Ethanol (E) Impairs Fetal Brain GSH Homeostasis by Inhibiting Excitatory Amino-Acid Carrier 1 (EAAC1)-Mediated Cysteine Transport

Abstract
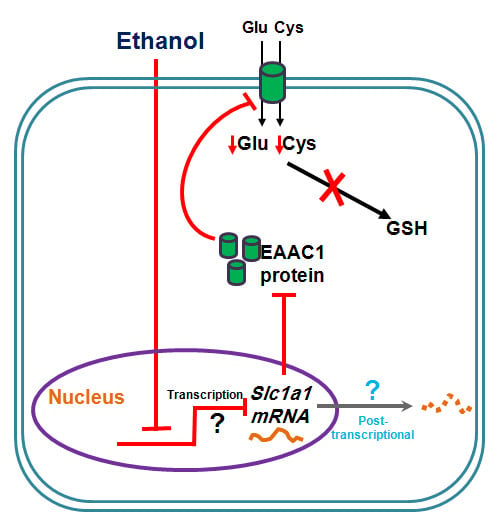
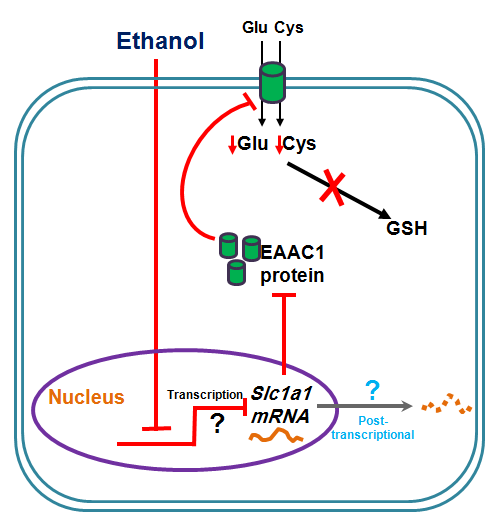
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
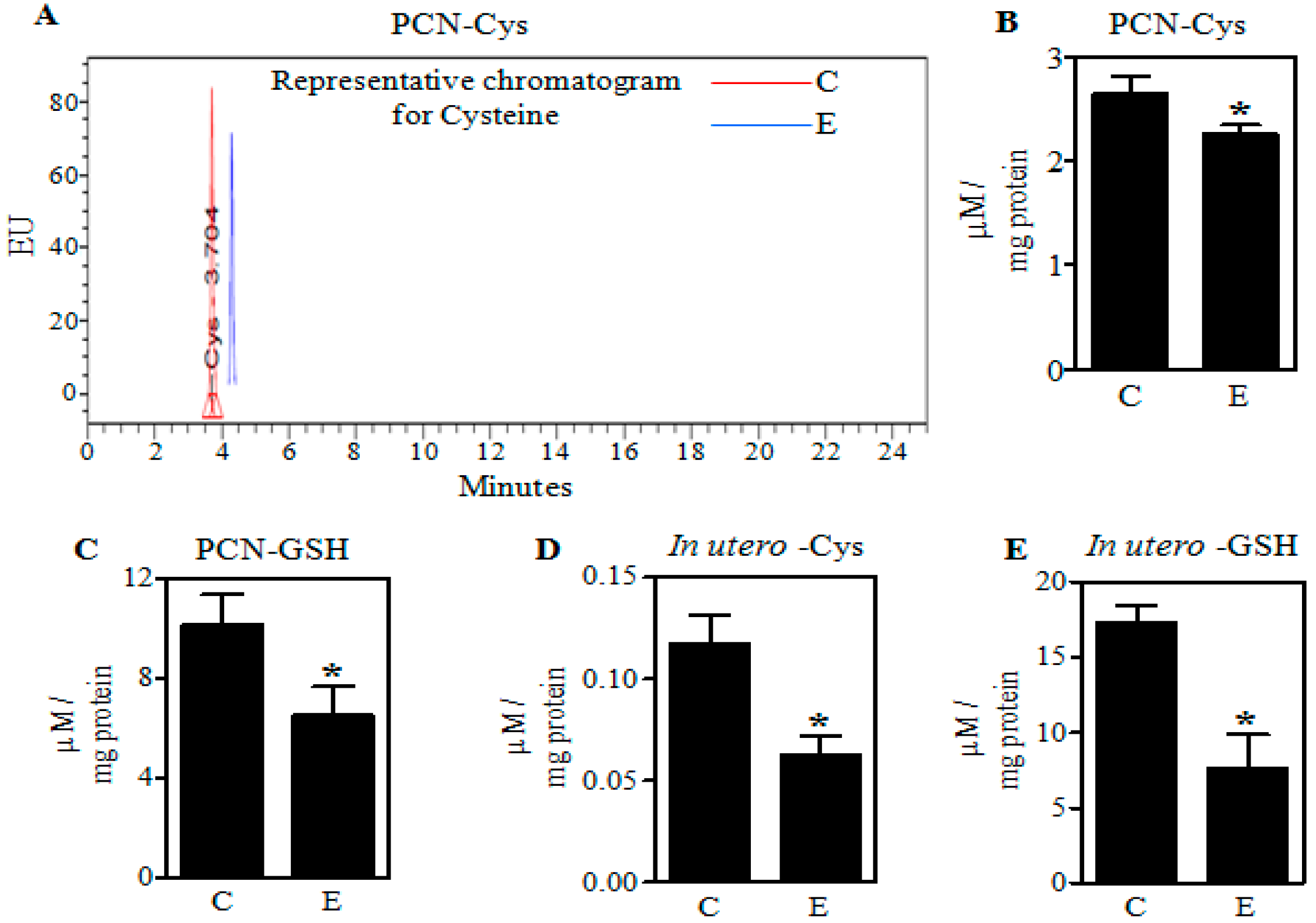
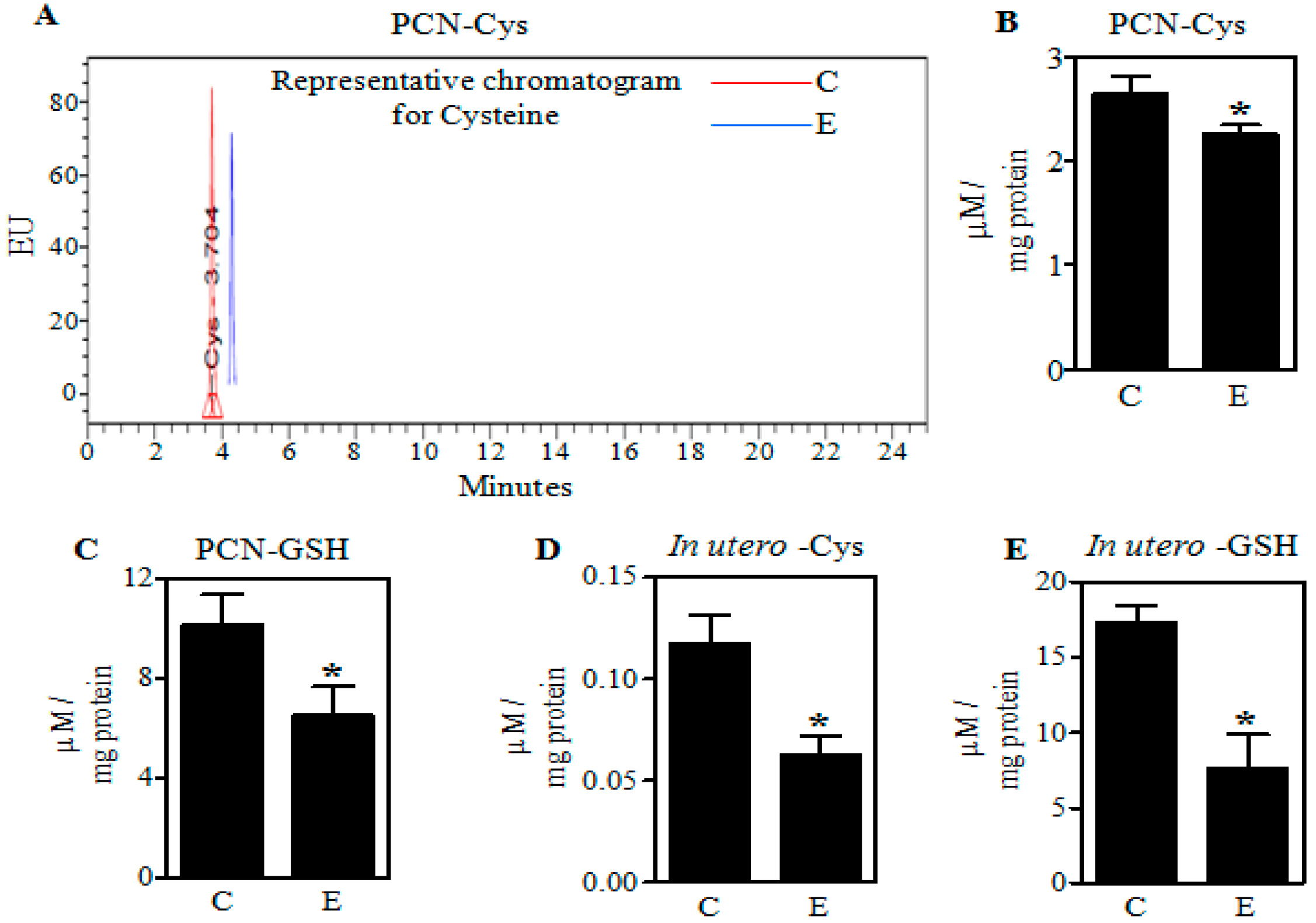
2.1. Ethanol Decreases Cysteine (Cys) and Glutathione (GSH) in Primary Cerebral Cortical Neurons (PCNs) and Fetal Brains
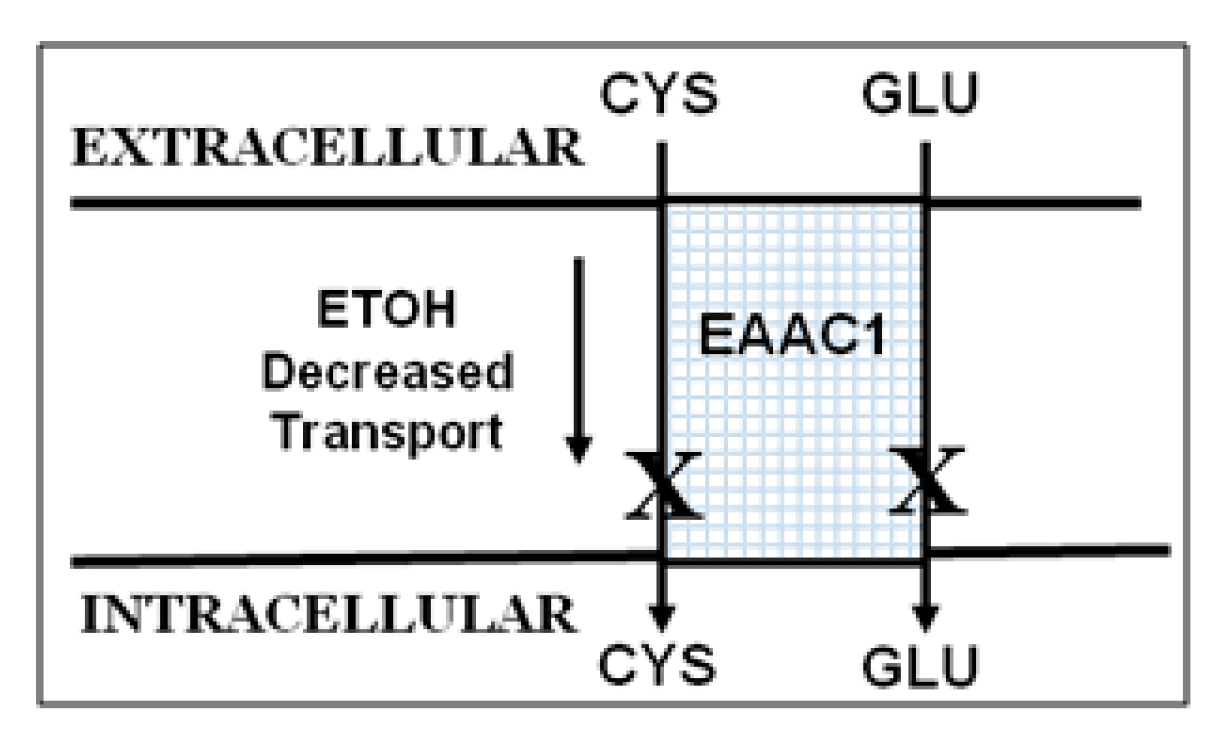
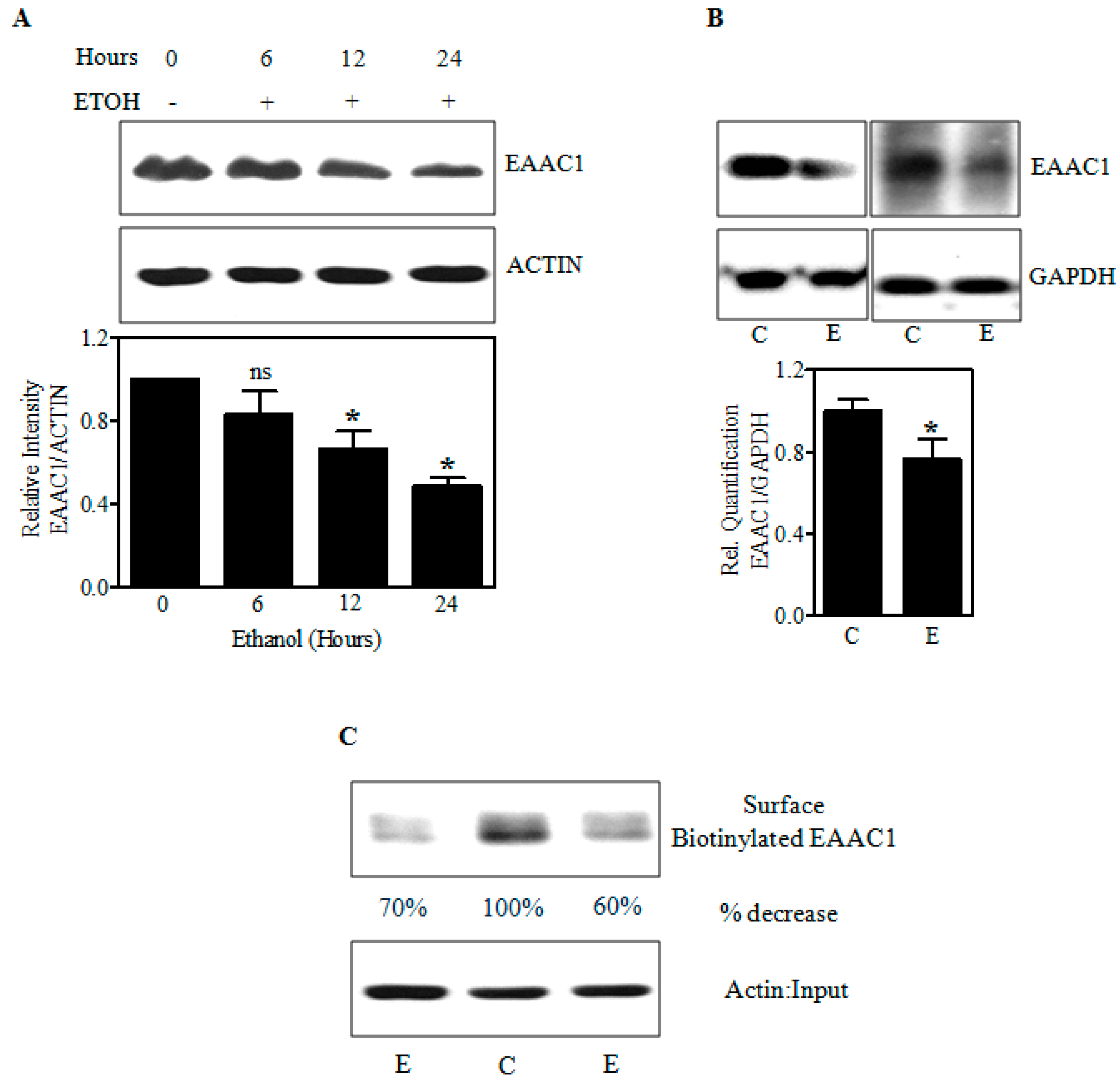
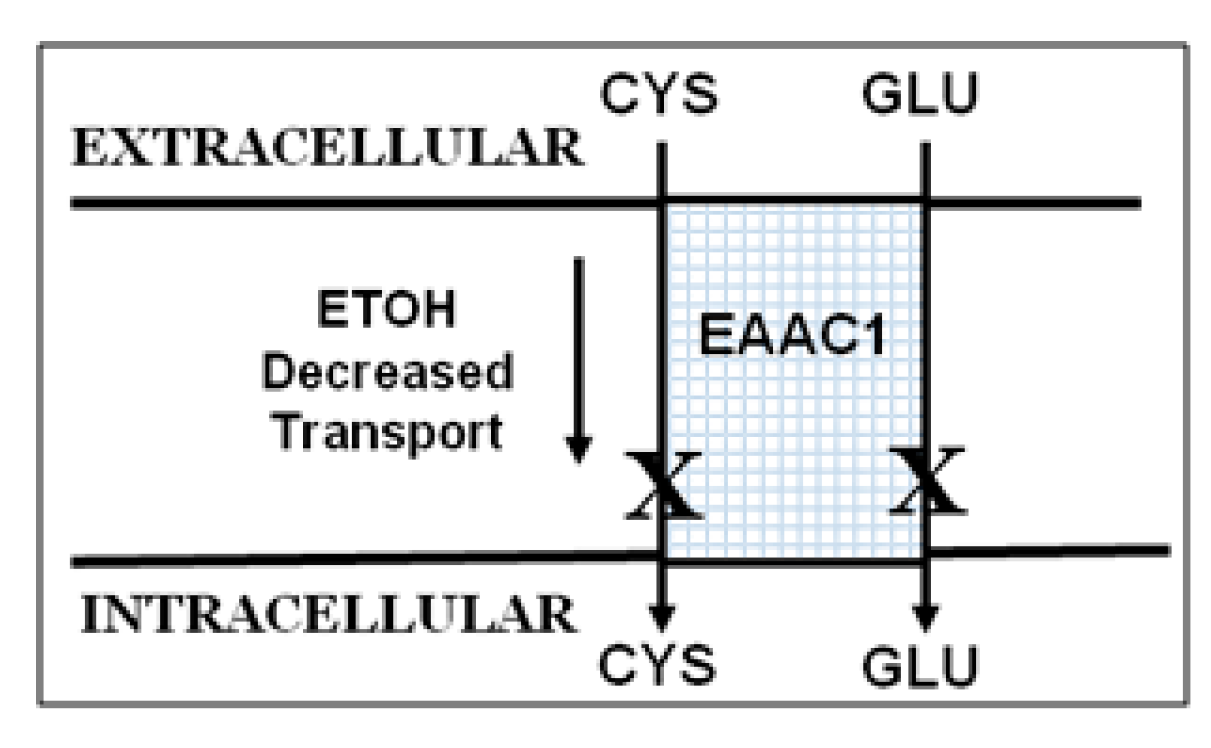
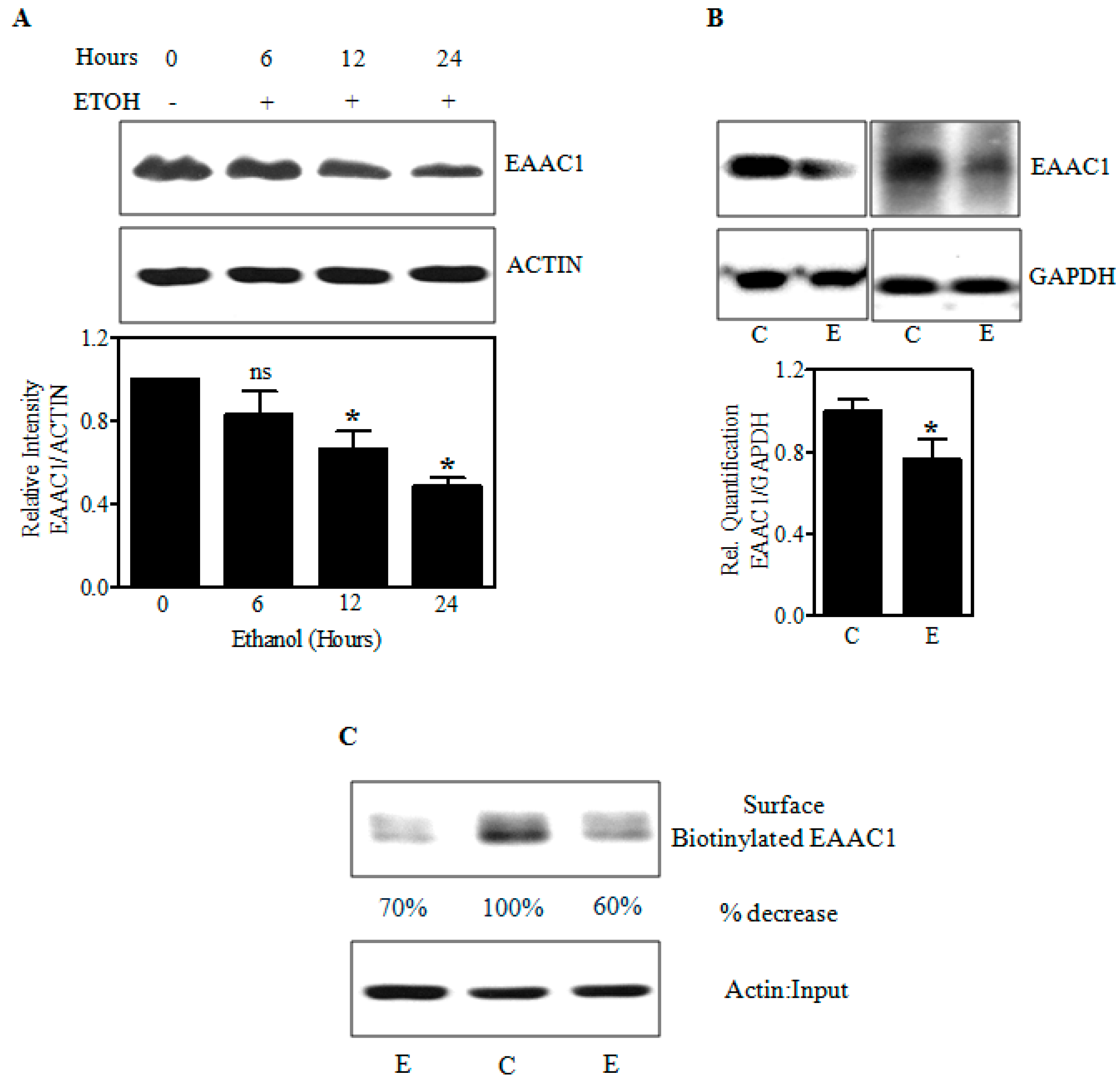
2.2. Ethanol Decreases Excitatory Amino Acid Carrier1 (EAAC1) Protein and Its Membrane Expression
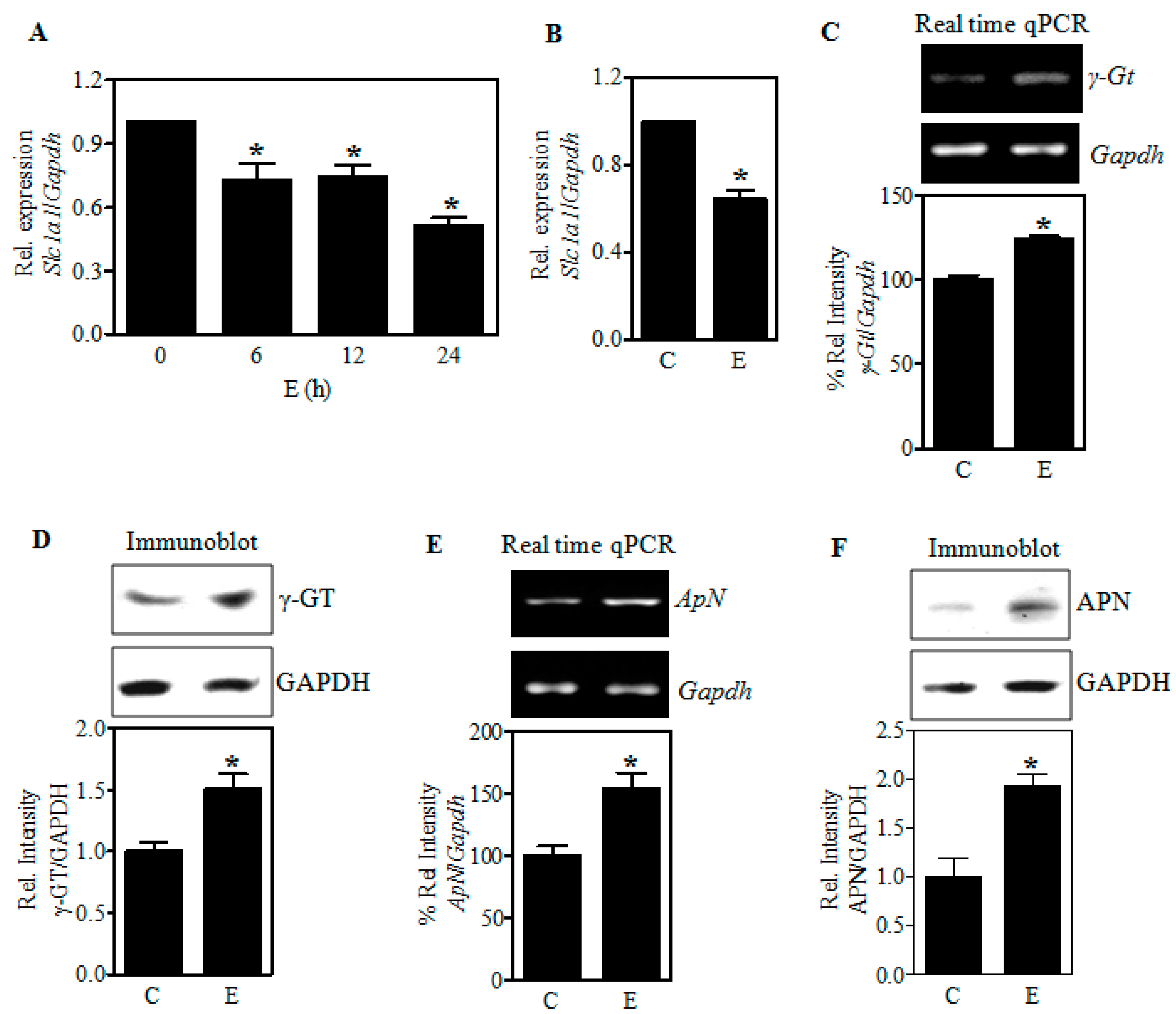
2.3. Ethanol-Induced Reduction of EAAC1 Protein Is Associated with a Decrease in Its Transcript Levels
2.4. Ethanol Does Not Restrict the Expression of γ-Glutamyl Transpeptidase (γGT) and Aminopeptidase N (APN), the Extracellular GSH-Cleaving Cysteine Homeostasis Factors
2.5. Inhibition of EAAC1 Expression (as by E) Reduces Neuron GSH Synthesis
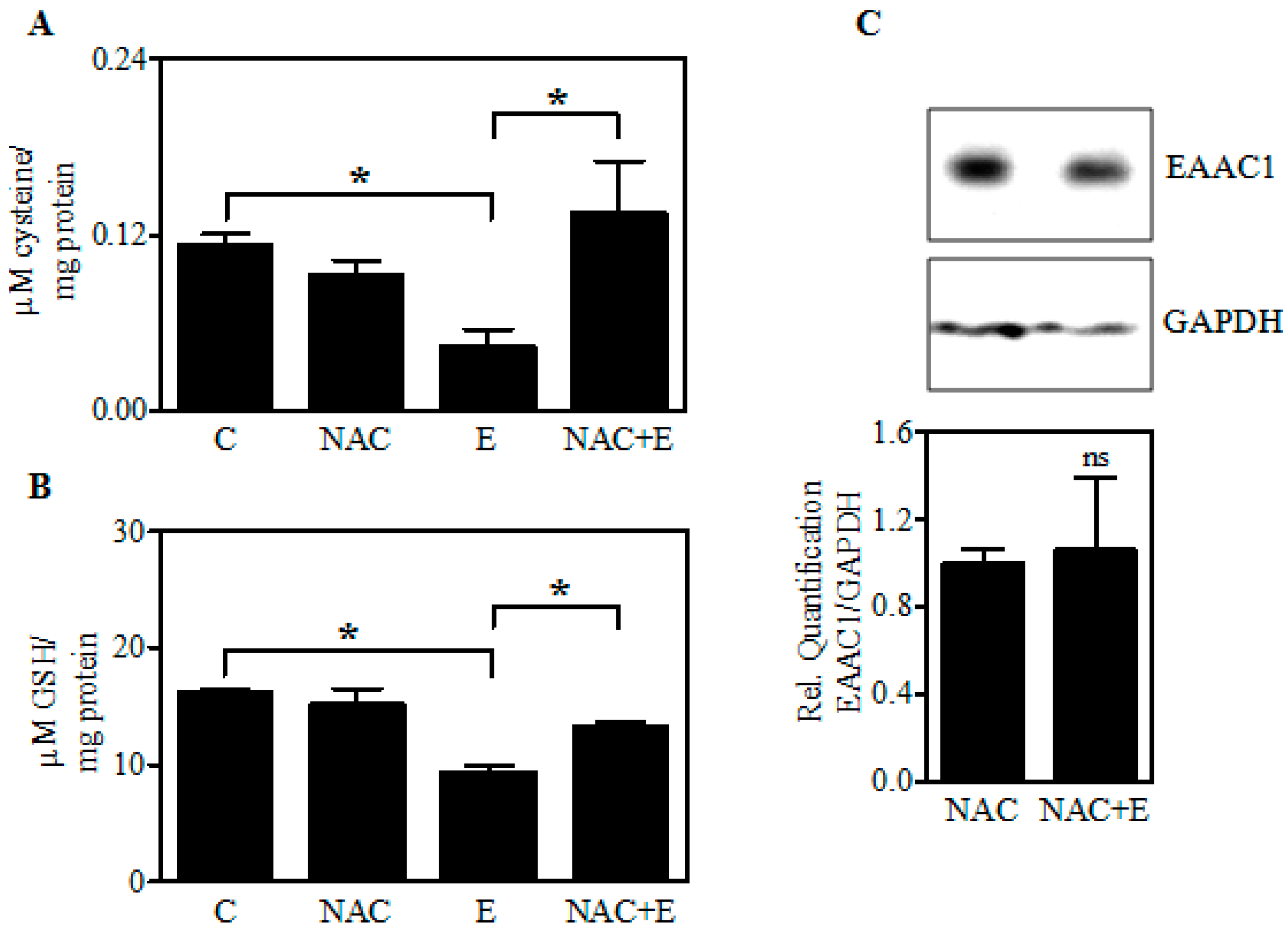
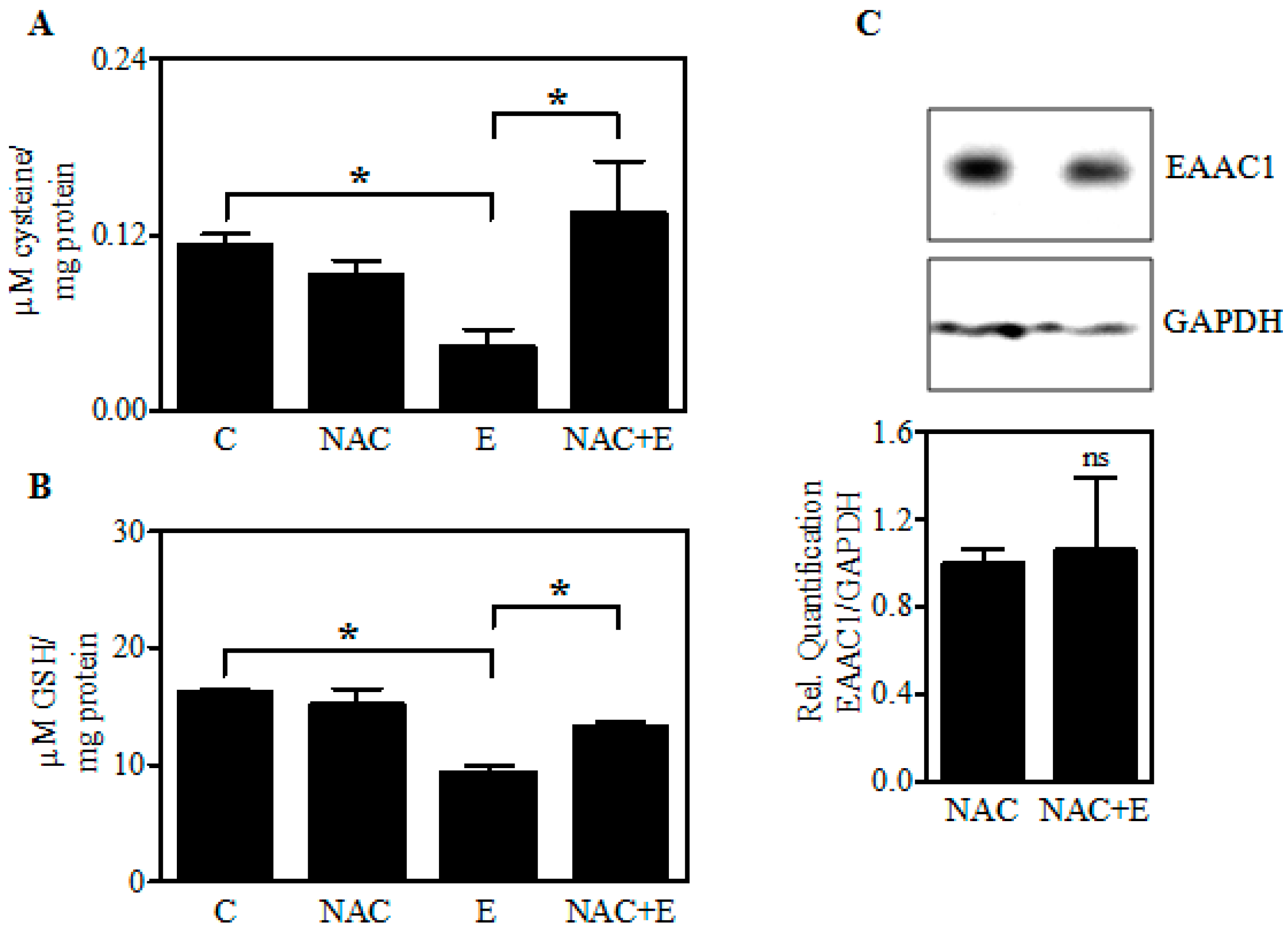
2.6. Increasing Fetal Brain Cys Prevents the Ethanol-Mediated GSH Reduction
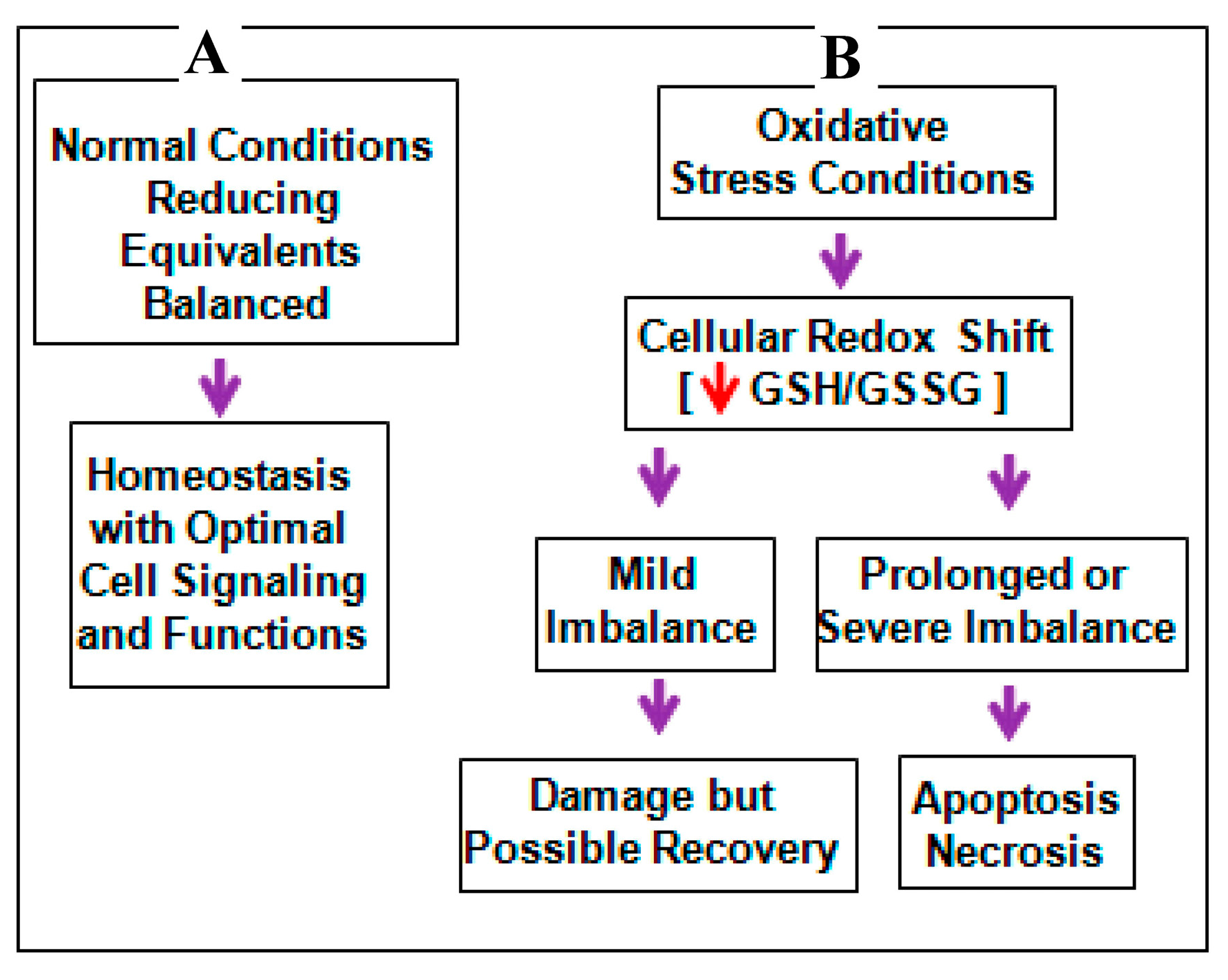
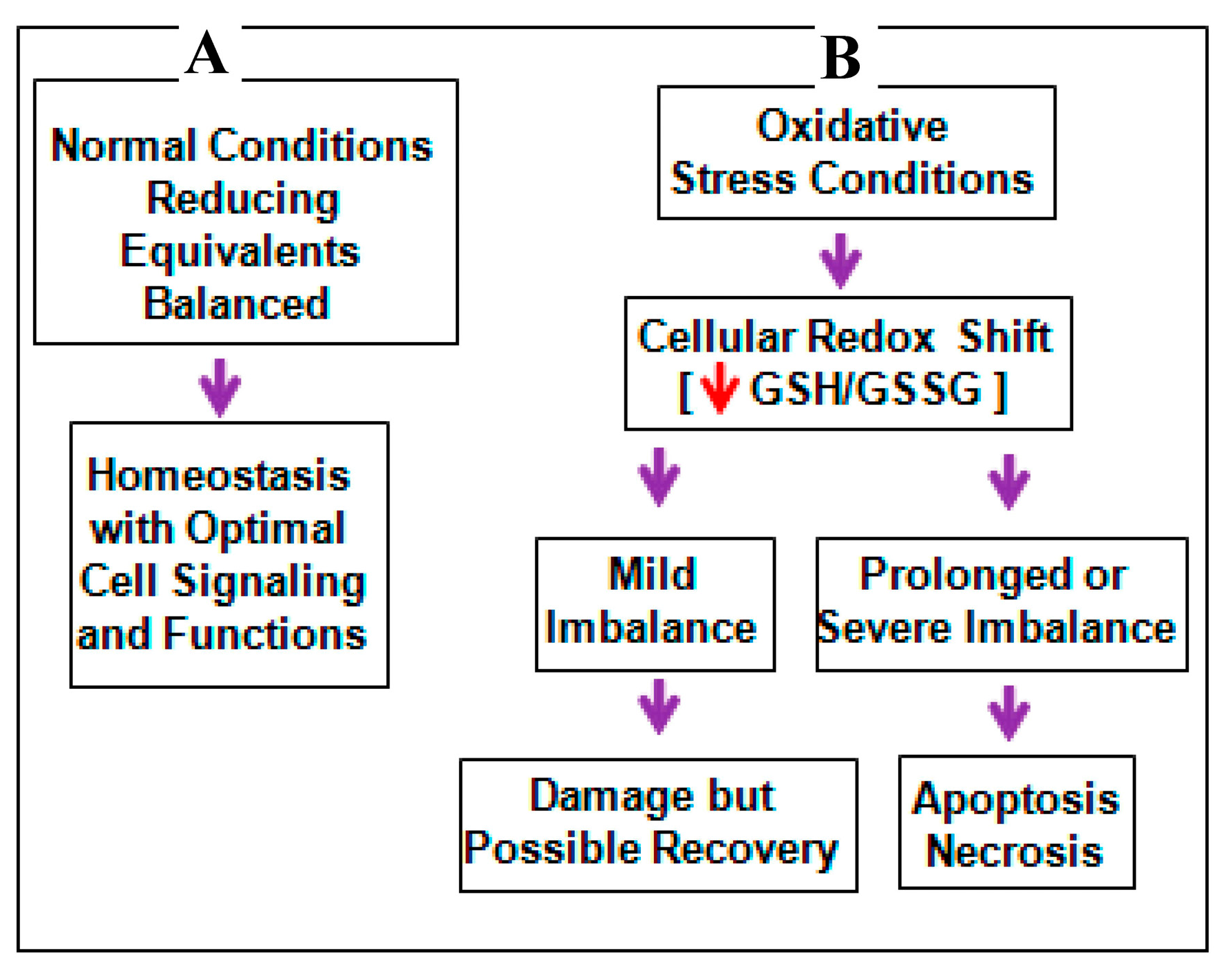
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Primary Cortical Neuron (PCN) Cultures and Ethanol Treatment
4.3. In Vivo Model
4.4. HPLC Based Determination of Cysteine and Total GSH
4.5. RNA Extraction and Real-Time qRT-PCR Analysis
4.6. Reverse Transcription-PCR Analysis
4.7. Gel Electrophoresis and Immunoblotting
4.8. In Vitro Biotinylation Assay for Cell Surface Determination of EAAC1
4.9. Small Interfering RNA (siRNA) Transfection
4.10. Fluorimetry Based Assay for Reduced and Oxidized Glutathione (GSH/GSSG)
4.11. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| GSH | Reduced Glutathione |
| GSSG | Oxidized Glutathione |
| Cys | Cysteine |
| EAAC1/EAAT3/Slc1a1 | Excitatory Amino Acid Carrier 1 |
| γGT | Gamma-glutamyl Transpeptidase |
| APN | Amionopeptidase N |
References
- Bearer, C.F.; Swick, A.R.; O’Riordan, M.A.; Cheng, G. Ethanol inhibits L1-mediated neurite outgrowth in postnatal rat cerebellar granule cells. J. Biol. Chem. 1999, 274, 13264–13270. [Google Scholar] [CrossRef] [PubMed]
- Goodlett, C.R.; Thomas, J.D.; West, J.R. Long-term deficits in cerebellar growth and rotarod performance of rats following “binge-like” alcohol exposure during the neonatal brain growth spurt. Neurotoxicol. Teratol. 1991, 13, 69–74. [Google Scholar] [CrossRef]
