonan syndrome associated with the Raf1(L613V) mutation. J. Clin. Investig. 2011, 121, 1009–1025. [Google Scholar] [CrossRef] [PubMed]
- Anastasaki, C.; Rauen, K.A.; Patton, E.E. Continual low-level MEK inhibition ameliorates cardio-facio-cutaneous phenotypes in zebrafish. Dis. Model. Mech. 2012, 5, 546–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Kim, E.; Wang, X.; Novitch, B.G.; Yoshikawa, K.; Chang, L.S.; Zhu, Y. ERK Inhibition Rescues Defects in Fate Specification of Nf1-Deficient Neural Progenitors and Brain Abnormalities. Cell 2012, 150, 816–830. [Google Scholar] [CrossRef] [PubMed]
- De la Croix Ndong, J.; Makowski, A.J.; Uppuganti, S.; Vignaux, G.; Ono, K.; Perrien, D.S.; Joubert, S.; Baglio, S.R.; Granchi, D.; Stevenson, D.A.; et al. Asfotase-alpha improves bone growth, mineralization and strength in mouse models of neurofibromatosis type-1. Nat. Med. 2014, 20, 904–910. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Kim, D.; Jang, M.J.; Han, Y.M. Variations in the epigenetic regulation of lineage-specific genes among human pluripotent stem cell lines. Biochem. Biophys. Res. Commun. 2012, 424, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, A.; Harkness, L.; Schroder, H.D.; Abdallah, B.M.; Kassem, M. Enhanced Differentiation of Human Embryonic Stem Cells to Mesenchymal Progenitors by Inhibition of TGF-beta/Activin/Nodal Signaling Using SB-431542. J. Bone Miner. Res. 2010, 25, 1216–1233. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.T.; Trinh, Q.M.; Lee, G.M.; Han, Y.M. Efficient Differentiation of Human Pluripotent Stem Cells into Mesenchymal Stem Cells by Modulating Intracellular Signaling Pathways in a Feeder/Serum-Free System. Stem Cells Dev. 2012, 21, 1165–1175. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Choi, J.; Han, K.M.; Lee, B.H.; Choi, J.H.; Yoo, H.W.; Han, Y.M. Impaired osteogenesis in Menkes disease-derived induced pluripotent stem cells. Stem Cell Res. Ther. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, J.-Y.; Han, K.-M.; Kim, D.; Lee, B.-H.; Yoo, H.-W.; Choi, J.-H.; Han, Y.-M. Impaired Osteogenesis of Disease-Specific Induced Pluripotent Stem Cells Derived from a CFC Syndrome Patient. Int. J. Mol. Sci. 2017, 18, 2591. https://doi.org/10.3390/ijms18122591
Choi J-Y, Han K-M, Kim D, Lee B-H, Yoo H-W, Choi J-H, Han Y-M. Impaired Osteogenesis of Disease-Specific Induced Pluripotent Stem Cells Derived from a CFC Syndrome Patient. International Journal of Molecular Sciences. 2017; 18(12):2591. https://doi.org/10.3390/ijms18122591
Chicago/Turabian StyleChoi, Jung-Yun, Kyu-Min Han, Dongkyu Kim, Beom-Hee Lee, Han-Wook Yoo, Jin-Ho Choi, and Yong-Mahn Han. 2017. "Impaired Osteogenesis of Disease-Specific Induced Pluripotent Stem Cells Derived from a CFC Syndrome Patient" International Journal of Molecular Sciences 18, no. 12: 2591. https://doi.org/10.3390/ijms18122591