- Kodali, V.N.; Jacobson, J.L.; Lindinger, N.M.; Dodge, N.C.; Molteno, C.D.; Meintjes, E.M.; Jacobson, S.W. Differential recruitment of brain regions during response inhibition in children prenatally exposed to alcohol. Alcohol. Clin. Exp. Res. 2017, 41, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Mooney, S.M.; Napper, R.M. Early postnatal exposure to alcohol reduces the number of neurons in the occipital but not the parietal cortex of the rat. Alcohol. Clin. Exp. Res. 2005, 29, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Maffi, S.K.; Rathinam, M.L.; Cherian, P.P.; Pate, W.; Hamby-Mason, R.; Schenker, S.; Henderson, G.I. Glutathione content as a potential mediator of the vulnerability of cultured fetal cortical neurons to ethanol-induced apoptosis. J. Neurosci. Res. 2008, 86, 1064–1076. [Google Scholar] [CrossRef] [PubMed]
- Maier, S.E.; West, J.R. Regional differences in cell loss associated with binge-like alcohol exposure during the first two trimesters equivalent in the rat. Alcohol 2001, 23, 49–57. [Google Scholar] [CrossRef]
- Narasimhan, M.; Mahimainathan, L.; Rathinam, M.L.; Riar, A.K.; Henderson, G.I. Overexpression of Nrf2 protects cerebral cortical neurons from ethanol-induced apoptotic death. Mol. Pharmacol. 2011, 80, 988–999. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, V.; Perez, A.; Chen, J.; Senthil, D.; Schenker, S.; Henderson, G.I. In utero ethanol exposure causes mitochondrial dysfunction, which can result in apoptotic cell death in fetal brain: A potential role for 4-hydroxynonenal. Alcohol. Clin. Exp. Res. 2001, 25, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, V.; Watts, L.T.; Maffi, S.K.; Chen, J.; Schenker, S.; Henderson, G. Ethanol-induced oxidative stress precedes mitochondrially mediated apoptotic death of cultured fetal cortical neurons. J. Neurosci. Res. 2003, 74, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Young, C.; Olney, J.W. Neuroapoptosis in the infant mouse brain triggered by a transient small increase in blood alcohol concentration. Neurobiol. Dis. 2006, 22, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Young, C.; Roth, K.A.; Klocke, B.J.; West, T.; Holtzman, D.M.; Labruyere, J.; Qin, Y.Q.; Dikranian, K.; Olney, J.W. Role of caspase-3 in ethanol-induced developmental neurodegeneration. Neurobiol. Dis. 2005, 20, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Heaton, M.B.; Paiva, M.; Madorsky, I.; Shaw, G. Ethanol effects on neonatal rat cortex: Comparative analyses of neurotrophic factors, apoptosis-related proteins, and oxidative processes during vulnerable and resistant periods. Brain Res. Dev. Brain Res. 2003, 145, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Heaton, M.B.; Paiva, M.; Mayer, J.; Miller, R. Ethanol-mediated generation of reactive oxygen species in developing rat cerebellum. Neurosci. Lett. 2002, 334, 83–86. [Google Scholar] [CrossRef]
- Narasimhan, M.; Rathinam, M.; Patel, D.; Henderson, G.; Mahimainathan, L. Astrocytes prevent ethanol induced apoptosis of Nrf2 depleted neurons by maintaining GSH homeostasis. Open J. Apoptosis 2012, 1. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.M.; Harris, C. Redox control of teratogenesis. Reprod. Toxicol. 2013, 35, 165–179. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.; Shuster, D.Z.; Roman Gomez, R.; Sant, K.E.; Reed, M.S.; Pohl, J.; Hansen, J.M. Inhibition of glutathione biosynthesis alters compartmental redox status and the thiol proteome in organogenesis-stage rat conceptuses. Free Radic. Biol. Med. 2013, 63, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P. Radical-free biology of oxidative stress. Am. J. Physiol. Cell Physiol. 2008, 295, C849–C868. [Google Scholar] [CrossRef] [PubMed]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Hamby-Mason, R.L.; Mason, P.A.; Schenker, S.; Henderson, G.I. Histochemical method for localization of hydrogen peroxide and oxygen radicals in the intact neonatal brain. Methods Find. Exp. Clin. Pharmacol. 1998, 20, 743–748. [Google Scholar] [PubMed]
- Watts, L.T.; Rathinam, M.L.; Schenker, S.; Henderson, G.I. Astrocytes protect neurons from ethanol-induced oxidative stress and apoptotic death. J. Neurosci. Res. 2005, 80, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Dennery, P.A. Effects of oxidative stress on embryonic development. Birth Defects Res. C Embryo Today 2007, 81, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Fantel, A.G. Reactive oxygen species in developmental toxicity: Review and hypothesis. Teratology 1996, 53, 196–217. [Google Scholar] [CrossRef]
- Wells, P.G.; Kim, P.M.; Laposa, R.R.; Nicol, C.J.; Parman, T.; Winn, L.M. Oxidative damage in chemical teratogenesis. Mutat. Res. 1997, 396, 65–78. [Google Scholar] [CrossRef]
- Wells, P.G.; McCallum, G.P.; Chen, C.S.; Henderson, J.T.; Lee, C.J.; Perstin, J.; Preston, T.J.; Wiley, M.J.; Wong, A.W. Oxidative stress in developmental origins of disease: Teratogenesis, neurodevelopmental deficits, and cancer. Toxicol. Sci. 2009, 108, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Watabe, M.; Nakaki, T. Regulation of neuronal glutathione synthesis. J. Pharmacol. Sci. 2008, 108, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.S.; Moore, W.R.; Meister, A. On the active site thiol of γ-glutamylcysteine synthetase: Relationships to catalysis, inhibition, and regulation. Proc. Natl. Acad. Sci. USA 1988, 85, 2464–2468. [Google Scholar] [CrossRef] [PubMed]
- Meister, A.; Anderson, M.E. Glutathione. Annu. Rev. Biochem. 1983, 52, 711–760. [Google Scholar] [CrossRef] [PubMed]
- Go, Y.M.; Jones, D.P. Thiol/disulfide redox states in signaling and sensing. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Nakaki, T. Neuroprotective properties of the excitatory amino acid carrier 1 (EAAC1). Amino Acids 2013, 45, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Watabe, M.; Nakaki, T. Modulation of neuronal glutathione synthesis by EAAC1 and its interacting protein GTRAP3-18. Amino Acids 2012, 42, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.M.; Wilson-Delfosse, A.L.; Mieyal, J.J. Dysregulation of glutathione homeostasis in neurodegenerative diseases. Nutrients 2012, 4, 1399–1440. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Suh, S.W.; Hamby, A.M.; Liu, J.; Chan, W.Y.; Chen, Y.; Swanson, R.A. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat. Neurosci. 2006, 9, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Zerangue, N.; Kavanaugh, M.P. Interaction of l-cysteine with a human excitatory amino acid transporter. J. Physiol. 1996, 493, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Berman, A.E.; Chan, W.Y.; Brennan, A.M.; Reyes, R.C.; Adler, B.L.; Suh, S.W.; Kauppinen, T.M.; Edling, Y.; Swanson, R.A. N-acetylcysteine prevents loss of dopaminergic neurons in the EAAC1-/- mouse. Ann. Neurol. 2011, 69, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Li, L.; Zuo, Z. N-acetylcysteine reverses existing cognitive impairment and increased oxidative stress in glutamate transporter type 3 deficient mice. Neuroscience 2012, 220, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Gressens, P.; Lammens, M.; Picard, J.J.; Evrard, P. Ethanol-induced disturbances of gliogenesis and neuronogenesis in the developing murine brain: An in vitro and in vivo immunohistochemical and ultrastructural study. Alcohol Alcohol. 1992, 27, 219–226. [Google Scholar] [PubMed]
- Won, S.J.; Yoo, B.H.; Brennan, A.M.; Shin, B.S.; Kauppinen, T.M.; Berman, A.E.; Swanson, R.A.; Suh, S.W. EAAC1 gene deletion alters zinc homeostasis and exacerbates neuronal injury after transient cerebral ischemia. J. Neurosci. 2010, 30, 15409–15418. [Google Scholar] [CrossRef] [PubMed]
- Escartin, C.; Won, S.J.; Malgorn, C.; Auregan, G.; Berman, A.E.; Chen, P.C.; Deglon, N.; Johnson, J.A.; Suh, S.W.; Swanson, R.A. Nuclear factor erythroid 2-related factor 2 facilitates neuronal glutathione synthesis by upregulating neuronal excitatory amino acid transporter 3 expression. J. Neurosci. 2011, 31, 7392–7401. [Google Scholar] [CrossRef] [PubMed]
- Fournier, K.M.; Gonzalez, M.I.; Robinson, M.B. Rapid trafficking of the neuronal glutamate transporter, EAAC1: Evidence for distinct trafficking pathways differentially regulated by protein kinase C and platelet-derived growth factor. J. Biol. Chem. 2004, 279, 34505–34513. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.I.; Susarla, B.T.; Fournier, K.M.; Sheldon, A.L.; Robinson, M.B. Constitutive endocytosis and recycling of the neuronal glutamate transporter, excitatory amino acid carrier 1. J. Neurochem. 2007, 103, 1917–1931. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Valencia, A.; Sapp, E.; Masso, N.; Alexander, J.; Reeves, P.; Kegel, K.B.; Aronin, N.; Difiglia, M. Aberrant Rab11-dependent trafficking of the neuronal glutamate transporter EAAC1 causes oxidative stress and cell death in Huntington’s disease. J. Neurosci. 2010, 30, 4552–4561. [Google Scholar] [CrossRef] [PubMed]
- Farr, S.A.; Poon, H.F.; Dogrukol-Ak, D.; Drake, J.; Banks, W.A.; Eyerman, E.; Butterfield, D.A.; Morley, J.E. The antioxidants alpha-lipoic acid and N-acetylcysteine reverse memory impairment and brain oxidative stress in aged SAMP8 mice. J. Neurochem. 2003, 84, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.; Won, S.J.; Park, Y.; Orr, A.; Jones, D.P.; Swanson, R.A.; Glass, G.A. Cerebrospinal fluid concentrations of N-acetylcysteine after oral administration in Parkinson’s disease. Parkinsonism Relat. Disord. 2015, 21, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Sen, C.K. Nutritional biochemistry of cellular glutathione. J. Nutr. Biochem. 1997, 8, 660–672. [Google Scholar] [CrossRef]
- Mazor, D.; Golan, E.; Philip, V.; Katz, M.; Jafe, A.; Ben-Zvi, Z.; Meyerstein, N. Red blood cell permeability to thiol compounds following oxidative stress. Eur. J. Haematol. 1996, 57, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.L.; Chipman, J.K. The role of glutathione in DNA damage by potassium bromate in vitro. Mutagenesis 2000, 15, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Cao, D.; Zhu, S.; Fu, G.; Wu, Q.; Liang, J.; Cao, M. Chronic exposure to morphine decreases the expression of EAAT3 via opioid receptors in hippocampal neurons. Brain Res. 2015, 1628 Pt A, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Harrington, E.P.; Moddel, G.; Najm, I.M.; Baraban, S.C. Altered glutamate receptor—Transporter expression and spontaneous seizures in rats exposed to methylazoxymethanol in utero. Epilepsia 2007, 48, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Vidensky, S.; Ruggiero, A.M.; Maier, S.; Sitte, H.H.; Rothstein, J.D. Reticulon RTN2B regulates trafficking and function of neuronal glutamate transporter EAAC1. J. Biol. Chem. 2008, 283, 6561–6571. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.Y.; Park, K.S.; Kim, J.H.; Do, S.H.; Zuo, Z. Propofol reverses oxidative stress-attenuated glutamate transporter EAAT3 activity: Evidence of protein kinase C involvement. Eur. J. Pharmacol. 2007, 565, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Borgstrom, L.; Kagedal, B.; Paulsen, O. Pharmacokinetics of N-acetylcysteine in man. Eur. J. Clin. Pharmacol. 1986, 31, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Olsson, B.; Johansson, M.; Gabrielsson, J.; Bolme, P. Pharmacokinetics and bioavailability of reduced and oxidized N-acetylcysteine. Eur. J. Clin. Pharmacol. 1988, 34, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Furuta, A.; Rothstein, J.D.; Martin, L.J. Glutamate transporter protein subtypes are expressed differentially during rat CNS development. J. Neurosci. 1997, 17, 8363–8375. [Google Scholar] [PubMed]
- Sims, K.D.; Robinson, M.B. Expression Patterns and Regulation of Glutamate Transporters in the Developing and Adult Nervous System. Crit. Rev. Neurobiol. 1999, 13, 169–197. [Google Scholar] [CrossRef] [PubMed]
- Freeman, M.R. Specification and morphogenesis of astrocytes. Science 2010, 330, 774–778. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, I.A.; Ohayon, D.; Li, H.; de Faria, J.P.; Emery, B.; Tohyama, K.; Richardson, W.D. Motor skill learning requires active central myelination. Science 2014, 346, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Shen, Q.; Goderie, S.K.; He, W.; Capela, A.; Davis, A.A.; Temple, S. Timing of CNS cell generation: A programmed sequence of neuron and glial cell production from isolated murine cortical stem cells. Neuron 2000, 28, 69–80. [Google Scholar] [CrossRef]
- Shanker, G.; Allen, J.W.; Mutkus, L.A.; Aschner, M. The uptake of cysteine in cultured primary astrocytes and neurons. Brain Res. 2001, 902, 156–163. [Google Scholar] [CrossRef]
- Watts, S.D.; Torres-Salazar, D.; Divito, C.B.; Amara, S.G. Cysteine transport through excitatory amino acid transporter 3 (EAAT3). PLoS ONE 2014, 9, e109245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kranich, O.; Hamprecht, B.; Dringen, R. Different preferences in the utilization of amino acids for glutathione synthesis in cultured neurons and astroglial cells derived from rat brain. Neurosci. Lett. 1996, 219, 211–214. [Google Scholar] [CrossRef]
- Wang, X.F.; Cynader, M.S. Astrocytes provide cysteine to neurons by releasing glutathione. J. Neurochem. 2000, 74, 1434–1442. [Google Scholar] [CrossRef] [PubMed]
- Nieoullon, A.; Canolle, B.; Masmejean, F.; Guillet, B.; Pisano, P.; Lortet, S. The neuronal excitatory amino acid transporter EAAC1/EAAT3: Does it represent a major actor at the brain excitatory synapse? J. Neurochem. 2006, 98, 1007–1018. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Swanson, R.A. The glutamate transporters EAAT2 and EAAT3 mediate cysteine uptake in cortical neuron cultures. J. Neurochem. 2003, 84, 1332–1339. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, M.L.; Watts, L.T.; Stark, A.A.; Mahimainathan, L.; Stewart, J.; Schenker, S.; Henderson, G.I. Astrocyte control of fetal cortical neuron glutathione homeostasis: Up-regulation by ethanol. J. Neurochem. 2006, 96, 1289–1300. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, C.; Aoyama, K.; Matsumura, N.; Kikuchi-Utsumi, K.; Watabe, M.; Nakaki, T. Rhythmic oscillations of the microRNA miR-96-5p play a neuroprotective role by indirectly regulating glutathione levels. Nat. Commun. 2014, 5, 3823. [Google Scholar] [CrossRef] [PubMed]
- Potenza, N.; Mosca, N.; Mondola, P.; Damiano, S.; Russo, A.; de Felice, B. Human miR-26a-5p regulates the glutamate transporter SLC1A1 (EAAT3) expression. Relevance in multiple sclerosis. Biochim. Biophys. Acta 2017, 1864, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, M.; Rathinam, M.; Riar, A.; Patel, D.; Mummidi, S.; Yang, H.S.; Colburn, N.H.; Henderson, G.I.; Mahimainathan, L. Programmed cell death 4 (PDCD4): A novel player in ethanol-mediated suppression of protein translation in primary cortical neurons and developing cerebral cortex. Alcohol. Clin. Exp. Res. 2013, 37, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.W. The drunkest drinking driver in Sweden: Blood alcohol concentration 0.545% w/v. J. Stud. Alcohol 1999, 60, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Page, A.; Paoli, P.P.; Hill, S.J.; Howarth, R.; Wu, R.; Kweon, S.M.; French, J.; White, S.; Tsukamoto, H.; Mann, D.A.; et al. Alcohol directly stimulates epigenetic modifications in hepatic stellate cells. J. Hepatol. 2015, 62, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Sulik, K.K.; Chen, S.Y. Nrf2-mediated transcriptional induction of antioxidant response in mouse embryos exposed to ethanol in vivo: Implications for the prevention of fetal alcohol spectrum disorders. Antioxid. Redox Signal. 2008, 10, 2023–2033. [Google Scholar] [CrossRef] [PubMed]
- Riar, A.K.; Narasimhan, M.; Rathinam, M.L.; Henderson, G.I.; Mahimainathan, L. Ethanol induces cytostasis of cortical basal progenitors. J. Biomed. Sci. 2016, 23, 6. [Google Scholar] [CrossRef] [PubMed]
- Henderson, G.I.; Devi, B.G.; Perez, A.; Schenker, S. In utero ethanol exposure elicits oxidative stress in the rat fetus. Alcohol. Clin. Exp. Res. 1995, 19, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.X.; Chen, Y.H.; Wang, H.; Zhao, L.; Wang, J.P.; Wei, W. Effect of N-acetylcysteine on lipopolysaccharide-induced intra-uterine fetal death and intra-uterine growth retardation in mice. Toxicol. Sci. 2005, 88, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Beloosesky, R.; Ginsberg, Y.; Khatib, N.; Maravi, N.; Ross, M.G.; Itskovitz-Eldor, J.; Weiner, Z. Prophylactic maternal N-acetylcysteine in rats prevents maternal inflammation-induced offspring cerebral injury shown on magnetic resonance imaging. Am. J. Obstet. Gyneol. 2013, 208, e1–e6. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, D.D.; Wiest, D.B.; Mulvihill, D.M.; Hlavacek, A.M.; Majstoravich, S.J.; Brown, T.R.; Taylor, J.J.; Buckley, J.R.; Turner, R.P.; Rollins, L.G.; et al. Fetal and neonatal effects of N-acetylcysteine when used for neuroprotection in maternal chorioamnionitis. J. Pediatr. 2016, 168, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Nolin, T.D.; McMenamin, M.E.; Himmelfarb, J. Simultaneous determination of total homocysteine, cysteine, cysteinylglycine, and glutathione in human plasma by high-performance liquid chromatography: Application to studies of oxidative stress. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 852, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R.; Gutterer, J.M.; Gros, C.; Hirrlinger, J. Aminopeptidase N mediates the utilization of the GSH precursor CysGly by cultured neurons. J. Neurosci. Res. 2001, 66, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Riar, A.K.; Narasimhan, M.; Rathinam, M.L.; Vedpathak, D.; Mummidi, S.; Henderson, G.I.; Mahimainathan, L. Ethanol-induced transcriptional activation of programmed cell death 4 (Pdcd4) is mediated by GSK-3beta signaling in rat cortical neuroblasts. PLoS ONE 2014, 9, e98080. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.N. Biotinylation of Cell Surface Proteins. Bio-protocol 2012, 2, 1. [Google Scholar] [CrossRef]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patel, D.; Mahimainathan, L.; Narasimhan, M.; Rathinam, M.; Henderson, G. Ethanol (E) Impairs Fetal Brain GSH Homeostasis by Inhibiting Excitatory Amino-Acid Carrier 1 (EAAC1)-Mediated Cysteine Transport. Int. J. Mol. Sci. 2017, 18, 2596. https://doi.org/10.3390/ijms18122596
Patel D, Mahimainathan L, Narasimhan M, Rathinam M, Henderson G. Ethanol (E) Impairs Fetal Brain GSH Homeostasis by Inhibiting Excitatory Amino-Acid Carrier 1 (EAAC1)-Mediated Cysteine Transport. International Journal of Molecular Sciences. 2017; 18(12):2596. https://doi.org/10.3390/ijms18122596
Chicago/Turabian StylePatel, Dhyanesh, Lenin Mahimainathan, Madhusudhanan Narasimhan, Marylatha Rathinam, and George Henderson. 2017. "Ethanol (E) Impairs Fetal Brain GSH Homeostasis by Inhibiting Excitatory Amino-Acid Carrier 1 (EAAC1)-Mediated Cysteine Transport" International Journal of Molecular Sciences 18, no. 12: 2596. https://doi.org/10.3390/ijms18122596